Week 2 HW: DNA Read, Write, and Edit

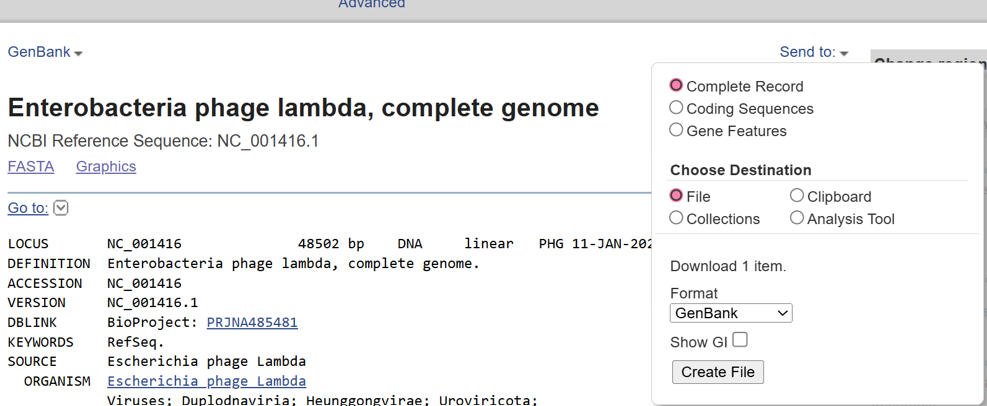
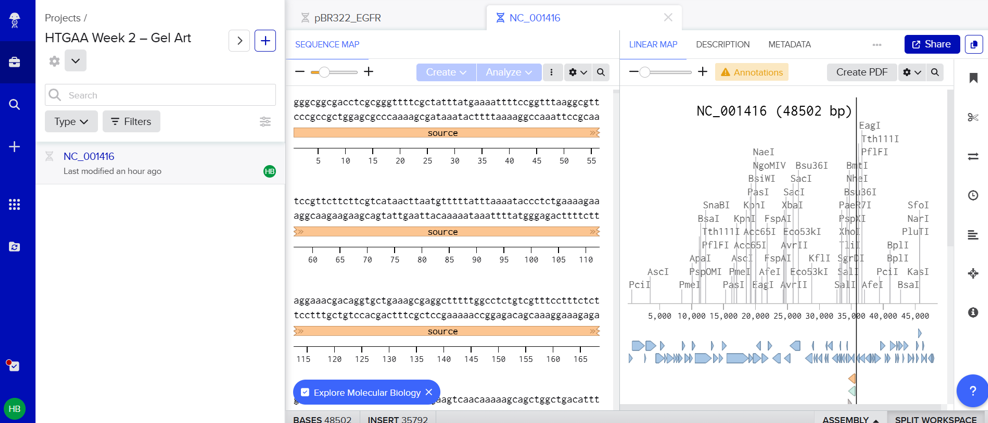
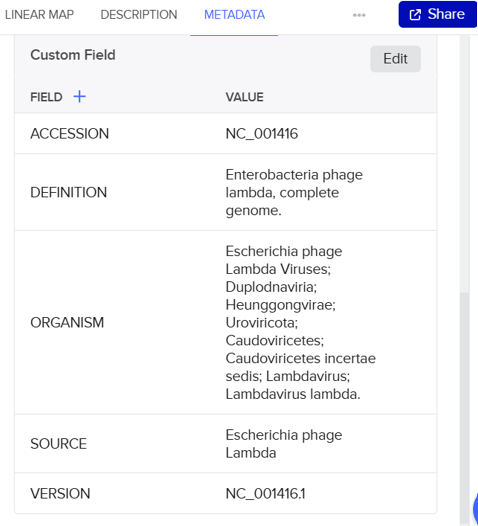
- In this part, I imported The complete 48,502 bp linear genome of bacteriophage lambda from NCBI GenBank into Benchling. This sequence corresponds to the Lambda DNA sold by NEB (N3011) and will be used for in-silico restriction digestion.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
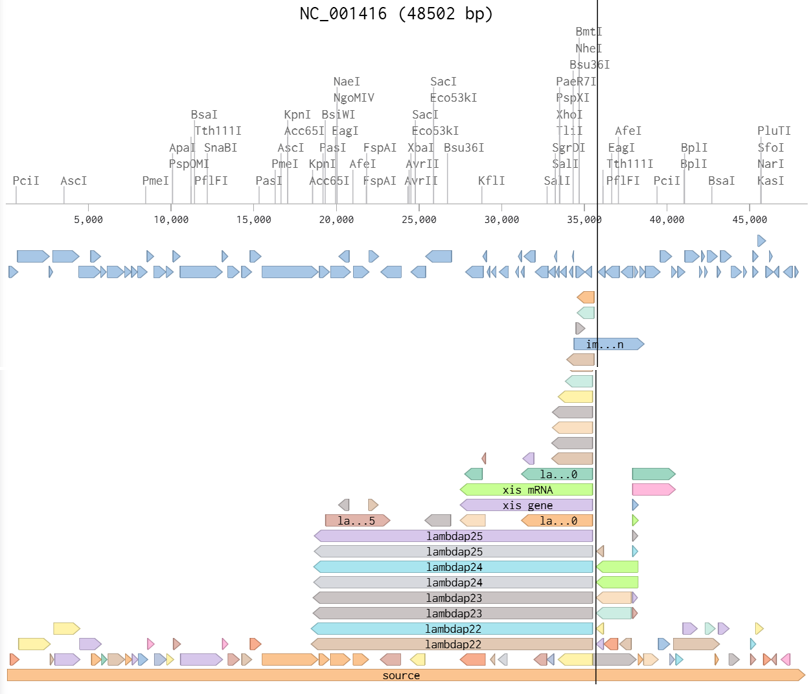
Then simulated restriction enzyme digestion using EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. By running in-silico gel electrophoresis . The resulting virtual gel shows discrete bands corresponding to these fragments, which demostrates how sequence information maps to physical separation in gel electrophoresis.

To create a pattern in the style of Paul Vanouse’s work, I experimented with different combinations of restriction enzymes to control the gel band patterns. By adjusting the number and length of the resulting DNA fragments, I explored how these parameters influence the final visual outcome. Through this process, I ultimately obtained a gel pattern resembling a butterfly shape.
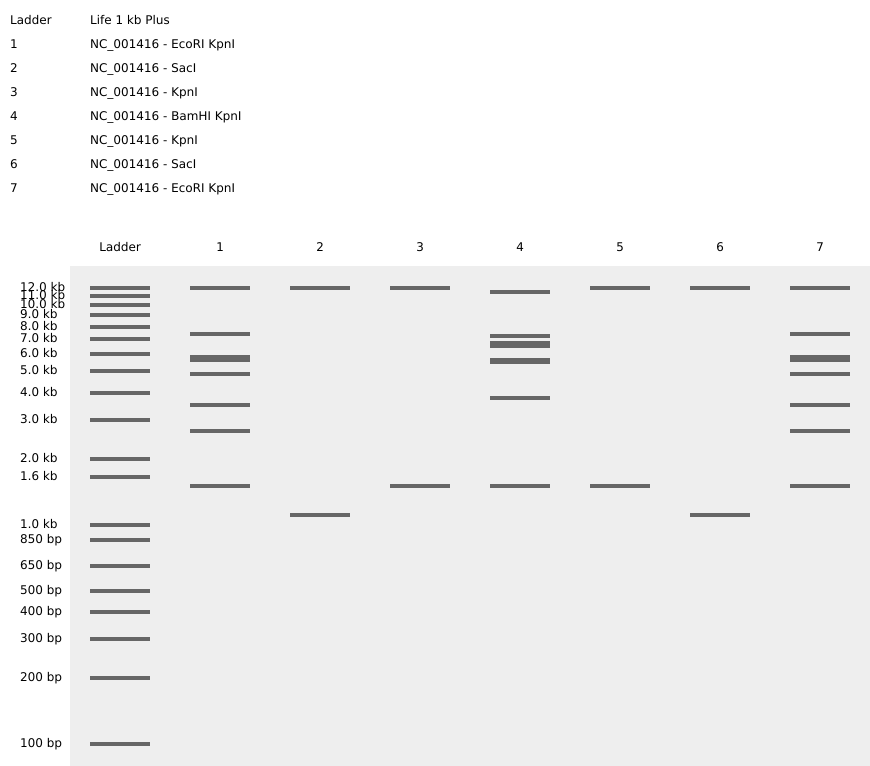
- This helped me understand how restriction digests and gels work before doing any real lab experiment. I treated this as both a technical exercise and a creative exploration, inspired by DNA gel art concepts.
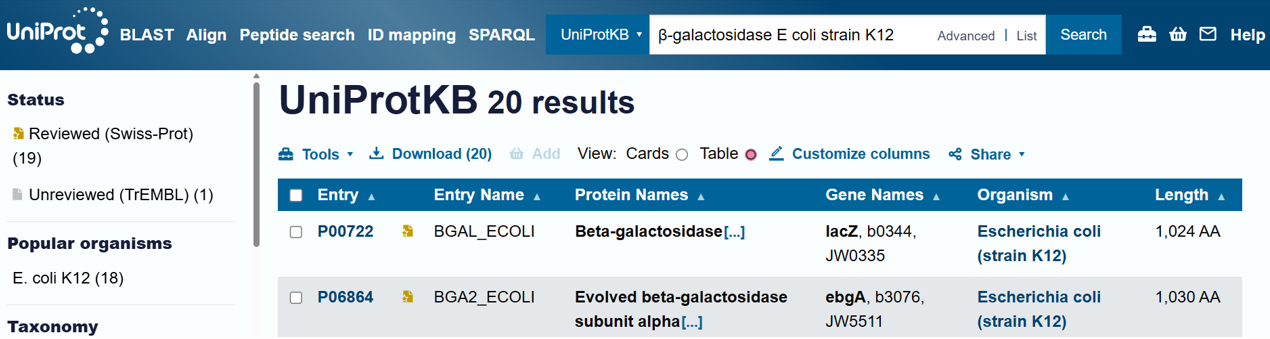
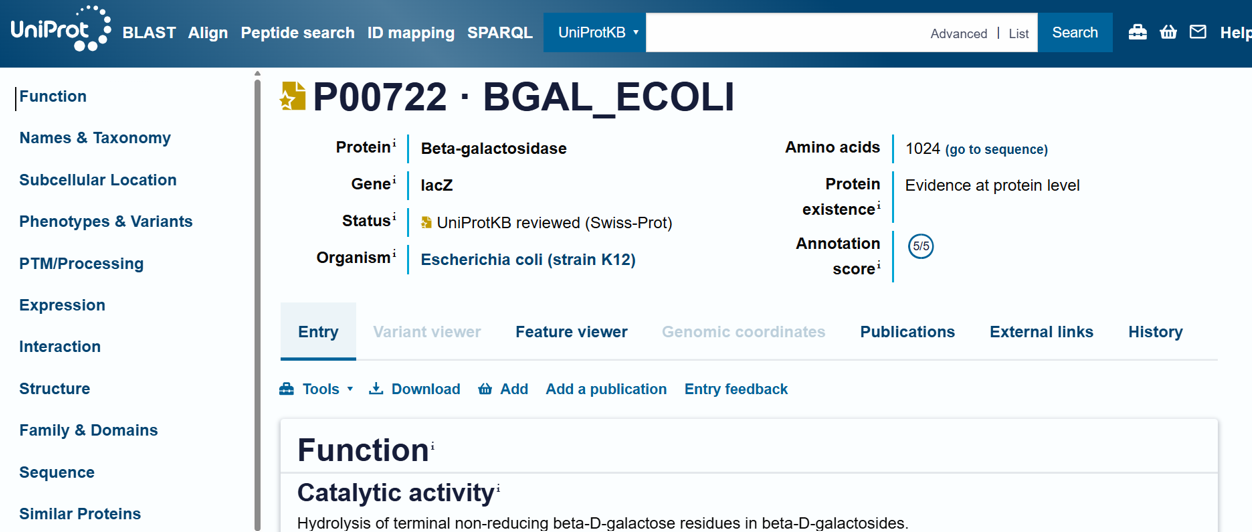
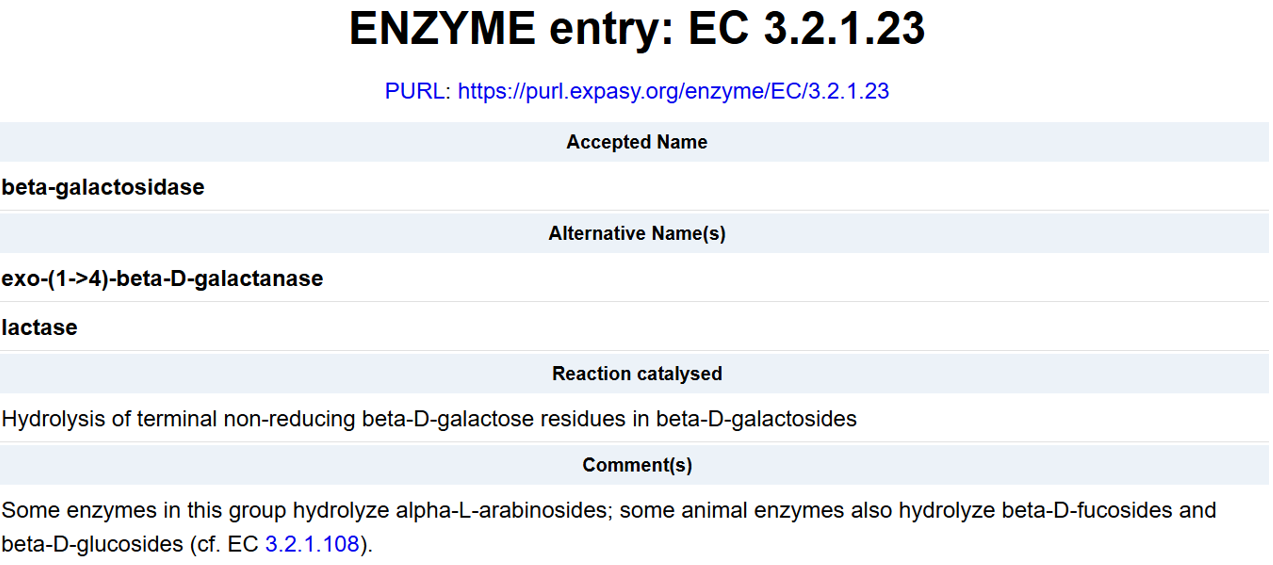
- For the DNA design challenge, I chose a protein related to my project interest in engineered probiotics and conditional enzyme release in the gut.The enzyme β-galactosidase is well-characterized and commonly expressed in Escherichia coli, making it an ideal candidate for computational DNA design and expression modeling.
- I first searched online database UniProt to obtain the amino acid sequence of the protein.
![]()
![]()
![]()
![]()
- the amino acid equence was as follow:
- After selecting the protein, I converted the amino acid sequence of β-galactosidase (1024 residues) into the corresponding DNA sequence using the Sequence Manipulation Suite Reverse Translate tool. Because the genetic code is degenerate, multiple codons can encode the same amino acid. The resulting 3072 bp DNA sequence represents one valid nucleotide sequence capable of encoding the β-galactosidase protein.
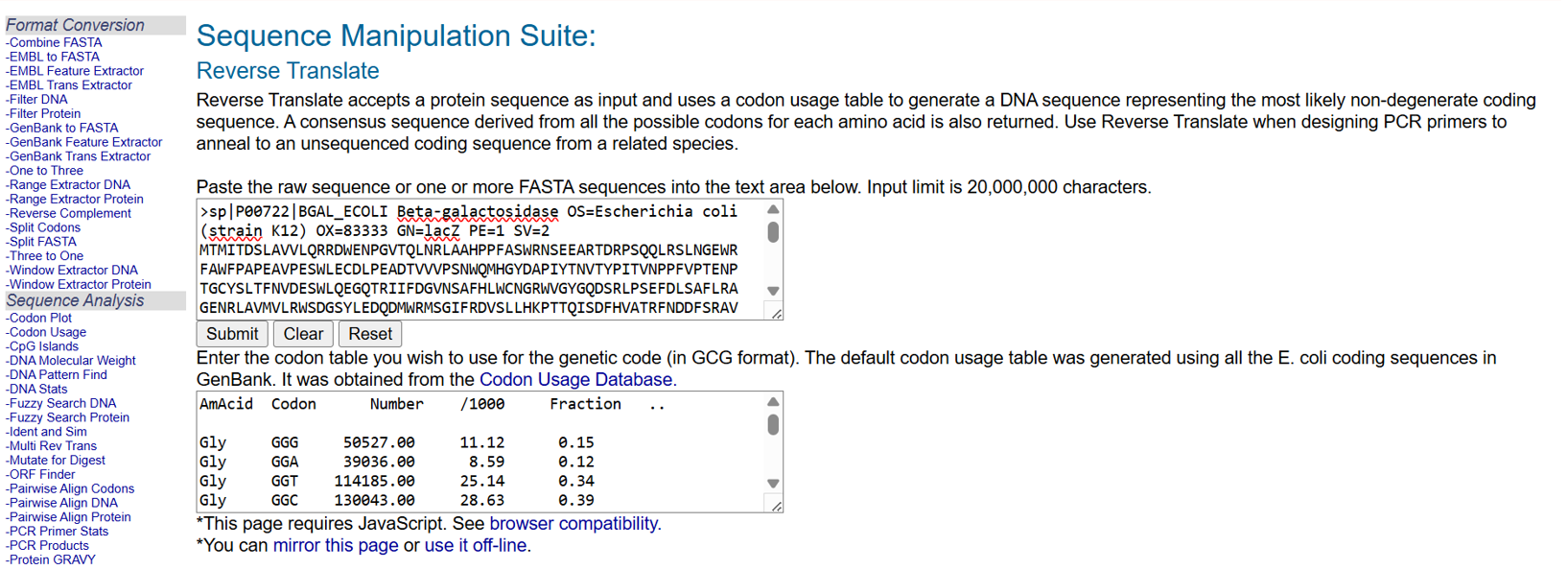
- the resulted DNA sequence was as follow:
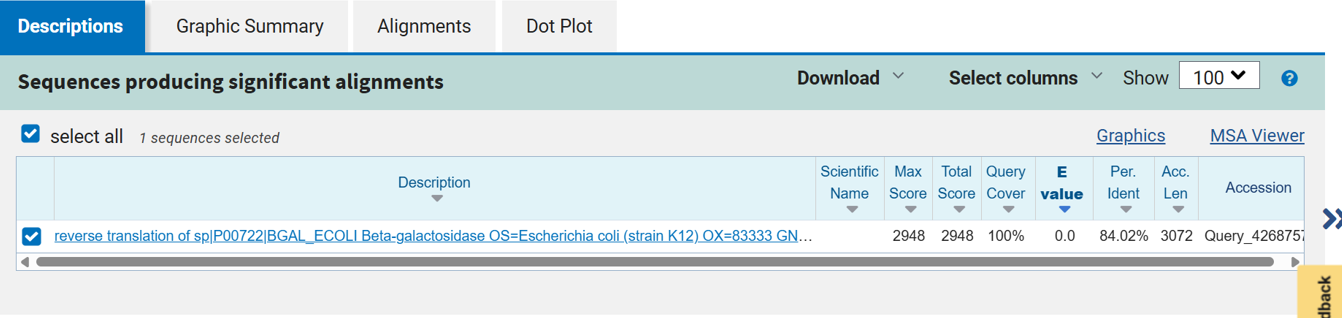
- After reverse translation, I verified the identity of the resulting nucleotide sequence by performing a BLASTn search against the reference lacZ gene from Escherichia coli K-12. The alignment showed 100% query coverage with an E-value of 0.0, confirming a highly significant match. The percent identity was ~84%, which is expected because reverse translation produces a synonymous DNA sequence that differs at the codon level while still encoding the same β-galactosidase protein. This result confirmed that the reverse-translated sequence correctly corresponds to the lacZ gene.
![]()
![]()
![]()
![]()
Next, I performed codon optimization of the sequence originates from E. coli K-12 to improve expression efficiency in a Lactobacillus probiotic strain (delbrueckii subsp. Bulgaricus), as this organism is the intended chassis for conditional lactase expression in the human gut, to ensure efficient translation in the final probiotic host organism. Codon optimization was performed using a host-specific algorithm using the Vector Builder codon orimisation tool that adjusts synonymous codon usage to match the preferred codons of L. delbrueckii while preserving the original amino acid sequence.
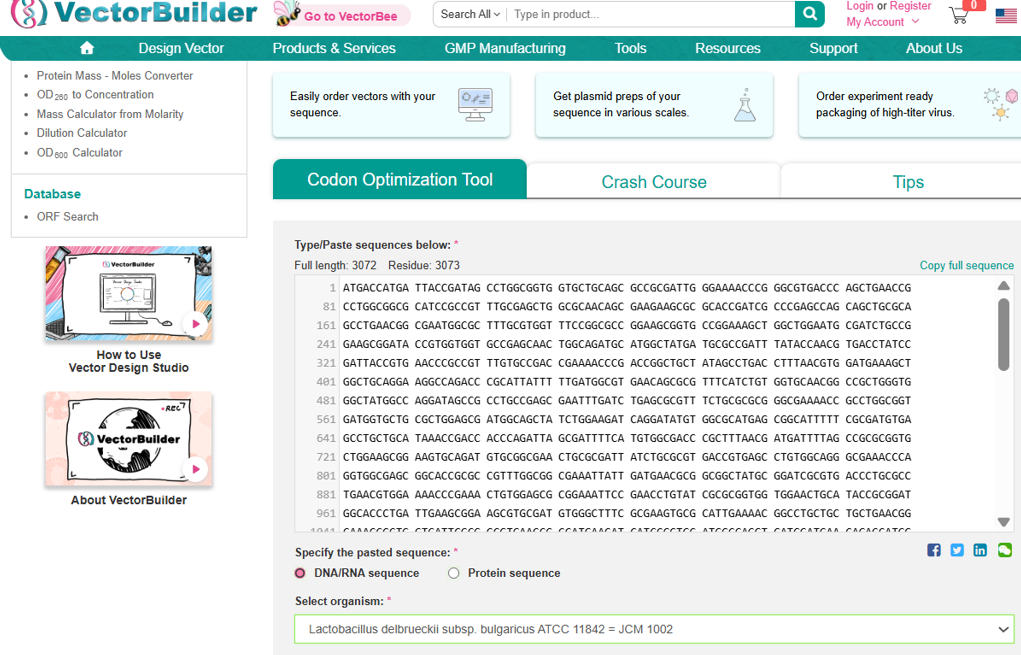
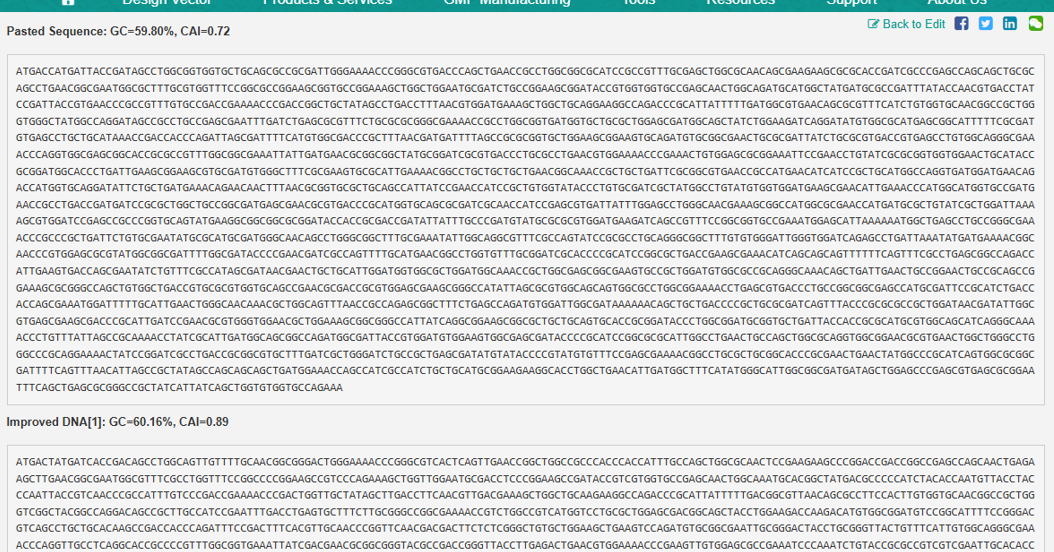
- the resulted optimised sequence is as following:
Host organism: Lactobacillus delbruekii susbsp. Bulgaricus ATCC 11842 = JCM 1002 Original Sequence: GC=59.80%, CAI=0.72 Optimized Sequence: GC=60.16%, CAI=0.89
- To produce the protein from this DNA sequence, I would use a cell-dependent expression system based on bacterial transformation and expression. In this approach, This gene is then placed into an expression cassette with the necessary regulatory elements so it can be used by a biological system.
- To produce the protein, I would use a cell-dependent expression system through bacterial cloning. The designed DNA sequence is inserted into a plasmid and introduced into a bacterial host by transformation. Inside the cell, the gene is transcribed into mRNA under the control of the selected promoter. The mRNA is then translated by ribosomes, which read the codons starting at the start codon and assemble the corresponding amino acids into the lactase protein. This approach follows the natural flow of genetic information (DNA to RNA to protein) and allows controlled production of the enzyme in living cells.
Part 4: Prepare a Twist DNA Synthesis Order
Assignees for the following sections MIT/Harvard students Required Committed Listeners Required This is a practice exercise, not necessarily your real Twist order!
4.1. Create a Twist account, and Benchling account
4.2. Build Your DNA Insert Sequence
For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
In Benchling, select New DNA/RNA sequence
Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
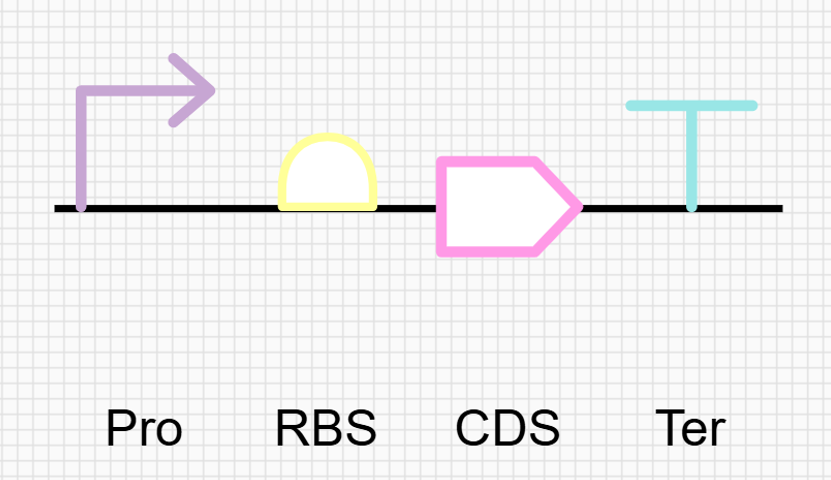
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is (see image below).
Promoter (e.g. BBa_J23106) TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
RBS (e.g. BBa_B0034 with spacers for optimal expression) CATTAAAGAGGAGAAAGGTACC
Start Codon ATG
Coding Sequence (your codon optimized DNA for a protein of interest, sfGFP for example)
AGCAAAGGAGAAGAACTTTTCACTGGAGTTGTCCCAATTCTTGTTGAATTAGATGGTGATGTTAATGGGCACAAATTTTCTGTCCGTGGAGAGGGTGAAGGTGATGCTACAAACGGAAAACTCACCCTTAAATTTATTTGCACTACTGGAAAACTACCTGTTCCGTGGCCAACACTTGTCACTACTCTGACCTATGGTGTTCAATGCTTTTCCCGTTATCCGGATCACATGAAACGGCATGACTTTTTCAAGAGTGCCATGCCCGAAGGTTATGTACAGGAACGCACTATATCTTTCAAAGATGACGGGACCTACAAGACGCGTGCTGAAGTCAAGTTTGAAGGTGATACCCTTGTTAATCGTATCGAGTTAAAGGGTATTGATTTTAAAGAAGATGGAAACATTCTTGGACACAAACTCGAGTACAACTTTAACTCACACAATGTATACATCACGGCAGACAAACAAAAGAATGGAATCAAAGCTAACTTCAAAATTCGCCACAACGTTGAAGATGGTTCCGTTCAACTAGCAGACCATTATCAACAAAATACTCCAATTGGCGATGGCCCTGTCCTTTTACCAGACAACCATTACCTGTCGACACAATCTGTCCTTTCGAAAGATCCCAACGAAAAGCGTGACCACATGGTCCTTCTTGAGTTTGTAACTGCTGCTGGGATTACACATGGCATGGATGAGCTCTACAAA
7x His Tag (Let’s add a 7×His tag at the C-terminus of the protein to enable protein purification from E. coli) CATCACCATCACCATCATCAC
Stop Codon TAA
Terminator (e.g. BBa_B0015) CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
Once you’ve completed this, click on Linear Map to preview the entire sequence. If you intend to have a TA review a sequence in the future, this is a good way to verify that all sections are annotated!
(Optional) Share your final sequence link with a TA for review!
This insert sequence you built is commonly referred to as an expression cassette in molecular biology (a sequence you can drop into any vector and it’ll perform its function). Go ahead and download the FASTA file for the sequence you made.
It’s helpful to visualize DNA designs using SBOL Canvas (Synthetic Biology Open Language) to convey your designs. Here’s an example of what you just annotated in Benchling: 
4.2. On Twist, Select The “Genes” Option
4.3. Select “Clonal Genes” option For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project. Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly. Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.4. Import your sequence You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
4.5. Choose Your Vector Since we’re ordering a clonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle! The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance. Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later. For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy. Click into your sequence and select download construct (GenBank) to get the full plasmid sequence: Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded. This is the plasmid you just built with your expression cassette included. Congratulations on building your first plasmid! Important For your final projects, remember to include:
- Fully annotated Benchling insert fragment
- Desired Twist cloning vector
- A lactose-inducible promoter was selected to enable conditional expression of lactase in response to lactose availability in the gut. The PlacA promoter region was extracted from the Lactococcus lactis lac operon upstream of the native ribosome binding site, with preserving lactose-responsive regulation.
![]()
![]()
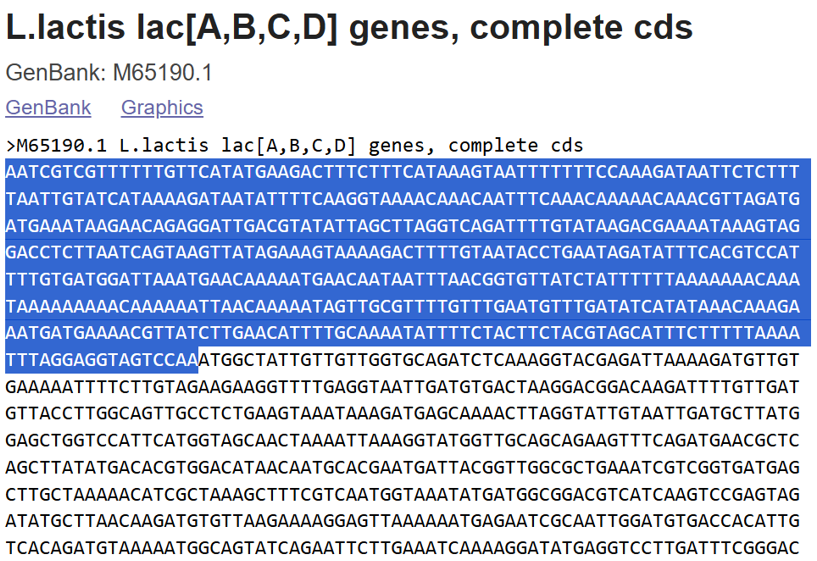
For the RBS, I chose to keep the native Lactococcus lactis ribosome binding site (RBS) derived from the lacA operon which is the region immediately upstream of the coding sequence (CDS) and preserved its original spacer length to ensure efficient translation initiation in the probiotic host. Maintaining native RBS spacing is critical in Gram-positive bacteria, as ribosome binding and translation efficiency are highly sensitive to the distance between the Shine–Dalgarno sequence and the start codon.
the RBS sequence is as follow:
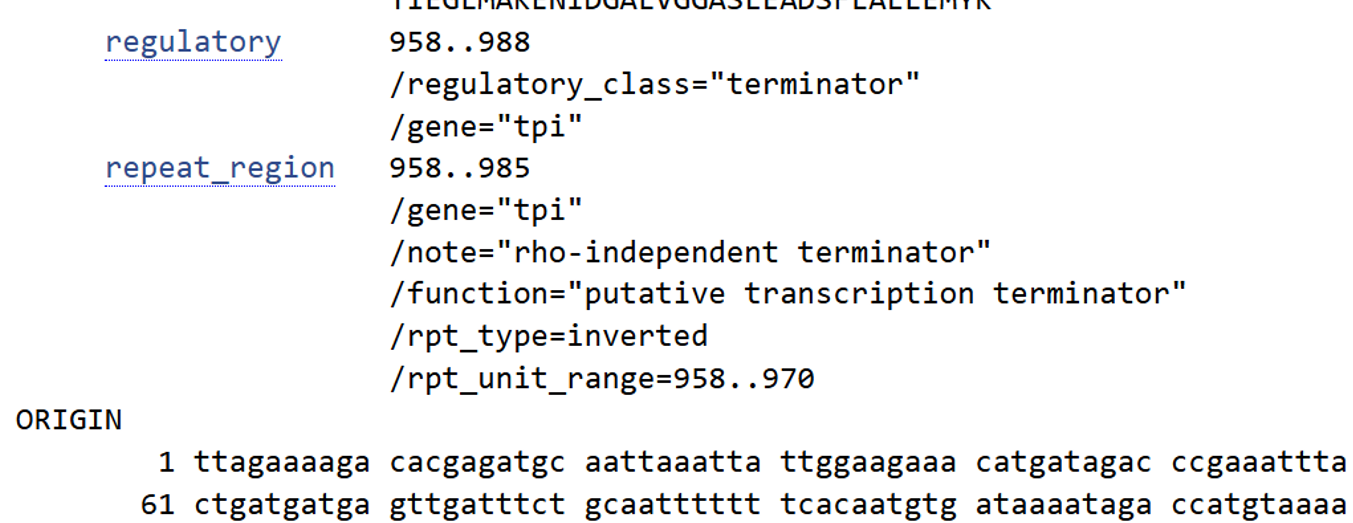
- I selected the transcription terminator from the tpi gene of Lactococcus lactis, a highly expressed native housekeeping gene, to ensure efficient and reliable transcription termination in the probiotic host. While two related annotations are present in GenBank for this region, both correspond to the same rho-independent transcription terminator. Therefore, I chose the complete annotated terminator region (positions 958–988), which includes both the inverted repeat and the downstream poly-T tract, to ensure proper formation of the termination hairpin and robust termination of transcription.
![]()
![]()
A transcription terminator was included downstream of the lactase coding sequence to ensure proper termination of transcription. This prevents transcriptional read-through into adjacent sequences and improves the stability and predictability of gene expression, independent of promoter regulation.
ATG used as start codon and AAG as stop codon
From the selected elements, I built a linear expression cassette in Benchling containing a lactose-regulated promoter, native LAB ribosome binding site, codon-optimized lacZ, and a native transcription terminator. I exported this sequence as a FASTA file. Cassette_link_to_Benchling
- When I first uploaded my expression cassette FASTA file to Twist Bioscience, I encountered an initial error related to the FASTA header name. The header exceeded the maximum allowed length (32 characters), which caused the sequence to be rejected. I fixed this issue by shortening the header name and re-uploading the file. After this correction, the sequence was accepted for further analysis.
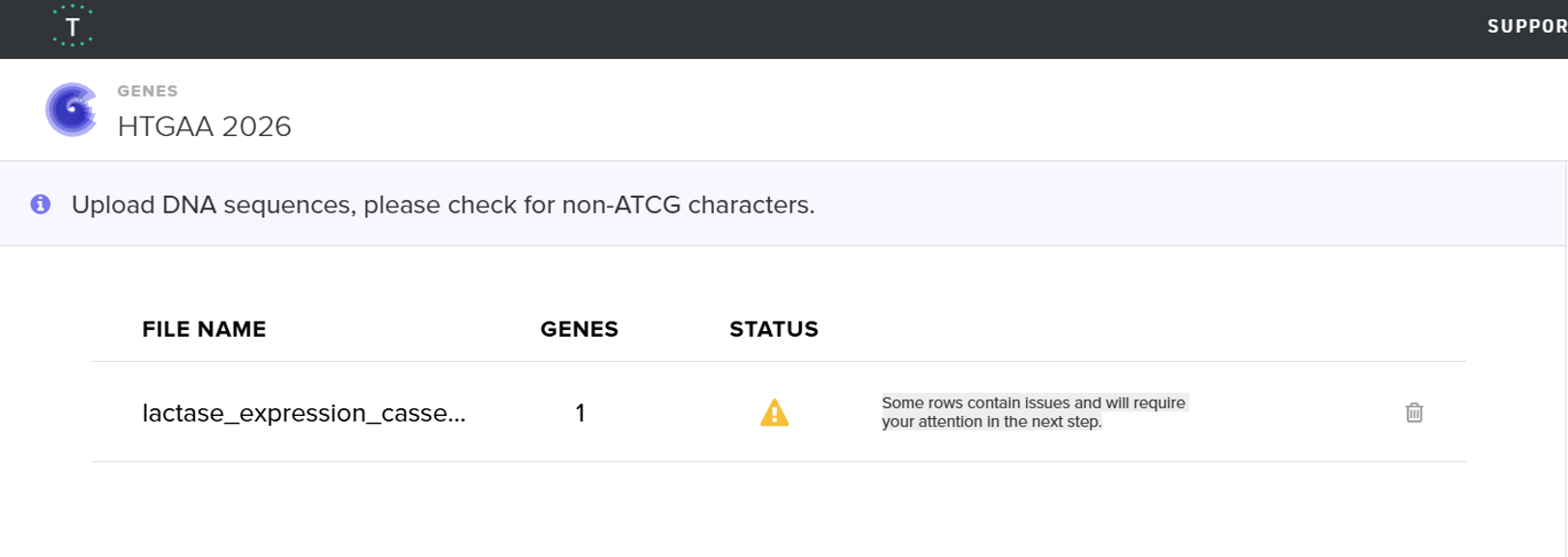
However, after re-uploading the corrected file, additional synthesis warnings appeared. These warnings were related to large GC content variation, repetitive regions, and overall sequence complexity. These issues are mainly due to the codon-optimized lacZ gene and the presence of multiple regulatory elements such as the ribosome binding site and transcription terminator. Twist flagged these features as potential manufacturability risks. Unfortunately, I was not able to resolve these additional issues at this stage. Fixing them would have required re-optimizing the enzyme sequence, possibly changing the host organism for codon optimization, and redesigning the regulatory architecture of the cassette. Due to time constraints and because this assignment focuses on learning the design and ordering workflow rather than producing a synthesis-ready construct, I chose not to redesign the sequence further.
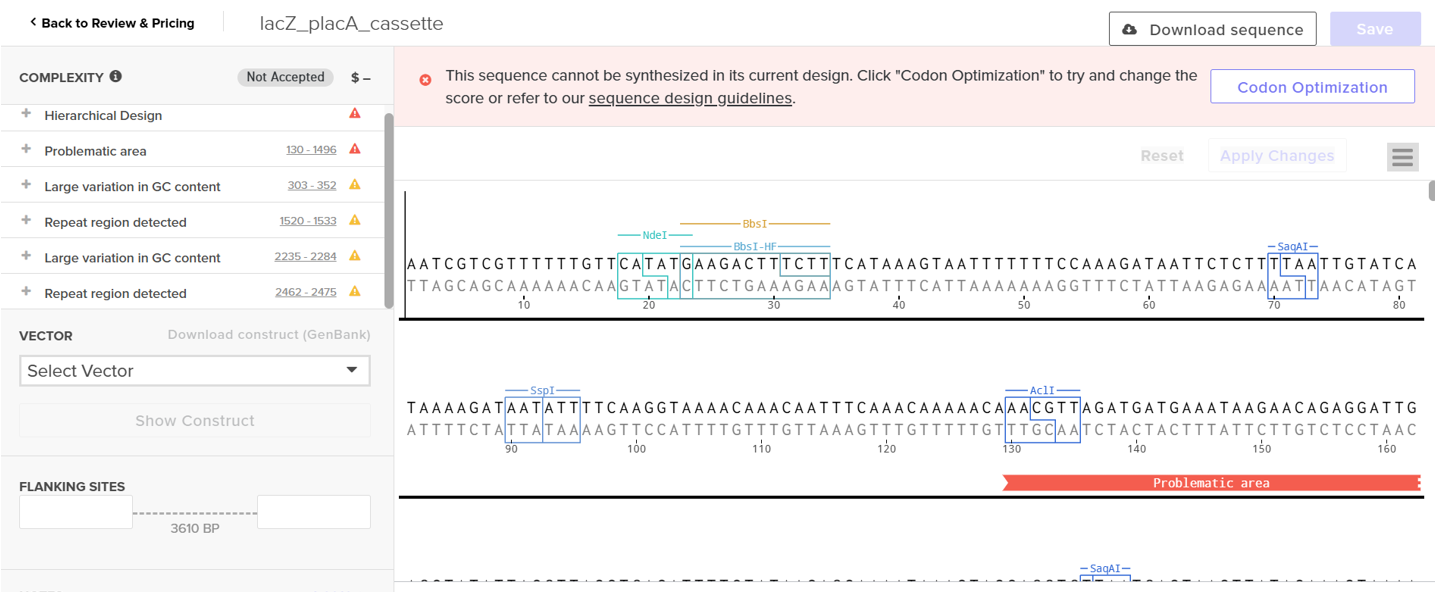
For this exercise, I proceeded by selecting a Twist clonal vector (pTwist Amp High Copy) to complete the plasmid design. Although the insert sequence still contained manufacturability warnings. However, In a real DNA synthesis order, additional sequence optimization would be required to reduce GC content extremes and repetitive regions to meet synthesis constraints.
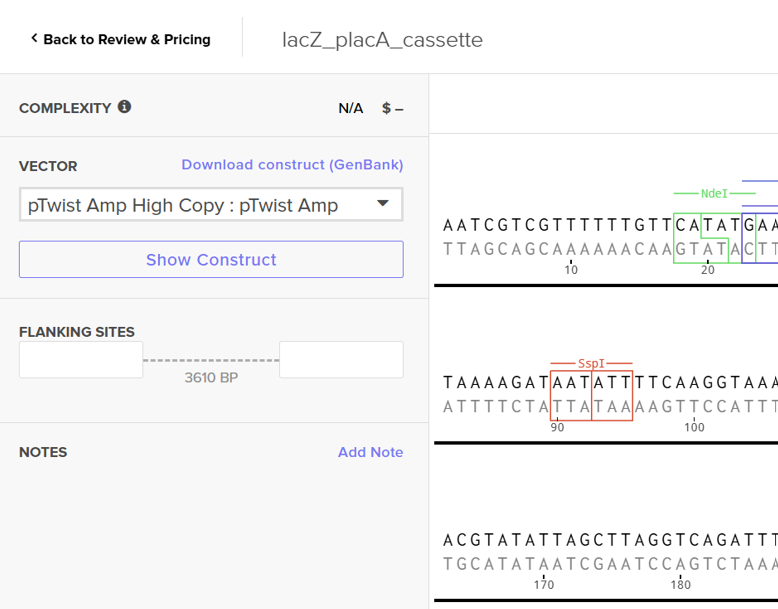
![]()
![]()
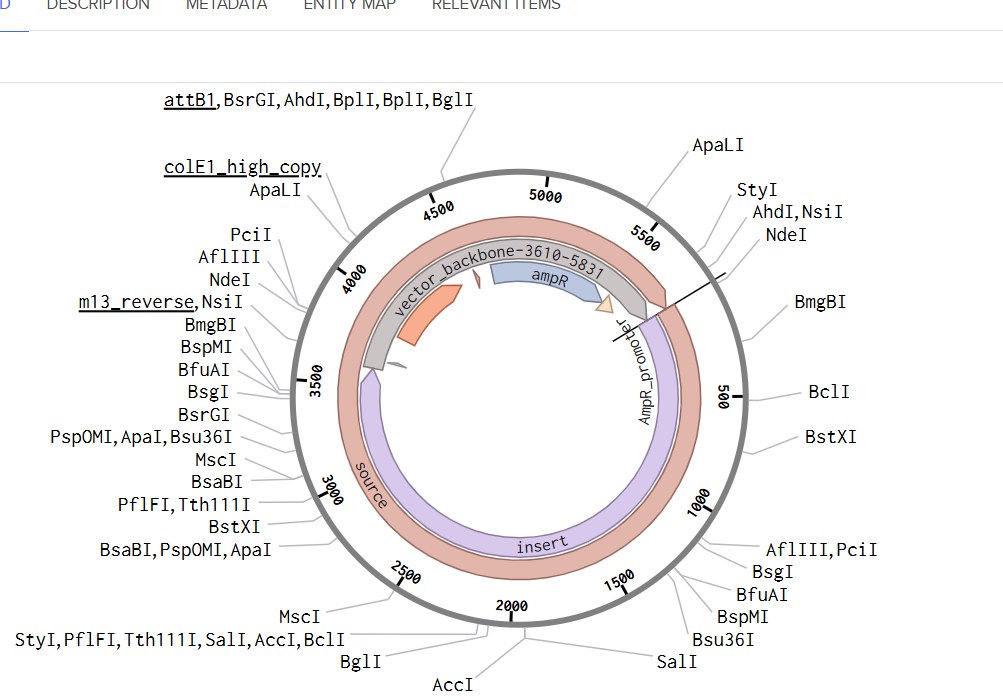
DNA read:
- I would want to sequence DNA used for digital data storage. In my knowledge, this technology enables the storage of digital information such as text, images, or files by encoding them into DNA sequences instead of being stored on hard drives. DNA is extremely stable and can store a huge amount of information in a very small space, which makes it interesting for long-term data storage. Reading this DNA by sequencing is necessary to retrieve the stored information and check that the data has not been damaged or changed over time.
- For this porpose, I would use Illumina sequencing because it is very accurate and well suited for reading short DNA fragments, which is how DNA data storage is usually organized. this strategy can be performed following 4 crusial steps: Image_adress
{kind=link}

Generation This method is a second-generation sequencing technology. It sequences millions of short DNA fragments in parallel, which makes it fast and reliable, but it cannot read very long DNA molecules in one piece.
Input and preparation The input is DNA that contains the encoded digital data. To prepare it: The DNA is fragmented into short pieces, Adapters are added to both ends of the fragments, The fragments are amplified using PCR, The prepared DNA is loaded onto a flow cell
How the technology reads DNA (base calling) Each DNA fragment is copied one base at a time using fluorescently labeled nucleotides. A camera records the color added at each step, and the machine translates these signals into DNA letters (A, T, C, G).
Output The output is a large number of short DNA sequence reads saved as digital files. These reads are then assembled and decoded to recover the original stored data.
DNA write:
I am particularly interested in the genes in human genomic DNA related to pharmacogenomics and pharmacogenetics. These fields study how genetic variation affects how people respond to drugs. So, I would want to synthesize genes encoding drug-metabolizing enzymes, like human cytochrome P450 enzymes. Since, these genes are central to pharmacogenetics as variations in them strongly influence how drugs are processed in the body. Synthesizing these genes allows them to be studied, expressed, and tested in controlled systems.
So in order to synthetizing them , I would use chemical DNA synthesis combined with gene assembly, which is the standard approach used by commercial DNA synthesis companies.
- Essential steps
DNA synthesis starts with the digital design of the DNA sequence. This is followed by the chemical synthesis of short oligonucleotides, which are then assembled into full-length genes (for example, using Gibson Assembly). The synthesized genes are cloned into plasmids and finally sequence-verified to confirm their accuracy before use.
This DNA synthesis method is easy to use and works well for many projects.However, it can sometimes make mistakes during the process. Parts of DNA that have lots of G and C letters or repeated sequences are harder to make. Very long DNA pieces also need to be built from many shorter fragments, which can be tricky and may cause errors.
DNA Edit:
I would want to edit DNA in human cell lines used for drug testing, focusing on genes that affect how drugs work. Changing these genes helps researchers see how different genetic variants influence drug effects and side effects, which is useful in pharmacogenomics.
The modification can be realised by CRISPR for editing because it allows precise and programmable changes to DNA. this stratigy works by using a guide RNA to find a specific DNA sequence. The Cas enzyme then makes a cut or nick, and the cell repairs it, introducing the change we want.
To use CRISPR, you need to design guide RNAs, prepare the CRISPR components (DNA, RNA, or protein), deliver them into cells, and then check which cells were correctly edited.
However, there are some limitations, like different editing efficiencies depending on cell type, and ethical or regulatory concerns when working with human cells.
in this homework, AI ChatGPT assisted me in organizing and clearly articulating my answers and descriptions, ensuring that the content is well-structured and easy to understand.
Sources:
- Ahmad, E., Mahapatra, V., M, V. V., & Nagaraja, V. (2022). Intrinsic and Rho-dependent termination cooperate for efficient transcription termination at 3’ untranslated regions (p. 2022.07.21.500918). bioRxiv. https://doi.org/10.1101/2022.07.21.500918
- Amin, A. A., Olama, Z. A., & Ali, S. M. (2023). Characterization of an isolated lactase enzyme produced by Bacillus licheniformis ALSZ2 as a potential pharmaceutical supplement for lactose intolerance. Frontiers in Microbiology, 14, 1180463. https://doi.org/10.3389/fmicb.2023.1180463 Bioinformatic Tools | VectorBuilder. (n.d.). Retrieved February 17, 2026, from https://en.vectorbuilder.com/tool/overview.html
- Coenen, T. M. M., Bertens, A. M. C., de Hoog, S. C. M., & Verspeek-Rip, C. M. (2000). Safety evaluation of a lactase enzyme preparation derived from Kluyveromyces lactis. Food and Chemical Toxicology, 38(8), 671–677. https://doi.org/10.1016/S0278-6915(00)00053-3
- De Jesus, L. C. L., Aburjaile, F. F., Sousa, T. D. J., Felice, A. G., Soares, S. D. C., Alcantara, L. C. J., & Azevedo, V. A. D. C. (2022). Genomic Characterization of Lactobacillus delbrueckii Strains with Probiotics Properties. Frontiers in Bioinformatics, 2, 912795. https://doi.org/10.3389/fbinf.2022.912795
- de Vrese, M., Stegelmann, A., Richter, B., Fenselau, S., Laue, C., & Schrezenmeir, J. (2001). Probiotics—Compensation for lactase insufficiency123. The American Journal of Clinical Nutrition, 73(2), 421s–429s. https://doi.org/10.1093/ajcn/73.2.421s
- How can I find the promoter sequence of a gene on NCBI? (n.d.). ResearchGate. Retrieved February 17, 2026, from https://www.researchgate.net/post/How_can_I_find_the_promoter_sequence_of_a_gene_on_NCBI
- Lactase—An overview | ScienceDirect Topics. (n.d.). Retrieved February 17, 2026, from https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/lactase
- Reverse Translate. (n.d.). Retrieved February 17, 2026, from https://www.bioinformatics.org/sms2/rev_trans.html
- Saqib, S., Akram, A., Halim, S. A., & Tassaduq, R. (2017). Sources of β-galactosidase and its applications in food industry. 3 Biotech, 7(1), 79. https://doi.org/10.1007/s13205-017-0645-5