week 04 HW: protein design-part-I

- Amino Acid Count in 500g Meat: Meat is roughly 20% protein by mass. (Human Nutrition - Protein, Vitamins, Minerals | Britannica, n.d.)
- 500g meat x 0.20 = 100g protein.
- Using an average mass of 100 Daltons (Da) per amino acid: 100g / 100 Daltons (or g/mol) = 1 moles of amino acids
- 1x 6.022 x 1023 = 6.022 x 1023 molecules /1 mole.
- Why we don’t become cows: When we eat protein, our digestive system breaks it down into individual amino acids. Our body then uses its own DNA information to reassemble those amino acids into human proteins. The information which is coded by the sequence of AA is destroyed, but the building blocks or AA are reused.
- Why only 20 amino acids: In nature, the use of 20 amino acids is often explained as a “frozen accident” that originated in the early RNA World. This set worked well very early in Earth’s history and then became fixed. These 20 amino acids were good enough to build strong and functional proteins. Even though many other amino acids exist, this small group provides enough variety to perform many functions while remaining simple, stable, and efficient for cells to use. (Doig, 2017)
- Non-natural amino acids: Yes, scientists can make non-natural (unnatural) amino acids. They do this using chemical methods and special genetic tools that allow new amino acids to be added to proteins. These new amino acids can give proteins new properties that natural amino acids do not have. (Young & Schultz, 2010) For example, A new amino acid could be made by taking a normal amino acid, like alanine, and adding a fluorine atom to its side chain. This fluorinated amino acid would make proteins more stable and less likely to break down, which is useful for drug design. (Adhikari et al., n.d.)
- Pre-life origins of amino acids: According to Gutiérrez-Preciado, Romero, and Peimbert (2010) Before enzymes and living organisms existed, amino acids were probably formed naturally on early Earth. Energy from lightning, UV light, and volcanic heat helped simple gases react to make amino acids. Some amino acids were also brought to Earth by meteorites and comets. Together, these processes created a “primordial soup” of basic organic molecules. (Amino Acids, Evolution | Learn Science at Scitable, n.d.)
- D-amino acid α-helix: In nature, L-amino acids form right-handed helices. If you used only D-amino acids, the stereochemistry would be mirrored, resulting in a left-handed $\alpha$-helix. (Zotti et al., n.d.)
- Additional helices: Yes, additional helical structures besides the standard α-helix can be found in proteins. Studies show that other types of helices occur in many proteins, but they are often overlooked or mistaken for small distortions in α-helices. These helices are especially common in membrane proteins and are found in a significant number of known protein structures.(Vieira-Pires & Morais-Cabral, 2010)
- Why right-handed helices: because this shape is the most stable for the natural building blocks of life. L-amino acids and D-sugars fit together best in a right-handed twist, which allows strong hydrogen bonds and reduces crowding between atoms. Left-handed helices are usually less stable or hard to form. (Right-Handed Alpha-Helix - an Overview | ScienceDirect Topics, n.d.)
- β -sheet aggregation: β-sheets tend to aggregate because their edges have exposed hydrogen-bonding groups that easily stick to other β-strands. The main driving forces are hydrogen bonding between strands and the hydrophobic effect, which together make the stacked β-sheet structure very stable and allow fibrils to form.(Gsponer & Vendruscolo, 2006)
- I chose the protein mCherry because it is a small red fluorescent protein that is easy to visualize and analyze using 3D protein visualization software. Its structure is well known and has a clear β-barrel shape, which makes it easy to study secondary structure, amino acid distribution, and surface features. This makes mCherry a good example protein for learning basic protein sequence and structure analysis.
- The mCherry protein analyzed here is the standard red fluorescent protein and does not function as a fluorescent timer. However, according to the fluorescent protein database (FPbase), mCherry is the parent fluorescent protein for several timer-based reporters, including the medium fluorescent timer planned for my final project. Therefore, mCherry is used in this assignment as a reference protein to understand the structure and sequence properties of fluorescent proteins before working with fluorescent timer variants.
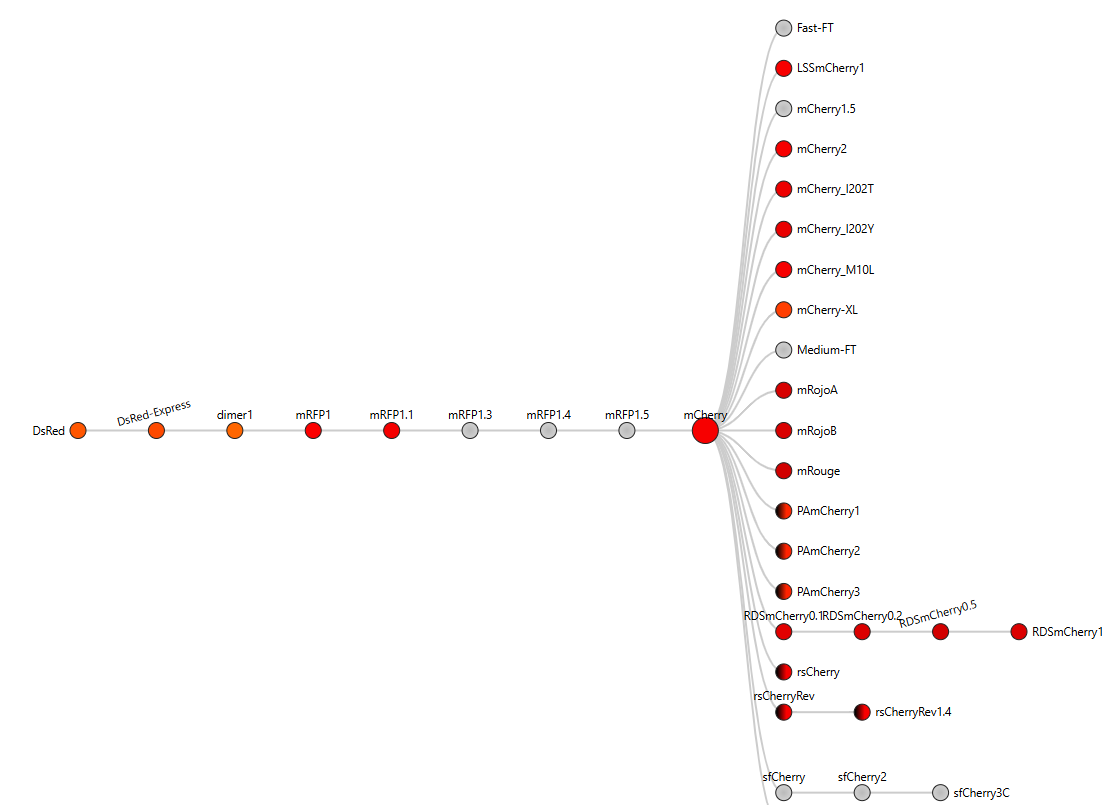
- I obtained the amino acid sequence of mCherry from the FPbase, which links laboratory fluorescent protein names to biological databases. FPbase provided the UniProt identifier X5DSL3, which is now stored in UniParc (UPI000046F63B) because the UniProtKB entry was removed. And also, the same database provided the genebank identifier for this protein AAV52164, which from where I got the sequence in fasta format.
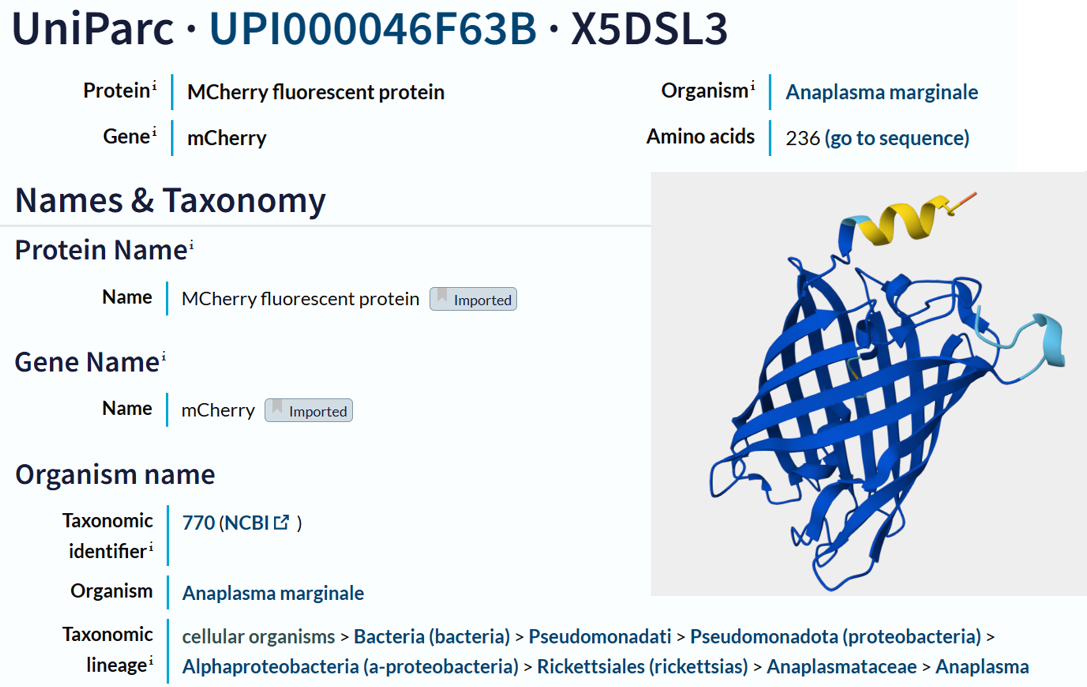
- This is the obtained sequence :
- The protein sequence is 236 amino acids long and a molecular mass of approximately 26.7 kDa. It has been confirmed at the protein level, although the UniProt entry is currently unreviewed (TrEMBL). Using the provided Colab notebook, I analyzed the amino acid composition of the sequence and found that glycine (G) is the most frequent amino acid, appearing 25 times.
Note
While analyzing the amino acid sequence of mCherry, I noticed a small difference between the sequence length reported by UniProt (236 amino acids) and the sequence obtained from the Colab notebook (241 amino acids). This discrepancy is likely due to the Colab sequence including extra residues from expression constructs, such as start codons, tags, or linkers, which are not part of the canonical protein. UniProt provides the biologically relevant, canonical sequence, which is what I used for further analysis and visualization in this homework.
To identify protein sequence homologs of mCherry, I used the BLAST tool available on UniProt.
Using the BLAST tool in UniProt, a total of 227 homologous protein sequences were identified for mCherry in the UniProtKB database. Among these results, 13 sequences are reviewed (Swiss-Prot) and 214 are unreviewed (TrEMBL). The homologs show a wide range of sequence identities, from about 23.6% up to 100%, with very low E-values (as low as 4.4 × 10⁻¹⁷⁵), indicating strong evolutionary relatedness.
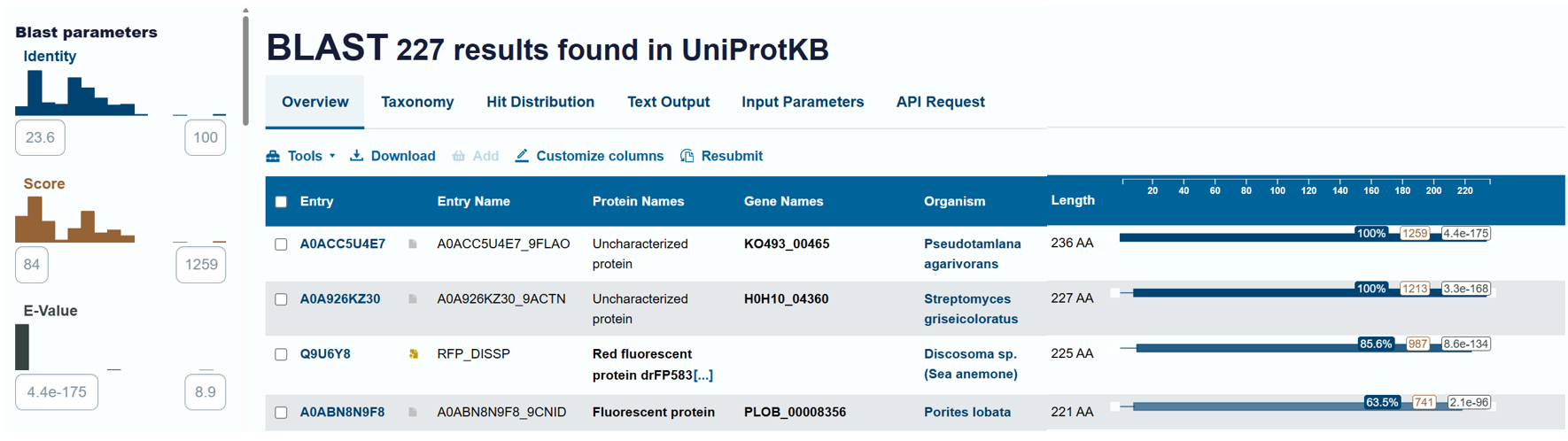
Most homologous proteins have sequence lengths between 200 and 400 amino acids, which is similar to mCherry (236 amino acids). Many homologs originate from marine organisms, especially corals and sea anemones such as Porites lobata, Pocillopora meandrina, and Discosoma species, which are known natural sources of GFP-like fluorescent proteins. Some homologs also appear in bacteria and other organisms, reflecting that mCherry is an engineered protein that has been widely introduced into different hosts for research purposes. Overall, these results confirm that mCherry belongs to a well-conserved GFP-like fluorescent protein family with broad biological and biotechnological use.
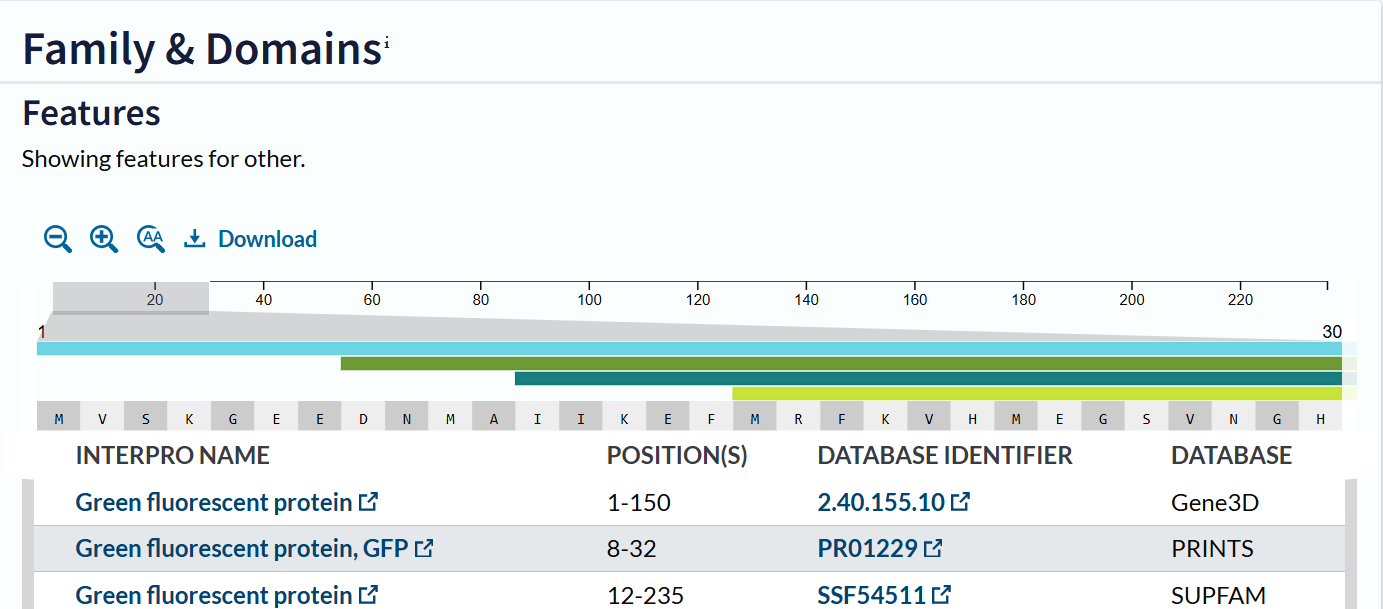
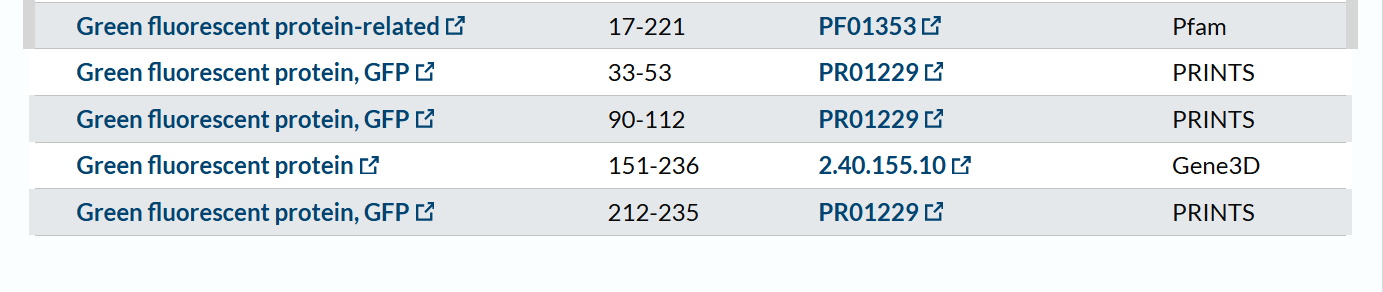
The mCherry protein belongs to a known protein family. According to UniProt family and domain analysis, mCherry is part of the green fluorescent protein (GFP)-like family , even though it emits red light. This classification is supported by several databases, including InterPro, Pfam, Gene3D, and PRINTS, all of which identify mCherry as a GFP or GFP-related protein. Proteins in this family share a conserved structure and chromophore-forming mechanism.
- The structure of the selected protein mCherry is available in the RCSB Protein Data Bank under the PDB ID 2H5Q, titled “Crystal structure of mCherry.” This structure represents the red fluorescent protein mCherry derived from Discosoma species and expressed in Escherichia coli. The structure was solved using X-ray diffraction and was deposited in May 2006 and released in August 2006.
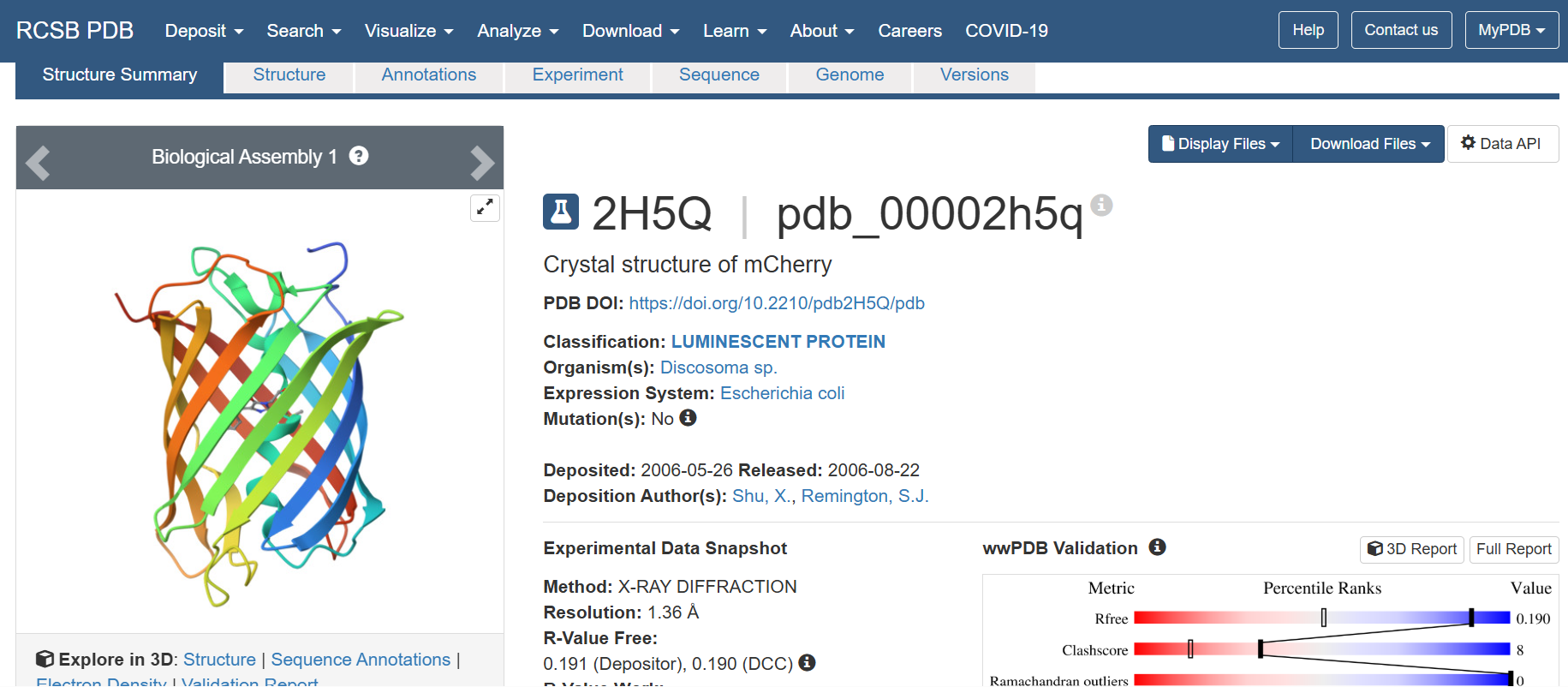
The quality of this structure is very high. It was solved at a resolution of 1.36 Å, which is much better than the 2.70 Å threshold typically used to define a good-quality structure. Lower resolution values indicate more detailed and accurate atomic positions, so a resolution of 1.36 Å means the structure is very reliable. In addition, the reported R-values (R-work ≈ 0.15 and R-free ≈ 0.19) further support that this is a well-refined and high-quality crystal structure.
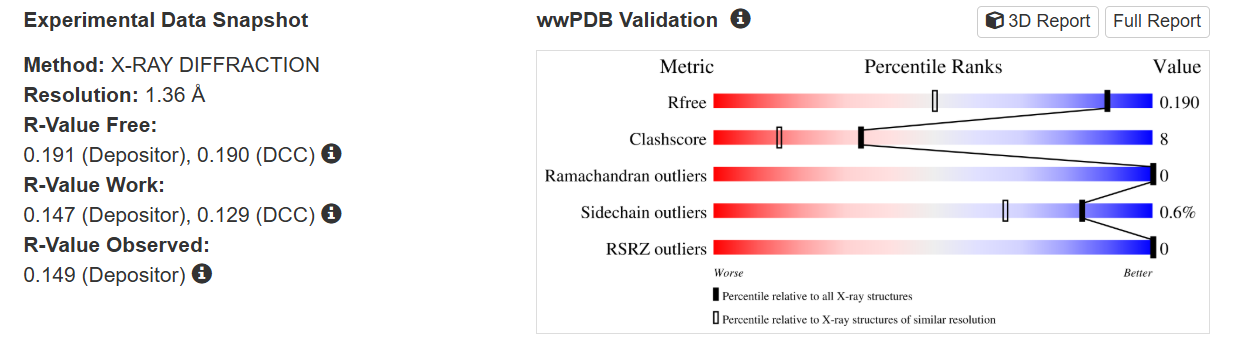
Besides the protein itself, the solved structure contains a modified residue that corresponds to the mature chromophore of mCherry. This chromophore is formed from amino acids within the protein chain and is responsible for fluorescence. No additional ligands, cofactors, or external small molecules are present. The biological assembly is a single monomer, which means that the protein functions as one chain and does not require binding to other protein subunits.
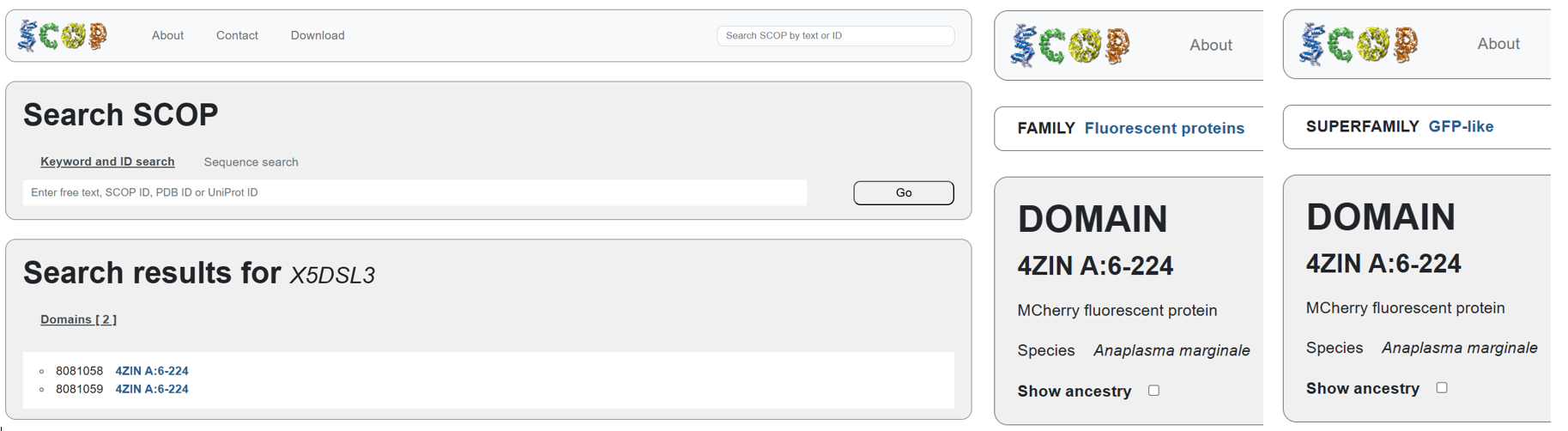
According to SCOP (Structural Classification of Proteins), mCherry is classified within the fluorescent protein family and the GFP-like superfamily. SCOP groups proteins based on their three-dimensional structure rather than their biological function or expression host. In this classification, mCherry contains a single domain (residues 6–224) that forms the characteristic β-barrel fold shared by GFP-like proteins. This confirms that mCherry belongs to the same structural superfamily as other green and red fluorescent proteins that use a similar fold to support fluorescence.
Note
The difference in the listed organism for mCherry between databases is not an error but is due to how engineered proteins are described. The Fluorescent Protein Database (FPbase) lists mCherry as originating from Discosoma species because mCherry was originally engineered from DsRed, a natural red fluorescent protein found in coral. FPbase focuses on the biological and evolutionary origin of fluorescent proteins. In contrast, UniProt lists mCherry under organisms such as Anaplasma marginale because the mCherry gene has been artificially inserted into this organism for experimental use. UniProt records the organism in which a protein sequence is present or expressed, even if the protein is not naturally produced by that organism. Therefore, both databases are correct and provide different but complementary information about the same engineered fluorescent protein.
- The protein was visualized using cartoon, ribbon, and ball-and-stick representations to examine overall fold and atomic details.
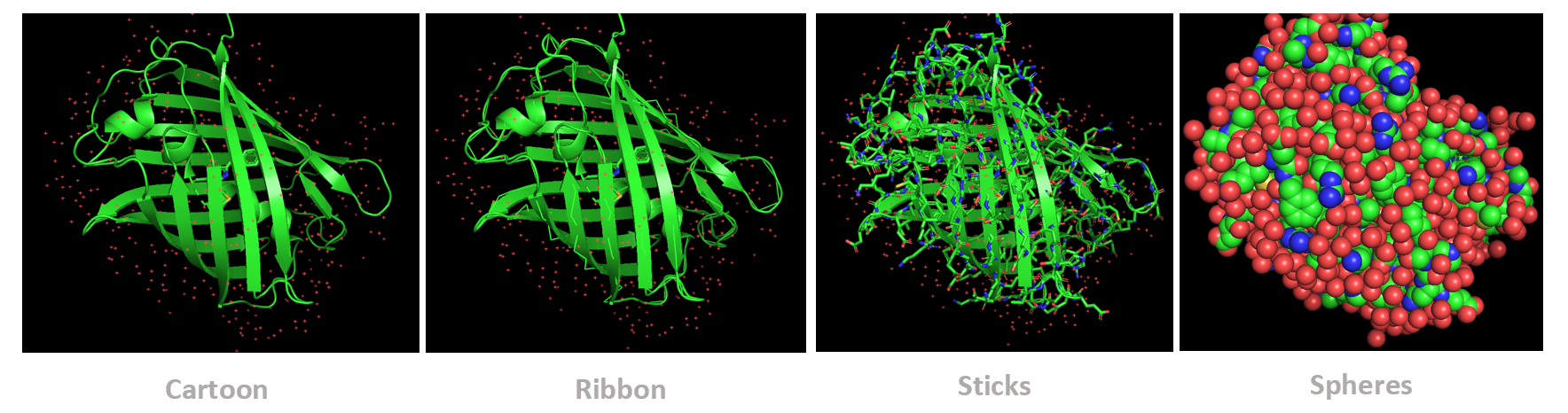
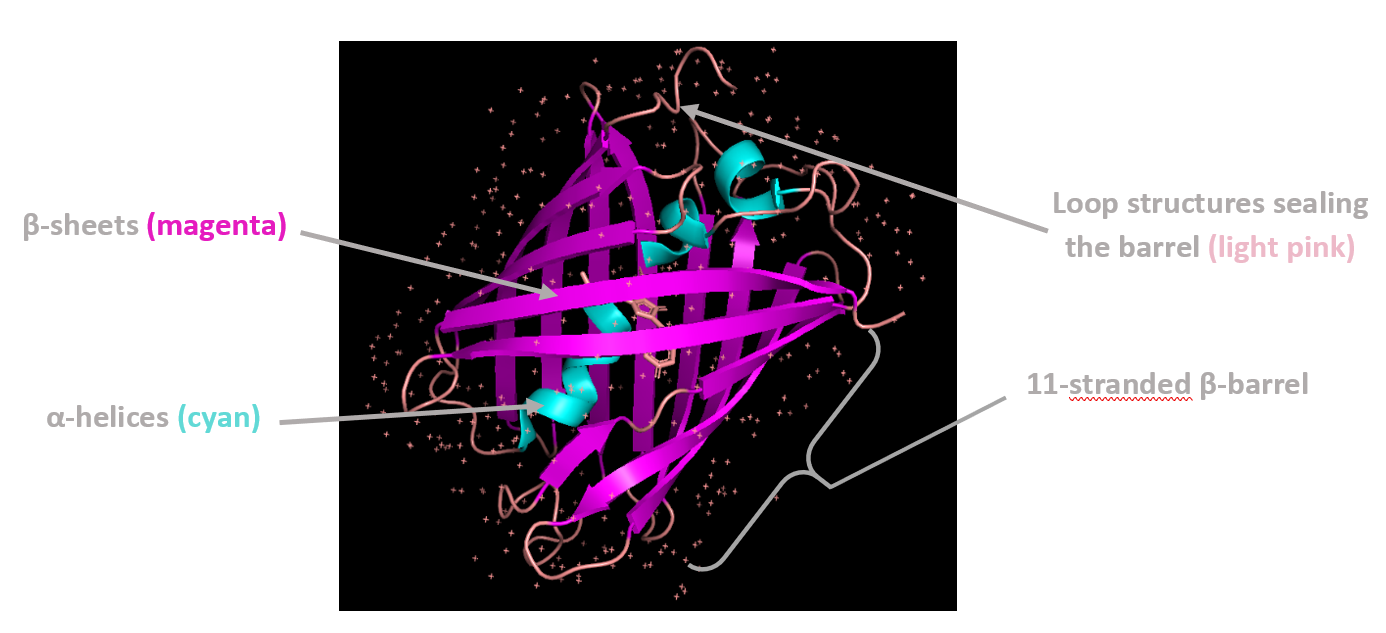
Coloring by secondary structure shows that mCherry contains many β-sheets about 11 β-sheets and very few α-helices (only 3 helices) . The protein is dominated by a β-barrel fold, which is typical for GFP-like fluorescent proteins.
Using the PyMOL command line, I colored the hydrophobic residues yellow and the hydrophilic residues red. The resulting structure shows a clear alternating pattern along the β-strands, where hydrophilic side chains face the exterior to interact with the aqueous environment (supported by the presence of surrounding water molecules), while hydrophobic side chains face the interior. This internal hydrophobic core effectively shields the chromophore from the solvent, which is essential for its fluorescence.

Based on the surface visualization of the mCherry protein (PDB: 2H5Q), the protein does not show any clear holes or binding pockets. The surface is compact and smooth, forming a closed β-barrel structure that surrounds the chromophore inside the protein. Although small bumps and grooves are visible on the surface due to amino acid side chains, there are no deep openings that lead into the protein core. This sealed structure is important for mCherry’s function, because it protects the internal chromophore from water or oxygen that could interfere with fluorescence. The closed surface therefore supports the role of mCherry as a stable fluorescent protein.

C1 Protein Language Modeling
- Deep Mutational Scans
To analyze how different mutations affect my protein, I used the ESM-2 protein language model to generate a deep mutational scan. The output is shown as a heatmap, where each color represents how favorable or unfavorable a specific mutation is. The score (z-value) reflects how likely the mutation is to be stable: positive values mean the mutation is well tolerated, while negative values suggest the mutation may damage the protein.
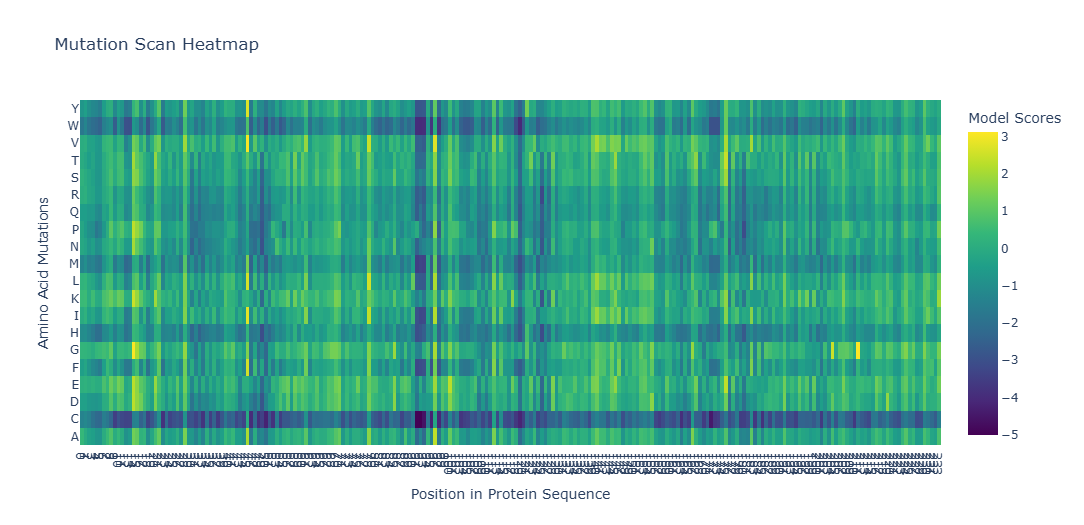
To Understand the Heatmap Colors, these are some exmples:
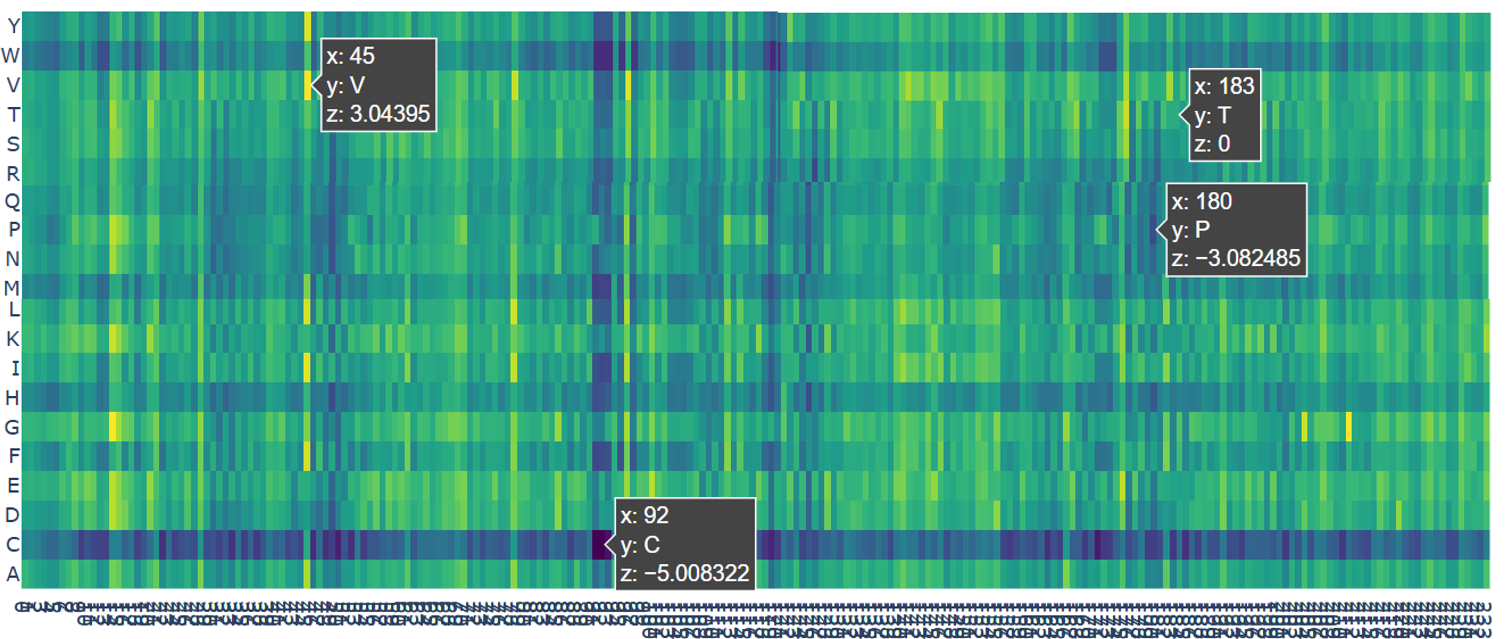
- The darkest color (black) represents the most harmful mutations. For example, the mutation at position 92 to Cysteine (C) has a very low score (z = −5.01). This position is buried deep inside the protein. Changing it to cysteine is predicted to strongly disrupt the protein, likely causing misfolding or aggregation.
- The dark blue color represents very risky mutations. An example is position 180 mutated to Proline (P) with a score of z = −3.08. This residue lies in a β-strand. Proline is known to break regular protein structures, so inserting it here would likely distort or break the β-barrel.
- The green color indicates neutral mutations. For example, position 183 mutated to Threonine (T) has a score of z = 0, meaning the model predicts little to no effect on protein stability.
- The yellow color represents favorable mutations. At position 45 mutated to Valine (V), the score is z = 3.04, suggesting this mutation may slightly improve protein stability compared to the original amino acid.
When looking at the entire heatmap, many positions appear as vertical dark bands. These positions do not tolerate most mutations and are therefore highly conserved. These residues usually form the hydrophobic core of the protein and point inward to build the β-barrel structure. Because mCherry has a tightly sealed β-barrel, mutations in these regions can disrupt proper folding or destabilize the barrel. If the β-barrel is damaged or becomes leaky, the chromophore inside can no longer be protected, which would stop the protein from fluorescing. So, this explains why mutations in these regions are strongly disfavored by the model.
My protein of interest is the Medium-FT variant, which is related to my final project and works as a protein “aging timer.” This behavior is controlled by specific mutations that change the chromophore chemistry without breaking the overall protein structure.
To explore the functional mutations in the parent protein mCherry (PDB: 2H5Q, the one I used to represent the heatmap), I focused on two important mutations: K69R, and A224S (F. V. Subach et al., 2009; O. M. Subach et al., 2022). So as indicated in the heatmap, they showed positive scores (z = 0.75 ; z = 1.08) respectively.
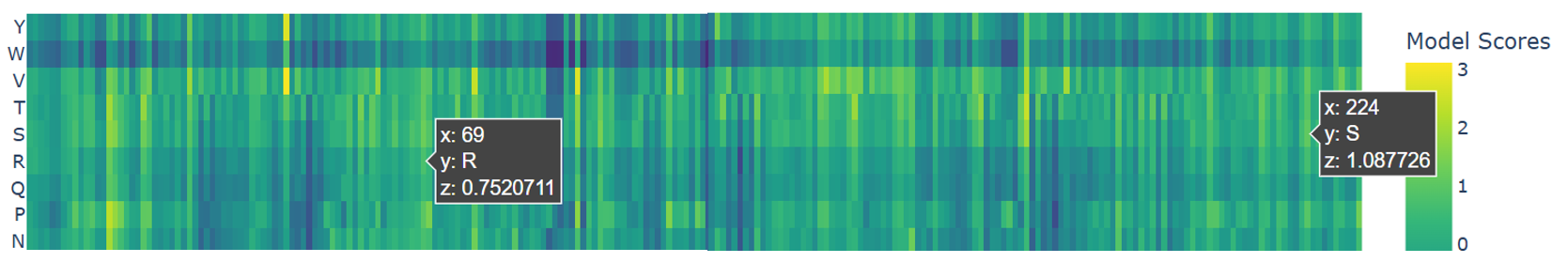
Both mutations appear as light green to yellow on the heatmap, meaning they are well tolerated. This confirms that these changes do not disrupt the β-barrel or overall stability. they adjust the protein’s function by slowing down fluorescence maturation while keeping the main structure intact.
- Latent Space Analysis
To perform latent space analysis, I used the provided dataset of protein sequences from the SCOP database and generated numerical embeddings for each sequence using the ESM-2 protein language model, which results in a three-dimensional map where each point represents one protein.
.png?width=300px)
When analyzing the resulting map, proteins do not appear randomly distributed. Instead, they form local neighborhoods where nearby points correspond to proteins with similar structural properties. These neighborhoods approximate similarities in protein fold and secondary structure rather than biological function. This shows that the language model organizes proteins based on shared “structural rules,” such as how alpha helices and beta sheets are arranged, even when the proteins come from different organisms or have different functions.
For example, the protein d2cw3a1 a.2.11.0 (A:4–90) from Perkinsus marinus has three closest neighbors that come from very different organisms, including Escherichia coli and cow. These neighboring proteins also have very different biological functions.
My protein of interest, mCherry (PDB: 2H5Q) which is represented by bleu dot, is located in a neighborhood dominated by proteins rich in β-sheet structures. Its closest neighbors include proteins such as the β-propeller domain of the enzyme PepX, the β-barrel domain of the chaperone protein Sis1, and other β-sheet–containing domains like transferrin-binding protein and latexin. Although these proteins perform very different biological roles, they share similar β-sheet-based structural architectures. The close proximity of mCherry to these proteins confirms that the ESM-2 model groups proteins based on structural similarity, correctly placing mCherry among other β-sheet and β-barrel-like proteins in the latent space.
.png?width=300px)
C2. Protein Folding
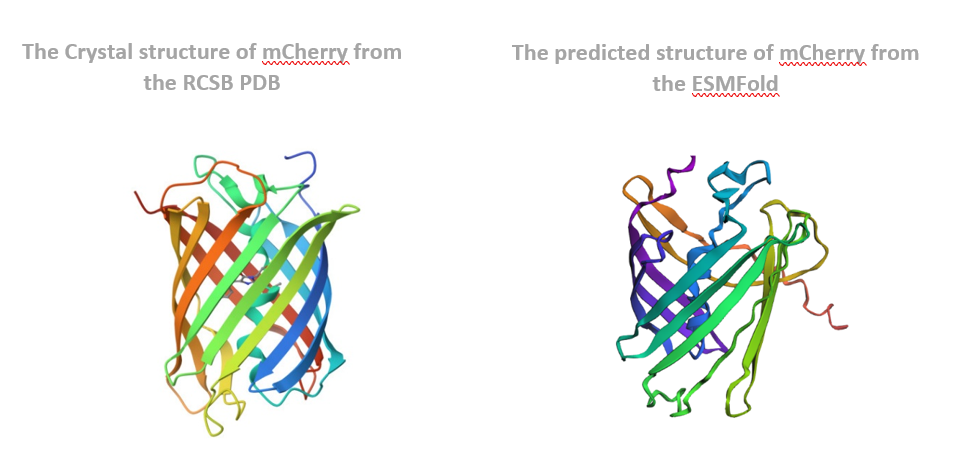
The predicted coordinates matched the original structure very well. The overall shape, especially the β-barrel structure, was preserved, and the folding pattern looked almost identical. This shows that ESMFold can accurately predict the structure of mCherry from its amino acid sequence.
Next, I changed the protein sequence by introducing several mutations, including small amino acid changes and changes spread across the sequence. After folding the mutated sequence with ESMFold, the structure showed noticeable changes compared to the original protein. While the general β-barrel shape was still present, some regions were slightly distorted. This indicates that mCherry is partly resilient to mutations, but too many or poorly placed mutations can affect proper folding and reduce structural stability.
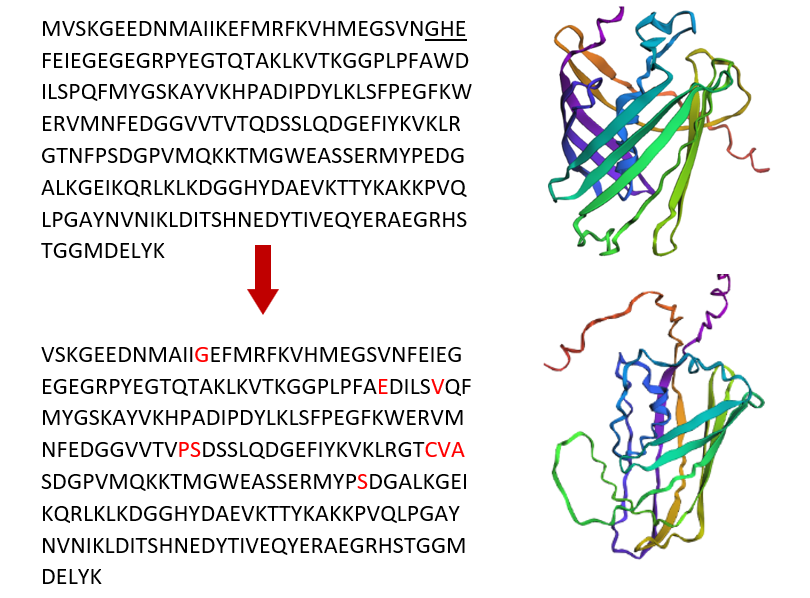
C3. Protein Generation
I used ProteinMPNN to do inverse folding on the mCherry protein (PDB: 2H5Q). I used the default settings and turned off the homomer option because this protein has only one chain. ProteinMPNN uses the 3D shape of the protein and suggests new amino acid sequences that can keep the same shape.
The output includes a probability heatmap, which shows the model’s confidence for each amino acid at every position in the sequence. In the heatmap, bright colors (yellow/green) indicate amino acids that are highly preferred at a specific position, while dark colors (blue/purple) indicate unlikely choices. Some positions show a strong preference for one amino acid, meaning they are important for maintaining the protein structure. Other positions show more flexibility, suggesting they can tolerate different amino acids without disrupting the fold.
.png?width=500px)
ProteinMPNN generated a new sequence candidate with a sequence recovery of about 47.93 %, meaning nearly half of the amino acids are identical to the original mCherry sequence. The designed sequence received a lower score (0.8107) compared to the native sequence score (1.3913). Because lower scores indicate a better statistical fit to the backbone, this suggests that the designed sequence is predicted to be highly compatible and stable for the 11-stranded β-barrel structure of mCherry.
- The native protein sequence and its score are shown below:
- The newly generated protein sequence and its evaluation metrics are shown below:
I attempted to refold the newly designed sequence using ESMFold in order to compare the predicted structure with the original mCherry structure. However, ESMFold requires GPU resources, and GPU access was not available at the time of execution. As a result, a direct structural comparison could not be performed. Despite this limitation, the strong sequence score and conserved structural regions indicate that the designed sequence would likely fold into a structure very similar to the original β-barrel if GPU resources were available.
Gemini AI tools integrated with Google Colab were used to help explain code errors, interpret the generated outputs such as heatmaps, and analyze the latent space by identifying the closest neighboring proteins through distance calculations between my protein and other sequences.
One-Page Proposal
in this homework, AI ChatGPT assisted me in organizing and clearly articulating my answers and descriptions, ensuring that the content is well-structured and easy to understand.
Sources:
- Adhikari, A., Bhattarai, B. R., Aryal, A., Thapa, N., KC, P., Adhikari, A., Maharjan, S., Chanda, P. B., Regmi, B. P., & Parajuli, N. (n.d.). Reprogramming natural proteins using unnatural amino acids. RSC Advances, 11(60), 38126–38145. https://doi.org/10.1039/d1ra07028b
- Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 Lysis of Escherichia coli Depends on Host Chaperone DnaJ. Journal of Bacteriology, 199(12), e00058-17. https://doi.org/10.1128/JB.00058-17
- Doig, A. J. (2017). Frozen, but no accident – why the 20 standard amino acids were selected. The FEBS Journal, 284(9), 1296–1305. https://doi.org/10.1111/febs.13982
- Gsponer, J., & Vendruscolo, M. (2006). Theoretical Approaches to Protein Aggregation. Protein & Peptide Letters, 13(3), 287–293. https://doi.org/10.2174/092986606775338407
- Human nutrition—Protein, Vitamins, Minerals | Britannica. (n.d.). Retrieved February 27, 2026, from https://www.britannica.com/science/human-nutrition/Meat-fish-and-eggs
- Mezhyrova, J., Martin, J., Börnsen, C., Dötsch, V., Frangakis, A. S., Morgner, N., & Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4), 28. https://doi.org/10.20517/mrr.2023.28
- Phage Therapy: Past, Present and Future. (n.d.). ASM.Org. Retrieved March 2, 2026, from https://asm.org:443/articles/2022/august/phage-therapy-past,-present-and-future
- Right-Handed Alpha-Helix—An overview | ScienceDirect Topics. (n.d.). Retrieved February 27, 2026, from https://www.sciencedirect.com/topics/chemistry/right-handed-alpha-helix
- Strathdee, S. A., Hatfull, G. F., Mutalik, V. K., & Schooley, R. T. (2023). Phage therapy: From biological mechanisms to future directions. Cell, 186(1), 17–31. https://doi.org/10.1016/j.cell.2022.11.017
- Subach, F. V., Subach, O. M., Gundorov, I. S., Morozova, K. S., Piatkevich, K. D., Cuervo, A. M., & Verkhusha, V. V. (2009). Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nature Chemical Biology, 5(2), 118–126. https://doi.org/10.1038/nchembio.138
- Subach, O. M., Tashkeev, A., Vlaskina, A. V., Petrenko, D. E., Gaivoronskii, F. A., Nikolaeva, A. Y., Ivashkina, O. I., Anokhin, K. V., Popov, V. O., Boyko, K. M., & Subach, F. V. (2022). The mRubyFT Protein, Genetically Encoded Blue-to-Red Fluorescent Timer. International Journal of Molecular Sciences, 23(6), 3208. https://doi.org/10.3390/ijms23063208
- Vieira-Pires, R. S., & Morais-Cabral, J. H. (2010). 310 helices in channels and other membrane proteins. The Journal of General Physiology, 136(6), 585–592. https://doi.org/10.1085/jgp.201010508
- Young, T. S., & Schultz, P. G. (2010). Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon. The Journal of Biological Chemistry, 285(15), 11039–11044. https://doi.org/10.1074/jbc.R109.091306