week-05-HW-protein-design-part-II
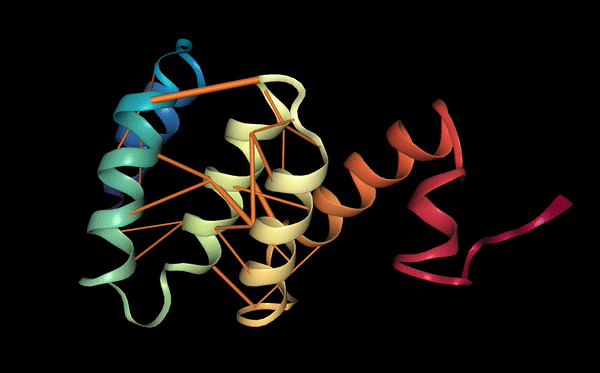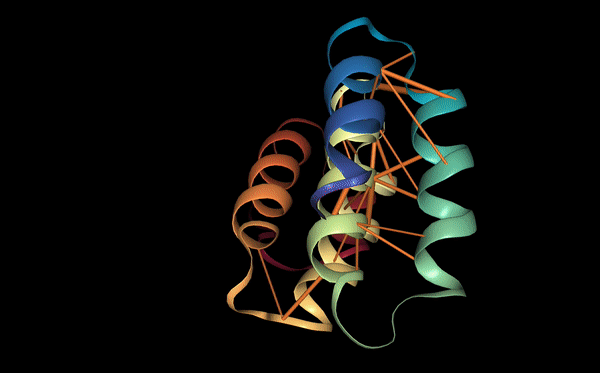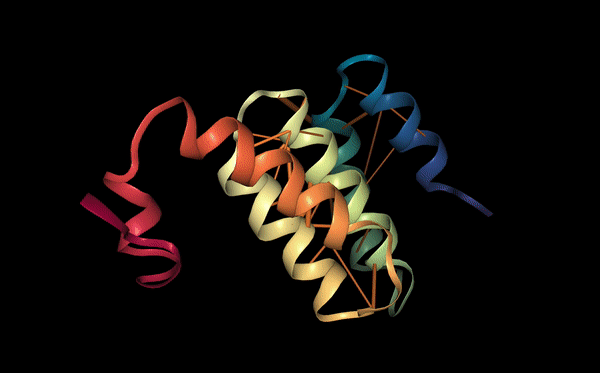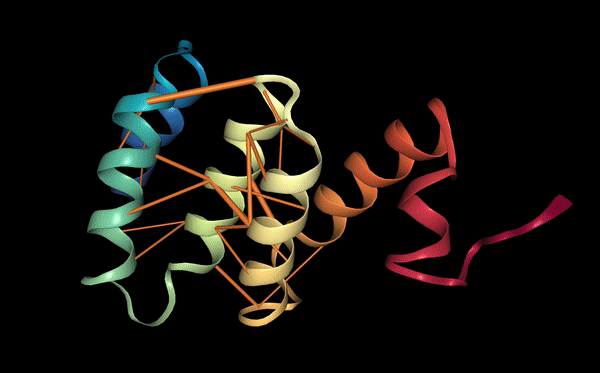
Part 1: Generate Binders with PepMLM
For the PepMLM analysis, the amino acid sequence of the normal Superoxide Dismutase 1 protein was obtained from UniProt using the accession number P00441.
 To simulate the disease-associated variant, the A4V mutation was then manually introduced into the sequence to generate the mutant form of the protein used for the peptide design experiments. This mutation corresponds to the substitution of alanine by valine at position 4 of the protein sequence.
To simulate the disease-associated variant, the A4V mutation was then manually introduced into the sequence to generate the mutant form of the protein used for the peptide design experiments. This mutation corresponds to the substitution of alanine by valine at position 4 of the protein sequence.
- Original Superoxide dismutase 1 (SOD1) sequence from Uniprot:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
- Mutant A4V sequence (A4V means Alanine → Valine at position 4):
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Note
The Problem: The A4V mutation you are studying is famous because it destabilizes the dimer interface. This causes the dimer to fall apart into monomers, which then misfold and aggregate into the toxic clumps seen in ALS patients.
After running the PepMLM model using mutant A4V sequence, I obtained four candidate peptide binder sequences. In the generated results, each sequence ended with the amino acid symbol ‘X’, which represents an undefined residue predicted by the model.
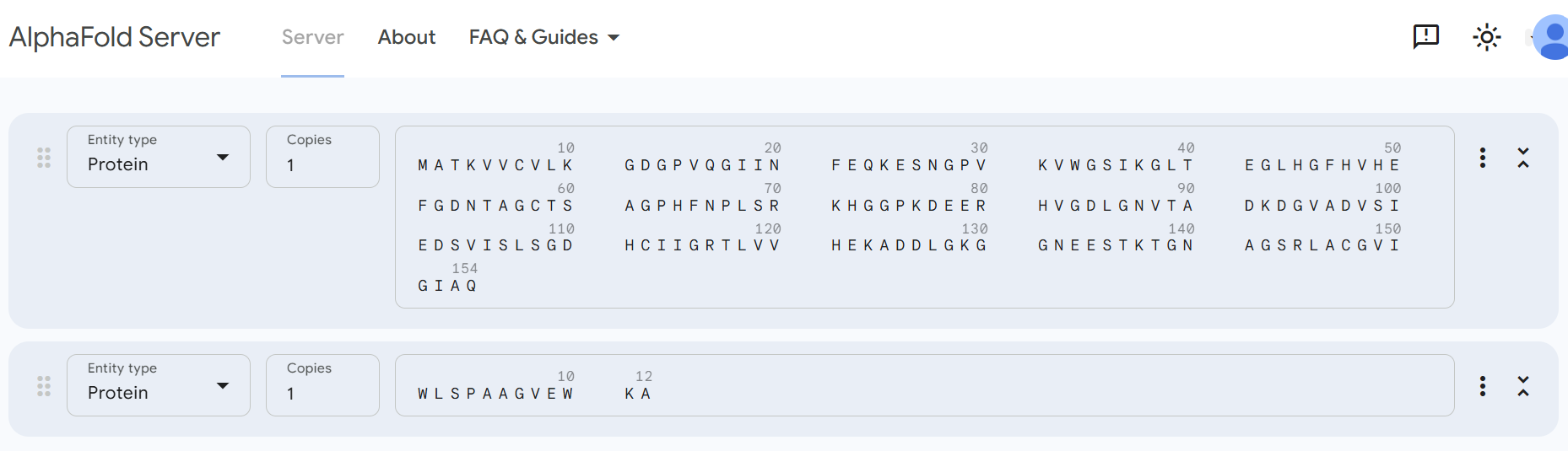
To proceed with the structural analysis and fold the predicted binders with the mutated SOD1 protein, I needed to assign a specific amino acid at this position. For this reason, I replaced the ‘X’ residue with alanine (A) in each sequence. I chose alanine because it is a small and neutral amino acid that generally has minimal effects on protein structure and interactions. This allowed me to obtain complete peptide sequences that could be used for the subsequent folding and interaction prediction with the A4V mutant SOD1 protein.
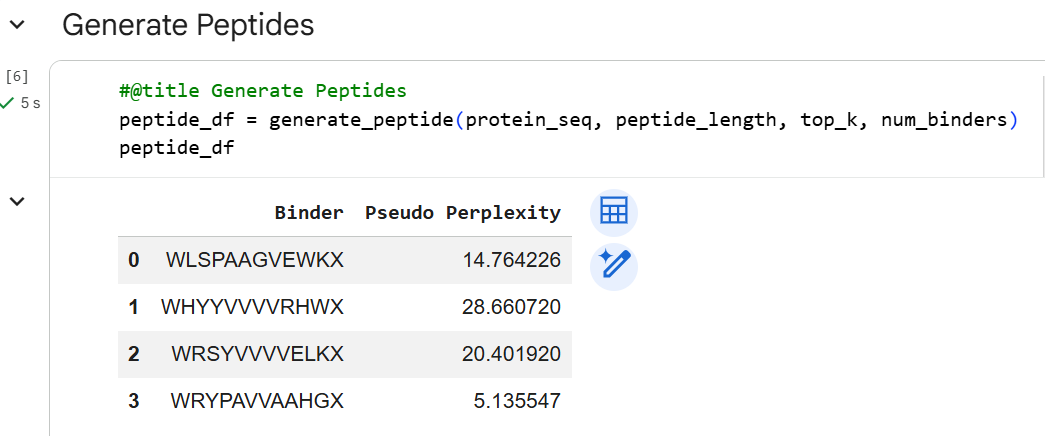
| Binder | Original Sequence | Modified Sequence | Perplexity |
|---|---|---|---|
| Known binder | FLYRWLPSRRGG | /// | /// |
| 1 | WLSPAAGVEWKX | WLSPAAGVEWKA | 14.764 |
| 2 | WHYYVVVVRHWX | WHYYVVVVRHWA | 28.661 |
| 3 | WRSYVVVVELKX | WRSYVVVVELKA | 20.402 |
| 4 | WRYPAVVAAHGX | WRYPAVVAAHGA | 5.136 |
Part 2: Evaluating Binders with AlphaFold3
The predicted peptide binders were evaluated using structural modeling. Each peptide was folded together with the A4V mutant of SOD1 to evaluate the potential protein–peptide interactions.

Note
pTM and ipTM scores: the predicted template modeling (pTM) score and the interface predicted template modeling (ipTM) score are both derived from a measure called the template modeling (TM) score. This measures the accuracy of the entire structure (Zhang and Skolnick, 2004; Xu and Zhang, 2010). A pTM score above 0.5 means the overall predicted fold for the complex might be similar to the true structure. ipTM measures the accuracy of the predicted relative positions of the subunits within the complex. Values higher than 0.8 represent confident high-quality predictions, while values below 0.6 suggest likely a failed prediction. ipTM values between 0.6 and 0.8 are a gray zone where predictions could be correct or incorrect. TM score is very strict for small structures or short chains, so pTM assigns values less than 0.05 when fewer than 20 tokens are involved; for these cases PAE or pLDDT may be more indicative of prediction quality.
The known binder (FLYRWLPSRRGG) showed a relatively low binding confidence, with an ipTM score of 0.28. The peptide binds mainly on the surface of the SOD1 β-barrel, close to the electrostatic loop and the zinc-binding loop. It does not bind near the N-terminus, where the A4V mutation is located, and it also does not interact with the dimer interface. The peptide remains mostly surface-bound rather than buried inside the protein structure. Several residues help stabilize the interaction. For example, Trp5 and Tyr3 can form aromatic contacts with the protein surface, while Arg8 and Arg9 may form hydrogen bonds with nearby residues of SOD1. However, the peptide does not form a strong or compact binding interface, which suggests that the interaction may be weak or transient.
Binder 1 (WLSPAAGVEWKA) showed a clear improvement compared with the known binder, with an ipTM score of 0.39. The peptide binds on a hydrophobic groove on the surface of the SOD1 β-barrel. In this interaction, Trp1 acts as an important anchoring residue, helping the peptide attach to a hydrophobic pocket on the protein surface. Other residues such as Ser3 and Pro4 help position the peptide backbone against the protein surface. In addition, Glu9 forms stabilizing hydrogen bonds with nearby residues on SOD1. Because of these interactions, the peptide forms a more compact and organized binding conformation than the known binder. Although the peptide still binds away from the A4V mutation site, the higher ipTM score and the stronger interaction network suggest that Binder 1 may represent a more promising peptide candidate.
Binder 2 (WHYYVVVVRHWA) showed a moderate interaction with SOD1, with an ipTM score of 0.33, which is higher than the known binder but lower than Binder 1. The peptide binds on a surface patch of the SOD1 β-barrel region. Several residues appear to contribute to this interaction. Trp1 participates in both hydrogen bonding and aromatic interactions with the protein surface, helping to anchor the peptide. Tyr3 and Arg9 also participate in hydrogen bonding that stabilizes the peptide orientation. In addition, the terminal residue Ala12 contributes to stabilizing the peptide backbone through hydrogen bonding with the protein surface. Compared with the known binder, Binder 2 shows a more localized and organized binding mode, although the peptide still binds mainly on the surface of the protein rather than deeply inside the structure.
Binder 3 (WRSYVVVVELKA) showed the lowest binding confidence among the designed peptides, with an ipTM score of 0.20, which is even lower than the known binder. The peptide still localizes on the surface of the SOD1 β-barrel, but the interaction appears weak and poorly defined. The interaction is mainly supported by Arg2 and Lys11, which can form hydrogen bonds with residues on the SOD1 surface. In addition, Tyr4 may contribute through aromatic interactions with the protein surface. However, the peptide forms only a limited number of stabilizing contacts, and the interaction appears less stable compared with Binder 1 and Binder 2. These results suggest that Binder 3 may not be a strong candidate for stable binding to the SOD1 mutant.
Binder 4 (WRYPAVVAAHGA) showed a moderate structural confidence, with an ipTM score of 0.33, similar to Binder 2 and higher than the known binder. The peptide binds on the surface of the SOD1 β-barrel region. Several residues contribute to this interaction. Trp3, Val6, and Gly11 appear to form hydrogen bonds with residues on the SOD1 surface, helping stabilize the interaction. In addition, an internal hydrogen bond between Val6 and His10 helps stabilize the peptide backbone and maintain its conformation. Compared with Binder 3, this peptide forms more defined interactions with the protein surface, which explains its higher predicted binding confidence. Although the peptide still binds away from the A4V mutation site, the interaction appears more organized and stable than the known binder.
To further explore whether peptide length influences binding stability, the same structural analysis was also performed using 11-residue versions of the peptides obtained by removing the final alanine that replaced the unknown residue X. For Binder 1, the ipTM score decreased from 0.39 (12 aa) to 0.27 (11 aa), indicating that the twelfth residue likely helps stabilize the interaction with the SOD1 surface. In contrast, Binder 2 showed a small increase in structural confidence, where the score changed from 0.33 (12 aa) to 0.35 (11 aa), suggesting that the slightly shorter peptide may adopt a somewhat better orientation on the protein surface. Binder 3 showed the strongest negative effect of shortening the peptide, with the score decreasing from 0.20 (12 aa) to 0.13 (11 aa), confirming that this peptide already forms weak interactions and becomes even less stable when shortened. Interestingly, Binder 4 showed the opposite trend, where the 11-residue version reached the highest score of all tested peptides (0.44) compared with 0.33 for the 12-residue version, suggesting that removing the last residue may allow the peptide to adopt a more favorable binding conformation. Overall, these exploratory results suggest that peptide length can influence binding stability, but the effect is sequence-dependent, since shortening the peptide reduced stability for some binders (Binder 1 and Binder 3) while improving it for others (Binder 2 and Binder 4).
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
I evaluated the therapeutic properties of all 12-residue peptide binders using PeptiVerse.
![]()
![]()
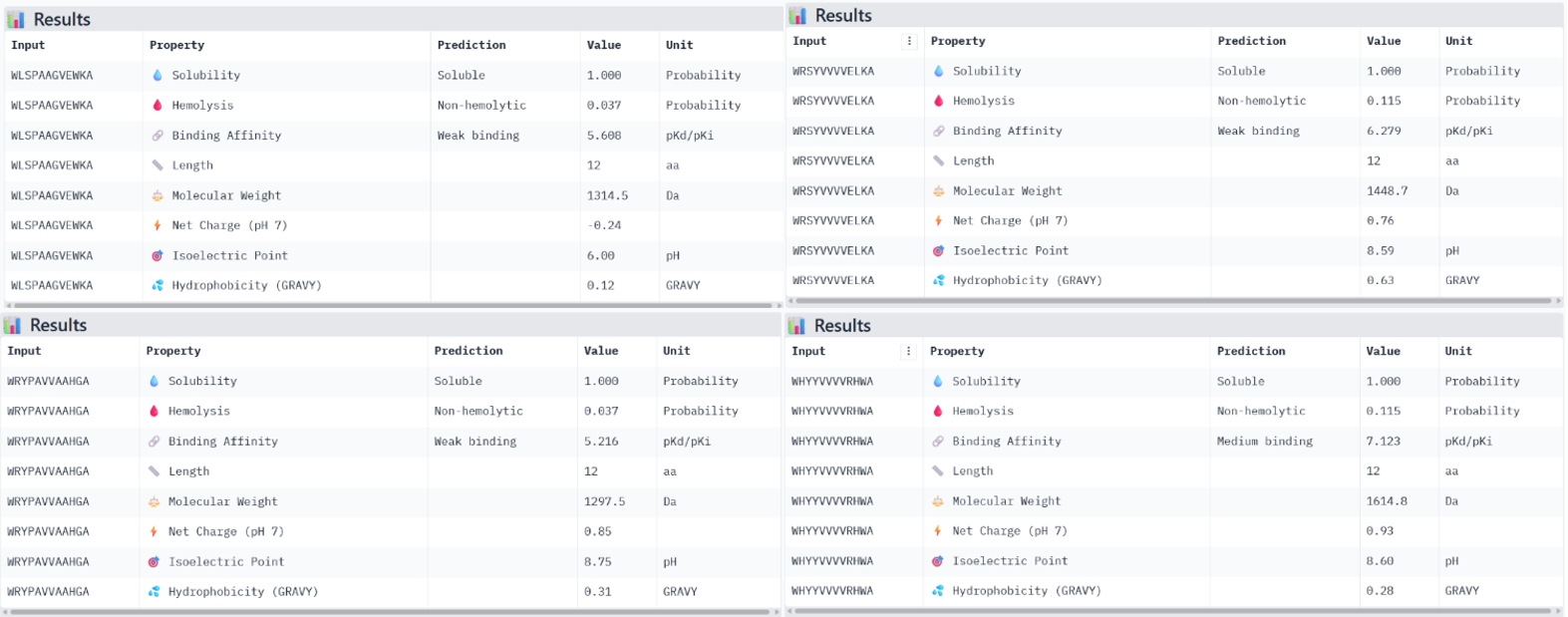
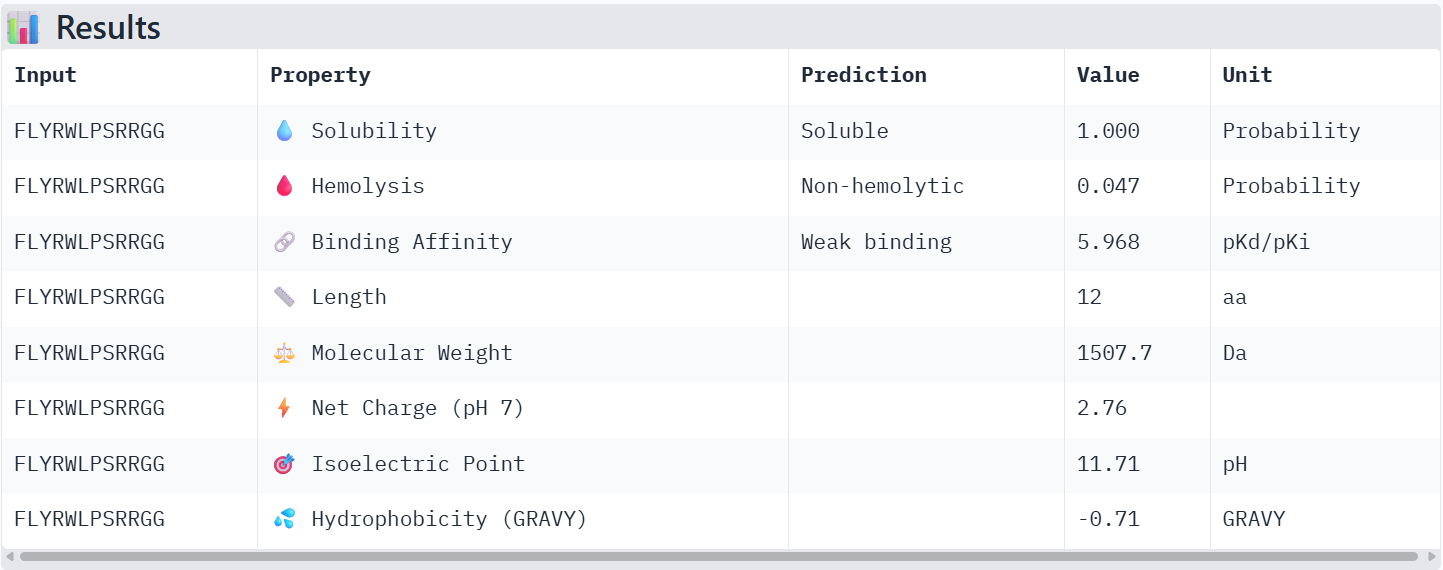 The known binder FLYRWLPSRRGG showed weak binding affinity (pKd 5.97), good solubility, very low hemolysis probability (0.047), and a positive net charge of 2.76. Among the PepMLM-generated peptides, Binder 1 (WLSPAAGVEWKA) had weak binding affinity (pKd 5.61), excellent solubility, very low hemolysis (0.037), and a near-neutral net charge (-0.24). Binder 2 (WHYYVVVVRHWA) exhibited medium binding affinity (pKd 7.12), fully soluble, non-hemolytic (0.115), and slightly positive net charge (0.93). Binder 3 (WRSYVVVVELKA) showed weak binding (pKd 6.28), soluble, non-hemolytic (0.115), and net charge 0.76. Binder 4 (WRYPAVVAAHGA) had weak binding (pKd 5.22), soluble, non-hemolytic (0.037), and net charge 0.85.
The known binder FLYRWLPSRRGG showed weak binding affinity (pKd 5.97), good solubility, very low hemolysis probability (0.047), and a positive net charge of 2.76. Among the PepMLM-generated peptides, Binder 1 (WLSPAAGVEWKA) had weak binding affinity (pKd 5.61), excellent solubility, very low hemolysis (0.037), and a near-neutral net charge (-0.24). Binder 2 (WHYYVVVVRHWA) exhibited medium binding affinity (pKd 7.12), fully soluble, non-hemolytic (0.115), and slightly positive net charge (0.93). Binder 3 (WRSYVVVVELKA) showed weak binding (pKd 6.28), soluble, non-hemolytic (0.115), and net charge 0.76. Binder 4 (WRYPAVVAAHGA) had weak binding (pKd 5.22), soluble, non-hemolytic (0.037), and net charge 0.85.
| Binder | Sequence | ipTM (AlphaFold) | Predicted Binding Affinity (pKd/pKi) | Solubility | Hemolysis Probability | Net Charge (pH 7) | Molecular Weight (Da) | Highlights |
|---|---|---|---|---|---|---|---|---|
| Known | FLYRWLPSRRGG | 0.28 | Weak (5.968) | Soluble (1) | 0.047 | 2.76 | 1507.7 | Surface-bound, low confidence; non-hemolytic |
| Binder 1 | WLSPAAGVEWKA | 0.39 | Weak (5.608) | Soluble (1) | 0.037 | -0.24 | 1314.5 | Highest ipTM, stable hydrophobic groove binding; non-hemolytic |
| Binder 2 | WHYYVVVVRHWA | 0.33 | Medium (7.123) | Soluble (1) | 0.115 | 0.93 | 1614.8 | Good binding, slightly lower ipTM than Binder 1; non-hemolytic |
| Binder 3 | WRSYVVVVELKA | 0.20 | Weak (6.279) | Soluble (1) | 0.115 | 0.76 | 1448.7 | Lowest ipTM, weak structural stability; non-hemolytic |
| Binder 4 | WRYPAVVAAHGA | 0.33 | Weak (5.216) | Soluble (1) | 0.037 | 0.85 | 1297.5 | Medium ipTM, non-hemolytic, good solubility |
Comparing these properties with the structural confidence from AlphaFold, we see that higher ipTM scores do not always directly match stronger predicted binding. For example, Binder 1 had the highest ipTM (0.39) but only weak predicted binding, while Binder 2 had slightly lower ipTM (0.33) but showed medium predicted binding. All generated peptides are soluble and non-hemolytic, which is favorable for therapeutic use. Considering both structural confidence and predicted properties, Binder 1 (WLSPAAGVEWKA) is the most promising overall: it has the highest structural stability on SOD1, is non-hemolytic, fully soluble, and has a near-neutral charge that may support safe and effective binding in a biological context.
Part 4: Generating Optimized Peptides with moPPIt
Mutations such as the A4V variant can destabilize the structure of Superoxide Dismutase 1, increasing the probability of protein misfolding, dissociation of the dimer, and toxic aggregation, which are processes associated with Amyotrophic Lateral Sclerosis (ALS). For this reason, the design strategy of this step focused on generating short peptides that can bind simultaneously to both monomers at the dimer interface, effectively acting as a molecular bridge that reconnects and stabilizes the two subunits. By reinforcing the interaction between the chains, these peptides may help restore a conformation closer to the native functional state of the SOD1 complex, while reducing the structural instability caused by the mutation.
To do so, several design parameters were selected before generating peptides. The peptide length was fixed at 12 amino acids. The motif position was focused on residues 3–10, meaning the central region of the peptide was encouraged to interact with the target protein. In addition, affinity guidance and solubility optimization were enabled, and hemolysis prediction was considered to reduce potential toxicity. These settings allow the model to design peptides that not only bind the protein but also have better therapeutic properties.
![]()
![]()
After generating the following peptides, their structures were evaluated using AlphaFold to predict how they interact with SOD1.
- The generated sequences:
| Optimized Binder | Sequence | IpTM Score | Binding Localization |
|---|---|---|---|
| 1 | KRQCEIFNQFMA | 0.91 | Interface between the two monomers |
| 2 | EKDNKWVITSQF | 0.86 | Interface between the two monomers |
| 3 | VCQFDYKTLFKK | 0.87 | Interface between the two monomers |
| 4 | GQQSLFKTKTLD | 0.89 | The outer surface of a single SOD1 monomer |
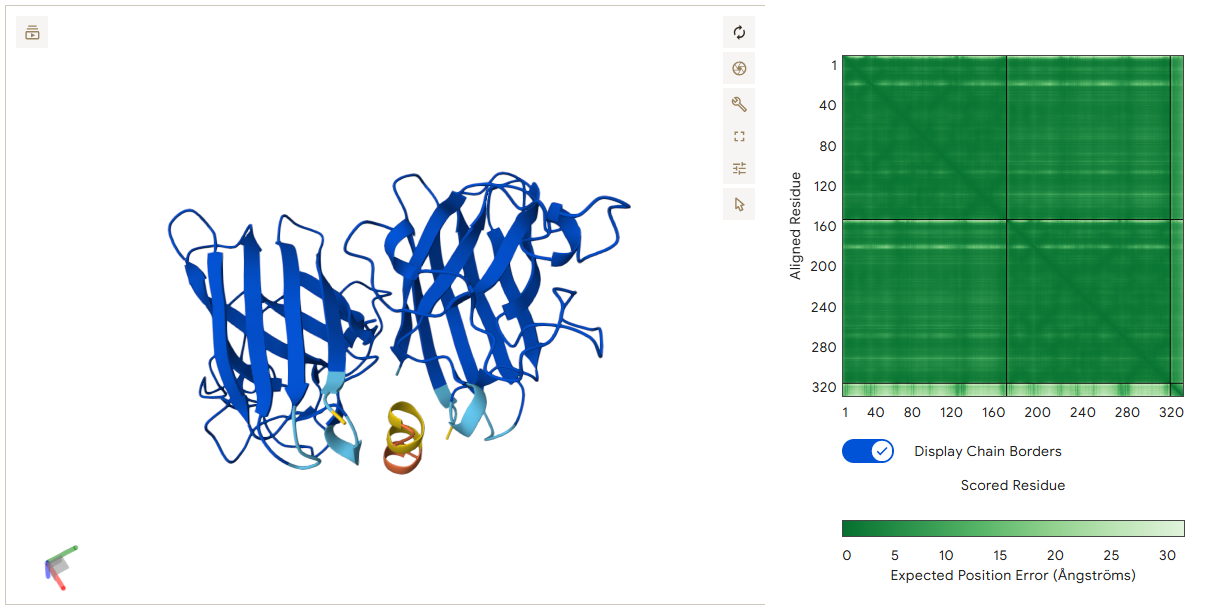
- Binder 1 – KRQCEIFNQFMA (ipTM: 0.91)
This peptide localizes at the dimer interface of the SOD1 homodimer and acts as a molecular bridge between the two monomers. Several residues of the peptide participate in stabilizing the interaction. Gln3 forms a hydrogen bond with residues on the first monomer, while Cys4 interacts with a cysteine residue on the second monomer. In addition, Asn8 forms multiple hydrogen bonds with residues on Chain A. These multiple contacts allow the peptide to connect both monomers simultaneously, which could help stabilize the dimer structure of SOD1.
- Binder 2 – EKDNKWVITSQF (ipTM: 0.86)
This peptide also binds at the dimer interface and connects the two monomers. The interaction is mainly driven by the N-terminal region of the peptide. Glu1 forms several hydrogen bonds with residues on Chain B, creating a strong anchoring point. In addition, Ser10 interacts with residues on Chain A. Through these interactions with both monomers, the peptide may help maintain the stability of the SOD1 dimer.
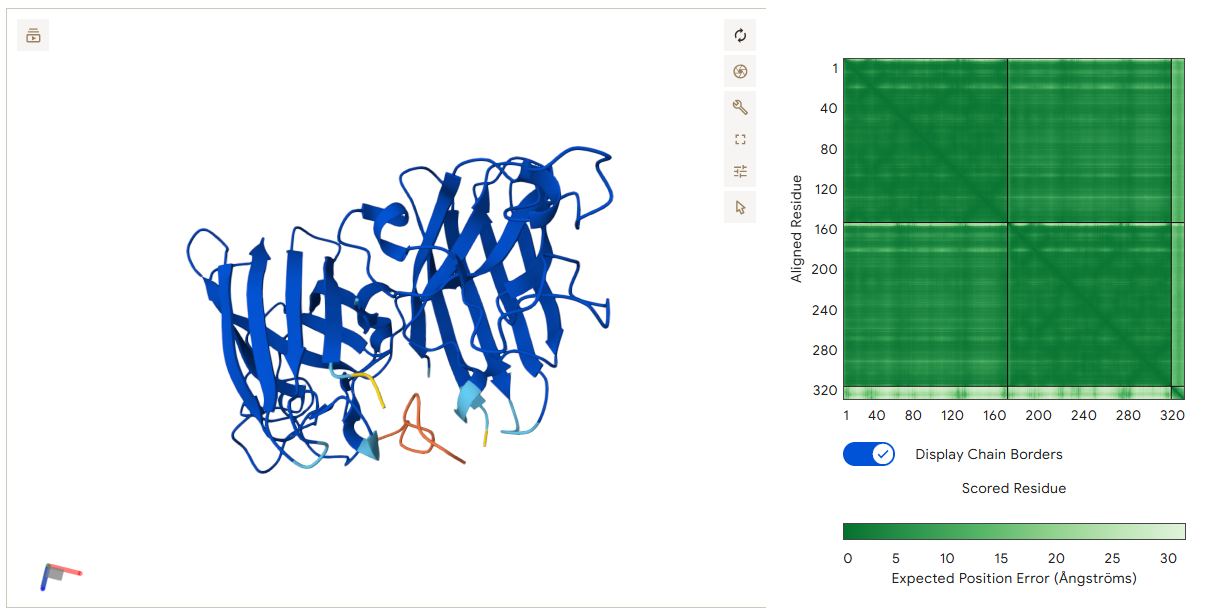
- Binder 3 – VCQFDYKTLFKK (ipTM: 0.87)
This peptide spans the interface between the two monomers, forming stabilizing contacts with both chains. Val1 forms a hydrogen bond with residues on Chain B, while Phe4 interacts with residues on Chain A. These interactions allow the peptide to bridge the two monomers and stabilize the interface region.
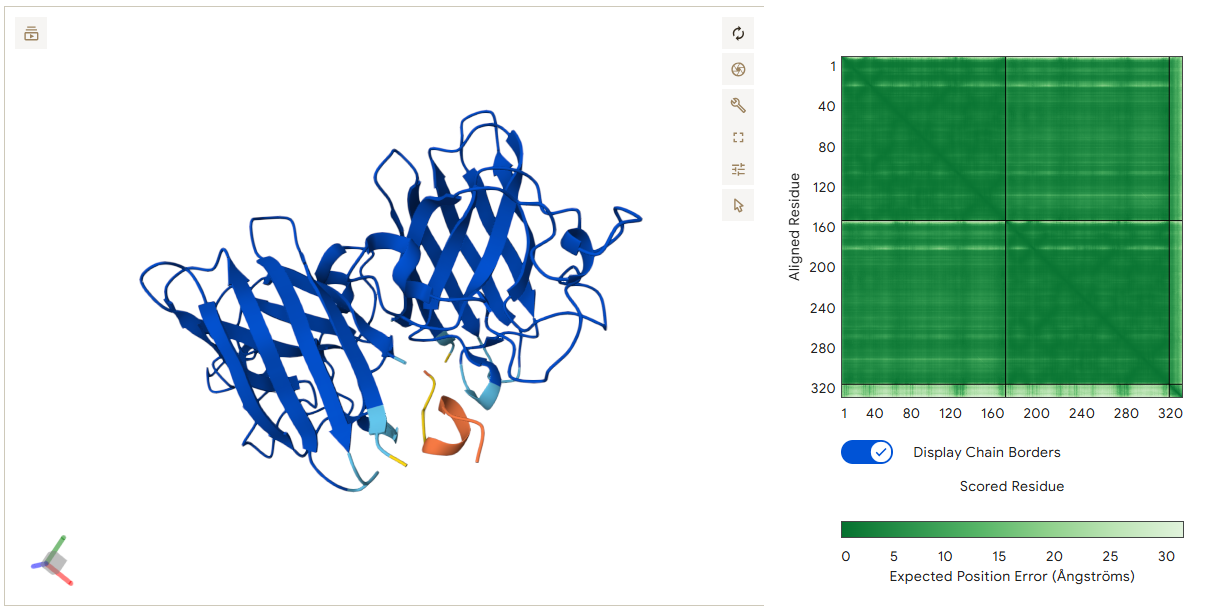
- Binder 4 – GQQSLFKTKTLD (ipTM: 0.89)
Unlike the previous peptides, this binder attaches to the outer surface of a single SOD1 monomer, particularly near the β-barrel structure. The interaction is mainly driven by residues near the C-terminus of the peptide. Thr10 forms a hydrogen bond with the monomer, while Asp12 forms two hydrogen bonds with residues on Chain A. Lys9 also contributes to stabilization by forming an additional hydrogen bond. This peptide does not bridge the dimer but instead stabilizes the surface structure of the monomer.
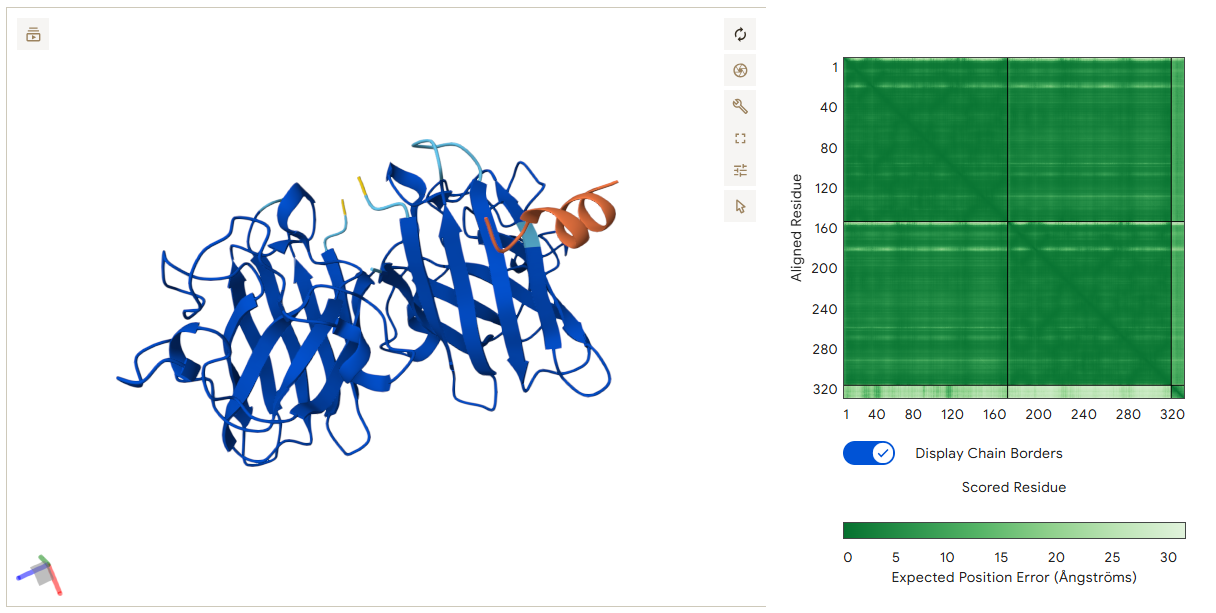 The four peptides show two different binding strategies:
The four peptides show two different binding strategies:
Three peptides (KRQCEIFNQFMA, EKDNKWVITSQF, and VCQFDYKTLFKK) bind at the dimer interface, where they interact with residues from both monomers. These peptides may help stabilize the SOD1 dimer by acting as a bridge between the two chains. In contrast, GQQSLFKTKTLD binds only to one monomer, specifically on the β-barrel surface. Instead of bridging the two chains, this peptide may stabilize the structure of the individual monomer.
Among the peptides, KRQCEIFNQFMA shows the highest ipTM score (0.91), suggesting the strongest predicted interaction with the protein complex. When comparing the peptides generated by PepMLM and moPPIt, the main difference lies in the design strategy. PepMLM mainly samples possible peptide sequences that could bind to the target protein based on patterns learned from protein sequence data. However, it does not allow the user to control exactly where the peptide should bind on the protein. As a result, the generated peptides are plausible binders, but their binding location and biochemical properties are not specifically optimized.
In contrast, moPPIt enables guided peptide design. In this approach, the user can select specific residues or regions on the protein where the peptide should bind, such as the dimer interface of Superoxide Dismutase 1 or regions near the A4V mutation. The model also optimizes several properties simultaneously, including binding affinity, solubility, hemolysis risk, and motif placement. Because of this multi-objective optimization, moPPIt peptides are designed to better satisfy several therapeutic requirements at the same time.
For this part, unfortunately, I was unable to access the BRD4 Drug Discovery Platform, as the access was not granted to me despite my request.
Option 1 : Improve autofolding and lysis efficiency
The goal of this part was to design mutations in the L-protein in order to improve its function. Two main objectives were considered. The first objective was to improve the autofolding ability of the L-protein so that it can fold correctly without strong dependence on host chaperones. The second objective was to improve the lysis efficiency of the protein by enhancing its ability to form pores in the E. coli membrane and promote faster or more efficient bacterial lysis.
To identify possible mutations, the provided mutation scoring notebook was used. This notebook evaluates all possible amino-acid substitutions in the L protein and assigns a score to each mutation.
After running the notebook, the resulted mutation predictions are presented in the following dataset:
| Position (DNA) | Position (Protein) | Wild Type AA | Mutation AA | LLR Score |
|---|---|---|---|---|
| 989 | 50 | K | L | 2.561468 |
| 574 | 29 | C | R | 2.395427 |
| 769 | 39 | Y | L | 2.241780 |
| 575 | 29 | C | S | 2.043150 |
| 173 | 9 | S | Q | 2.014325 |
| 573 | 29 | C | Q | 1.997049 |
| 572 | 29 | C | P | 1.971029 |
| 569 | 29 | C | L | 1.960646 |
| 987 | 50 | K | I | 1.928801 |
| 1049 | 53 | N | L | 1.864932 |
| 1209 | 61 | E | L | 1.818098 |
| 1029 | 52 | T | L | 1.813968 |
| 984 | 50 | K | F | 1.802069 |
| 576 | 29 | C | T | 1.797247 |
| 568 | 29 | C | K | 1.795878 |
| 93 | 5 | F | Q | 1.795244 |
| 94 | 5 | F | R | 1.659717 |
| 560 | 29 | C | A | 1.648656 |
| 534 | 27 | Y | R | 1.628061 |
| 434 | 22 | F | R | 1.602028 |
| 92 | 5 | F | P | 1.596891 |
| 997 | 50 | K | V | 1.594576 |
| 995 | 50 | K | S | 1.574557 |
| 96 | 5 | F | T | 1.559024 |
| 95 | 5 | F | S | 1.556417 |
| 889 | 45 | A | L | 1.539248 |
| 775 | 39 | Y | S | 1.517457 |
| 535 | 27 | Y | S | 1.497053 |
| 789 | 40 | V | L | 1.477630 |
| 529 | 27 | Y | L | 1.474637 |
| 435 | 22 | F | S | 1.423358 |
| 563 | 29 | C | E | 1.383281 |
| 760 | 39 | Y | A | 1.364999 |
| 571 | 29 | C | N | 1.362601 |
| 980 | 50 | K | A | 1.357795 |
| 567 | 29 | C | I | 1.344121 |
| 89 | 5 | F | L | 1.332615 |
| 334 | 17 | N | R | 1.323651 |
| 767 | 39 | Y | I | 1.320103 |
| 776 | 39 | Y | T | 1.302804 |
| 514 | 26 | D | R | 1.268762 |
| 566 | 29 | C | H | 1.246107 |
| 764 | 39 | Y | F | 1.245851 |
| 777 | 39 | Y | V | 1.244390 |
| 454 | 23 | K | R | 1.236555 |
| 494 | 25 | E | R | 1.229350 |
| 474 | 24 | H | R | 1.227779 |
| 996 | 50 | K | T | 1.222131 |
| 533 | 27 | Y | Q | 1.218851 |
| 536 | 27 | Y | T | 1.215567 |
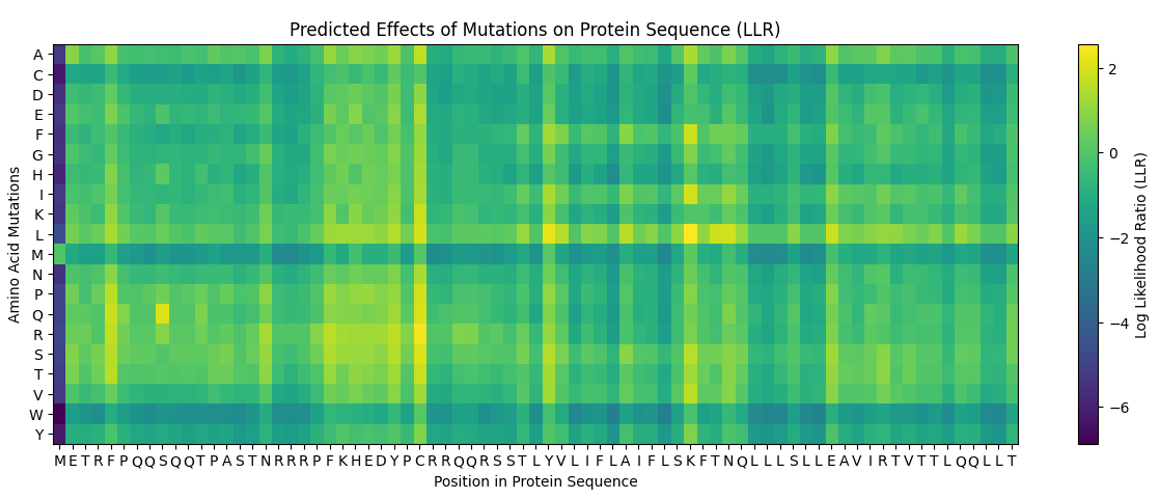 The predicted mutations were compared with the experimental dataset of L-protein mutants provided in the course material. This dataset contains mutations that were experimentally tested and their effect on lysis activity.
The predicted mutations were compared with the experimental dataset of L-protein mutants provided in the course material. This dataset contains mutations that were experimentally tested and their effect on lysis activity.
The goal of this comparison was to determine whether mutations with high prediction scores correspond to mutations that show improved lysis in experimental studies. This step helps evaluate the reliability of the prediction model. The results of this comparison revealed a limited overlap between predicted beneficial mutations and experimentally tested mutations. Two mutations, C29R and K50I, appeared in both datasets. However, experimental data indicated that these substitutions did not improve lysis activity. This suggests that, while the protein language model captures sequence compatibility, it does not fully predict functional outcomes such as lysis efficiency. For this raison, experimental validation remains essential to confirm computational predictions.
To avoid mutations that could disrupt essential protein functions, sequence conservation analysis was performed. Multiple sequences related to the MS2 L protein obtained from the BLAST results provided in the course folder were uploaded to Clustal Omega and aligned.
 The conserved regions of the L protein were identified after analyzing the multiple sequence alignment results. Highly conserved residues, which are the same across all sequences, were marked with stars (*) in the alignment output. while colon (:) indicates residues with strongly similar chemical properties. These positions were considered critical for protein function, so mutations at these residues were avoided. The remaining positions, which showed variability among sequences, were classified as non-conserved and were selected as potential sites for mutation. This approach ensured that the chosen mutations would minimize disruption of essential protein structure and function.
The conserved regions of the L protein were identified after analyzing the multiple sequence alignment results. Highly conserved residues, which are the same across all sequences, were marked with stars (*) in the alignment output. while colon (:) indicates residues with strongly similar chemical properties. These positions were considered critical for protein function, so mutations at these residues were avoided. The remaining positions, which showed variability among sequences, were classified as non-conserved and were selected as potential sites for mutation. This approach ensured that the chosen mutations would minimize disruption of essential protein structure and function.
 Mutations were selected using the resulted mutation scoring predictions and evolutionary conservation analysis. Only residues located in non-conserved positions were chosen in order to reduce the risk of disrupting essential protein functions. The selected mutations (F5Q, S9Q, F22S, Y27L, and A45L) -as represented in the following table- are distributed between the N-terminal region, the central region, and the transmembrane domain of the L protein. This distribution allows the exploration of potential effects on protein autofolding and membrane activity, while maintaining the overall structural integrity of the protein.
Mutations were selected using the resulted mutation scoring predictions and evolutionary conservation analysis. Only residues located in non-conserved positions were chosen in order to reduce the risk of disrupting essential protein functions. The selected mutations (F5Q, S9Q, F22S, Y27L, and A45L) -as represented in the following table- are distributed between the N-terminal region, the central region, and the transmembrane domain of the L protein. This distribution allows the exploration of potential effects on protein autofolding and membrane activity, while maintaining the overall structural integrity of the protein.

| Mutation | LLR Score* | Protein Region | AA Property Change | Mutation Type | Conserved Residue? | Structural Risk | Rationale for Selection |
|---|---|---|---|---|---|---|---|
| S9Q | ~2.01 | N-terminal region | Small polar → Polar amide | Conservative | Unconserved | Low | Similar polarity; minimal structural disruption while potentially altering hydrogen bonding |
| F5Q | ~1.80 | N-terminal region | Hydrophobic aromatic → Polar amide | Moderate | Unconserved | Moderate | Introduces polarity which may affect folding and interaction with cytoplasmic environment |
| A45L | ~1.54 | Transmembrane helix | Small hydrophobic → Larger hydrophobic | Conservative | Unconserved | Low | Maintains hydrophobic nature; may stabilize helix packing in membrane |
| Y27L | ~1.47 | Near transmembrane region | Aromatic → Hydrophobic aliphatic | Moderate | Unconserved | Moderate | Maintains hydrophobicity but removes aromatic ring; could affect membrane insertion |
| F22S | ~1.42 | Cytoplasmic / near TM region | Hydrophobic aromatic → Small polar | Moderate | Unconserved | Moderate | Reduces hydrophobicity; may influence membrane interaction and folding |
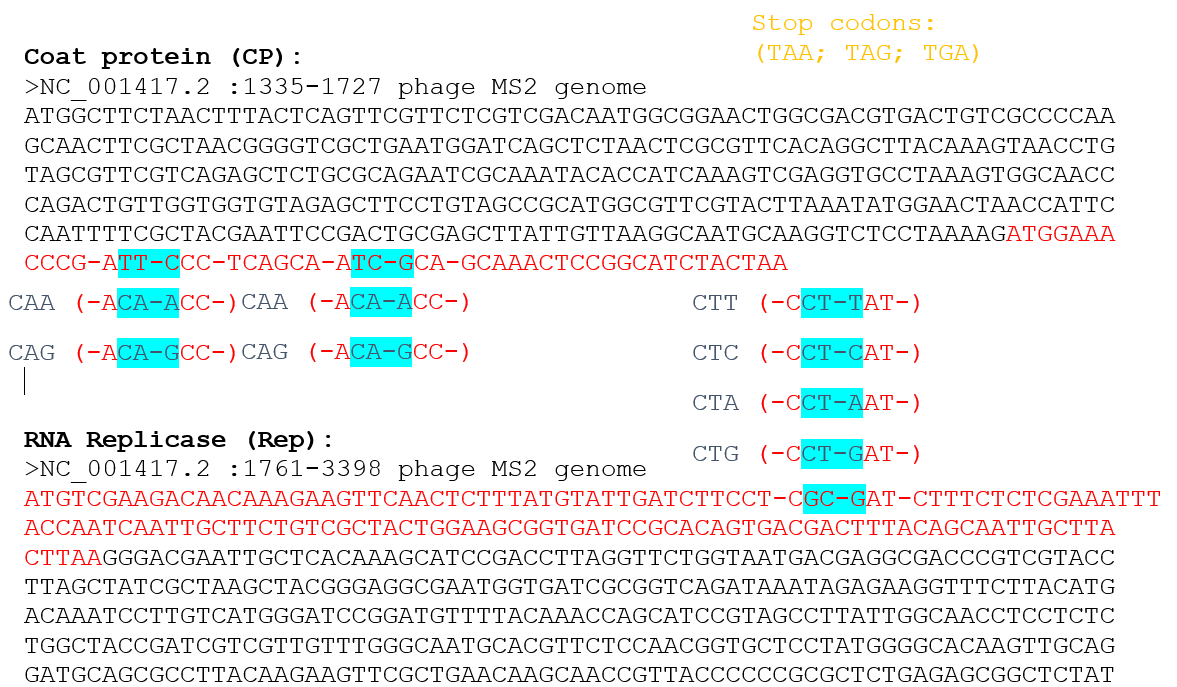
Because the L gene overlaps with other genes in the MS2 genome, the nucleotide changes corresponding to the selected mutations were checked to ensure that they do not introduce stop codons in the overlapping reading frames.
![]()
![]()
 The mutations F5Q and S9Q are located in the region overlapping with the coat protein (CP) gene, near its C-terminal end, while the mutation A45L is located in the region overlapping with the replicase (Rep) gene, near its N-terminal region.
For each mutation, the possible codon substitutions were examined and confirmed not to generate stop codons in the overlapping genes. Therefore, these mutations are considered compatible with the genome organization of MS2.
The mutations F5Q and S9Q are located in the region overlapping with the coat protein (CP) gene, near its C-terminal end, while the mutation A45L is located in the region overlapping with the replicase (Rep) gene, near its N-terminal region.
For each mutation, the possible codon substitutions were examined and confirmed not to generate stop codons in the overlapping genes. Therefore, these mutations are considered compatible with the genome organization of MS2.
Option 2: Achieve DnaJ independence
Here the goal was to reduce or eliminate the dependence of the L-protein on the host chaperone DnaJ. By designing mutations in the soluble N-terminal domain of the L-protein, i aimed to weaken its interaction with DnaJ while maintaining proper folding. This approach could potentially allow the phage to function even if DnaJ is mutated or absent in the host.
To study the interaction, i used the AlphaFold2-Multimer notebook in ColabFold to co-fold the soluble domain of the L-protein with the full sequence of E. coli DnaJ. The sequences used were:
- DnaJ sequence:
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
- The soluble domain of Lysis protein (N terminal Domain):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSS
The first co-folding run generated five ranked models with the following parameters. Here, pLDDT reflects the confidence in the predicted structure of the L–DnaJ complex, pTM indicates the overall predicted quality of the complex, and ipTM estimates the predicted strength of the interaction between L-protein and DnaJ:
Rank_004_alphafold2_multimer_v3_model_1: pLDDT=79.3 pTM=0.567 ipTM=0.198
![]()
![]() Rank_005_alphafold2_multimer_v3_model_2 : pLDDT=78.9 pTM=0.573 ipTM=0.161
Rank_005_alphafold2_multimer_v3_model_2 : pLDDT=78.9 pTM=0.573 ipTM=0.161
![]()
![]() Rank_003_alphafold2_multimer_v3_model_3: pLDDT=78.4 pTM=0.572 ipTM=0.265
Rank_003_alphafold2_multimer_v3_model_3: pLDDT=78.4 pTM=0.572 ipTM=0.265
![]()
![]()
Rank_001_alphafold2_multimer_v3_model_4: pLDDT=78.9 pTM=0.583 ipTM=0.373
![]()
![]()
Rank_002_alphafold2_multimer_v3_model_5: pLDDT=77.9 pTM=0.572 ipTM=0.287
![]()
![]()
- Plots for L_DnaJ_complex
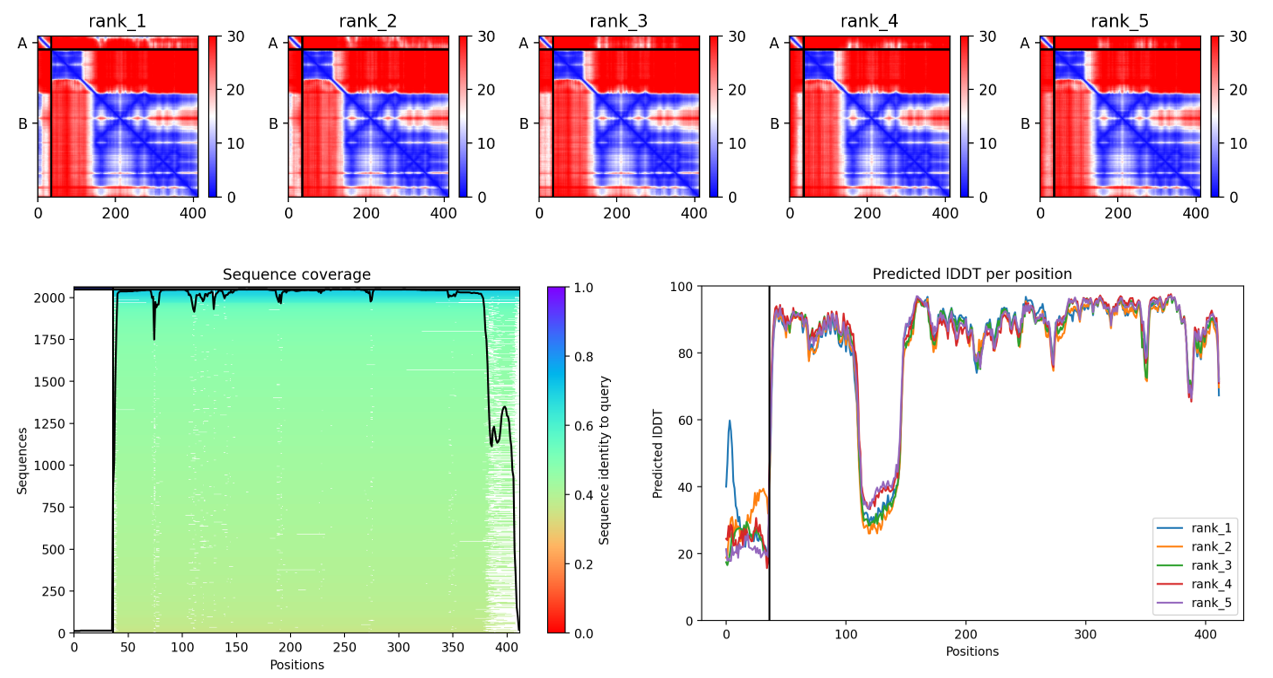
By comparing the different predicted models, the fourth model was identified as the best-ranked model because it showed the highest ipTM score, indicating the strongest predicted interaction between the L-protein and DnaJ.
![]()
![]() Using PyMol I Analysed the predicted L–DnaJ complex of the best predicted model (rank 4) and the results revealed multiple interaction residues located in the N-terminal region of the L protein as summarized in the following table:
Using PyMol I Analysed the predicted L–DnaJ complex of the best predicted model (rank 4) and the results revealed multiple interaction residues located in the N-terminal region of the L protein as summarized in the following table:
| Residue | Type | Contacts with DnaJ | Typical Interaction Role |
|---|---|---|---|
| Met 1 | Hydrophobic (non-polar) | ASP116, ARG113 | Hydrophobic contact |
| GLU2 | Negatively charged | ALA115, ASP116 | Electrostatic / salt bridge |
| THR3 | Polar (uncharged) | ASP116, LEU117, ARG118 | Hydrogen bonding |
| ARG4 | Positively charged | ALA115, ARG118, LEU117, GLU233, ASP116 | Electrostatic / salt bridge |
| PHE5 | Hydrophobic aromatic | ASN120, LEU117, ARG118 | Hydrophobic packing |
| PRO6 | Hydrophobic (rigid) | LEU117, ARG118, ASN120, TYR119, ASP116 | Structural / hydrophobic contact |
| GLN8 | Polar (uncharged) | ASN120, ARG118, TYR119 | Hydrogen bonding |
| SER15 | Polar (uncharged) | GLN252, GLU122, LYS251 | Hydrogen bonding |
| ASN17 | Polar (uncharged) | GLN252 | Hydrogen bonding |
| ARG18 | Positively charged | VAL250, GLN252, GLN249, GLU122, LYS251 | Electrostatic interaction |
| ARG19 | Positively charged | GLN252 | Electrostatic interaction |
| ARG20 | Positively charged | GLN252 | Electrostatic interaction |
| PRO21 | Hydrophobic | GLN252, GLU257 | Structural / hydrophobic contact |
| PHE22 | Hydrophobic aromatic | PRO254, GLU266, GLU257 | Hydrophobic contact |
| LYS23 | Positively charged | GLU266 | Electrostatic interaction |
| HIS24 | Positively charged / polar | VAL326, ARG324, GLU266 | Electrostatic / hydrogen bond |
| GLU25 | Negatively charged | GLU266 | Electrostatic interaction |
| ASP26 | Negatively charged | ARG324, GLU266, VAL326 | Electrostatic interaction |
| TYR27 | Aromatic polar | VAL327, THR329, GLU328 | Hydrophobic + H-bond |
Key residues such as Arg4, Thr3, Pro6, Phe5, Arg18, Lys23, His24, and Tyr27 were found to interact with several residues of DnaJ, including Asp116, Leu117, Arg118, Glu122, and Glu266. These interactions involve a combination of electrostatic, hydrophobic, and hydrogen-bond contacts. Residues forming multiple contacts were considered potential targets for mutagenesis aimed at reducing the dependence of the L protein on the DnaJ chaperone.
Two hydrophobic residues (Pro6 and Phe22), two positively charged residues (Arg4 and Arg18), and two negatively charged residues (Glu2 and Asp26) were selected for mutational analysis. These residues participate in multiple contacts with DnaJ and represent different physicochemical interaction types involved in stabilizing the L–DnaJ interface.
To evaluate the contribution of different interaction types at the L–DnaJ interface, selected residues were substituted with alanine using an alanine-scanning approach in order to remove their side-chain interactions while minimizing structural perturbation.
| Original Residue | Mutation | Reason |
|---|---|---|
| PRO6 | P6A | removes rigid hydrophobic contact |
| PHE22 | F22A | removes aromatic hydrophobic interaction |
| ARG4 | R4A | removes positive charge |
| ARG18 | R18A | removes strong electrostatic interaction |
| GLU2 | E2A | removes negative charge |
| ASP26 | D26A | removes negative charge |
The resulting N-terminal sequence of the lysis protein was used to re-predict the interaction with the DnaJ protein in order to evaluate whether the introduced mutations could reduce the dependence of the lysis protein on the host chaperone:
MATAFAQQSQQTPASTNARRPAKHEAYPCRRQQRSS
The mutated L-protein was co-folded again with DnaJ using AlphaFold2-Multimer. The five ranked models obtained were:
| Rank | pLDDT | pTM | ipTM |
|---|---|---|---|
| 3 | 78.7 | 0.579 | 0.291 |
| 4 | 78.4 | 0.574 | 0.235 |
| 5 | 77.1 | 0.569 | 0.233 |
| 2 | 79.1 | 0.581 | 0.219 |
| 1 | 79.4 | 0.568 | 0.206 |
When we compared the new models with the wild-type complex, we can see clearly that the ipTM values were slightly lower. In the wild-type prediction, the best model showed an ipTM value of 0.373, while after mutation the highest ipTM value decreased to 0.291. Since ipTM reflects the predicted strength of interaction between two proteins, this decrease suggests that the interaction between the L-protein and DnaJ became weaker after the mutations were introduced. This reduction is consistent with the mutation strategy, where several key residues involved in hydrophobic and electrostatic contacts were replaced with alanine in order to remove their side-chain interactions.
Despite these changes, the overall structural confidence of the models (pLDDT values were 78.9 to 78.6) remained similar to the wild-type predictions, indicating that the L-protein is still likely to fold correctly. Therefore, these results suggest that the designed mutations may reduce the dependence of the L-protein on the DnaJ chaperone while maintaining a stable protein structure. This computational approach demonstrates how targeted mutagenesis combined with AlphaFold2-Multimer predictions can be used to design L-protein variants with potentially lower chaperone dependency.
in this homework, AI ChatGPT assisted me in organizing and clearly articulating my answers and descriptions, ensuring that the content is well-structured and easy to understand.