Week 06 HW: genetic circuits part-I

Answer 01:
image_ref
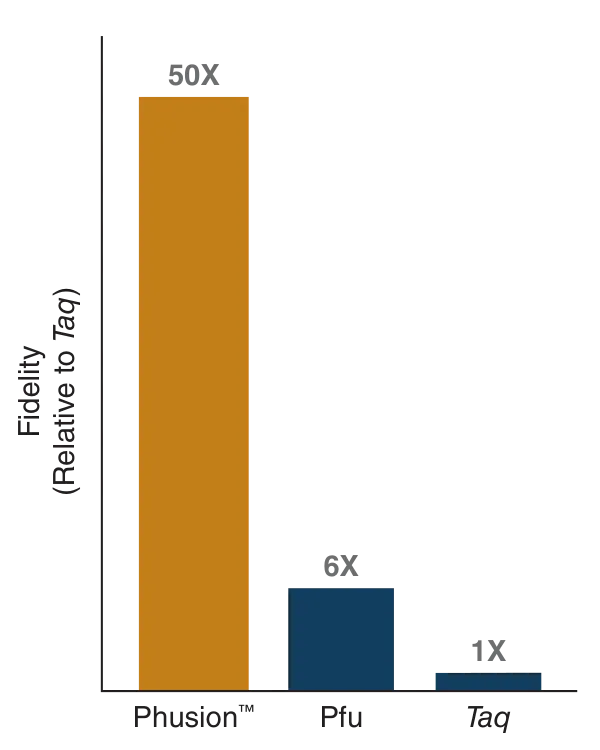
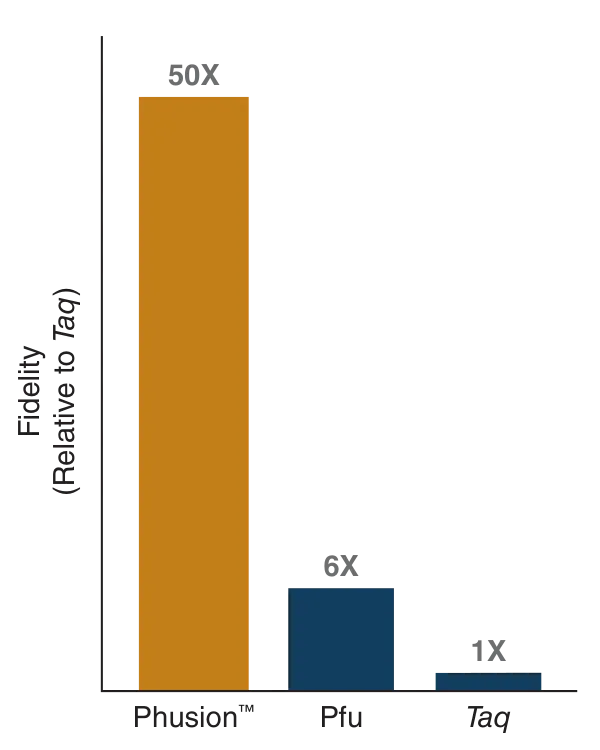
 The Phusion High-Fidelity PCR Master Mix is a ready-to-use solution used for PCR amplification with high accuracy. It already contains the main components needed for DNA amplification.
image_ref
The Phusion High-Fidelity PCR Master Mix is a ready-to-use solution used for PCR amplification with high accuracy. It already contains the main components needed for DNA amplification.
image_ref
{kind=link}
{kind=link}
- Phusion DNA Polymerase
Phusion DNA polymerase is the enzyme that copies the DNA during PCR. It synthesizes new DNA strands using the template DNA. This polymerase has proofreading activity, which helps detect and correct errors during DNA synthesis, making the amplification very accurate.
- Reaction Buffer
The reaction buffer provides the optimal chemical environment for the polymerase to function properly. It maintains the correct pH and salt conditions needed for efficient DNA amplification. Different buffers can be used depending on the DNA template, such as HF buffer for standard templates or GC buffer for GC-rich DNA.
- MgCl₂ (Magnesium Chloride)
Magnesium ions are essential cofactors for DNA polymerase activity. They help stabilize the interaction between the enzyme, the primers, and the DNA template during DNA synthesis.
- dNTPs (Deoxynucleotide Triphosphates)
dNTPs are the building blocks used to synthesize new DNA strands. They include dATP, dTTP, dCTP, and dGTP. During PCR, the polymerase adds these nucleotides to the growing DNA strand according to complementary base-pairing rules.
- Additional additives (e.g., DMSO)
Some reactions may include additives such as DMSO, which helps improve amplification of GC-rich DNA by reducing secondary structures and improving primer binding.
Answer 02:
The primer annealing temperature (Ta) is the temperature at which primers bind to the DNA template during PCR. It mainly depends on the primer melting temperature (Tm). In practice, the annealing temperature is usually set about 3–5 °C lower than the lowest primer Tm so that the primers can bind correctly to the DNA template.
Important factors (from the lab and lecture)
-Primer melting temperature (Tm):
image_ref
 The melting temperature is the temperature at which 50 % of the primer–DNA duplex separates into single strands. It is the main factor used to determine the annealing temperature.
The melting temperature is the temperature at which 50 % of the primer–DNA duplex separates into single strands. It is the main factor used to determine the annealing temperature.
-Primer length (18–22 nucleotides):
 image_ref
Primers are usually designed with a length of 18–22 bases. This length provides good specificity and stable binding to the template DNA.
image_ref
Primers are usually designed with a length of 18–22 bases. This length provides good specificity and stable binding to the template DNA.
-GC content (40–60 %)
Primers should contain about 40–60 % GC bases. GC pairs form stronger bonds than AT pairs, which increases the stability of primer binding.
-GC clamp (≤3 GC bases at the 3′ end)
image_ref
A small number of GC bases at the 3′ end of the primer (called a GC clamp) helps the primer bind more strongly to the template DNA and improves PCR efficiency.
-Primer secondary structures
image_ref
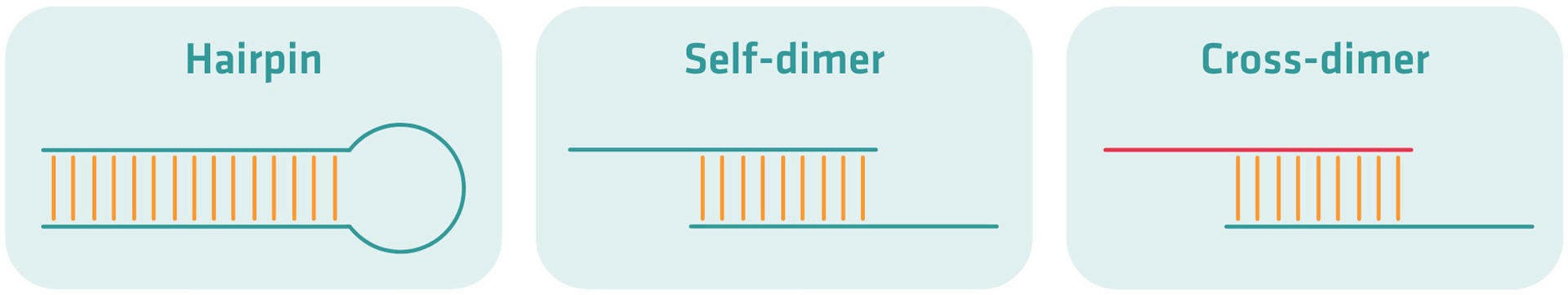 Primers should avoid forming hairpins, self-dimers, or cross-dimers. These structures prevent primers from binding properly to the template DNA.
Recommended primer Tm range (52–58 °C).
Primers should avoid forming hairpins, self-dimers, or cross-dimers. These structures prevent primers from binding properly to the template DNA.
Recommended primer Tm range (52–58 °C).
Primers are usually designed to have a melting temperature between 52 °C and 58 °C, which allows efficient and specific amplification.
-GC sequence composition
Primers with higher GC content bind more strongly because GC base pairs form three hydrogen bonds, while AT pairs form only two.
Additional factors
-Ionic environment (Mg²⁺ and salt concentration)
Ions such as Mg²⁺ and other salts stabilize the DNA double strand and influence primer binding. Changes in these concentrations can affect the optimal annealing temperature.
-Primer concentration
Higher primer concentrations increase the probability that primers will bind to the DNA template, which can influence the optimal annealing temperature.
-Optimization using gradient PCR
In many experiments, scientists perform gradient PCR to test different annealing temperatures and find the best one for efficient and specific amplification.
Answer 03:
PCR (Polymerase Chain Reaction) and restriction enzyme digestion are two common molecular biology methods used to produce linear DNA fragments, but they work in very different ways. PCR works by amplifying a specific DNA sequence, while restriction enzymes cut existing DNA at specific recognition sites. Both techniques are widely used in cloning and DNA assembly experiments, including methods such as Gibson Assembly.
image_ref
 PCR is a technique used to copy a specific region of DNA many times. It starts with a small amount of template DNA and uses specific primers that bind to the target sequence. A heat-stable DNA polymerase enzyme (such as Taq or Phusion polymerase) then synthesizes new DNA strands. The reaction takes place in a thermal cycler, which repeatedly changes the temperature through three main steps: denaturation, where the DNA strands separate; annealing, where primers bind to the template DNA; and extension, where the polymerase enzyme copies the DNA. After many cycles, PCR produces large amounts of a specific linear DNA fragment. One advantage of PCR is that researchers can design primers to add new sequences to the ends of the fragment, such as restriction sites or overlapping regions for Gibson Assembly.
image_ref
PCR is a technique used to copy a specific region of DNA many times. It starts with a small amount of template DNA and uses specific primers that bind to the target sequence. A heat-stable DNA polymerase enzyme (such as Taq or Phusion polymerase) then synthesizes new DNA strands. The reaction takes place in a thermal cycler, which repeatedly changes the temperature through three main steps: denaturation, where the DNA strands separate; annealing, where primers bind to the template DNA; and extension, where the polymerase enzyme copies the DNA. After many cycles, PCR produces large amounts of a specific linear DNA fragment. One advantage of PCR is that researchers can design primers to add new sequences to the ends of the fragment, such as restriction sites or overlapping regions for Gibson Assembly.
image_ref
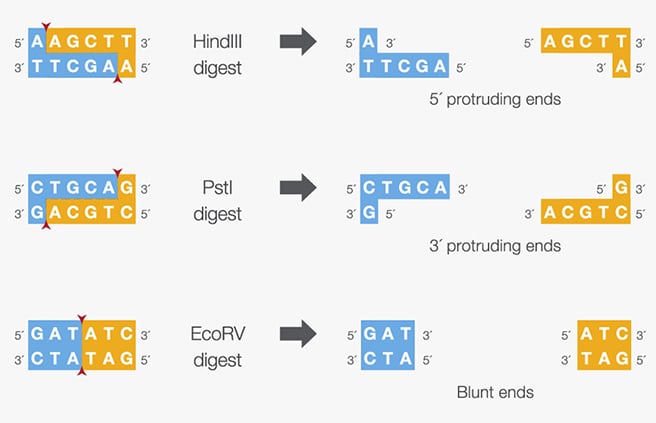 Restriction enzyme digestion works differently. Instead of amplifying DNA, it cuts existing DNA molecules at specific short sequences called recognition sites. Restriction enzymes recognize these sequences and cut the DNA at or near those locations. In a typical protocol, the DNA (for example a plasmid) is mixed with the restriction enzyme and a specific buffer, and the reaction is incubated at a constant temperature, usually around 37 °C, for about one hour. The enzyme then cuts the DNA to produce linear fragments. Depending on the enzyme, the cut DNA can produce sticky ends (overhangs) or blunt ends, which can be used for cloning.
Restriction enzyme digestion works differently. Instead of amplifying DNA, it cuts existing DNA molecules at specific short sequences called recognition sites. Restriction enzymes recognize these sequences and cut the DNA at or near those locations. In a typical protocol, the DNA (for example a plasmid) is mixed with the restriction enzyme and a specific buffer, and the reaction is incubated at a constant temperature, usually around 37 °C, for about one hour. The enzyme then cuts the DNA to produce linear fragments. Depending on the enzyme, the cut DNA can produce sticky ends (overhangs) or blunt ends, which can be used for cloning.
These two methods are used in different situations depending on the goal of the experiment. PCR is preferred when the DNA is present in low concentration, because it can amplify a very small amount of template into large quantities. PCR is also useful when researchers want to introduce new sequences, mutations, or overlaps into the DNA fragment. For example, primers can be designed to add restriction sites, promoter sequences, or homologous overlaps needed for Gibson Assembly. PCR is also commonly used when scientists want to isolate a specific gene or region from genomic DNA.
Restriction enzyme digestion is more suitable when the DNA is already available in large quantities, such as a purified plasmid. It is commonly used when researchers want to cut DNA at precise and known locations to isolate fragments or prepare a plasmid for cloning. Restriction enzymes are also often used for diagnostic analysis, such as verifying plasmid identity or checking the size of DNA fragments through restriction mapping.
Answer 04:
To ensure that DNA fragments produced by PCR or restriction digestion are suitable for Gibson Assembly, several preparation and verification steps must be followed. Gibson Assembly joins DNA fragments that contain overlapping homologous sequences, so the fragments must be designed carefully and purified before the assembly reaction.
The first and most important step is primer design. Primers used in PCR should include overlapping sequences of about 20–40 base pairs that match the ends of the neighboring DNA fragment. These overlaps allow the fragments to align and assemble correctly during the Gibson reaction. The overlapping regions should have similar melting temperatures (Tm) to allow stable annealing during the isothermal reaction. It is also important to design overlaps with a balanced GC content and to avoid strong secondary structures such as hairpins, because these structures can reduce assembly efficiency.
Another important step is using a high-fidelity DNA polymerase, such as Phusion or Q5 polymerase, during PCR amplification. These enzymes have proofreading activity and reduce the number of mutations introduced during amplification. This is important because Gibson Assembly is often used to construct precise DNA sequences or multi-fragment plasmids.
After PCR amplification, the DNA fragments should be verified using agarose gel electrophoresis to confirm that the fragments have the expected size. The correct DNA bands are then purified from the gel to remove primers, nucleotides, enzymes, and non-specific products that might interfere with the assembly reaction.
To reduce background contamination from the original template plasmid, PCR products can be treated with the restriction enzyme DpnI, which digests methylated template DNA but does not affect the newly synthesized PCR fragments.
If a plasmid backbone is used, the vector must be completely linearized before Gibson Assembly. This can be done by restriction enzyme digestion or PCR. When restriction enzymes are used, it is important to ensure that the digestion is complete so that no circular plasmid remains, because this could produce unwanted background colonies during transformation.
Another important step is DNA quantification. The concentration of each DNA fragment should be carefully measured using methods such as fluorometric quantification (for example Qubit) or gel analysis. The correct molar ratio of vector to insert fragments, often about 1:2 or 1:3, helps improve assembly efficiency.
Finally, after Gibson Assembly and bacterial transformation, the resulting plasmid constructs are usually verified by DNA sequencing to confirm that the fragments assembled correctly and that no mutations were introduced during PCR.
Answer 05:
Plasmid DNA enters Escherichia coli cells during a process called bacterial transformation. In this process, the bacterial cells must first be made competent, meaning their membranes become temporarily able to allow DNA molecules to enter.
image_ref
 In the most common method, called chemical transformation, the cells are treated with a solution containing calcium chloride (CaCl₂). The calcium ions (Ca²⁺) play an important role because they neutralize the negative charges on both the plasmid DNA and the phospholipids of the bacterial membrane. Normally, DNA and the membrane repel each other because they are both negatively charged. The calcium ions reduce this repulsion and allow the plasmid DNA to attach to the surface of the bacterial cell.
In the most common method, called chemical transformation, the cells are treated with a solution containing calcium chloride (CaCl₂). The calcium ions (Ca²⁺) play an important role because they neutralize the negative charges on both the plasmid DNA and the phospholipids of the bacterial membrane. Normally, DNA and the membrane repel each other because they are both negatively charged. The calcium ions reduce this repulsion and allow the plasmid DNA to attach to the surface of the bacterial cell.
After mixing the plasmid DNA with the competent cells, the mixture is kept on ice (around 0 °C) for a short time. The cells are then exposed to a brief heat shock, usually at about 42 °C for 30–60 seconds. This sudden temperature change creates a strong thermal gradient between the cold cells and the warm environment. As a result, the bacterial membrane becomes temporarily destabilized and small pores form, allowing the plasmid DNA to pass into the cell.
Immediately after the heat shock, the cells are placed back on ice. This rapid cooling helps close the pores and stabilize the membrane again. The cells are then transferred into a nutrient recovery medium and incubated for a short period. During this recovery step, the cells repair their membranes and begin expressing the antibiotic resistance gene carried by the plasmid.
Finally, the bacteria are plated on agar plates containing the appropriate antibiotic. Only the cells that successfully received the plasmid DNA will survive and form colonies.
Another alternative method used to introduce plasmid DNA into E. coli is electroporation. In this method, competent bacterial cells are mixed with plasmid DNA and placed in a special electroporation cuvette. A short electrical pulse is then applied using an electroporator. The electrical pulse temporarily creates small pores in the bacterial cell membrane, allowing the plasmid DNA to pass directly into the cell.
After the pulse, the membrane quickly reseals and the cells recover in a nutrient medium. Electroporation is often more efficient than chemical transformation and is commonly used when transforming difficult DNA constructs or when very high transformation efficiency is required.
Answer 06:
Another DNA assembly method is Golden Gate Assembly, which allows several DNA fragments to be joined together in a single reaction. This technique uses special restriction enzymes called Type IIS restriction enzymes, such as BsaI or BsmBI, together with T4 DNA Ligase. Unlike traditional restriction enzymes, Type IIS enzymes cut outside their recognition sequence, which allows scientists to design custom 4-base pair overhangs at the ends of DNA fragments. These overhangs are designed so that fragments can only join with the correct neighboring fragment, ensuring the correct order and orientation of the assembled DNA. During the reaction, the restriction enzyme cuts the DNA fragments and creates the overhangs, and the DNA ligase joins the fragments together. The recognition sites of the restriction enzyme are removed during assembly, which means the final DNA construct cannot be cut again by the same enzyme. The digestion and ligation steps occur in the same tube using alternating temperatures, making Golden Gate Assembly a very efficient method for assembling multiple DNA fragments, especially in synthetic biology and modular cloning experiments.
image_ref
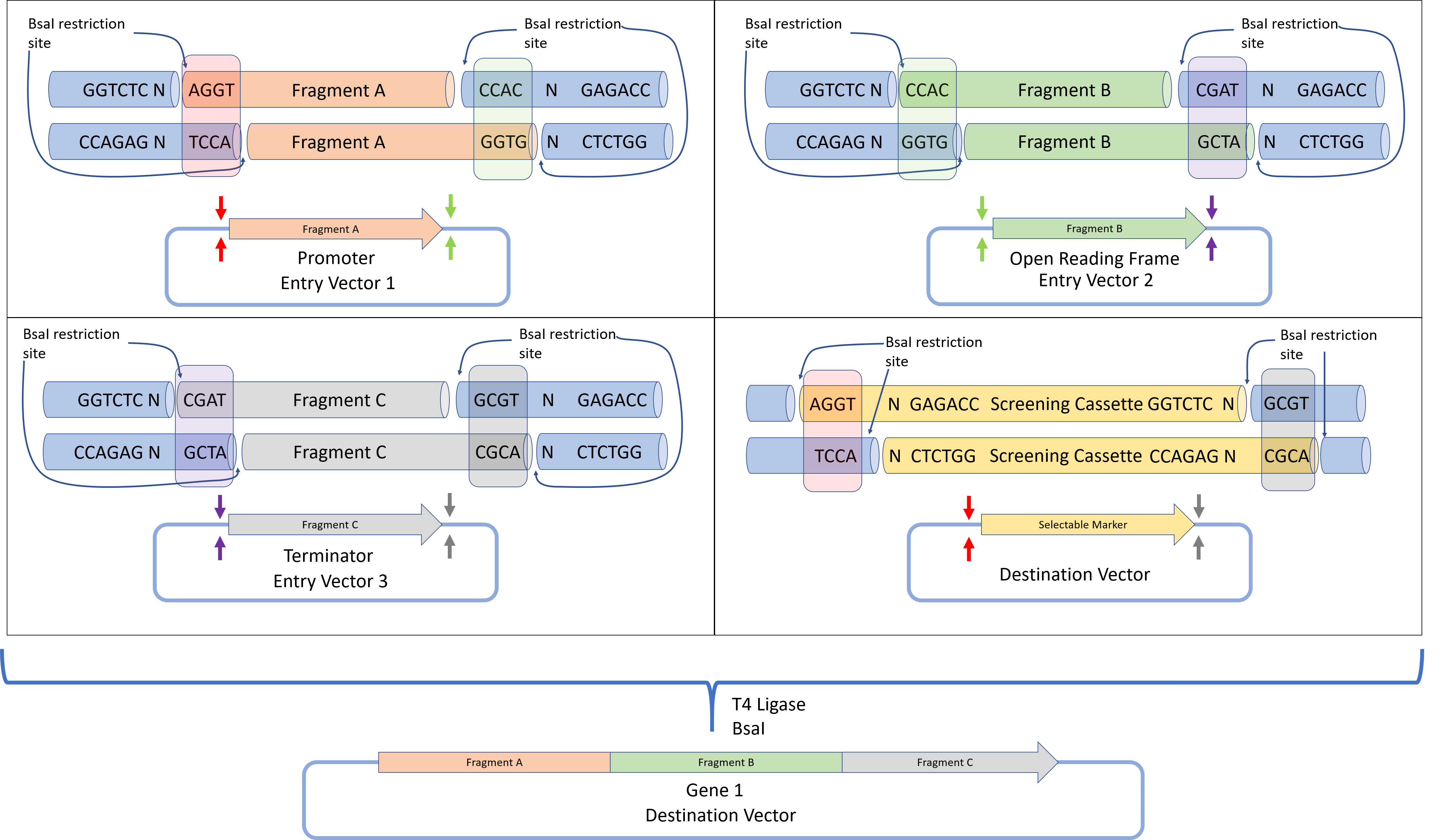
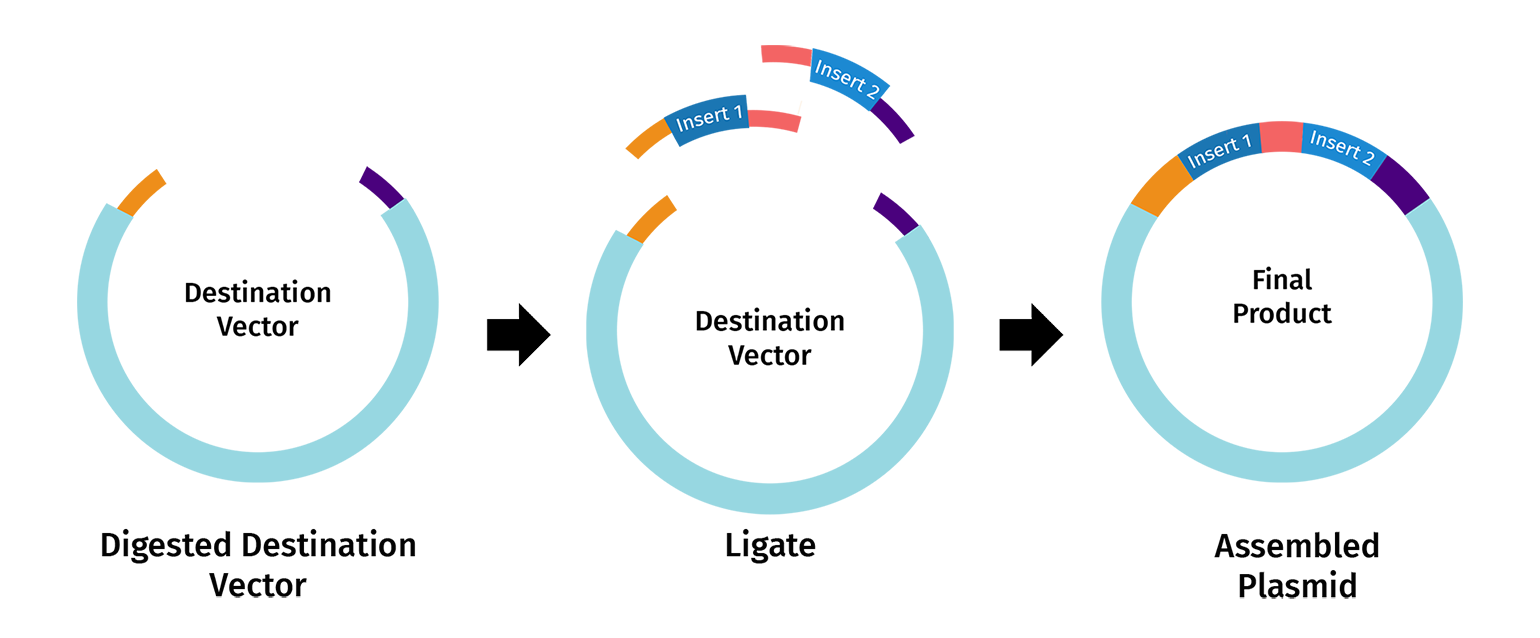
This diagram is a clear example of Golden Gate Assembly, a cloning method that joins several DNA fragments in one reaction. In the example, three DNA parts — Promoter (Fragment A), ORF (Fragment B), and Terminator (Fragment C) — are assembled into a final plasmid called the destination vector. The process uses the Type IIS restriction enzyme BsaI together with T4 DNA Ligase.
In the first step, each fragment is present in an entry vector that contains the BsaI recognition site (GGTCTC). Unlike classical restriction enzymes, BsaI cuts outside of its recognition site, generating specific 4-base pair sticky ends (overhangs). Because the cut occurs outside the recognition sequence, the recognition site is removed during assembly and does not remain in the final DNA construct.
The fragments are designed with specific overhangs so they connect in the correct order. For example, Fragment A ends with the overhang CCAC, which matches the beginning of Fragment B. Fragment B ends with CGAT, which matches the start of Fragment C. These complementary overhangs act like puzzle pieces, ensuring that the fragments assemble correctly and in the proper orientation.
All fragments, the destination vector, BsaI, and T4 DNA Ligase are mixed in a single tube. During the reaction, BsaI cuts the DNA fragments to create sticky ends, and T4 DNA ligase joins fragments with matching overhangs. The reaction cycles between temperatures that allow DNA digestion and ligation, gradually assembling the correct construct.
Once fragments are ligated together, the BsaI recognition sites are no longer present, so the final product cannot be cut again by the enzyme. This makes the process efficient and irreversible, allowing the formation of a seamless DNA construct containing Fragment A + Fragment B + Fragment C in the destination plasmid.
image_ref
- Modeling Golden Gate Assembly in Benchling
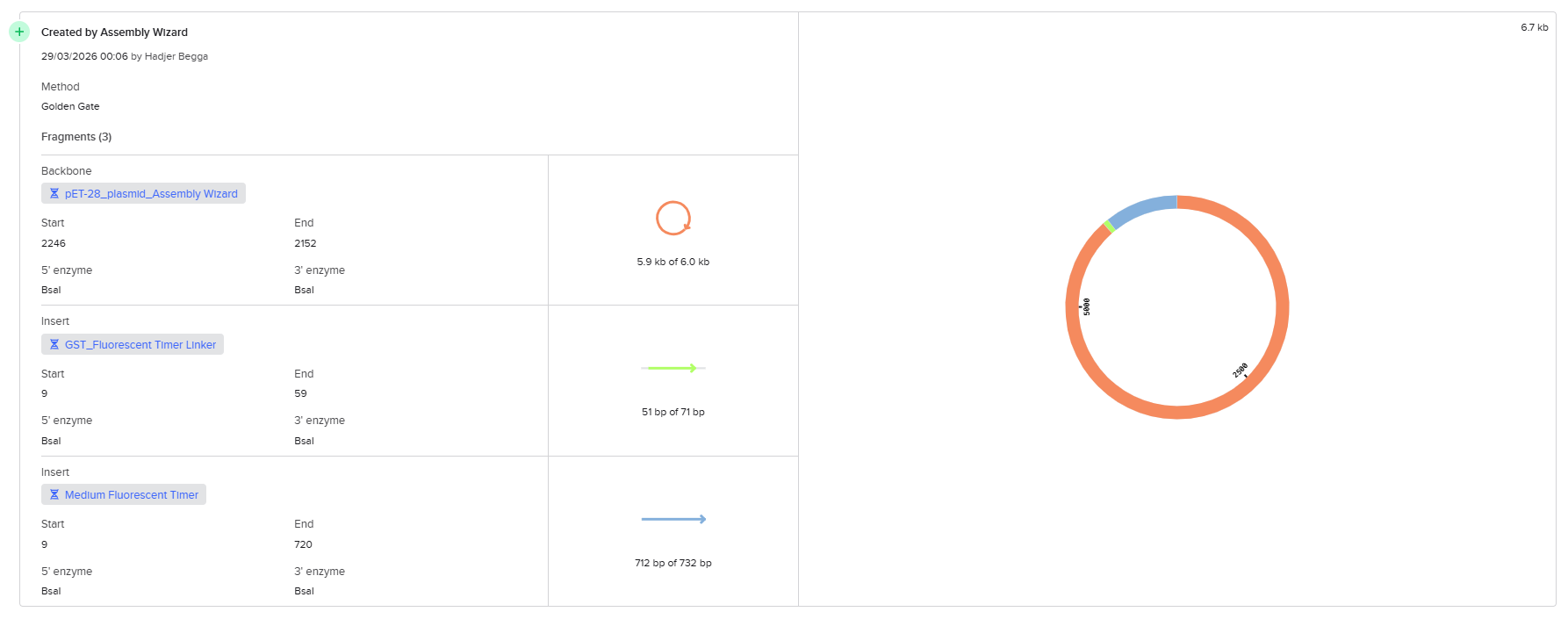
In this part, I modeled a Golden Gate Assembly to construct a genetic circuit for my second project, which is the engineering of an Escherichia coli reporter strain to monitor protein aging using a fluorescent timer protein.
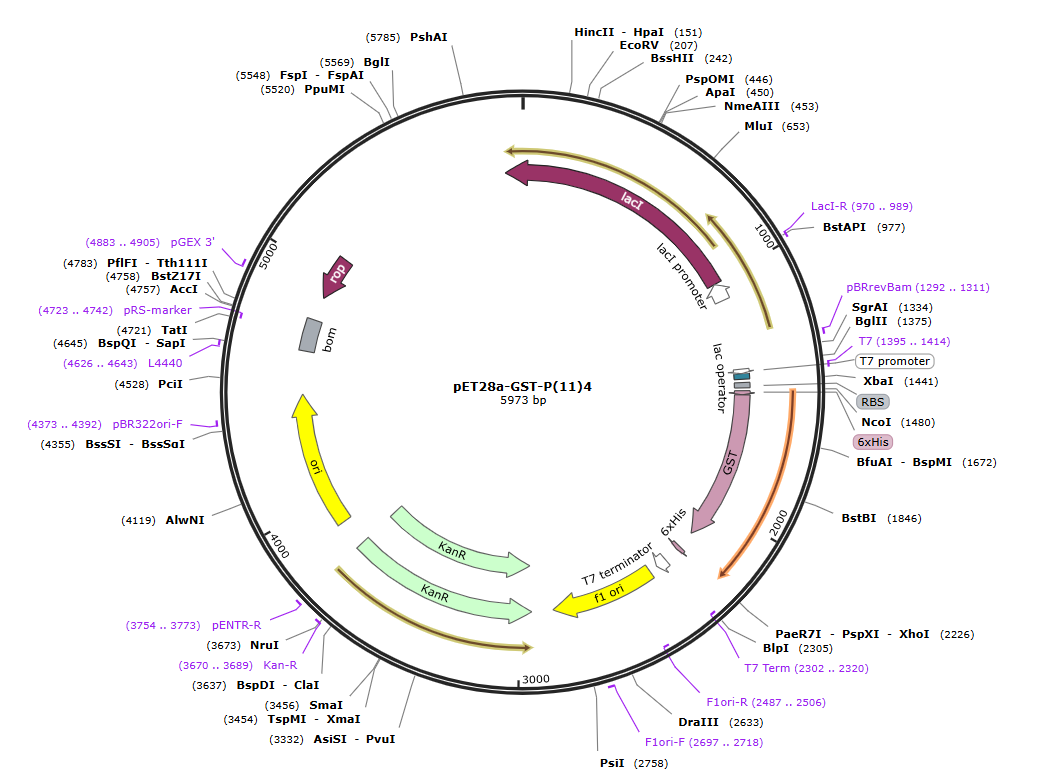
First, I selected all the genetic elements needed for my construct. The backbone plasmid was obtained from Addgene, and it already contains a T7 promoter, a ribosome binding site (RBS), and a T7 terminator, which are very suitable for strong expression of the inserted gene. This vector also includes the GST (Glutathione S-Transferase from Schistosoma japonicum), which I used as the protein of interest because it has stable folding and is suitable for initial testing of my genetic system.
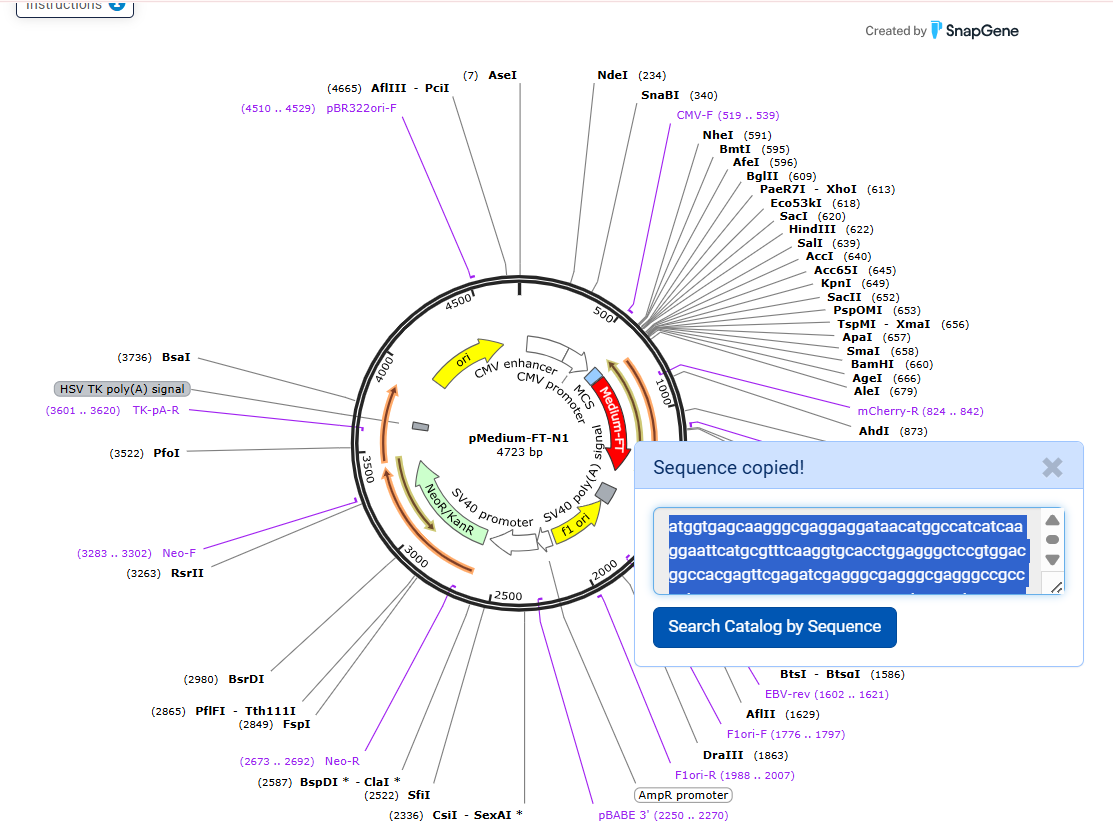
Then, I designed two additional fragments: a flexible linker (Gly₄Ser)₃ and a fluorescent timer (FT) protein (Medium FT). Their sequences were also obtained from Addgene. The linker allows proper folding between the GST protein and the fluorescent timer, while the FT protein provides a signal that changes over time, allowing estimation of protein age inside the cell.
- The full sequence of the pET28a-GST-P(11)4:
- Linker sequence (Gly₄Ser)₃ :
- Medium-FT sequence:
At the beginning, I manually designed the overhangs based on the coding sequence. I assumed that the last four nucleotides of the GST sequence (GAAG) would serve as the correct overhang to connect with the next fragment. Based on this assumption, I designed the linker fragment to have a compatible overhang (GAAG, GGTA). Similarly, I defined the overhangs between the linker and the fluorescent timer protein (GGTA) in order to maintain a continuous reading frame. During this step, I also verified that no frameshift was introduced at the junctions and that the coding sequence remained in frame across all fragments as indicated in the following table:
| Junction | DNA Sequence | Resulting Amino Acids | Status |
|---|---|---|---|
| GST to Linker | ...AAG GGT... | Lys - Gly | In Frame |
| Linker to FT | ...TCT CCG GTA ATG... | Ser - Pro - Val - Met | In Frame |
| FT to 6xHis | ...AAG AAG CAC... | Lys - Lys - His | In Frame |
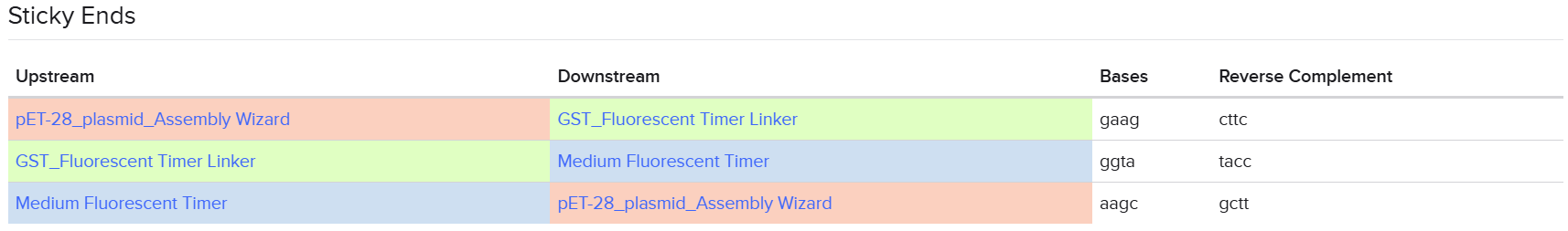
In addition, I checked that all BsaI restriction enzyme recognition sites were positioned outside of the fragments that would be recovered after digestion, ensuring that the internal sequences of the inserts would not be disrupted during the assembly process. the designed overhangs are as the following:

The designed overhangs are supposed to orient the assembly in the following order: the linker is placed immediately after the GST sequence, and the Medium FT is positioned just before the C-terminal His tag, as indicated in the following diagram:
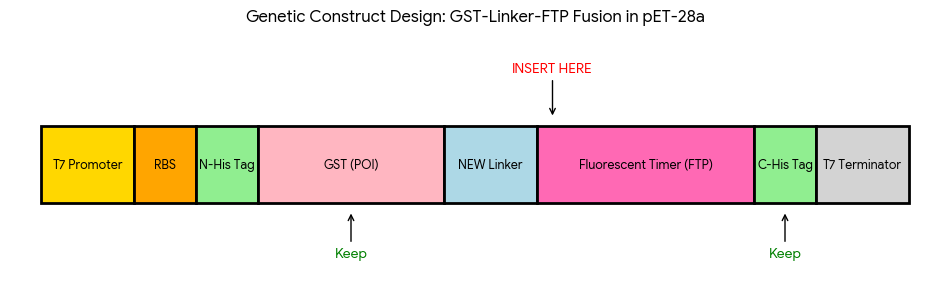
After preparing the vector and all fragments, the designed vector digestion cuts were defined as follows:
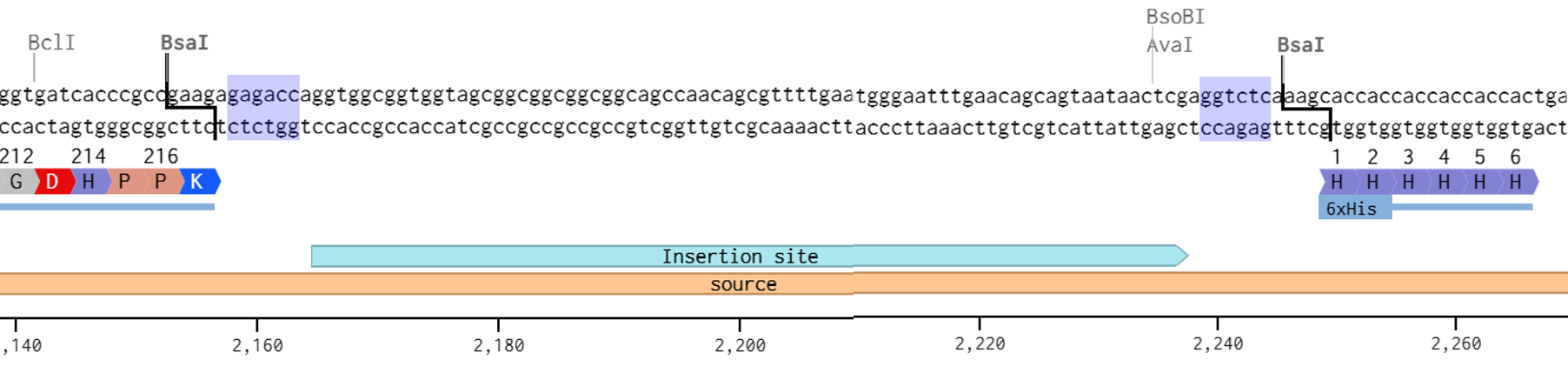 The designed linker fragment sticky ends were defined as follows:
The designed linker fragment sticky ends were defined as follows:
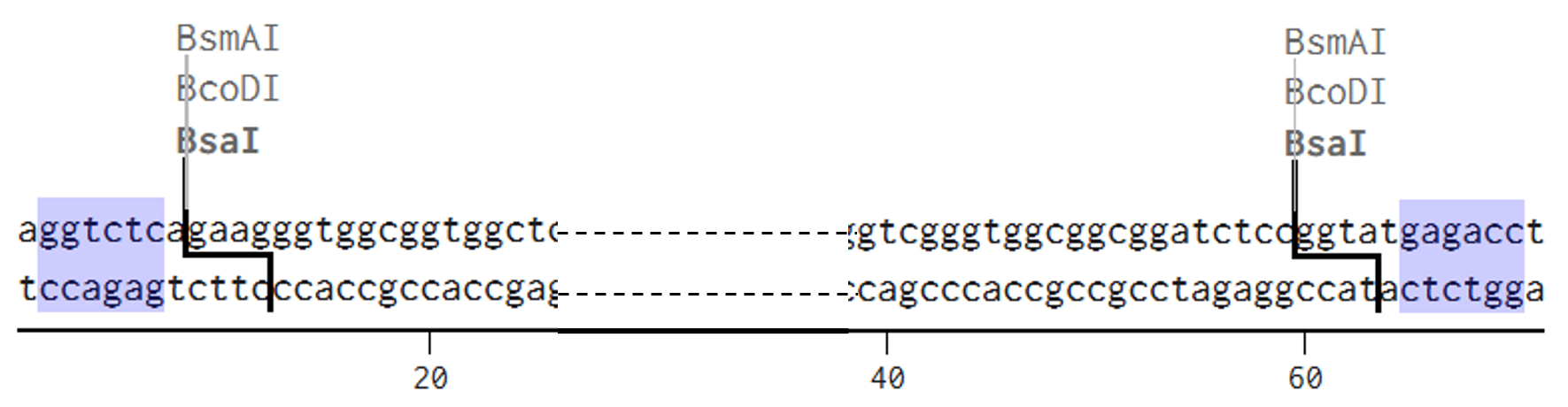 The designed Medium FT sticky ends were defined as follows:
The designed Medium FT sticky ends were defined as follows:
Be careful !! A critical point to consider during the design is the correct placement of BsaI restriction enzyme recognition sites. For the inserted fragments, the BsaI sites must be located outside of the sequences of interest so that they are removed during digestion and do not remain in the final construct. In contrast, for the backbone vector, the BsaI sites must be positioned within the region to be replaced, so that digestion removes this segment and allows the insertion of the designed fragments.
It is also essential to ensure that the BsaI recognition sites are oriented correctly (inverted orientation) to generate the desired overhangs and to cut the backbone precisely at the intended insertion site. Any incorrect placement or orientation of these sites can lead to incompatible sticky ends and result in assembly failure.
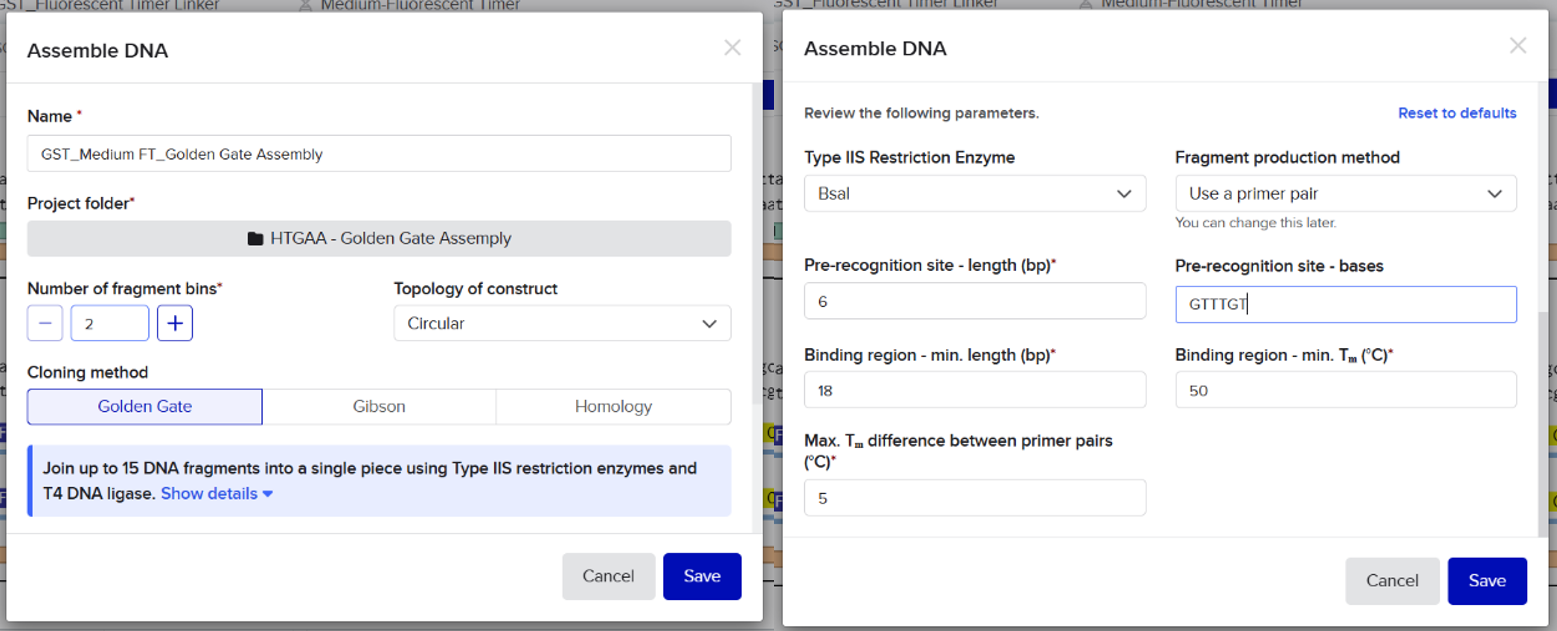
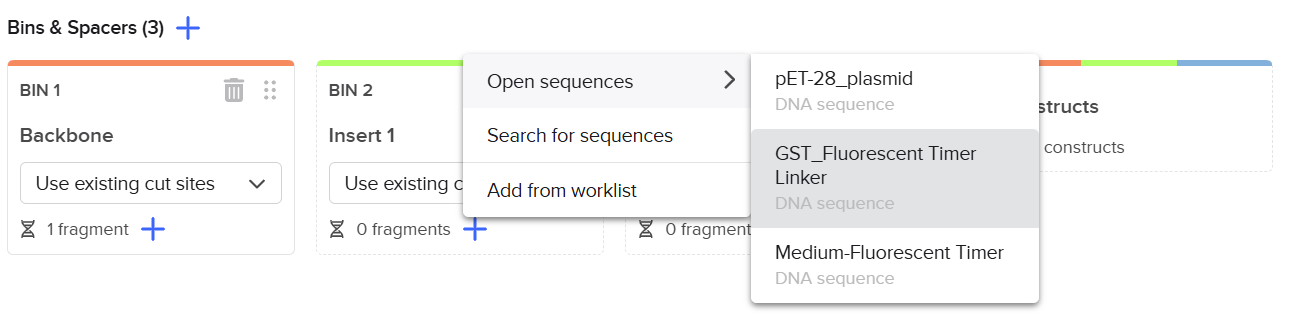
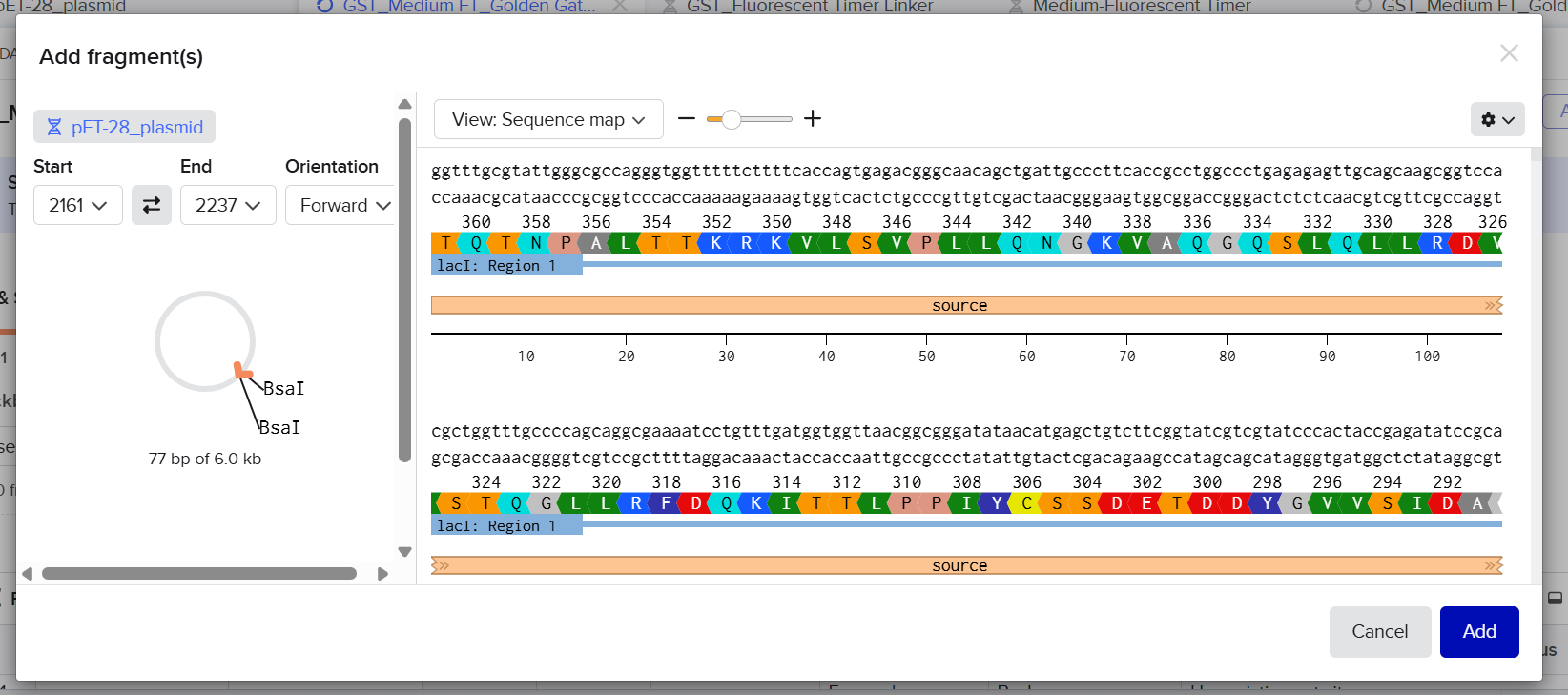
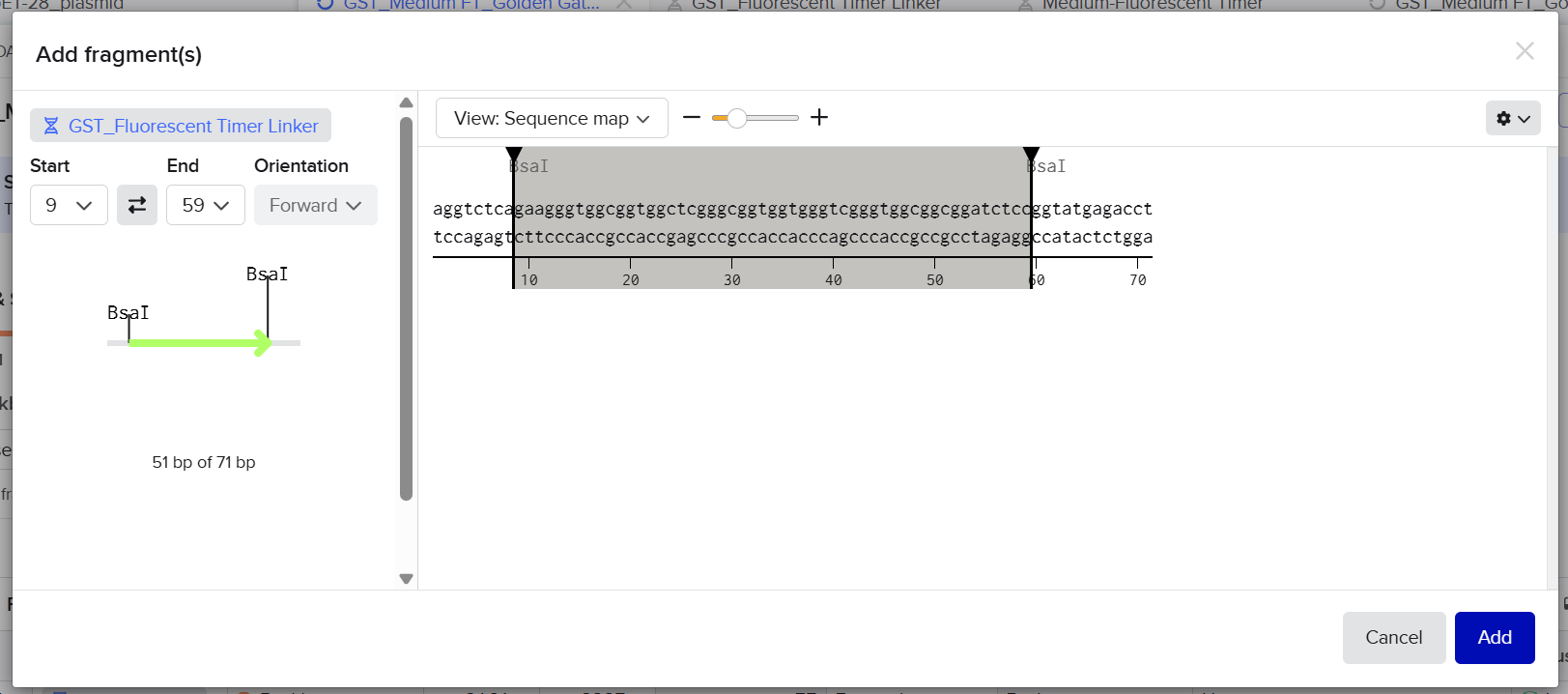
I imported all sequences into Benchling and created a new assembly using the Golden Gate cloning option. I selected the pET-28 plasmid as the backbone and added the designed fragments, including the linker and the fluorescent timer protein, as inserts. I specified the use of the BsaI restriction enzyme and defined the final construct as circular. Since all sequences were already designed with appropriate BsaI recognition sites, I selected the option to use existing restriction sites for fragment generation. I then attempted to run the assembly.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
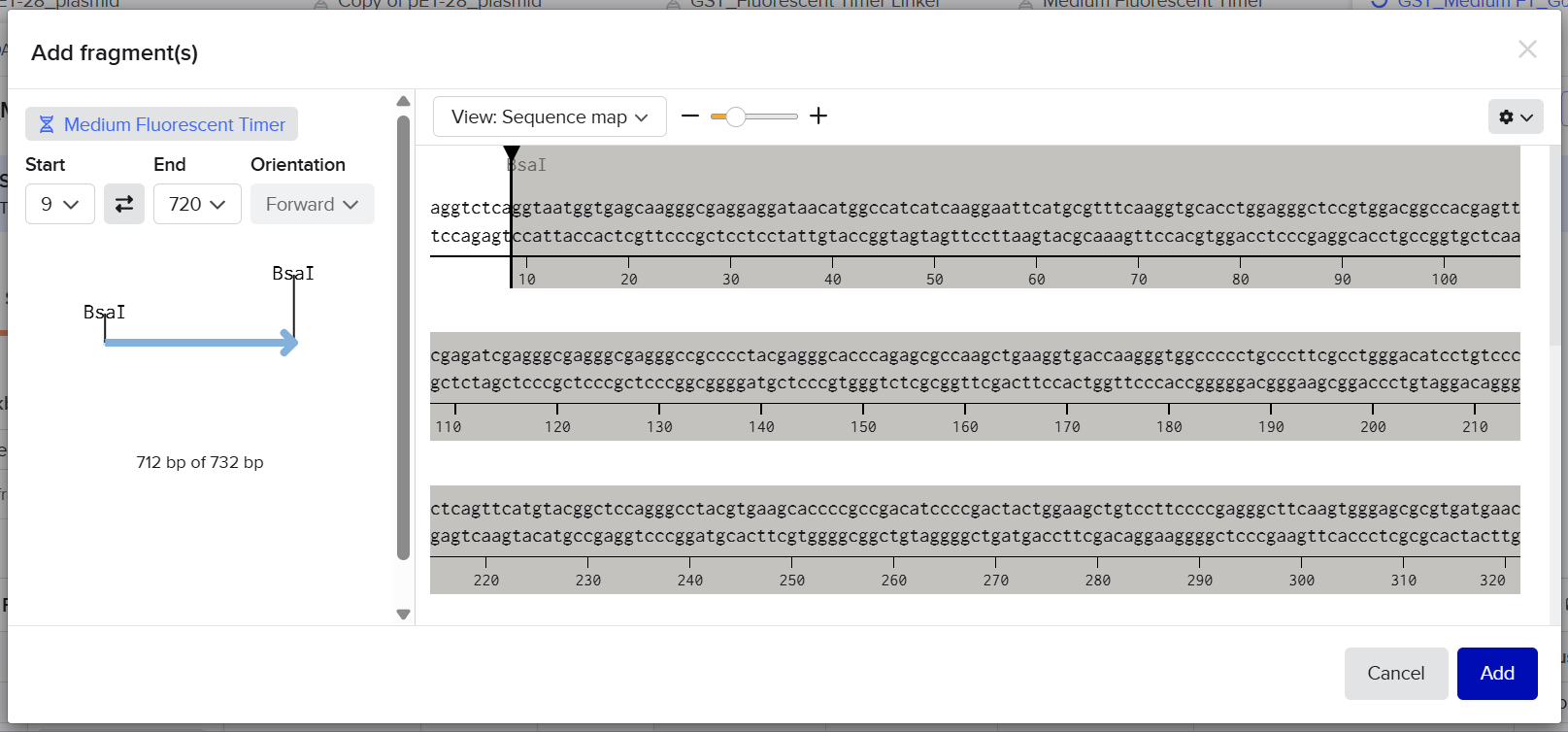
However, the assembly failed, and Benchling returned an error indicating that the sticky ends were incompatible. Specifically, the system showed a mismatch between the overhangs “AAGC” (from the vector) and “GAAG” (from the insert). This result indicated that the fragments could not ligate properly.
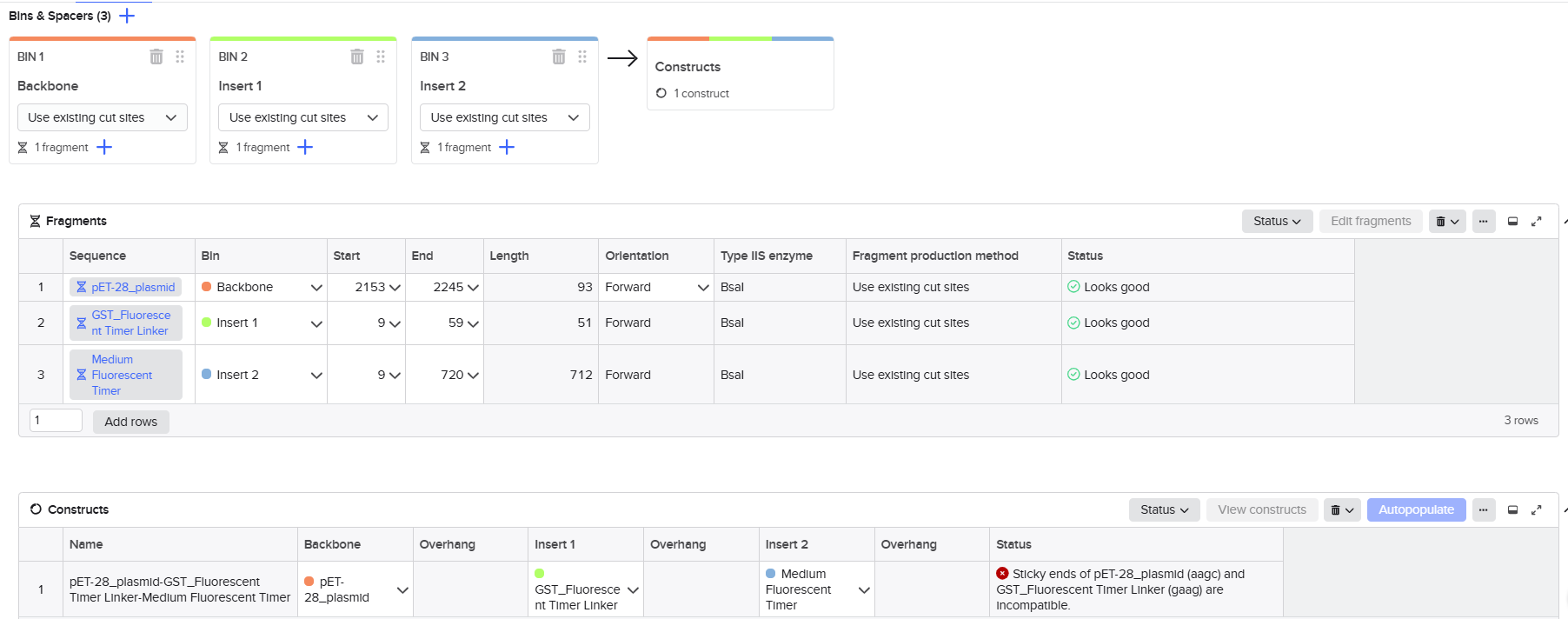
After analyzing this issue, I realized that the mistake came from misunderstanding how Golden Gate Assembly works. I initially assumed that the overhang corresponds directly to visible nucleotides in the sequence. In reality, the overhang is determined by the position of the BsaI cutting site, not simply by the sequence at the end of the gene. Since BsaI cuts outside of its recognition site, the actual generated overhang in the vector was “AAGC” and not “GAAG” as I had expected.
This mismatch between expected and real overhangs caused the failure of the assembly. Additionally, the cloning workflow in Benchling does not automatically correct or reinterpret overhangs; it strictly checks for compatibility. Therefore, any small design error leads to a complete assembly failure.
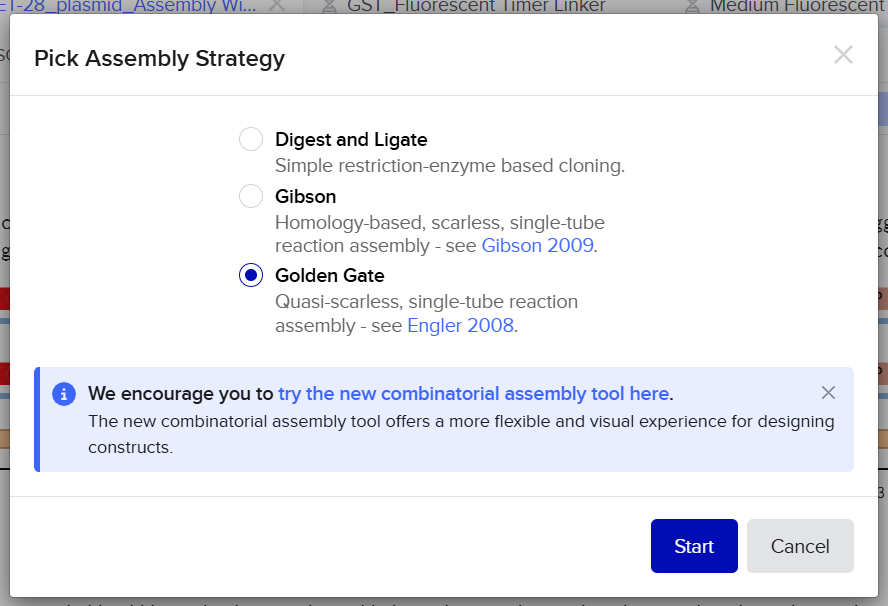
In order to overcome the limitations encountered in the first approach, I tried another method available in Benchling by using the Assembly tool dedicated to multi-fragment cloning. This method is specifically designed to simulate Golden Gate Assembly in a more automated and flexible way, allowing better handling of fragment compatibility and overhang generation.
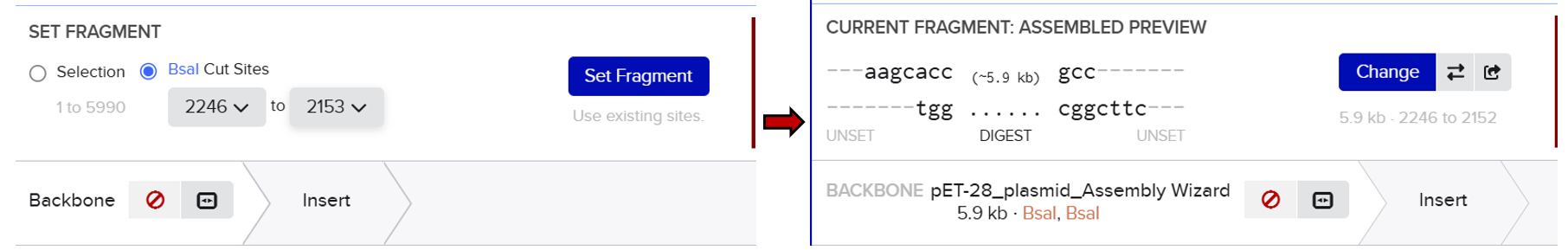
First, I opened the Assembly tool from the bottom toolbar and created a new assembly. I then added all the required DNA sequences, including the pET-28 plasmid as the backbone and the designed fragments (linker and Medium FT) as inserts. After that, I selected the BsaI restriction enzyme as the Type IIS restriction enzyme used for the assembly.

![]()
![]()
![]()
![]()
![]()
![]()
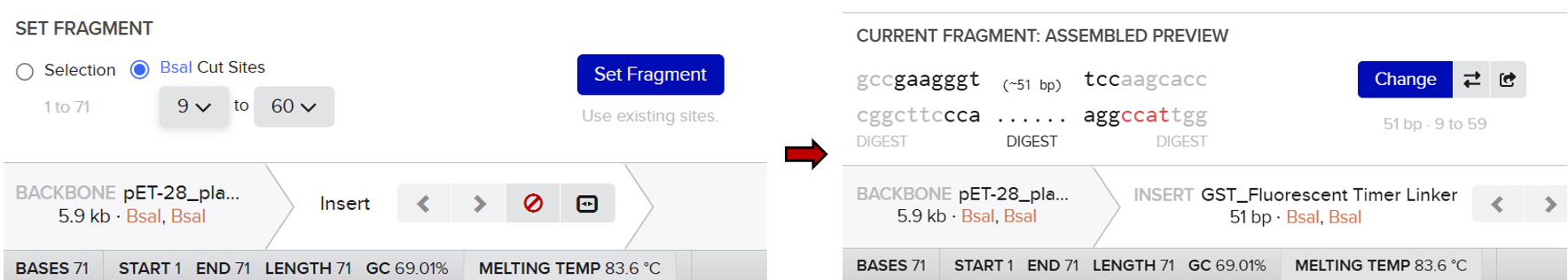
![]()
![]()
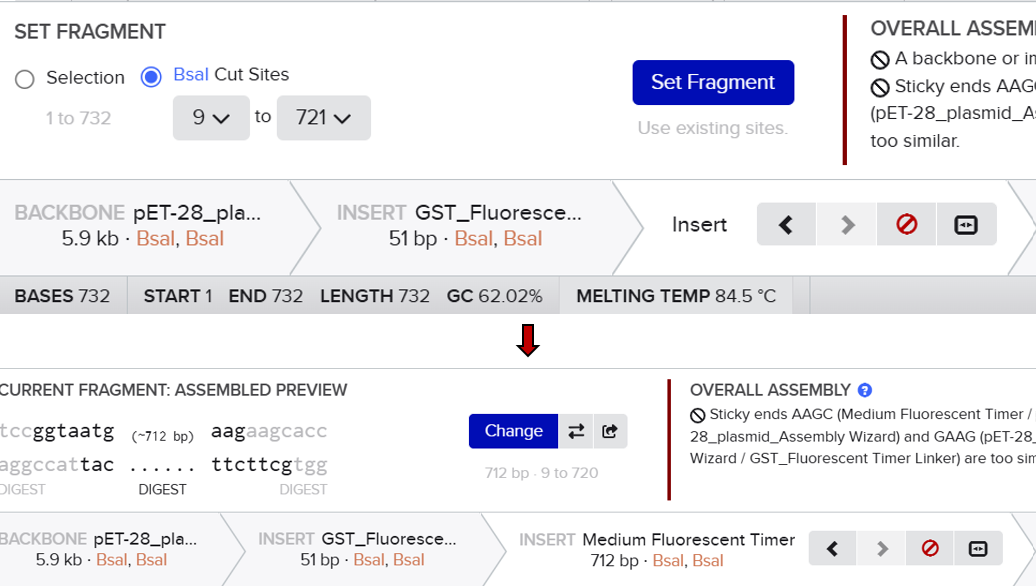
Unlike the previous method, this approach automatically analyzed the positions of the BsaI recognition sites and simulated the digestion process. It generated the correct sticky ends based on the actual cutting positions of the enzyme and evaluated the compatibility between fragments. This allowed the system to correctly align and assemble the different parts according to their matching overhangs.
After running the assembly, the construct was successfully generated as a circular plasmid. I carefully verified that all fragments were assembled in the correct order and orientation. I also confirmed that no frameshift was introduced across the junctions and that the reading frame was maintained from the GST sequence through the linker and into the fluorescent timer protein. In addition, I checked that no unwanted BsaI sites remained inside the final construct and that all restriction sites had been properly removed during the assembly process.
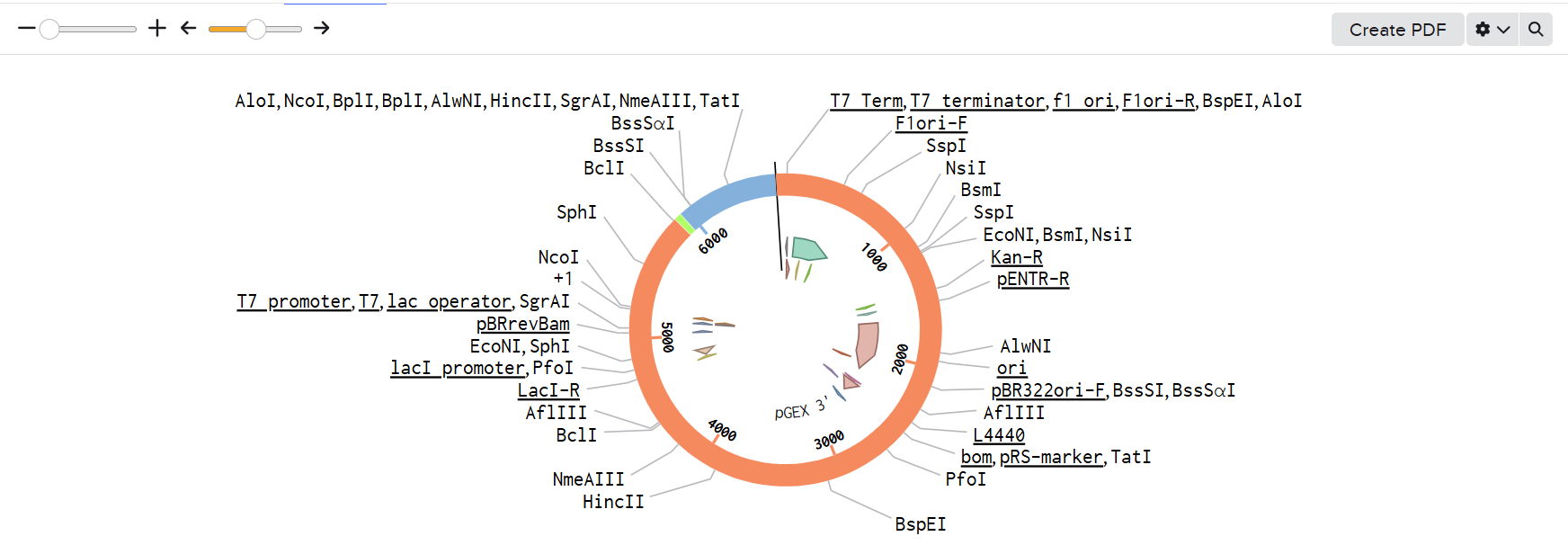
![]()
![]()
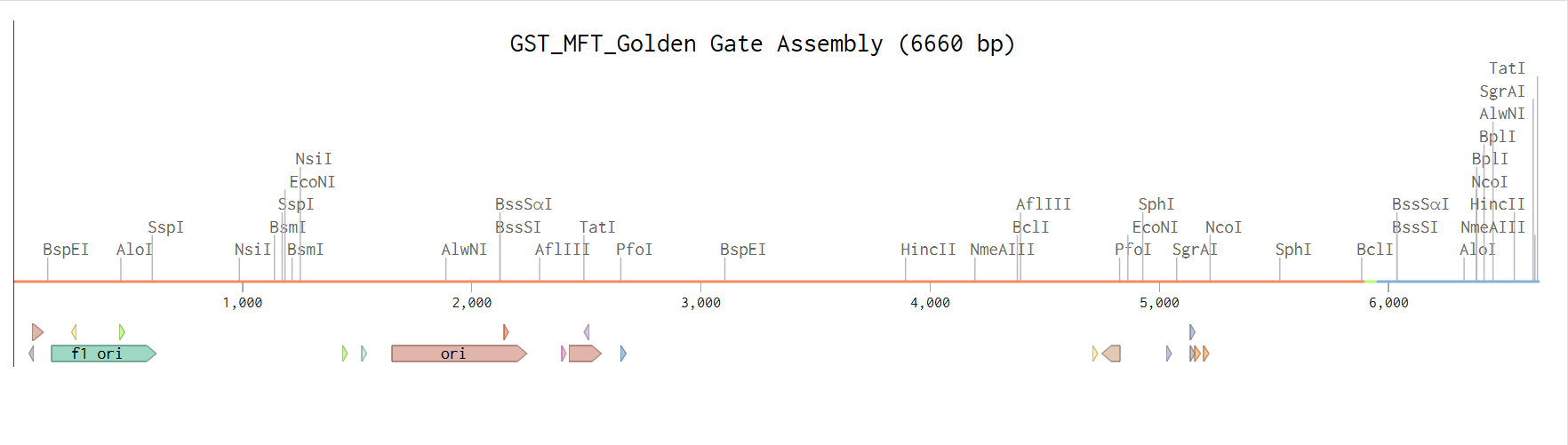
![]()
![]()
![]()
![]()
this is the direct link to benchling for this assembly: using the assembly tool
in this homework, AI ChatGPT assisted me in organizing and clearly articulating my answers and descriptions, ensuring that the content is well-structured and easy to understand.
Sources:
- Activity 3-2—Primer Design and Barcoding via Bioinformatics. (2025, March 8). Biology LibreTexts. https://bio.libretexts.org/Courses/Irvine_Valley_College/Lab_manual%3A_Molecular_biology_and_Genetic_Engineering_(Biot_275)/11%3A_Activity_3-2_-_Primer_Design_and_Sequence_Verification_Using_Bioinformatics
- Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2002). Isolating, Cloning, and Sequencing DNA. In Molecular Biology of the Cell. 4th edition. Garland Science. https://www.ncbi.nlm.nih.gov/books/NBK26837/
- Biocat.com/bc/files/Gibson_Guide_V2_101417_web_version_8.5_x_11_FINAL.pdf. (n.d.). Retrieved March 30, 2026, from https://www.biocat.com/bc/files/Gibson_Guide_V2_101417_web_version_8.5_x_11_FINAL.pdf
- Bird, J. E., Marles-Wright, J., & Giachino, A. (2022). A User’s Guide to Golden Gate Cloning Methods and Standards. ACS Synthetic Biology, 11(11), 3551–3563. https://doi.org/10.1021/acssynbio.2c00355
- Competent Cells: Principle, Methods & Functions Explained. (n.d.). VEDANTU. Retrieved March 30, 2026, from https://www.vedantu.com/biology/competent-cells
- DNA Quantification with a Plate Reader | BMG LABTECH. (n.d.). Retrieved March 30, 2026, from https://www.bmglabtech.com/en/blog/dna-quantification/
- Erjavec, M. S. (2019). Annealing Temperature of 55°C and Specificity of Primer Binding in PCR Reactions. In Synthetic Biology—New Interdisciplinary Science. IntechOpen. https://doi.org/10.5772/intechopen.85164
- Froger, A., & Hall, J. E. (2007). Transformation of Plasmid DNA into E. coli Using the Heat Shock Method. Journal of Visualized Experiments : JoVE, (6), 253. https://doi.org/10.3791/253
- Golden Gate Assembly—Snapgene. (n.d.). Retrieved March 30, 2026, from https://www.snapgene.com/guides/golden-gate-assembly
- Hoseini, S. S., & Sauer, M. G. (2015). Molecular cloning using polymerase chain reaction, an educational guide for cellular engineering. Journal of Biological Engineering, 9, 2. https://doi.org/10.1186/1754-1611-9-2
- How are competent bacterial cells transformed with a plasmid? (n.d.). Retrieved March 30, 2026, from https://worldwide.promega.com/resources/pubhub/enotes/how-are-competent-bacterial-cells-transformed-with-a-plasmid/
- Jacob Elmer. (2019, August 29). Chemical Transformation of E. coli [Video recording]. https://www.youtube.com/watch?v=xcc2ywDASag Med.unc.edu/pharm/sondeklab/wp-content/uploads/sites/868/2019/10/gibson-cloning.pdf. (n.d.). Retrieved March 30, 2026, from https://www.med.unc.edu/pharm/sondeklab/wp-content/uploads/sites/868/2019/10/gibson-cloning.pdf
- Molecular Cloning Methods – Benchling. (n.d.). Retrieved March 30, 2026, from https://help.benchling.com/hc/en-us/articles/9684255457805-Molecular-Cloning-Methods
- Molecular cloning using Gibson assembly. (n.d.). EPFL. Retrieved March 30, 2026, from https://www.epfl.ch/labs/lpbs/internal/general/molecular-cloning-using-gibson-assembly/
- PCR conditions | Primer annealing specificity | PCR buffers. (n.d.). Retrieved March 30, 2026, from https://www.qiagen.com/us/knowledge-and-support/knowledge-hub/bench-guide/pcr/introduction/pcr-conditions
- PCR Using Phusion® High-Fidelity PCR Master Mix with HF Buffer (NEB #M0531) | NEB. (n.d.). Retrieved March 30, 2026, from https://www.neb.com/en/protocols/protocol-phusion-high-fidelity-pcr-master-mix-with-hf-buffer-m0531
- (PDF) Study of Transformation in Escherichia coli. (n.d.). ResearchGate. https://doi.org/10.13140/RG.2.2.22413.45285 Polymerase Chain Reaction—An overview | ScienceDirect Topics. (n.d.). Retrieved March 30, 2026, from https://www.sciencedirect.com/topics/food-science/polymerase-chain-reaction
- Protocol for PhusionTM High-Fidelity PCR Master Mix with GC Buffer | NEB. (n.d.). Retrieved March 30, 2026, from https://www.neb.com/en/protocols/protocol-for-phusion-high-fidelity-pcr-master-mix-with-gc-buffer-m0532
- Restriction Enzyme Digestion | NEB. (n.d.). Retrieved March 30, 2026, from https://www.neb.com/en/applications/cloning-and-synthetic-biology/dna-preparation/restriction-enzyme-digestion
- SnapGene. (2022, June 30). Introduction à l’assemblée de Golden Gate [Video recording]. https://www.youtube.com/watch?v=aBcqev1NMMo Sorida, M., & Bonasio, R. (2023). An efficient cloning method to expand vector and restriction site compatibility of Golden Gate Assembly. Cell Reports Methods, 3(8), 100564. https://doi.org/10.1016/j.crmeth.2023.100564
- Staff, L. in the L. (2025, May 29). Gibson Assembly 101: Expert Cloning Tips You Need to Know. Life in the Lab. https://www.thermofisher.com/blog/life-in-the-lab/gibson-assembly-101-expert-cloning-tips-you-need-to-know/
- The Different Types of PCR Methods | Pipette.com. (n.d.). Retrieved March 30, 2026, from https://pipette.com/blog/types-of-pcr Universal Annealing Temperature in PCR and its Impact on Amplification Results. (n.d.).