Projects
Final projects:
- My Individual Final Project Documention



This document presents the complete final project report, including the design strategy, construct engineering workflow, structural analyses, and in silico validation steps. For a more interactive and visually detailed presentation including animated rotating views of the predicted protein structures, enhanced figures, additional simulations, and direct access to all Benchling design files and cloning maps, please refer to the project documentation webpages associated with this final project.
These resources provide a more comprehensive visualization of the project beyond the static figures included in this PDF document.
Project Title: Engineering Houseplants for Atmospheric Carbon Monoxide Capture: Chloroplast-Targeted Expression of the Bacterial CODH Enzyme Complex in Nicotiana tabacum

The Problem This Project Addresses
Carbon monoxide (CO) is a colorless, odorless, tasteless toxic gas that cannot be detected by human senses. It is produced whenever something burns incompletely — gas heaters, stoves, car engines, fireplaces, and wood-burning appliances all release CO. Indoors, CO accumulates silently and can reach dangerous or fatal concentrations before anyone notices. The current standard of protection is a battery-powered electrochemical CO detector. These devices are excellent at detecting CO and sounding an alarm , but they cannot remove the gas from the air. Once the alarm sounds, the occupants must evacuate and ventilate the space manually. Furthermore, CO detectors require regular battery replacement and eventually need to be replaced entirely. In low-income households worldwide, detectors are frequently absent, have dead batteries, or are past their useful lifespan.
–> This project proposes a fundamentally different approach: instead of detecting CO, make the plant remove it.
The Core Idea
Certain bacteria ,particularly Oligotropha carboxidovorans, have evolved the ability to use CO as a food source. They do this using an enzyme called Carbon Monoxide Dehydrogenase (CODH), which converts CO into CO₂ according to this reaction:
CO + H₂O → CO₂ + 2 electrons + 2 protons
The CO₂ produced by this reaction is not harmful at the quantities involved and supposed to be reused by a plant’s own photosynthesis through the Calvin cycle.
This project proposes to take the bacterial CODH system out of the bacterium and introduce it into a plant, specifically targeting it to the chloroplast (the organelle where photosynthesis happens). By placing CODH inside the chloroplast, two elegant outcomes occur simultaneously:
The scientific foundation for this idea is already established in the literature. Duffus et al. (2018) demonstrated that the complete CODH complex can be functionally expressed in Escherichia coli –> proving heterologous expression is achievable. South et al. (2019) demonstrated in Science that bacterial enzymes introduced into tobacco chloroplasts producing CO₂ directly in the stroma increased plant biomass by up to 40% –> proving that chloroplast-produced CO₂ is efficiently captured by photosynthesis. This project extends this logic to a new substrate: atmospheric CO.
The Complete Genetic System Required
The CODH enzyme from O. carboxidovorans is not a single protein. It is a complex system requiring seven genes organized into two functional groups:
coxL –> the large catalytic subunit (~88 kDa) where CO is actually oxidized. Contains the unique [CuSMoO₂] active site coxM –> the medium subunit (~30 kDa) containing FAD, responsible for electron transfer coxS –> the small subunit (~18 kDa) containing [2Fe-2S] iron-sulfur clusters, part of the electron relay chain
These three proteins assemble into a (CoxL·CoxM·CoxS)₂ heterohexamer — a complex of six protein subunits working together.
coxD –> an AAA+ ATPase chaperone that acts as a “maturation protein,” responsible for the post-translational insertion of copper and the essential bridging sulfur into the apo-enzyme, converting it to active holo-enzyme. coxE, coxF and coxG –> “final processing” and “sulfur addition” are part of a complex pathway. According to research, coxF plays a role in copper acquisition/mobilization, and coxE and coxG are involved in the maturation pathway that leads to the properly sulfurated and copper-inserted active site. The exact individual functions of coxE and coxG are still being elucidated, though their role in the maturation complex is essential.
Overview of the Three Aims

AIM 1 — Computational Design and Validation of the Complete Genetic System
In simple terms: Design the complete genetic blueprint for the CO-capturing plant system on a computer, verify every element computationally, and produce a synthesis-ready design.
The seven bacterial genes cannot simply be pasted into a plant. They need to be comprehensively redesigned for plant expression:
All of this is done computationally using Benchling, A codon optimization tool, ChloroP 1.1, Boltz, and the Asimov Kernel –> producing a complete verified design ready for DNA synthesis through Twist Biosciences.
AIM 2 — Wet Lab Transformation and Functional Validation (The next step — beyond this course)
In simple terms: Actually build the constructs in the lab, put them into tobacco plants, and prove the enzyme works. Aim 2 begins where Aim 1 ends. The Twist-synthesized multicassettes fragments are assembled into the pCAMBIA vectors using Gibson Assembly. The constructs are introduced into Nicotiana tabacum via Agrobacterium tumefaciens-mediated leaf disc transformation , the standard method for introducing genes into tobacco. Transgenic plants are selected on dual antibiotic medium (hygromycin + kanamycin, confirming both constructs integrated).
The experimental progression follows strict logic — each step must succeed before the next begins:
for more details, please take a look on part I of week 10 homework.
AIM 3 — Optimization, Transfer to Houseplants, and Real-World Deployment(The long-term vision)
In simple terms: Assuming Aim 2 succeeds, optimize the system, transfer it to real houseplants, and develop it toward real-world deployment. If Aim 2 demonstrates functional CO oxidation in tobacco, Aim 3 pursues three parallel directions:
Direction 1 — Transfer to real houseplants: The validated genetic architecture from tobacco is adapted for transformation into Epipremnum aureum (Pothos) and Spathiphyllum wallisii (Peace Lily) — widely kept, hardy, aesthetically acceptable houseplants. Agrobacterium-mediated transformation protocols established for tobacco are adapted for these species.
Direction 2 — System optimization: Several improvements are pursued to increase CO removal efficiency and operational range:
A CO-responsive inducible promoter system replaces constitutive promoters, activating CODH expression only when CO is present and saving plant energy otherwise Constitutively open stomata engineering to maintain CO uptake during nighttime hours when CO poisoning risk is highest Expression levels are optimized based on the quantitative CO removal model to increase per-plant removal capacity
Direction 3 — Safety, containment, and deployment:
Genetic Use Restriction Technology (GURT): To prevent seed viability and uncontrolled environmental spread, I will implement Genetic Use Restriction Technology (GURT). This ensures that any engineered plants cannot reproduce outside controlled environments. Additional containment strategy — chloroplast genome integration:
As an alternative or complement to GURT, I can integrate the transgenes into the chloroplast genome instead of the nuclear genome. Chloroplast DNA is maternally inherited in most flowering plants, including tobacco (Nicotiana tabacum). This means the transgenes are not transmitted via pollen, virtually eliminating the risk of gene flow to wild relatives. This is a well-established biosafety strategy for plant synthetic biology.
Regulatory pathway planning begins under USDA APHIS (Regulation of genetically engineered plantsand) EPA (Regulation of plants producing pesticidal substances (if applicable))frameworks.
The deployment target is refined based on the quantitative CO removal analysis: rather than acute emergency protection in homes (which requires too many plants), the primary application is chronic CO reduction in high-exposure industrial and semi-industrial environments like workshops, garages, underground parking facilities, and developing-world indoor cooking spaces where CO concentrations are higher and more sustained.
The ethical framework for commercial deployment ,including informed consent, false assurance prevention, equity of access, and environmental risk, is fully developed and integrated into regulatory submissions.

Sources:
To obtain the gene sequences, I used the accession number GenBank CP002827.1, which corresponds to the genome of Oligotropha carboxidovorans. I accessed this record through the National Center for Biotechnology Information platform.
Within the genome page, I used the graphical genome viewer to locate the genes of interest. I specifically identified the structural genes (coxL, coxM, coxS) and the maturation genes (coxD, coxE, coxF, coxG) involved in the CO dehydrogenase (CODH) system.
For each gene, I clicked on its corresponding feature in the graphical map, opened its detailed annotation page, and selected the FASTA format option. This allowed me to retrieve the nucleotide sequence of each gene individually.
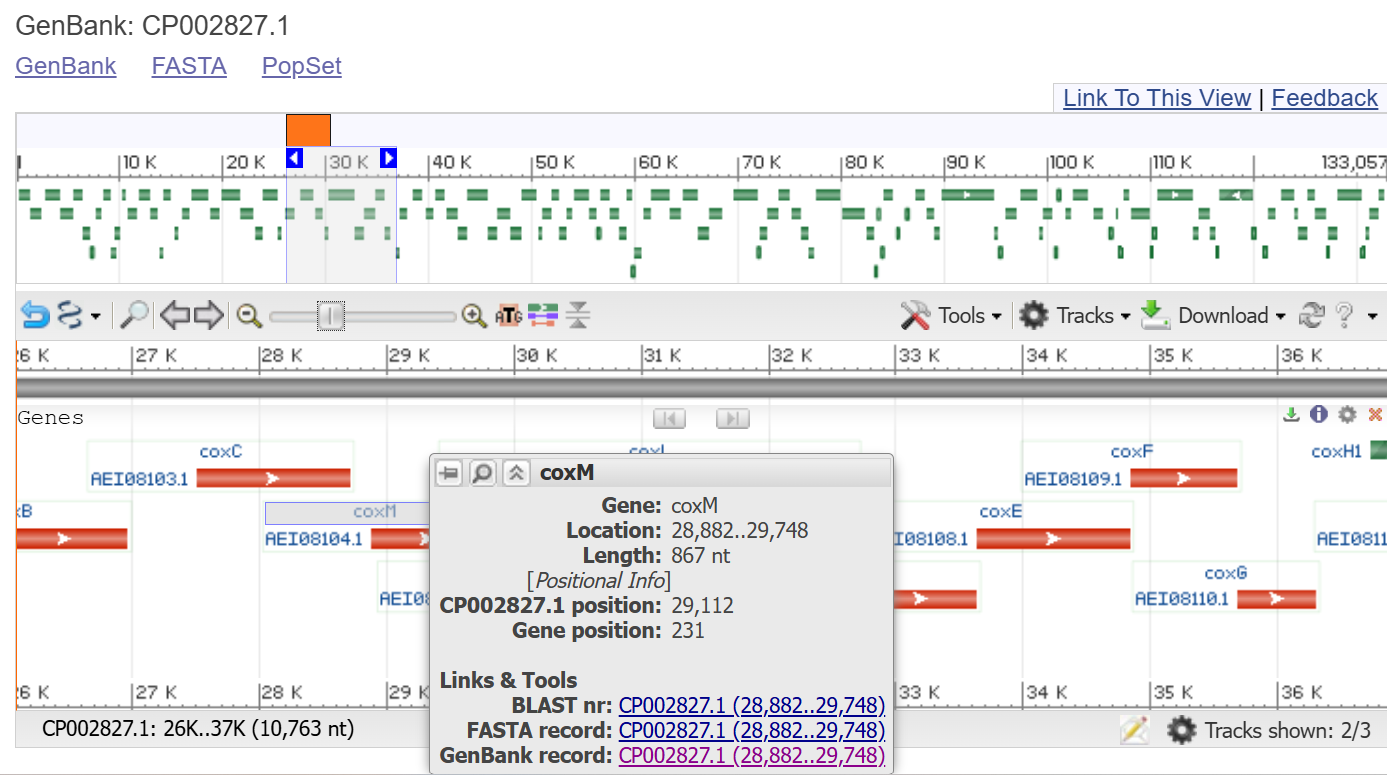 All sequences were downloaded separately in FASTA format and then compiled for further analysis and use in my project.
All sequences were downloaded separately in FASTA format and then compiled for further analysis and use in my project.
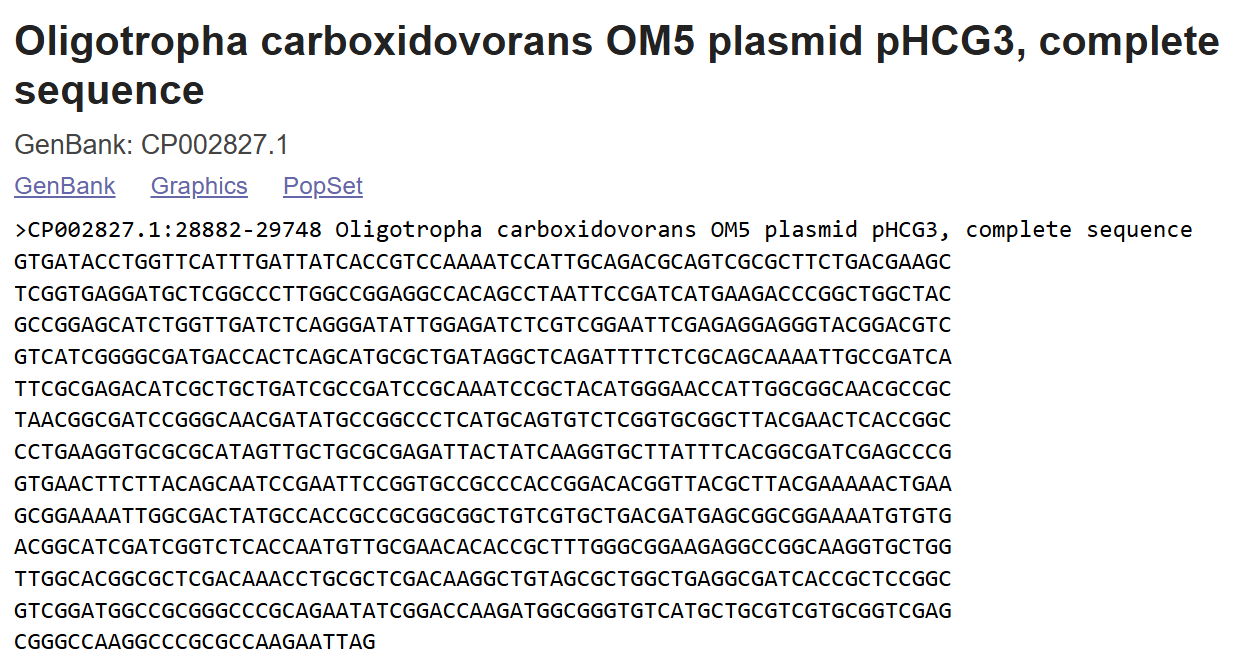
CoxL structural subunit sequence:
Cox M structural subunit sequence:
Cox S structural subunit sequence:
Cox D structural subunit sequence:
Cox E structural subunit sequence:
Cox F structural subunit sequence:
Cox G structural subunit sequence:
| Gene | Genomic Coordinates (NCBI) | Protein ID | Biological Role | Assigned Construct |
|---|---|---|---|---|
| coxL | CP002827.1 (30264–32693) | AEI08106.1 | Catalytic subunit responsible for CO oxidation | Construct 1 (Structural) |
| coxM | CP002827.1 (28882–29748) | AEI08104.1 | FAD-binding subunit involved in electron transfer | Construct 1 (Structural) |
| coxS | CP002827.1 (29767–30267) | AEI08105.1 | Fe-S cluster-containing subunit for electron relay | Construct 1 (Structural) |
| coxD | CP002827.1 (32748–33635) | AEI08107.1 | Molybdenum cofactor insertion and enzyme maturation | Construct 2 (Maturation) |
| coxE | CP002827.1 (33637–34836) | AEI08108.1 | Assists in Mo-cofactor biosynthesis and assembly | Construct 2 (Maturation) |
| coxF | CP002827.1 (34840–35682) | AEI08109.1 | Active site processing and enzyme activation | Construct 2 (Maturation) |
| coxG | CP002827.1 (35682–36299) | AEI08110.1 | Sulfur ligand incorporation into the active site | Construct 2 (Maturation) |
TobUbi.U4 proximal promoter:
The 263 bp proximal promoter region of the Ubi.U4 gene from Nicotiana tabacum was obtained based on the study by Genschik et al., (1994)This region corresponds to the sequence spanning −263 to −1 relative to the transcription start site (TSS) and contains key cis-regulatory elements involved in transcriptional regulation.
The transcription start site (TSS, +1) was not directly annotated in the GenBank entry. Therefore, it was determined based on the promoter analysis presented in the original publication by Genschik et al. (1994), where the TSS was experimentally identified and illustrated in Figure 3.
The nucleotide sequence was retrieved from the GenBank database (accession: X77456.1), corresponding to positions 575–837 of the N. tabacum Ubi.U4 gene.

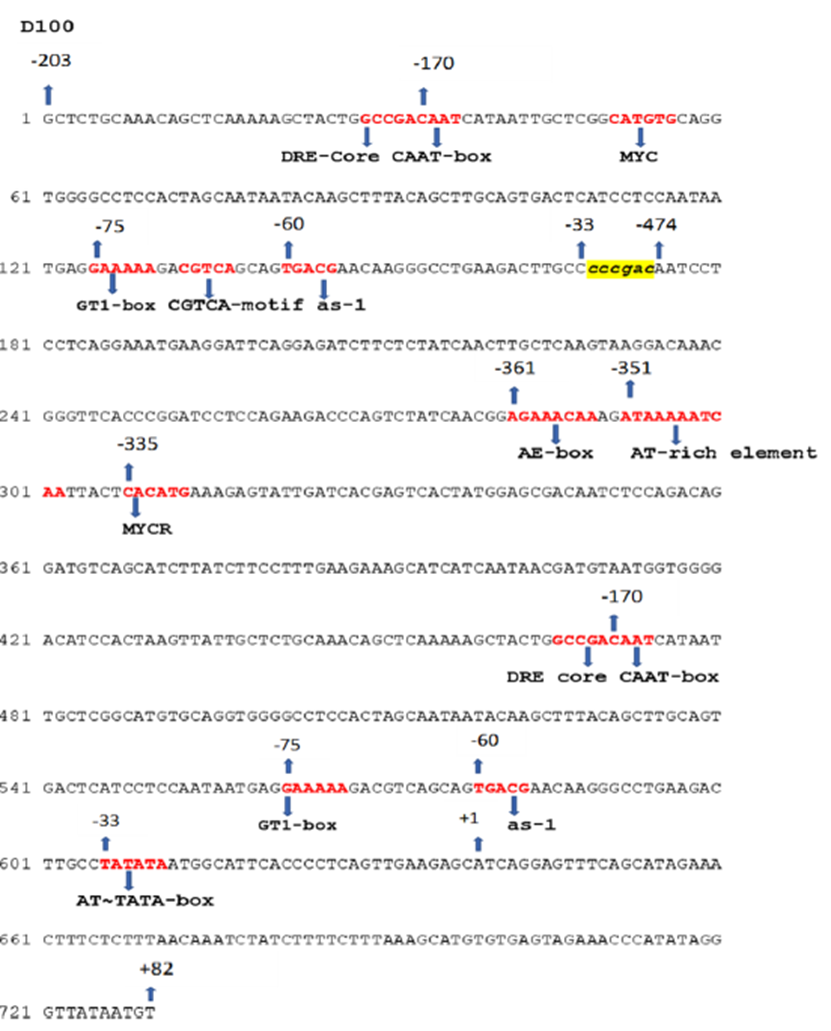
D100 chimeric promoter (Dahlia mosaic virus - DaMV):
The D100 promoter is a synthetic construct derived from the Dahlia mosaic virus (DaMV) genome, as described by (Khadanga et al., 2021)based on the work of (Sahoo et al., 2015). It is designed by combining an upstream activation sequence with a core promoter region to enhance transcriptional activity.
The following sequences were extracted based on coordinate mapping:
DaMV14UAS (−203 to −33):
DaMV4CP (−474 to +82):
Initially, the promoter sequence was reconstructed using GenBank coordinates. However, slight discrepancies were observed when compared to the promoter structure illustrated in the published figure.
 Therefore, the final D100 promoter sequence was generated using an Gemini AI tool based on the figure from Khadanga et al. (2021), as it accurately reflects the reported experimental construct:
Therefore, the final D100 promoter sequence was generated using an Gemini AI tool based on the figure from Khadanga et al. (2021), as it accurately reflects the reported experimental construct:
S100 chimeric promoter (Soybean vein clearing virus, SVBV):
The S100 promoter is a synthetic chimeric construct derived from the Soybean vein clearing virus (SVBV), as described by Khadanga et al., (2021)based on Pattanaik et al., (2004). It is designed by combining an upstream activation sequence with a core promoter region to enhance transcriptional activity.
The S100 promoter sequence was directly extracted from Figure 1 of Pattanaik et al. (2004), where the nucleotide sequence is explicitly provided in text format, and assembled in this order [SV10UAS] + [CCCGAC linker] + [SV10CP]:
DaMVFLt4 promoter (556 pb):
The DaMV4CP fragment corresponds to a natural promoter region derived from the Dahlia mosaic virus (DaMV). It consists of a 556 bp sequence spanning positions −474 to +82 relative to the transcription start site (TSS) according to Sahoo et al., (2014) study.
This fragment was directly extracted from the DaMV genome available in the GenBank database (accession: JX272320.1), corresponding to genomic coordinates 6579–7134.
SM chimeric hybrid promoter (SUAS + MUAS fusion):
The SM promoter is a synthetic chimeric hybrid promoter constructed by combining regulatory elements from two plant viruses, as described by Kumari et al., (2024). It integrates an upstream activation sequence from Sugarcane bacilliform virus with an enhancer domain from Mirabilis mosaic virus to enhance transcriptional activity.
To find the first fragment SUAS, I first mapped both boundaries of the 839 bp SCBV promoter using the SCBV-F primer anchor (ATTGAATGG) and the complement of the SCBV-R primer (GAATTACACCTTTCCGCA) against the Sugarcane bacilliform virus (SCBV) Ireng Maleng isolate sequence (accession AJ277091). This allowed me to confirm the full span of the mother fragment from relative coordinate −770 to +69
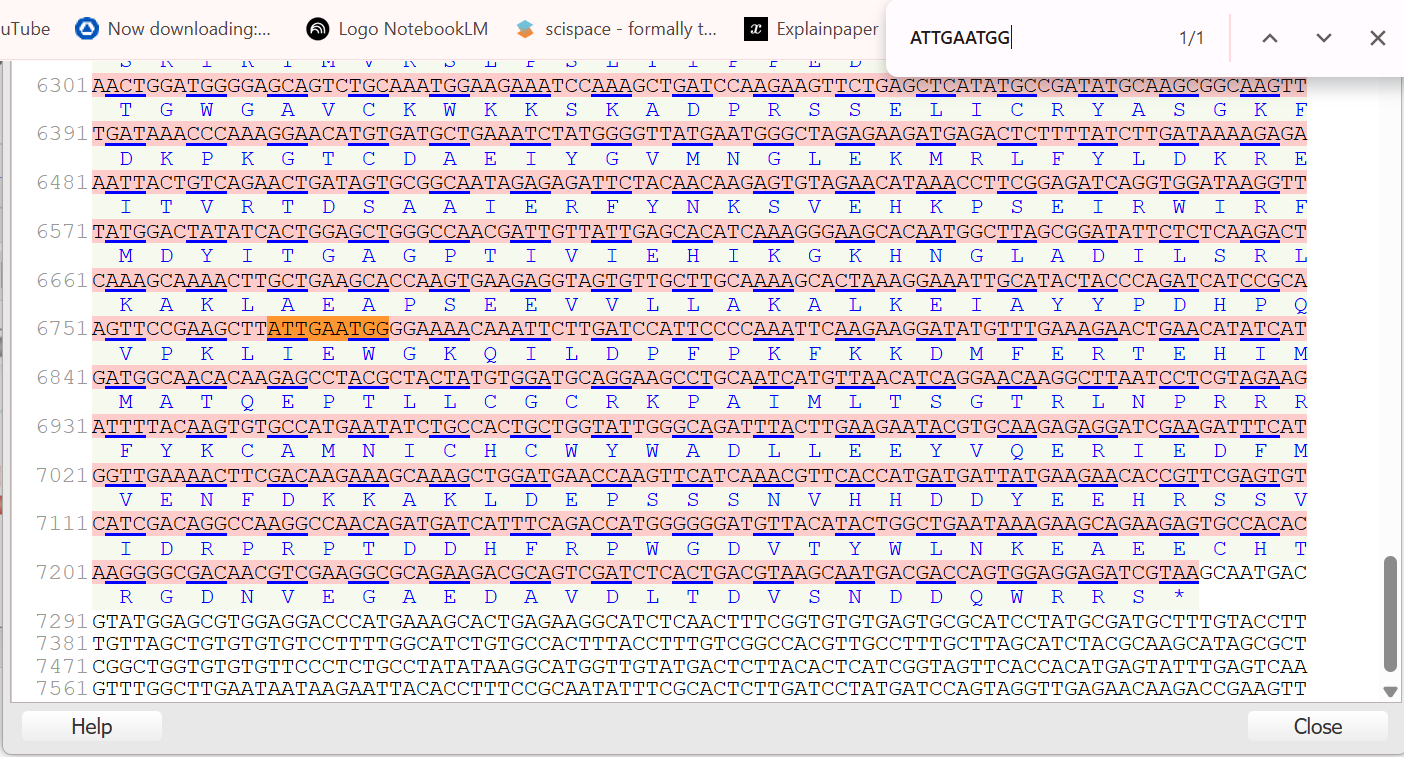
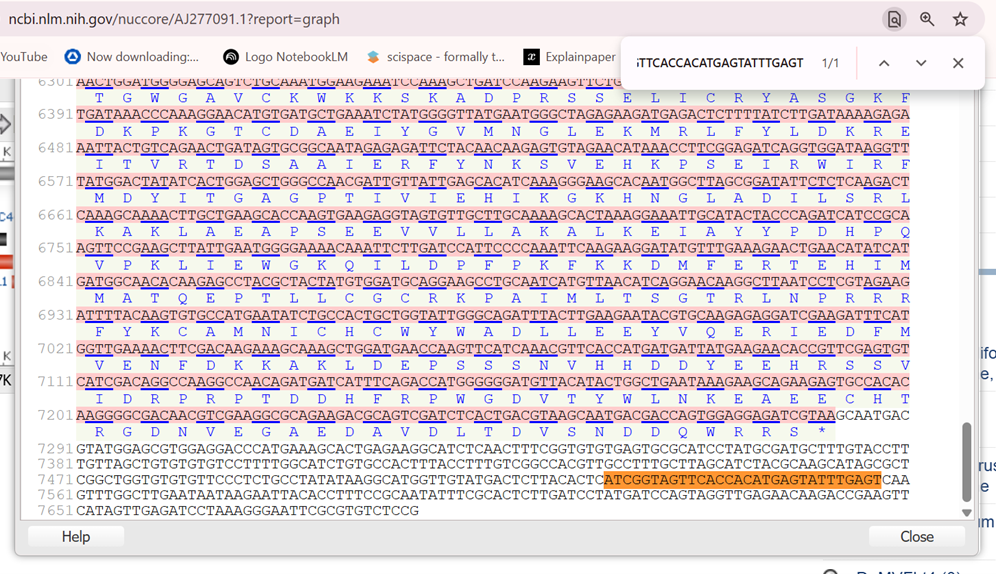
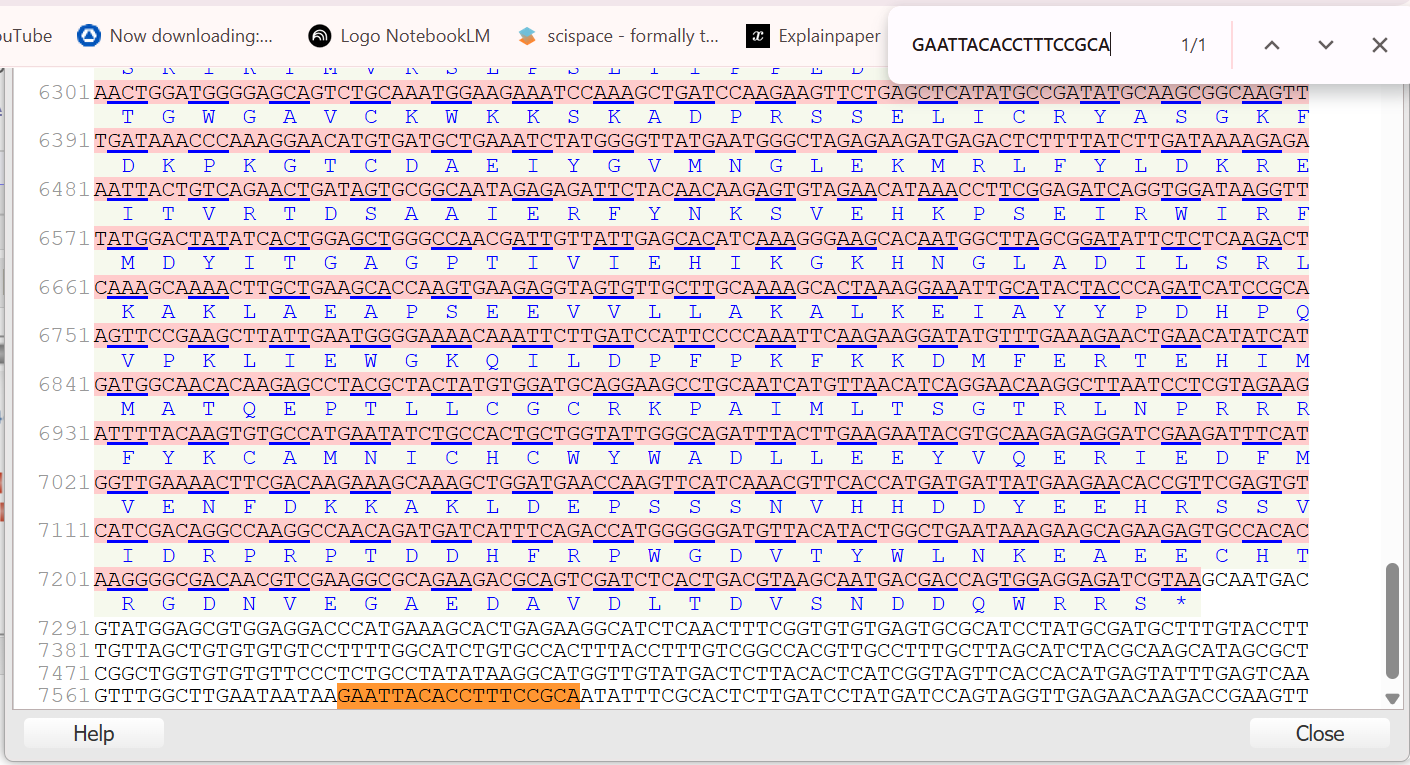 Next, I identified the Transcription Start Site (TSS) based on the underlined leader sequence reported in the Figure 2 from the Davies (2014) study. I could identify the TSS (+1) as the 7528th nucleotide in the Sugarcane bacilliform virus (SCBV) Ireng Maleng isolate sequence:
7528 ATC GGTAGTTCAC CACATGAGTA TTTGAGTCAA 7560
Next, I identified the Transcription Start Site (TSS) based on the underlined leader sequence reported in the Figure 2 from the Davies (2014) study. I could identify the TSS (+1) as the 7528th nucleotide in the Sugarcane bacilliform virus (SCBV) Ireng Maleng isolate sequence:
7528 ATC GGTAGTTCAC CACATGAGTA TTTGAGTCAA 7560
 To isolate the specific SUAS domain for the SM promoter, which the sources define as the segment from relative coordinates −434 to −153, I calculated the internal absolute indices within the 839 bp mother fragment. By mapping these relative coordinates back from the TSS, I determined the exact 282 bp enhancer sequence required to be joined directly to the MMV core promoter to build the chimeric SM promoter:
To isolate the specific SUAS domain for the SM promoter, which the sources define as the segment from relative coordinates −434 to −153, I calculated the internal absolute indices within the 839 bp mother fragment. By mapping these relative coordinates back from the TSS, I determined the exact 282 bp enhancer sequence required to be joined directly to the MMV core promoter to build the chimeric SM promoter:
To find the second fragment MUAS, I first identified the source as the Mirabilis mosaic virus (MMV) full-length transcript (FLt) promoter from the Dey and Maiti (1999) article. Because the original study provided the literal nucleotide sequence in Figure 1 rather than a GenBank accession number, I used the printed sequence obtained from Gemini AI tool as my primary reference.
 I then established the Transcription Start Site (TSS or +1) as the anchor point, which the researchers mapped via primer extension to a guanidine (G) residue located 24 nucleotides downstream of the TATATAA box. To isolate the specific MUAS fragment, which spans the relative coordinates −297 to −38, I counted upstream from the TSS to locate the nucleotide at position −297 and extracted the sequence through to the nucleotide at position −38. This process provided the 259 bp enhancer domain required for the construction of the SM and BM chimeric promoters:
I then established the Transcription Start Site (TSS or +1) as the anchor point, which the researchers mapped via primer extension to a guanidine (G) residue located 24 nucleotides downstream of the TATATAA box. To isolate the specific MUAS fragment, which spans the relative coordinates −297 to −38, I counted upstream from the TSS to locate the nucleotide at position −297 and extracted the sequence through to the nucleotide at position −38. This process provided the 259 bp enhancer domain required for the construction of the SM and BM chimeric promoters:
The SM promoter was generated by directly fusing the SUAS fragment upstream of the MUAS enhancer sequence, as described by (Kumari et al., 2024a) based on the source sequence described in Dey & Maiti, (1999) study:
BM chimeric hybrid promoter (BUAS + MUAS fusion):
The BM promoter is a synthetic chimeric hybrid promoter constructed by the fusion of two regulatory elements, as described by (Kumari et al., 2024a). It combines an upstream activation sequence from Banana streak virus with an enhancer domain from Mirabilis mosaic virus to enhance transcriptional efficiency.
To find the first fragment BUAS, I first identified the source as the Banana streak virus (BSV) Cavendish isolate, which corresponds to GenBank accession AF215815. Although the current database entry for this accession may show a length of 1,287 bp, I noted that the sources utilize a 1,304 bp synthesized version of this isolate spanning from relative coordinates −1,150 to +154.
Next, I used the BSV-F primer anchor sequence (GGTTGCATGGAAGG) to locate the beginning of the promoter region within the GenBank file. By finding this exact sequence at the very start of the file, I established that Nucleotide 1 of the GenBank entry corresponds to the relative coordinate −1,150.
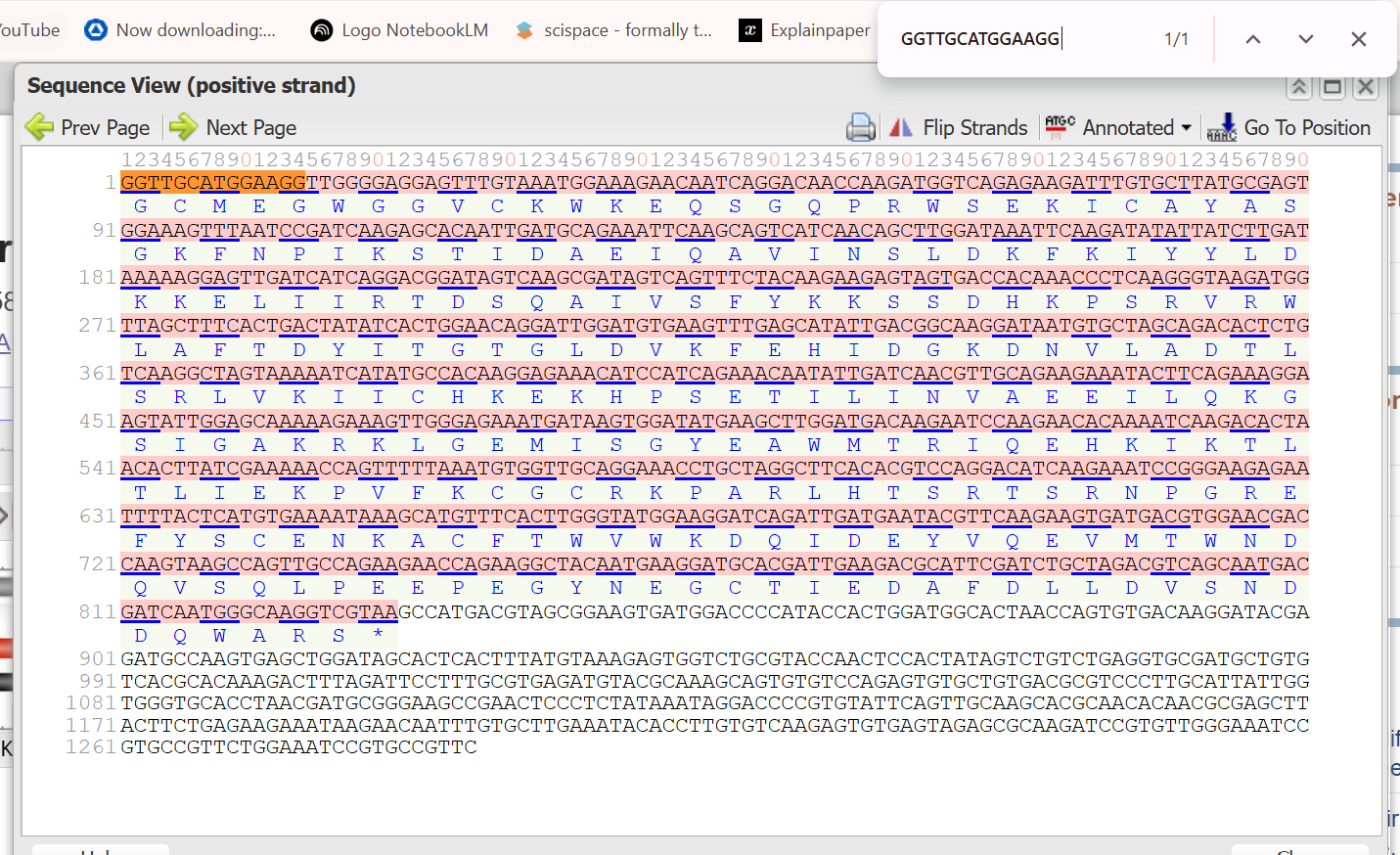 I then determined the Transcription Start Site (TSS or +1) by mapping the relative coordinates to the absolute indices of the 1,304 bp sequence. Since there are 1,150 bases upstream of the start site, the TSS is located at Nucleotide 1151. To isolate the specific BUAS domain, which the sources define as the segment from −1,150 to −33, I calculated the end index by subtracting 33 from the TSS (1151−33=1118).
Finally, I extracted the sequence from Nucleotide 1 to Nucleotide 1118, which provided the approximately 1,117 bp (mathematically 1,118 bp) enhancer fragment required to construct the BM chimeric promoter:
I then determined the Transcription Start Site (TSS or +1) by mapping the relative coordinates to the absolute indices of the 1,304 bp sequence. Since there are 1,150 bases upstream of the start site, the TSS is located at Nucleotide 1151. To isolate the specific BUAS domain, which the sources define as the segment from −1,150 to −33, I calculated the end index by subtracting 33 from the TSS (1151−33=1118).
Finally, I extracted the sequence from Nucleotide 1 to Nucleotide 1118, which provided the approximately 1,117 bp (mathematically 1,118 bp) enhancer fragment required to construct the BM chimeric promoter:
The BM promoter was generated by directly fusing the BUAS fragment upstream of the MUAS enhancer sequence, as described by Kumari et al., (2024):
MSD3 chimeric deletion-hybrid promoter (MUAS + SD3):
The MSD3 promoter is a “deletion-hybrid” construct composed of the following two fragments joined directly together as described in the study of (Kumari et al., 2024b):
The final MSD3 promoter was obtained by direct assembly of the MUAS enhancer upstream of the SD3 core promoter fragment:
M24 synthetic promoter (MMV-derived):
The M24 promoter is a synthetic high-expression promoter derived from the Mirabilis mosaic virus (MMV), as described by (Sahoo et al., 2014). It was engineered to enhance transcriptional activity in plant systems. Based on the full-length transcript (FLt) promoter of MMV, the promoter was enhanced by duplication of upstream enhancer domains, leading to a significant increase in transcriptional strength.
The M24 promoter sequence was retrieved from the binary vector pSiM24 available in GenBank (accession: KF032933.1). The promoter corresponds to the region spanning positions 235–860 of the vector sequence.
PClSV FLt promoter (Peanut chlorotic streak caulimovirus):
The PClSV FLt promoter is a constitutive plant promoter derived from the Peanut chlorotic streak caulimovirus. It is composed of a basic full-length transcript (FLt) promoter region and upstream enhancer elements, which can be arranged in single or duplicated configurations to modulate transcriptional strength.
The promoter elements were identified from the PClSV genome (GenBank accession: U13988.1) as follows:
The assembled PClSV FLt promoter [Enhancer] + [Core promoter] sequence:
Double enhancer PCisV FLt promoter:
Based on (Maiti & Shepherd, 1998), the double enhancer configuration was constructed by duplicating the enhancer region upstream of the core promoter: [Enhancer] + [Enhancer] + [Core promoter] (~428 bp)
The PClSV FLt promoter sequence was reconstructed from GenBank (U13988.1) and assembled in a double enhancer configuration based on the design described by Maiti & Shepherd (1998):
The double enhancer configuration of the PClSV FLt promoter results in an approximately threefold increase in transcriptional activity compared to the single enhancer version. Overall, this promoter exhibits strong constitutive expression in transgenic plants, with activity levels reported to be comparable to the FLt promoter of the Figwort mosaic virus and functionally similar to the widely used CaMV 35S promoter, making it a robust alternative for high-level gene expression in plant systems.
CVP1 and CVP2 promoters (Cassava vein mosaic virus, CsVMV):
The CVP1 and CVP2 promoters are constitutive plant promoters derived from the Cassava vein mosaic virus (CsVMV), as described by Verdaguer et al., (1996) and Verdaguer et al., (1998) based on the reference genome reported by Calvert et al., (1995). These promoters correspond to two fragments of different lengths within the viral genome and differ in their regulatory strength.
Both fragments contain core promoter elements, including the TATA box and upstream regulatory motifs, with CVP2 retaining additional upstream sequences that enhance transcriptional activity.
The sequences were directly retrieved from the CsVMV reference genome (GenBank accession: U20341.1) using the genomic coordinates reported in the original studies:
CPV 1 :
CPV 2 :
Functional analyses have demonstrated that CVP2 exhibits expression levels comparable to the enhanced CaMV 35S promoter (e35S), whereas CVP1 shows approximately half of this activity, indicating that CVP2 is about twofold more active than CVP1. These results highlight the importance of additional upstream regulatory sequences in driving stronger gene expression in plant systems.
FMV Sgt (34S) promoter (Figwort mosaic virus):
The Sgt (34S) promoter is a subgenomic promoter derived from the Figwort mosaic virus (FMV). It is located between ORF V and ORF VI and is responsible for driving the expression of ORF VI via a subgenomic transcript.
According to (Bhattacharyya et al., 2002) , a 301 bp fragment spanning −270 to +31 relative to the transcription start site (TSS) provides maximal promoter activity.
The promoter sequence was extracted from the published figure using an AI tool (Gemini), as it was only available in image format:
.png?width=500px)
PTSB1 promoter (Arabidopsis thaliana):
The PTSB1 promoter is a constitutive plant promoter I derived from the Arabidopsis thaliana tryptophan synthase β-subunit gene (TSB1). I identified this as a powerful alternative to the CaMV 35S promoter for high-level gene expression in tobacco (Shirasawa-Seo et al. 2002).
I retrieved this promoter from GenBank accession M23872, corresponding to a 1.5 kb fragement. I defined the exact boundaries of this fragment by mapping the reported PCR primers directly onto the reference sequence (Shirasawa-Seo et al. 2002):
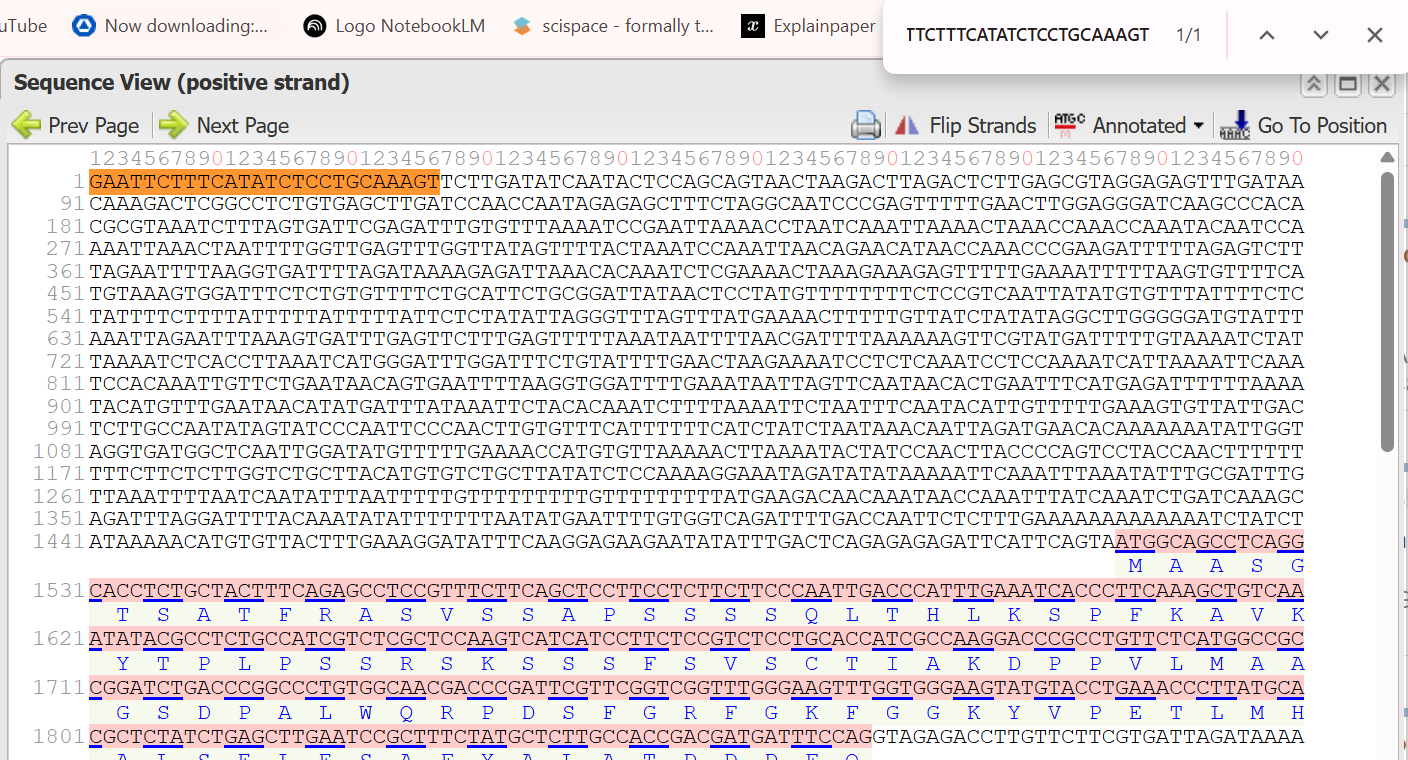
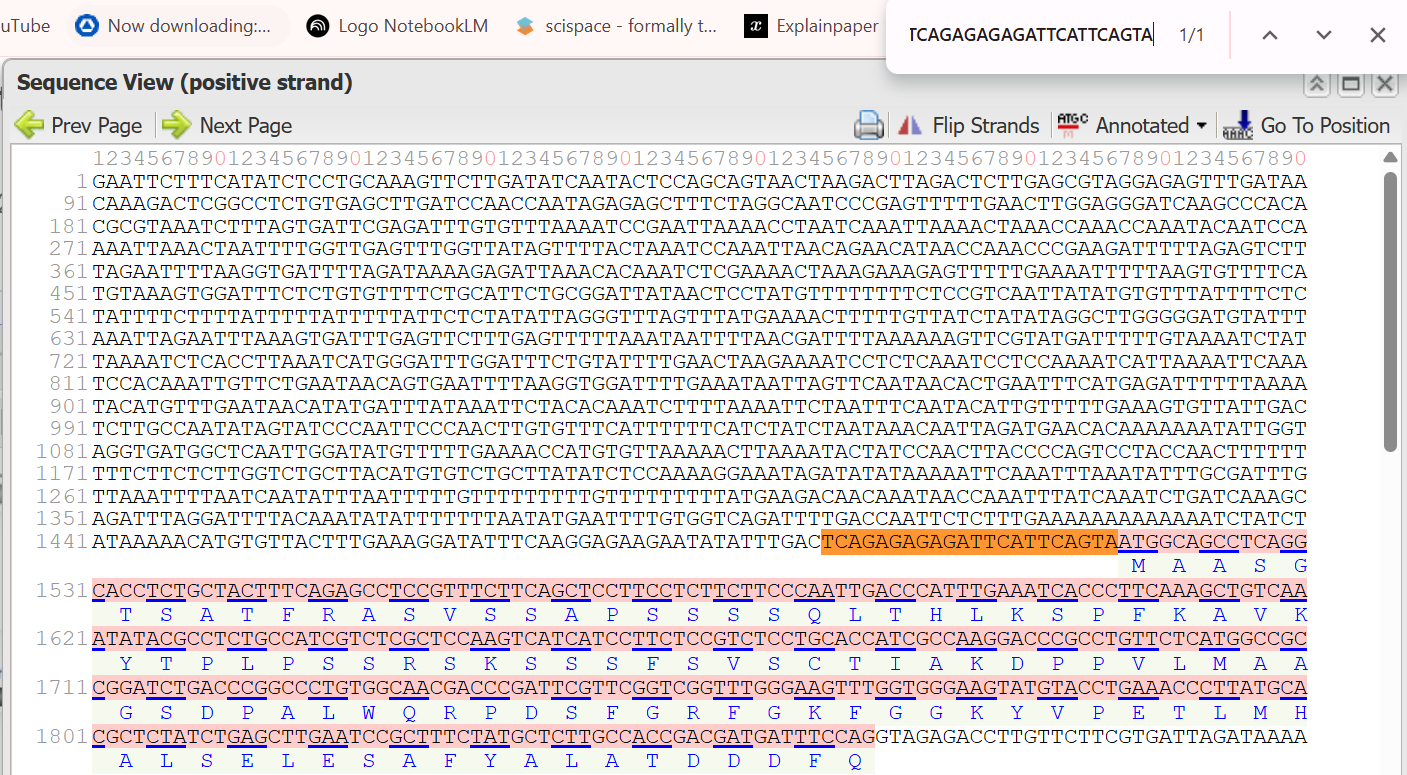 The resulted extracted sequence of PTSB1 promoter:
The resulted extracted sequence of PTSB1 promoter:This region contains the core promoter and upstream regulatory elements responsible for its strong constitutive activity. This promoter exhibited approximately 2.4-fold higher expression than the CaMV 35S promoter in mature tobacco leaves, with activity increasing in lower leaf positions (Shirasawa-Seo et al. 2002).
PPHYB promoter (Arabidopsis thaliana):
The PPHYB promoter is a constitutive promoter derived from the Arabidopsis thaliana phytochrome B (PHYB) gene (Goosey et al. 1997; Shirasawa-Seo et al. 2002).
I retrieved this sequence from GenBank accession L09262, which corresponds to a 2.3 kb fragment. The promoter boundaries were defined by mapping the experimentally reported primers onto the sequence (Shirasawa-Seo et al. 2002):
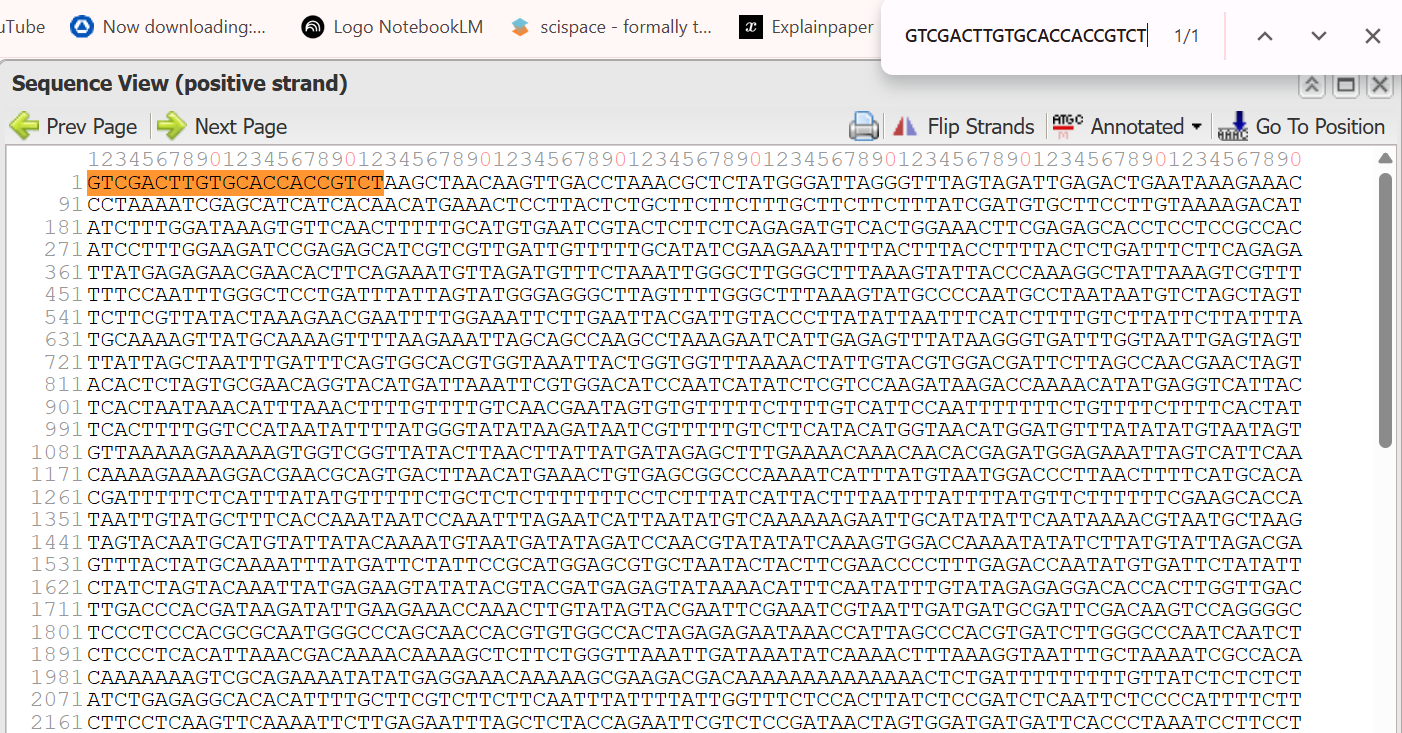
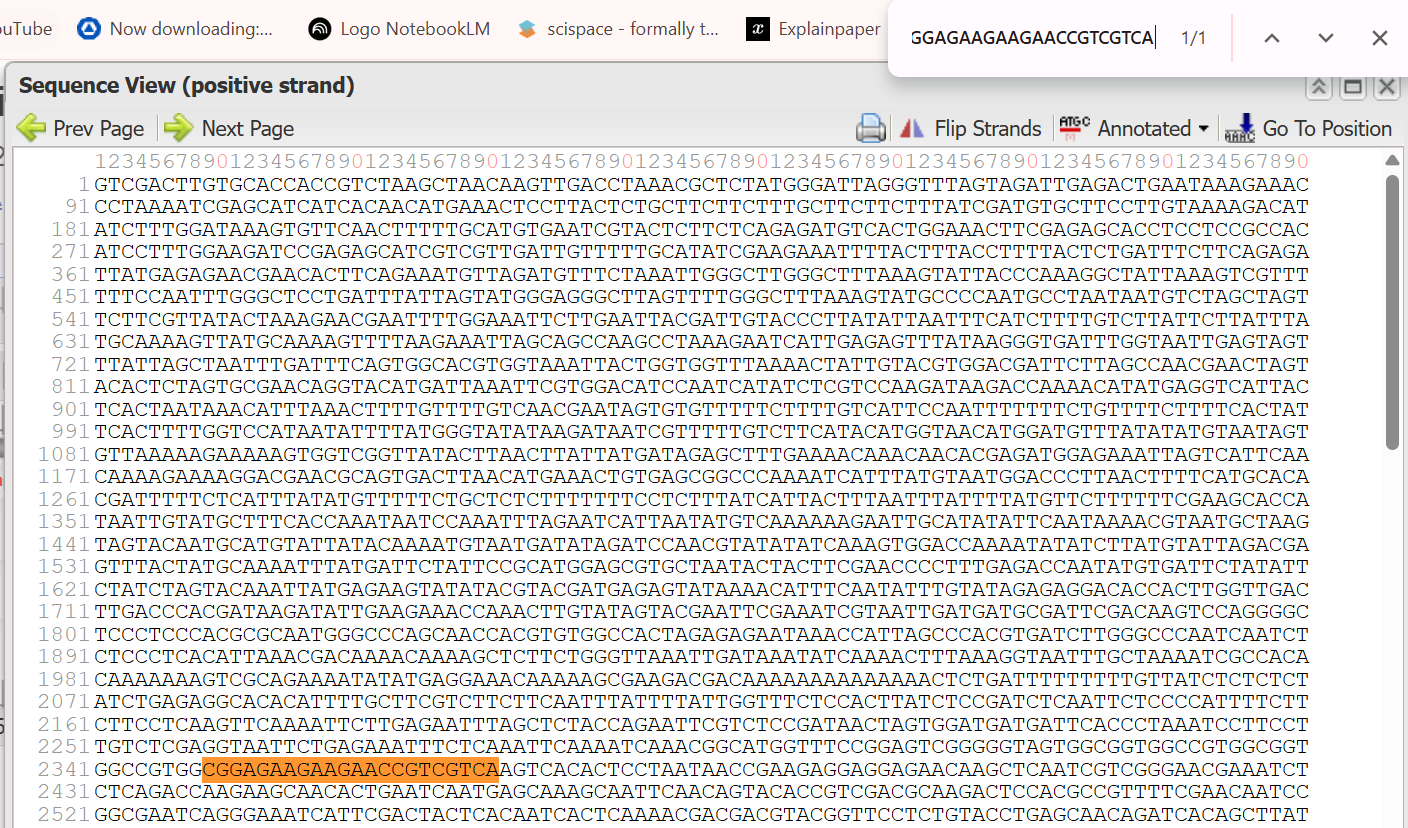 The resulted extracted sequence of PPHYB promoter:
The resulted extracted sequence of PPHYB promoter:This fragment includes the core promoter and regulatory regions required for stable expression. Functionally, PPHYB provides approximately 1.5-fold higher expression than the CaMV 35S promoter in mature tobacco leaves, with a more uniform expression pattern across leaf positions compared to PTSB1 (Shirasawa-Seo et al. 2002).
PNCR promoter (Soybean chlorotic mottle virus):
The PNCR promoter is a viral-derived constitutive promoter isolated from the large noncoding region of the Soybean chlorotic mottle virus (Conci et al. 1993). Based on the reported genome size (~8,175 bp), I identified the corresponding genomic sequence and retrieved it from GenBank accession X15828.2. I then defined the functional ~486 bp promoter fragment by mapping the reported PCR primers onto the genome (Conci et al. 1993):
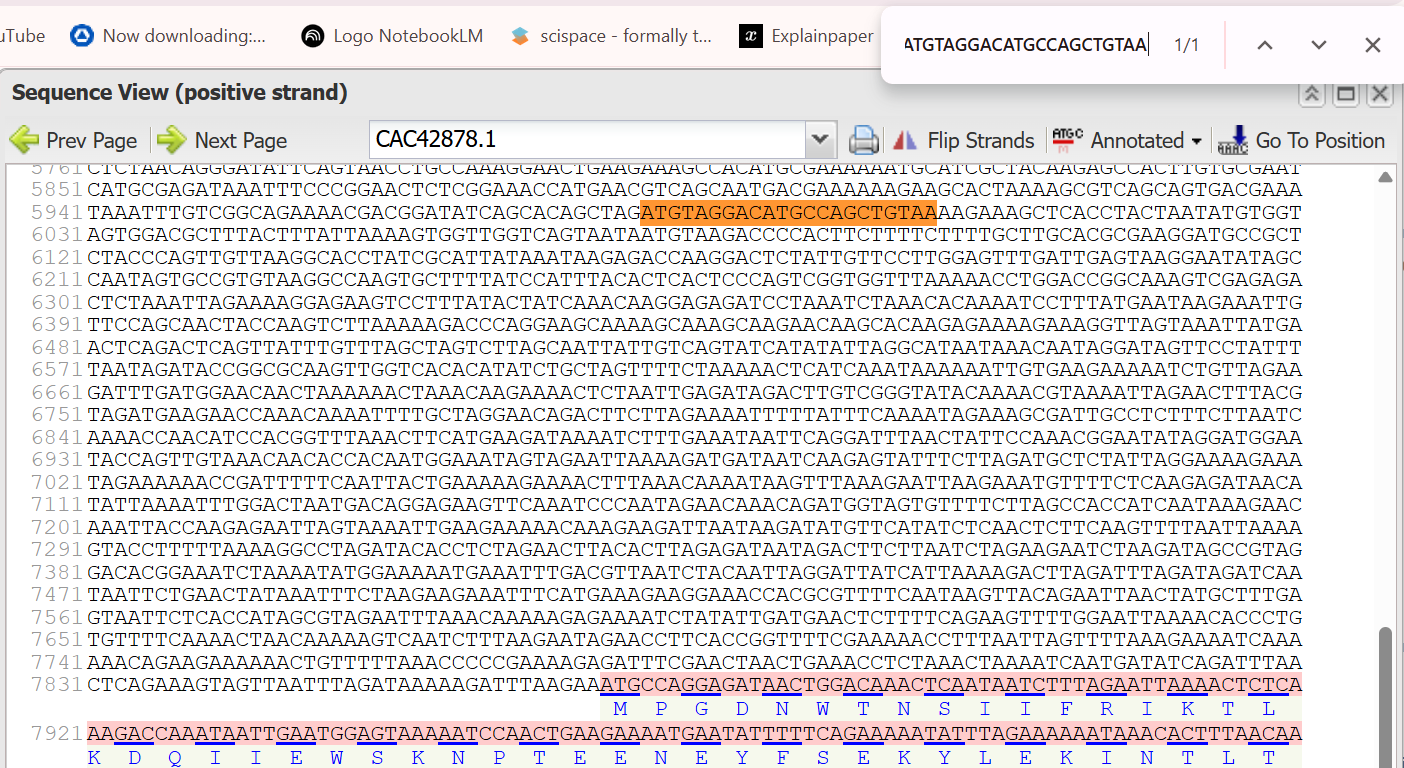
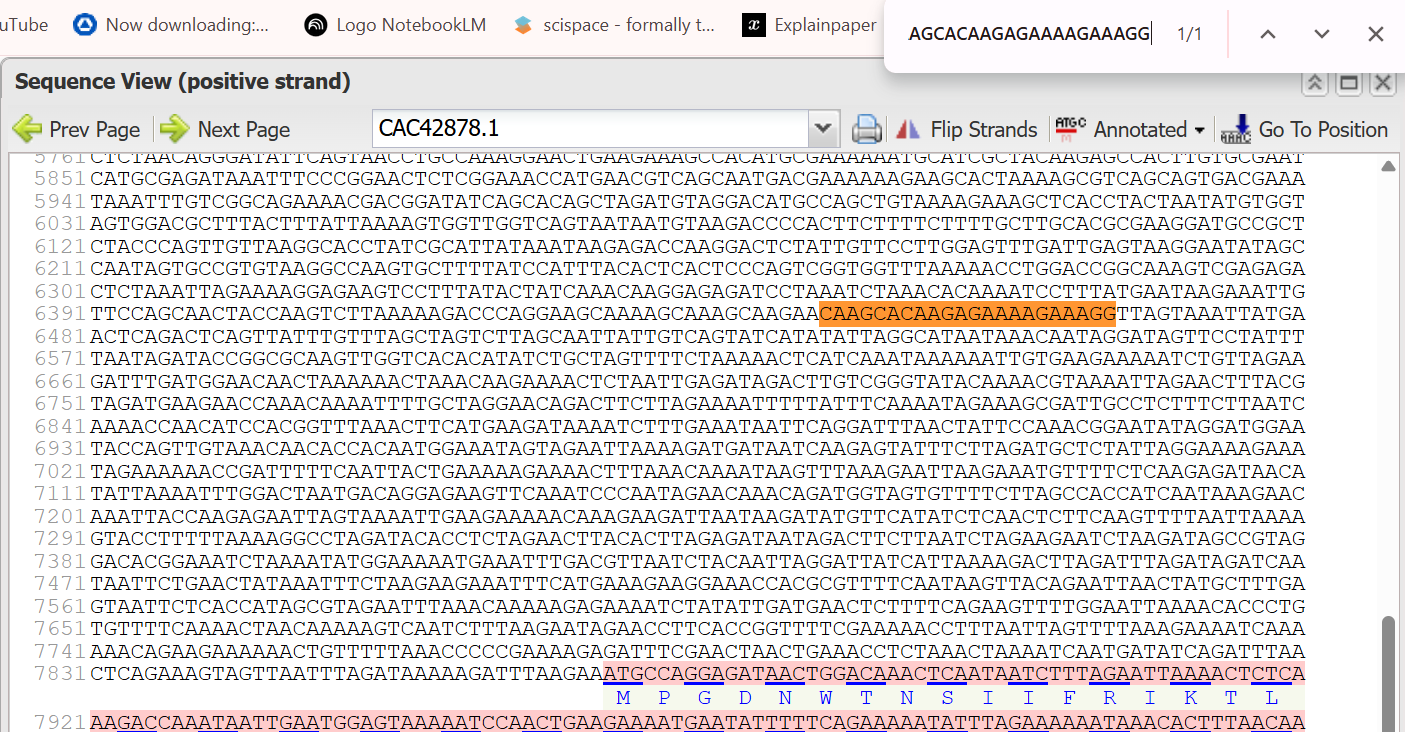
This region contains key regulatory features including a TATA box, CAAT-like motifs, and multiple enhancer-related elements. Functionally, this promoter exhibits approximately five-fold higher expression than the CaMV 35S promoter in tobacco protoplasts (Conci et al. 1993), while showing moderate constitutive activity (~67% of P35S) in mature leaves (Shirasawa-Seo et al. 2002).
FMV promoter (Figwort mosaic virus):
The FMV promoter is a constitutive viral promoter derived from the Figwort mosaic virus genome. In this work, I used the promoter sequence obtained directly from the supplementary Benchling file provided in (Shakhova et al., 2022):
To verify its genomic origin, I performed a BLAST analysis using the NCBI nblast, and obtained a 100% sequence match corresponding to coordinates 6358 to 6955 of the reference genome (GenBank accession NC_003554.1), confirming the exact location of the promoter fragment within the FMV genome. According to (Shakhova et al., 2022), the FMV promoter exhibited lower activity compared to the CaMV 35S promoter under their experimental conditions, indicating that while it remains a functional constitutive promoter, it is not as strong as p35S in this specific system.
p35S (CAMV 35S promoter):
The p35S promoter is a canonical constitutive promoter derived from the Cauliflower mosaic virus and is one of the most widely used regulatory elements in plant biotechnology.
In my study, I used the specific p35S sequence provided in the supplementary Benchling file of (Shakhova et al., 2022):
pAtUBQ10 promoter (Arabidopsis thaliana):
The pAtUBQ10 promoter (version 0.8) is a strong constitutive plant promoter derived from the Arabidopsis thaliana ubiquitin-10 gene (At4g05320). In this work, I used the exact ~800 bp upstream fragment as characterized in (Shakhova et al., 2022).
I obtained the sequence directly from the supplementary Benchling file provided in the study, ensuring that the construct corresponds precisely to the experimentally validated version used for expression analysis:
This fragment represents the regulatory region immediately upstream of the translation start site and includes key cis-regulatory elements responsible for its constitutive activity.
Functionally, in Nicotiana systems, this promoter provides high and stable expression levels, outperforming several endogenous plant promoters such as pAtAct2, pAtTCTP, and pAtPD7 (Shakhova et al., 2022). Although its activity is lower than the viral Cauliflower mosaic virus 35S promoter, it shows comparable expression strength to other viral promoters such as Figwort mosaic virus (FMV) and Cotton leaf curl Multan virus (CmYLCV), making it a reliable and predictable option for high-level gene expression in both Nicotiana benthamiana leaves and tobacco BY-2 cell packs.
pAtAct2 promoter (Arabidopsis thaliana):
The pAtAct2 promoter is a constitutive plant promoter derived from the Arabidopsis thaliana actin 2 gene (AT3G18780). In this work, I used the specific version characterized in (Shakhova et al., 2022).
I obtained the sequence directly from the supplementary Benchling file provided in the study, ensuring that the construct corresponds exactly to the experimentally tested version. In this configuration, the native promoter was fused to the 5′UTR omega sequence of the Tobacco mosaic virus (TMV), a common modification used to enhance translation efficiency in Nicotiana expression systems:
Functionally, although pAtAct2 is historically described as a strong constitutive promoter in Arabidopsis, the results of (Shakhova et al., 2022) show that it exhibits relatively low activity in tobacco systems. When compared to the 0.4 kb version of the Cauliflower mosaic virus 35S promoter (p35S) used as the reference in this study, pAtAct2 ranks among the weakest promoters in the tested set. This indicates that, despite its native strength in Arabidopsis, pAtAct2 behaves as a moderate-to-low strength promoter in Nicotiana, even after optimization via the TMV omega 5′UTR fusion.
NOS promoter (Agrobacterium tumefaciens nopaline synthase):
The NOS promoter is a constitutive plant promoter derived from the nopaline synthase (nos) gene of Agrobacterium tumefaciens, and is widely used in plant transformation vectors for moderate gene expression.
In this work, I retrieved the NOS promoter sequence from GenBank entry AF485783.1, corresponding to the binary vector pBI121, using the coordinates 2519 to 2825. This fragment represents the regulatory region upstream of the nos gene as commonly implemented in plant expression constructs.
The sequence was directly extracted from the annotated GenBank record, ensuring consistency with a well-established and experimentally validated vector backbone frequently used in plant biotechnology.
Functionally, the NOS promoter is considered a moderate-to low strength constitutive promoter, typically weaker than strong viral promoters such as the Cauliflower mosaic virus 35S promoter, but valued for its stable and reliable expression across different plant tissues.
| Promoter | Origin | Relative Strength vs. CaMV 35S | Key Advantage / Note | Source |
|---|---|---|---|---|
| TobUbi.u4 | Nicotiana tabacum (polyubiquitin) | ~7× stronger | Native to tobacco; excellent stability for long-term expression | Genschik et al., 1994 (GenBank: X77456.1) |
| D100 | Synthetic (Dahlia mosaic virus) | ~2.2× stronger | One of the strongest synthetic promoters validated in tobacco | Khadanga et al., 2021; Sahoo et al., 2015 |
| MSD3 | Synthetic chimeric (MMV + SCBV) | ~1.15× stronger | Works in both monocots and dicots; stable in tobacco | Kumari et al., 2024; Dey & Maiti, 1999 |
| DaMVFLt4 | Dahlia mosaic virus | ~5× stronger | Very high activity in protoplasts and transgenic plants | Sahoo et al., 2014; GenBank: JX272320.1 |
| M24 | MMV-derived | ~10× stronger | Extremely strong promoter with enhanced duplicated domains | Sahoo et al., 2014 |
| S100 | Synthetic (Strawberry vein banding virus) | ~1.8× stronger | Strong synthetic alternative; slightly weaker than D100 | Khadanga et al., 2021; Pattanaik et al., 2004 |
| SM | Synthetic chimeric (SCBV + MMV) | ~2.1× stronger | Highly effective in dicots like tobacco | Kumari et al., 2024; Davies et al., 2014 |
| BM | Synthetic chimeric (BSV + MMV) | ~1.72× stronger | Good alternative synthetic promoter for dicots | Kumari et al., 2024; Remans et al., 2005 |
| FMV 34S | Figwort mosaic virus | ~2× stronger | Widely used constitutive promoter in dicots | Bhattacharyya et al., 2002 |
| CaMV 35S | Cauliflower mosaic virus | 1× (reference) | Gold standard promoter for plant expression | Odell et al., 1985; Shakhova et al., 2022 |
| PTSB1 | Arabidopsis thaliana (TSB1) | ~2.4× stronger | Very strong in mature leaves; tissue-dependent variation | Shirasawa-Seo et al., 2002 |
| PPHYB | Arabidopsis thaliana (PHYB) | ~1.5× stronger | Uniform expression across tissues | Shirasawa-Seo et al., 2002; Goosey et al., 1997 |
| PNCR | Soybean chlorotic mottle virus | ~5× (protoplasts), moderate in plants | Strong viral promoter distinct from CaMV and FMV | Conci et al., 1993; Shirasawa-Seo et al., 2002 |
| PCisV | PClSV FLt promoter | ~2× stronger | Strong constitutive promoter comparable to FMV | Maiti & Shepherd, 1998 |
| dPCisV | Double enhancer PCisV | ~6× stronger | Highly powerful promoter due to enhancer duplication | Maiti & Shepherd, 1998 |
| CPV1 | Cassava vein mosaic virus | ~0.5× of CPV2 | Moderate activity; tissue-specific expression | Verdaguer et al., 1996; Calvert et al., 1995 |
| CPV2 | Cassava vein mosaic virus | ~1× (similar to e35S) | Stronger version; high activity in vascular tissues | Verdaguer et al., 1998 |
| pFMV | Figwort mosaic virus | <1 (weaker than 35S) | Common alternative but weaker in this system | Shakhova et al., 2022 |
| AtUBQ10 (0.8) | Arabidopsis thaliana | <1 (similar to pFMV) | Stable expression across tissues | Shakhova et al., 2022 |
| AtAct2 | Arabidopsis thaliana | Moderate to low | Constitutive but weak in tobacco system | Shakhova et al., 2022 |
| P-Nos | Agrobacterium tumefaciens | Weak to moderate | Commonly used for selectable marker genes | GenBank: AF485783 |
| Terminator | Origin | Relative Performance | Key Characteristics | Sequence Source |
|---|---|---|---|---|
| tOCS | Agrobacterium tumefaciens (octopine synthase) | Highest (Top performer) | Most stable and strongest expression in Nicotiana systems; best overall choice | Shakhova et al., 2022 (supplementary Benchling file) |
| tHSP18.2 | Arabidopsis thaliana (heat shock protein 18.2) | Very high (slightly below tOCS) | Strong expression; highly efficient but slightly less than tOCS in tobacco | Shakhova et al., 2022 (supplementary Benchling file) |
| tATPase | Solanum lycopersicum (ATPase gene) | High | Robust and consistent performance; comparable to tHSP18.2 | Shakhova et al., 2022 (supplementary Benchling file) |
| tAtAct2 | Arabidopsis thaliana (actin 2) | Low | Weak expression in Nicotiana; not suitable for high-expression constructs | Shakhova et al., 2022 (supplementary Benchling file) |
| tRBCS3C | Solanum lycopersicum (Rubisco small subunit 3C) | Low | Limits transcription efficiency; weakest among tested terminators | Shakhova et al., 2022 (supplementary Benchling file) |
| T-35S | Cauliflower mosaic virus | Moderate to high | Widely used standard terminator; reliable polyadenylation signal | GenBank: GQ497234.1 (pEAQ-HT vector) |
| T-E9 | Pisum sativum (Rubisco small subunit) | High | Efficient transcription termination and mRNA stabilization in plants | GenBank: HM036220.1 (pKM24KH vector) |
The three chloroplast transit peptides (RbcS CTP, Ferredoxin-2 CTP, and RecA CTP) were identified from Arabidopsis thaliana proteins using the UniProt database. For each protein, I first retrieved the corresponding entry (accessions P10795, P16972, and Q39199), then examined the “Features” section, specifically under PTM/Processing, to locate the annotated transit peptide regions.



The CTP sequences were directly extracted from the annotated transit peptide segments, which correspond to the N-terminal targeting signals responsible for directing proteins to the chloroplast. This approach ensures that the selected sequences match experimentally curated annotations and represent functional chloroplast-targeting peptides.
The extracted sequences are:
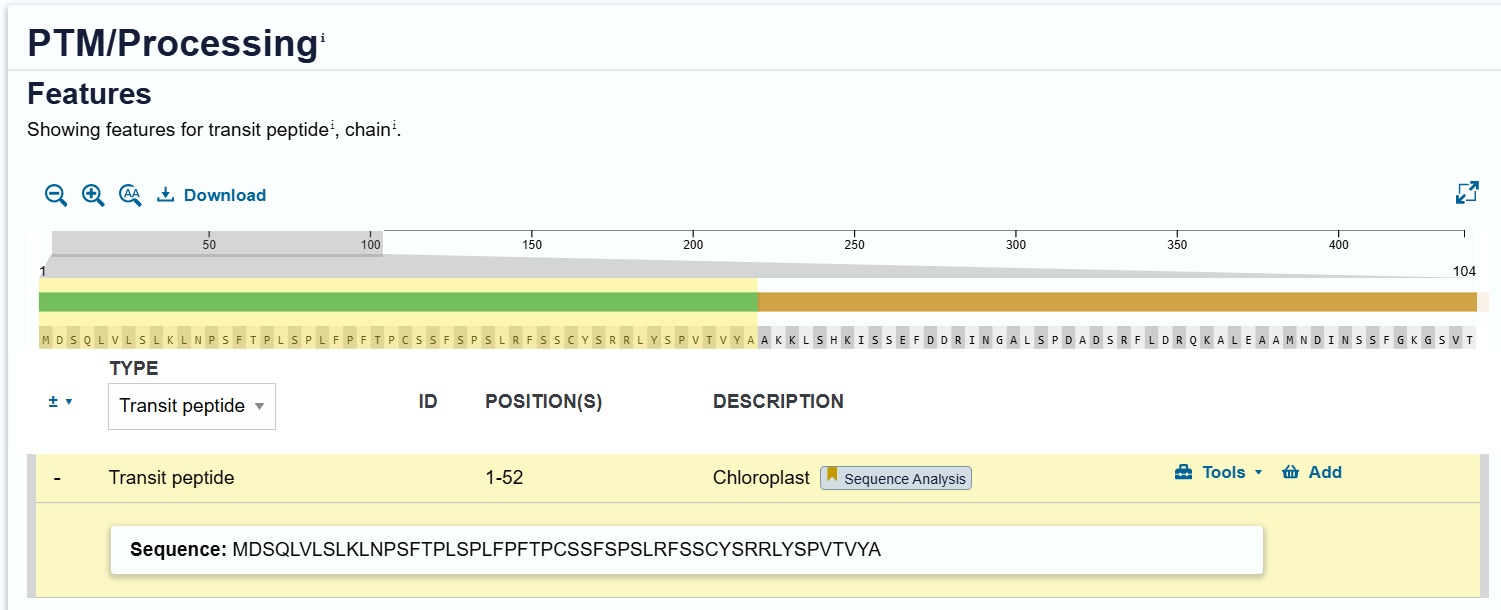
RbcS CTP (P10795):
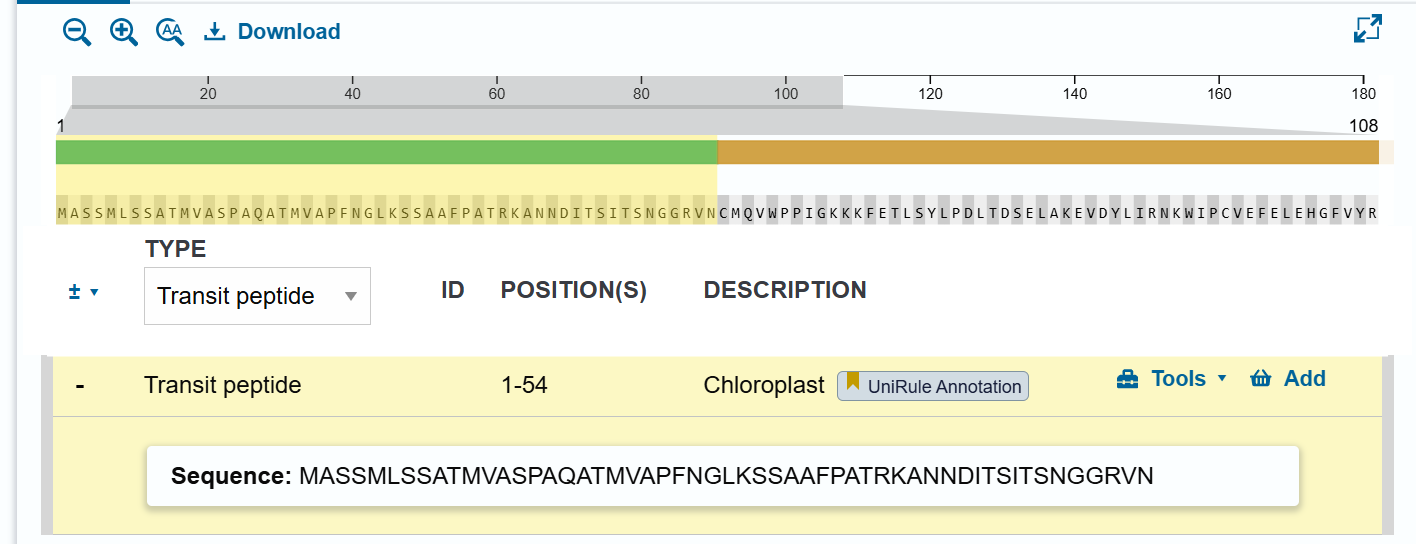
Ferredoxin-2 CTP (P16972):
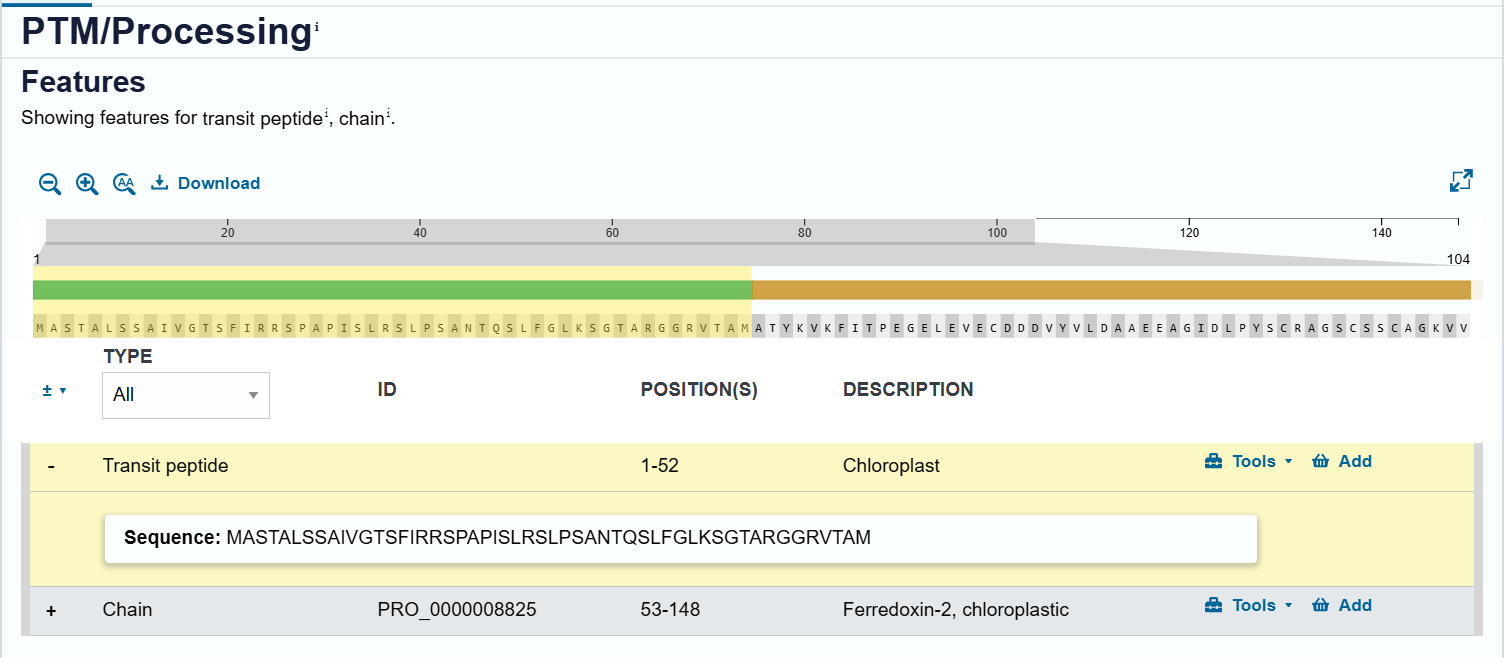
RecA CTP (Q39199):
These sequences were selected to provide alternative chloroplast targeting signals with potentially different import efficiencies, enabling flexibility in construct design.
| CTP | Source Protein | Organism | UniProt Accession | Length (aa) | Key Function |
|---|---|---|---|---|---|
| RbcS CTP | Ribulose-1,5-bisphosphate carboxylase/oxygenase small subunit | Arabidopsis thaliana | P10795 | 57 | Targets proteins to chloroplast stroma (photosynthetic pathway) |
| Ferredoxin-2 CTP | Ferredoxin-2 (chloroplastic) | Arabidopsis thaliana | P16972 | 53 | Directs proteins to chloroplast electron transport system |
| RecA CTP | DNA repair protein RecA homolog 1 | Arabidopsis thaliana | Q39199 | 57 | Targets proteins to chloroplast nucleoids (DNA maintenance) |
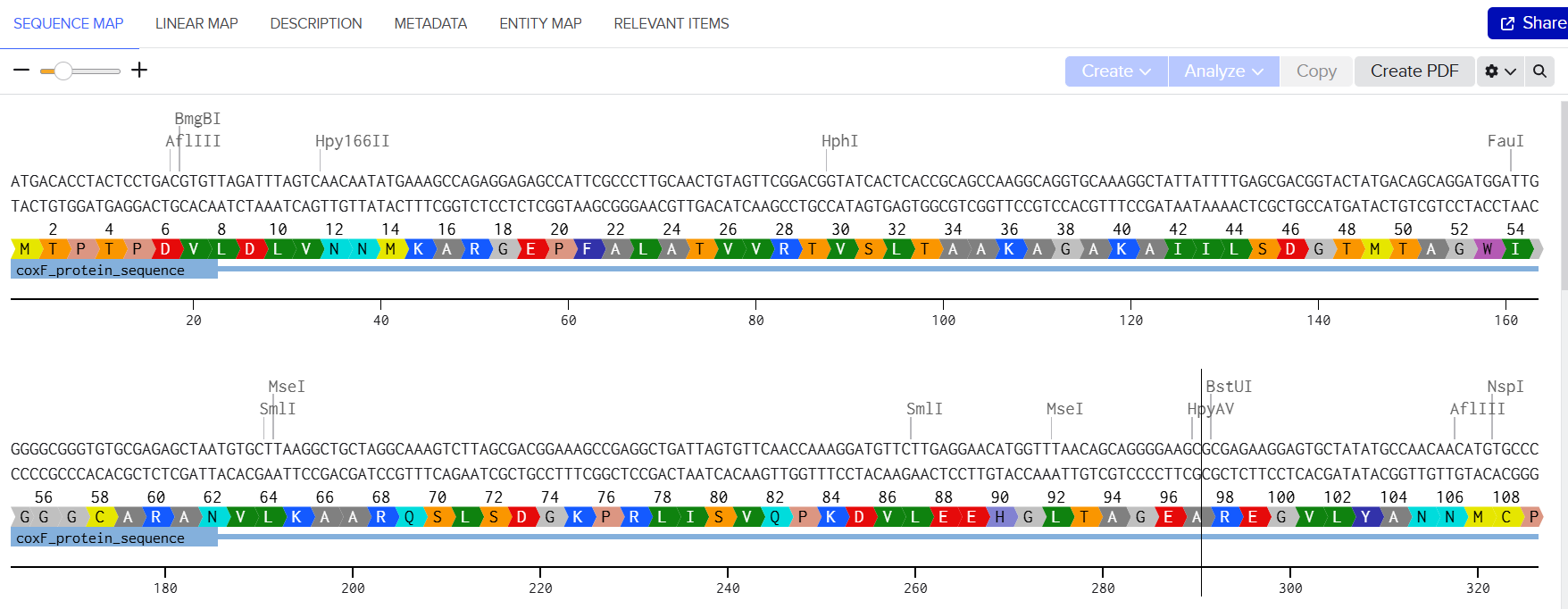
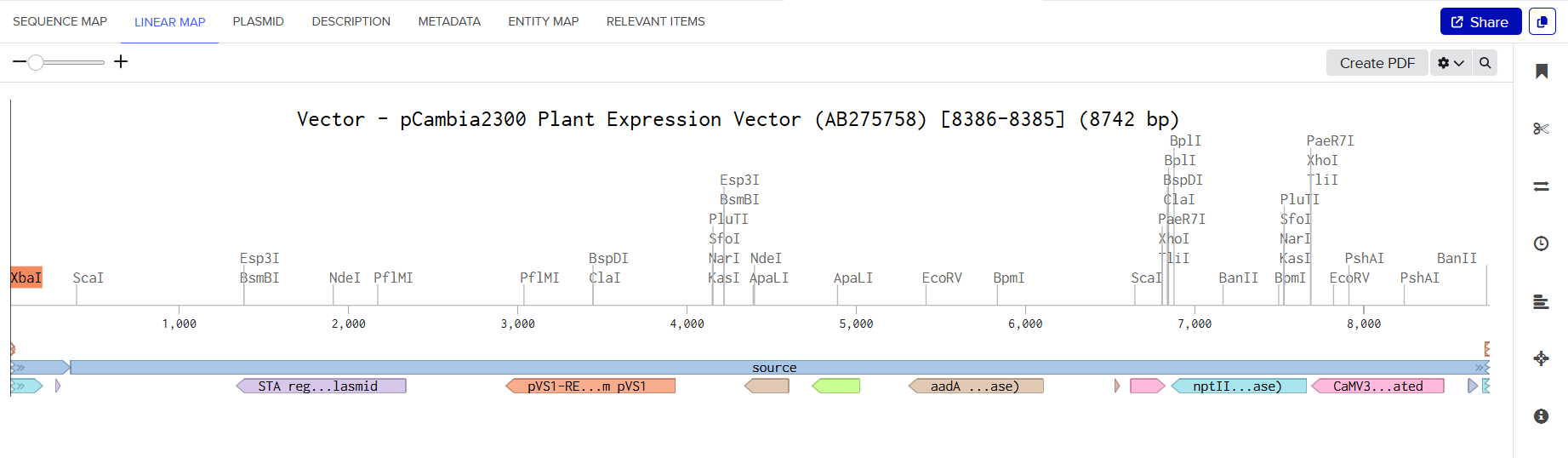
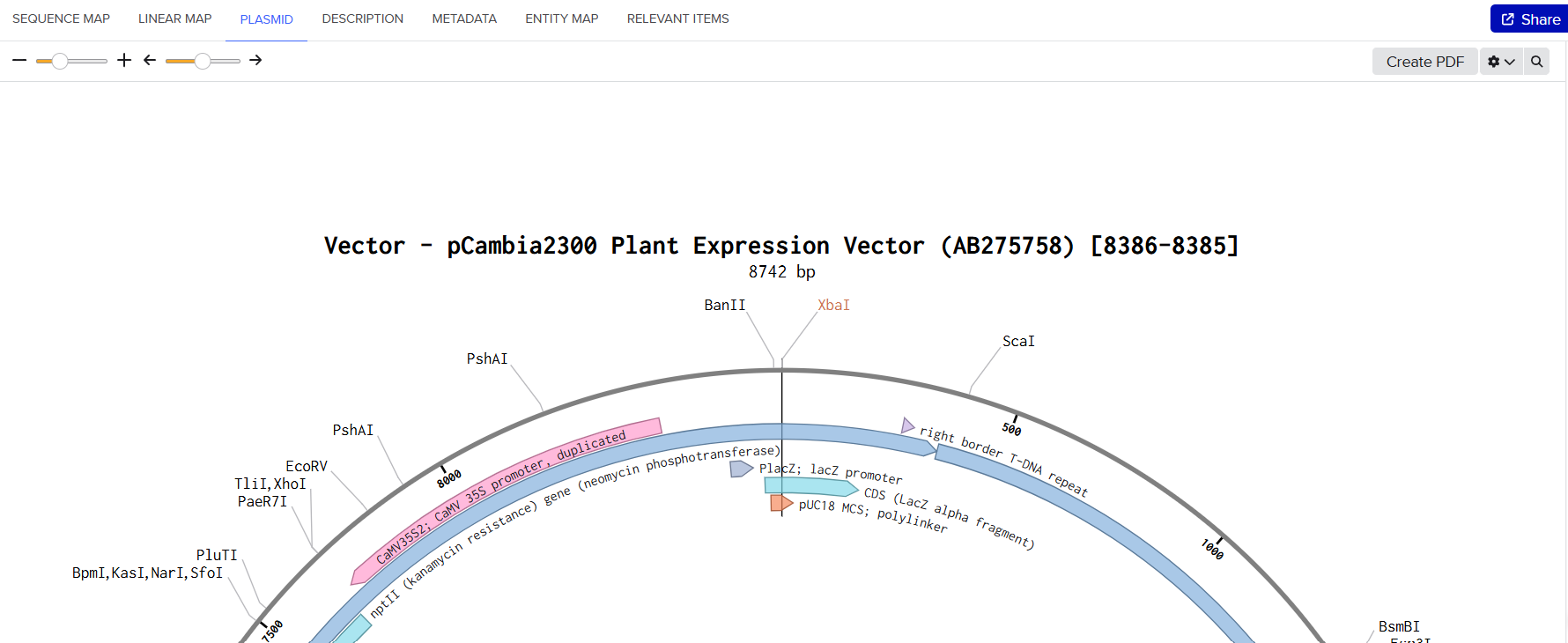
pCAMBIA2300 (Construct 1: Structural genes – coxL, M, S)
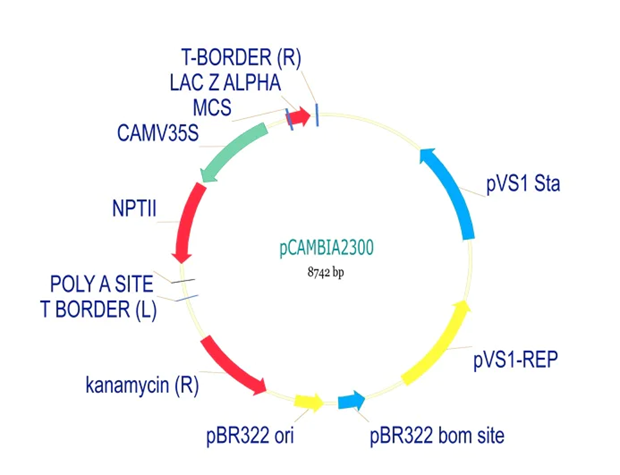
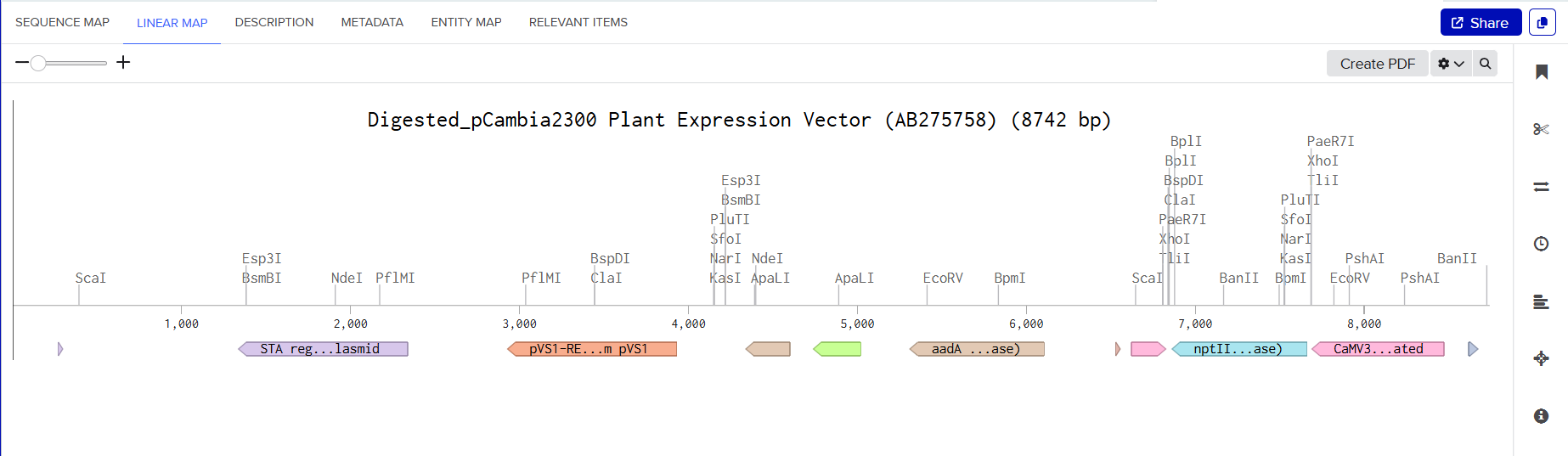
The pCAMBIA2300 vector (GenBank accession AF234315.1) was used as the backbone for the structural gene construct. It is a binary plant expression vector with an approximate size of 8.7 kb, designed as an empty cloning system without any reporter gene, allowing full customization of inserted expression cassettes.
This vector carries the nptII gene, which confers kanamycin resistance in plants, making it suitable for selecting transformants expressing the structural genes (coxL, coxM, coxS). For bacterial propagation, it also includes a kanamycin resistance marker, enabling selection in E. coli prior to Agrobacterium transformation.
The cloning region consists of a pUC18-derived multiple cloning site (MCS) containing standard restriction sites. Additionally, the presence of the pVS1 origin of replication ensures high plasmid stability in Agrobacterium. This vector is well-suited for accommodating multi-cassette inserts, such as the structural gene assembly used in this project.
pCAMBIA1300 (Construct 2: Maturation genes – coxD, E, F, G)
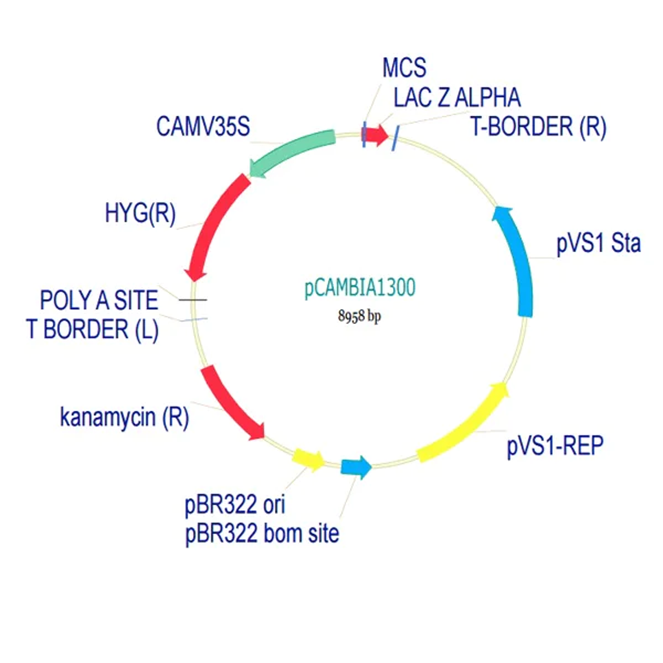
The pCAMBIA1300 vector (GenBank accession AF234296.1) was selected as the backbone for the maturation gene construct. Similar to pCAMBIA2300, it is an empty binary vector (~8.9 kb) designed for flexible insertion of custom genetic elements.
Its key feature is the presence of a hygromycin resistance gene (HygR) for plant selection, which complements the kanamycin resistance used in pCAMBIA2300. This enables the implementation of a dual-selection strategy for identifying co-transformed plants carrying both constructs.
For bacterial selection, pCAMBIA1300 also carries a kanamycin resistance marker, allowing propagation in E. coli. The vector includes a standard pUC18-derived MCS, suitable for inserting large DNA fragments such as the multi-gene maturation cassette (coxD, coxE, coxF, coxG).
Dual-Vector Strategy and Considerations
The combined use of pCAMBIA2300 and pCAMBIA1300 allows efficient co-expression of multiple genes through independent constructs:
| Construct | Genes | Vector | Plant Selection |
|---|---|---|---|
| Structural | coxL, coxM, coxS | pCAMBIA2300 | Kanamycin |
| Maturation | coxD, coxE, coxF, coxG | pCAMBIA1300 | Hygromycin |
This dual-selection system enables reliable identification of plants carrying both constructs. An important technical consideration is that both vectors use kanamycin for bacterial selection, which prevents simultaneous selection of both plasmids in E. coli. Therefore, each construct must be cloned and verified independently before being introduced into Agrobacterium. Co-transformation can then be achieved, followed by selection at the plant level using both antibiotics.
Plant Expression Vectors: pCAMBIA2300 and pCAMBIA1300
For my plant transformation system, I selected two complementary binary vectors: pCAMBIA2300 and pCAMBIA1300, enabling the independent construction and co-expression of structural and maturation gene cassettes. Detailed technical specifications for both vectors can be found in their respective datasheets provided by Abcam for pCAMBIA1300 and pCAMBIA2300.
| Feature | pCAMBIA2300 | pCAMBIA1300 |
|---|---|---|
| Construct Use | Structural genes (coxL, coxM, coxS) | Maturation genes (coxD, coxE, coxF, coxG) |
| Approx. Size | ~8.7 kb | ~8.9 kb |
| Plant Selection Marker | Kanamycin (nptII) | Hygromycin (HygR) |
| Bacterial Selection | Kanamycin | Kanamycin |
| Reporter Gene | None (empty vector) | None (empty vector) |
| Cloning Site | pUC18-derived MCS | pUC18-derived MCS |
| Replication in Agrobacterium | pVS1 origin (high stability) | pVS1 origin (high stability) |
| Insert Capacity | Suitable for large multi-cassette inserts | Suitable for large multi-cassette inserts |
| Main Advantage | Compatible with kanamycin-based plant selection | Enables dual selection with hygromycin |
Sequence Selection and Modification Strategy
The AMV RNA4 enhancer sequence was selected based on the work of Jobling & Gehrke (1987), which demonstrated that this viral leader sequence can strongly enhance translation efficiency in plant systems.
The original viral RNA sequence reported in the article was:
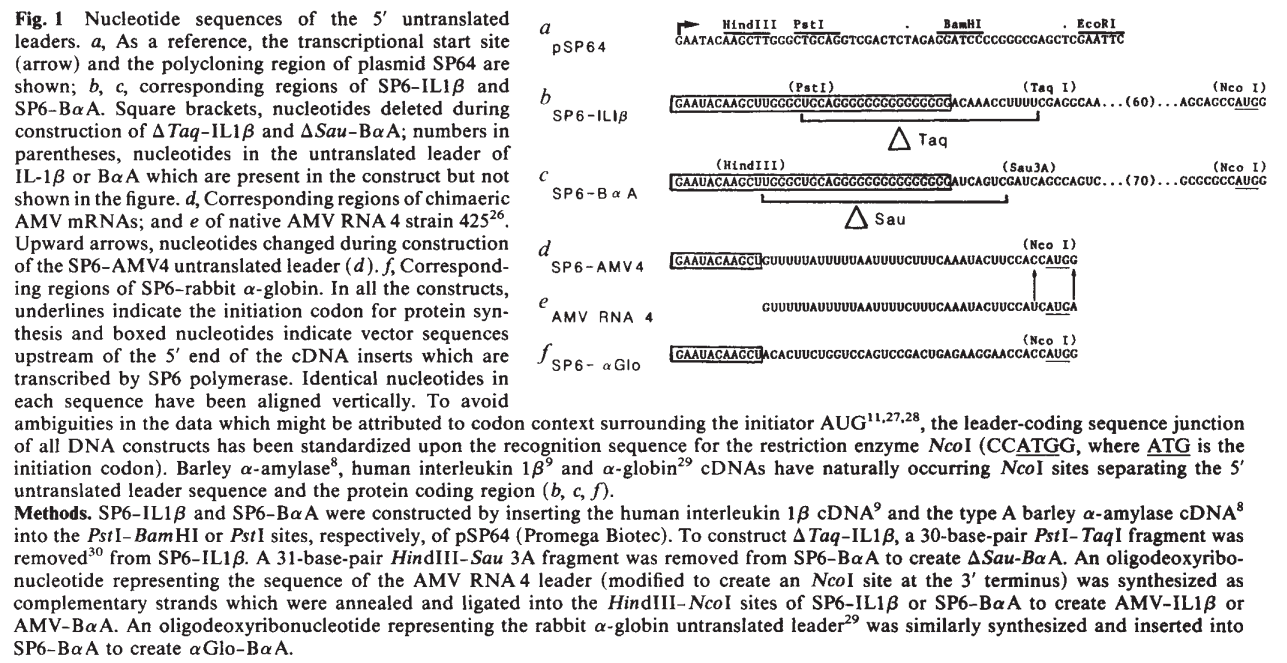
Because the enhancer naturally exists as RNA, the sequence was converted into its complementary DNA (cDNA) equivalent for incorporation into the double-stranded DNA constructs designed for Twist Bioscience synthesis:
During sequence analysis, the native terminal ATG codon present at the 3′ end of the enhancer was identified as a potential problem. If retained, this endogenous ATG could initiate translation before the intended chloroplast transit peptide coding sequence, potentially producing non-functional proteins or frame-shifted translation products.
To prevent this issue, the terminal ATG codon was manually removed, generating the final modified enhancer sequence:
This modification ensured that the first translation initiation codon encountered by the ribosome corresponded to the optimized start codon of the chloroplast transit peptide fusion construct.
Sequence Verification and Validation
Several validation steps were performed after enhancer modification.
First, restriction enzyme screening was conducted to verify that problematic restriction sites such as EcoRI, BamHI, HindIII and XbaI were not unintentionally introduced into the final fused constructs. This step was important for preserving compatibility with downstream cloning verification and diagnostic digestion workflows.
Next, the modified enhancer sequence was evaluated for secondary structure formation to ensure that removal of the terminal ATG did not generate stable hairpins or inhibitory RNA structures that could interfere with ribosome binding or translation initiation.
The final modified AMV enhancer sequence remained structurally suitable for efficient translational enhancement and integration into the multi-cassette CODH system.
To improve protein production from the engineered CODH expression cassettes, the 5′ untranslated region (UTR) of Alfalfa Mosaic Virus (AMV) RNA4 was incorporated as a translational enhancer upstream of each coding sequence. The objective of this element was to increase translational efficiency and improve ribosome recruitment in Nicotiana tabacum cells.
Spacer Design Strategy and Selection
To minimize unwanted interactions between adjacent expression cassettes, neutral spacer sequences were introduced between transcriptional units in the final multi-gene constructs.
Rather than reusing the same spacer repeatedly, four different spacer sequences were designed for the different cassette junctions (Spacer 1–4). Using identical spacer sequences multiple times is generally discouraged because repeated DNA regions can increase the probability of homologous recombination during bacterial cloning or after plant transformation, potentially leading to construct rearrangement or partial deletion.
For this reason, unique spacer sequences were selected for each junction to improve structural stability of the final constructs.
To generate biologically neutral spacers, fragments derived from the genome of Enterobacteria phage lambda NC_001416.1) were used. Lambda phage DNA is commonly utilized in synthetic biology as inert “stuffer DNA” because it lacks known regulatory activity in plant cells, contains no plant-specific coding regions, is well characterized, and minimizes unintended interactions within eukaryotic systems.
Each spacer was designed to be approximately 100 bp long. Although this represents the minimal recommended spacer size, it was considered sufficient to physically separate neighboring transcriptional units, reduce transcriptional and steric interference between cassettes, and improve overall construct organization during multi-cassette assembly.
Spacer Validation and Optimization
Before final selection, several validation steps were performed to ensure that the spacer sequences were suitable for stable multi-cassette assembly and plant expression.
First, all spacer sequences were designed to be different from one another in order to reduce repeated DNA regions and minimize the risk of homologous recombination within the construct.
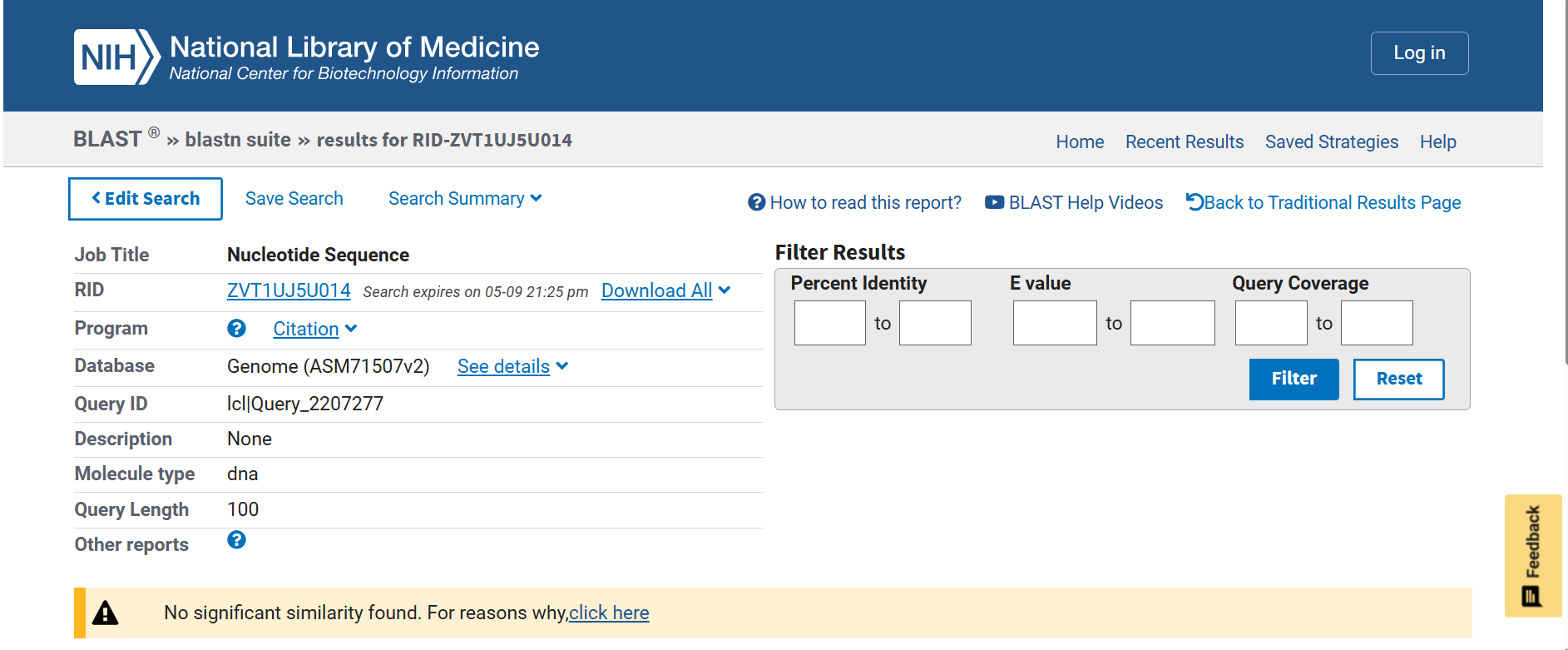
Next, each spacer was analyzed against the Nicotiana tabacum reference genome GCF_000715075.1) using BLASTn to verify genome neutrality. The analysis confirmed the absence of significant similarity with endogenous tobacco genes or regulatory regions, reducing the risks of off-target recombination, post-transcriptional gene silencing (PTGS), and unintended genomic interactions.
![]()
![]()
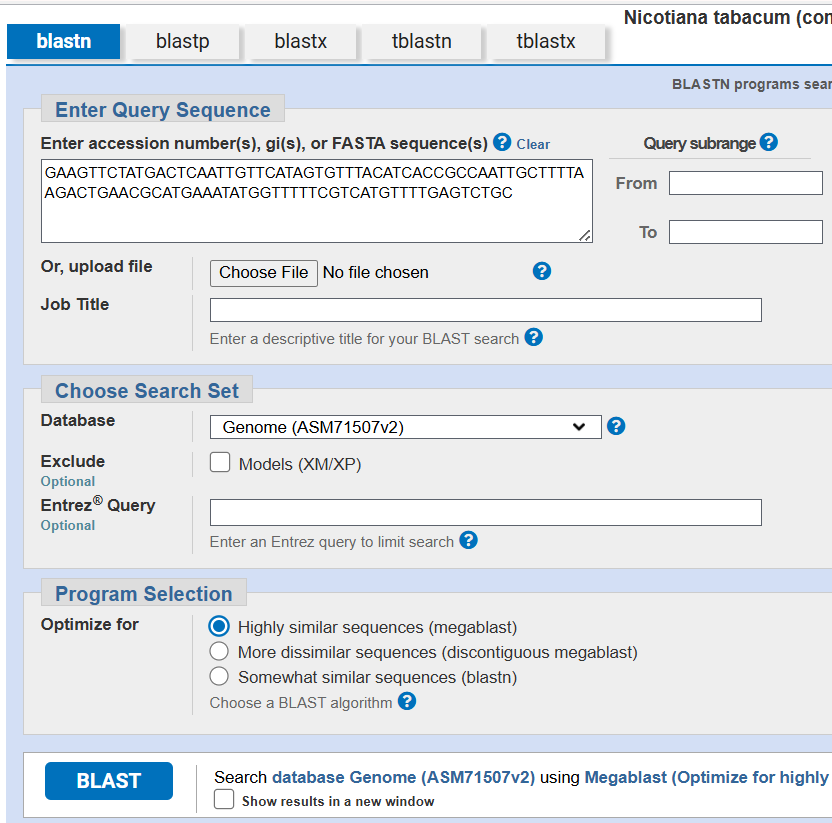
![]()
![]()
The spacer sequences were also screened to avoid problematic restriction enzyme recognition sites that could interfere with downstream cloning and Gibson Assembly workflows.
Finally, GC content was maintained within moderate ranges (~37–48%) to avoid extremely AT-rich or GC-rich regions that could negatively affect DNA synthesis stability, PCR amplification, or secondary structure formation.
The final validated spacer sequences are presented below:
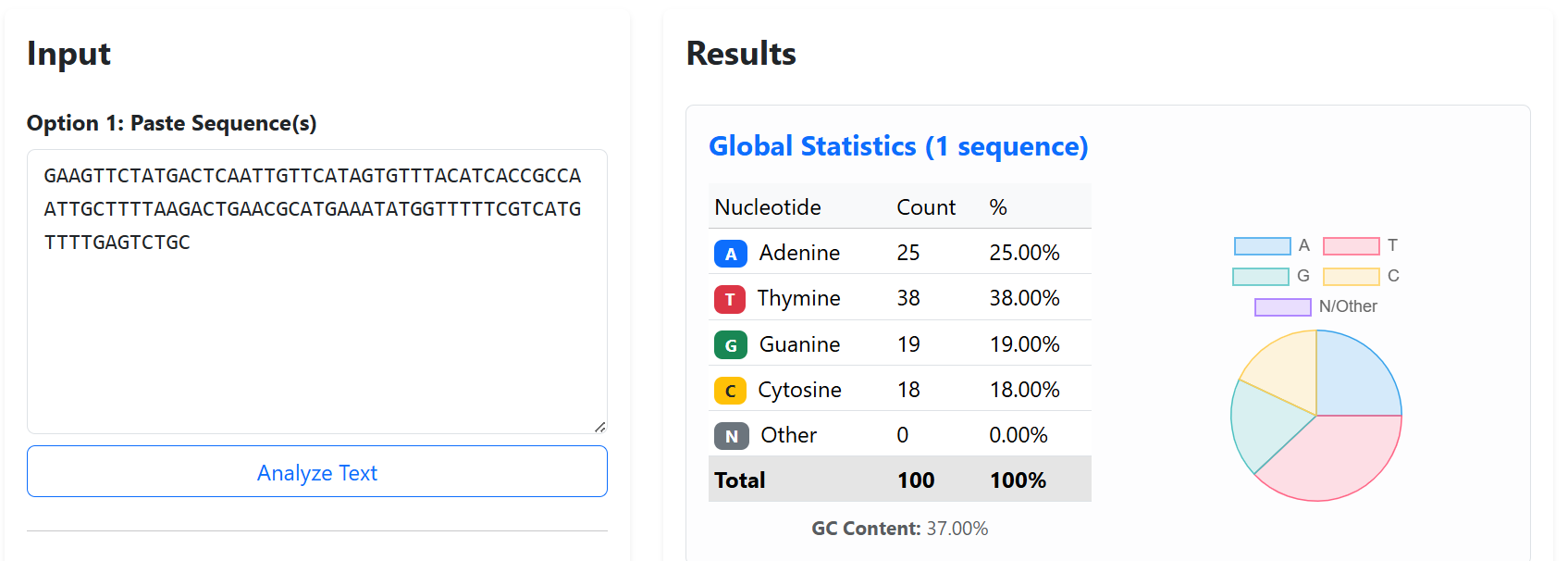
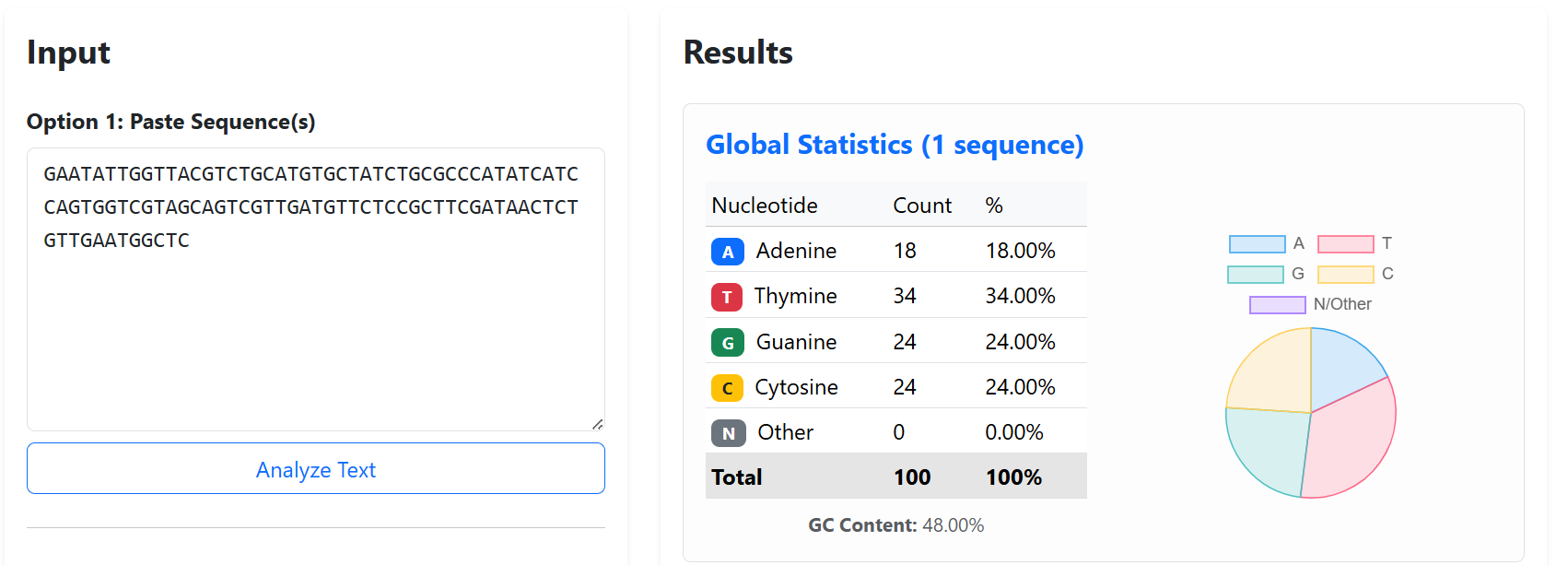
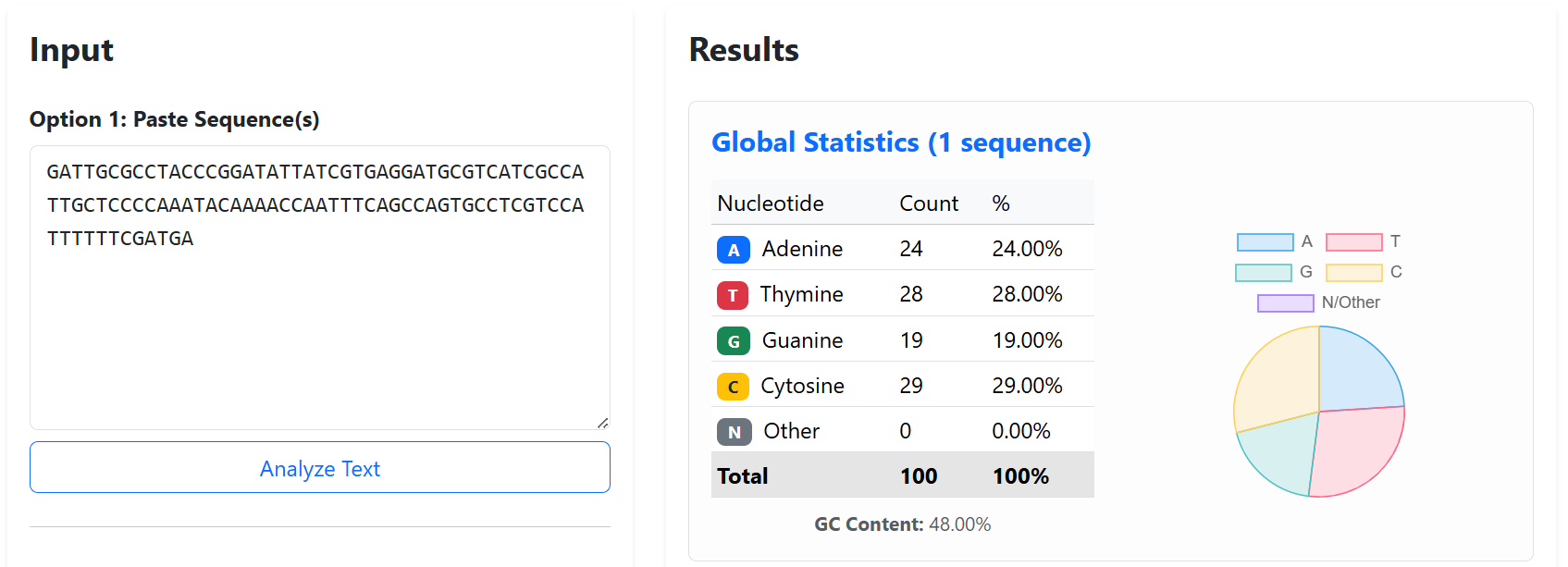
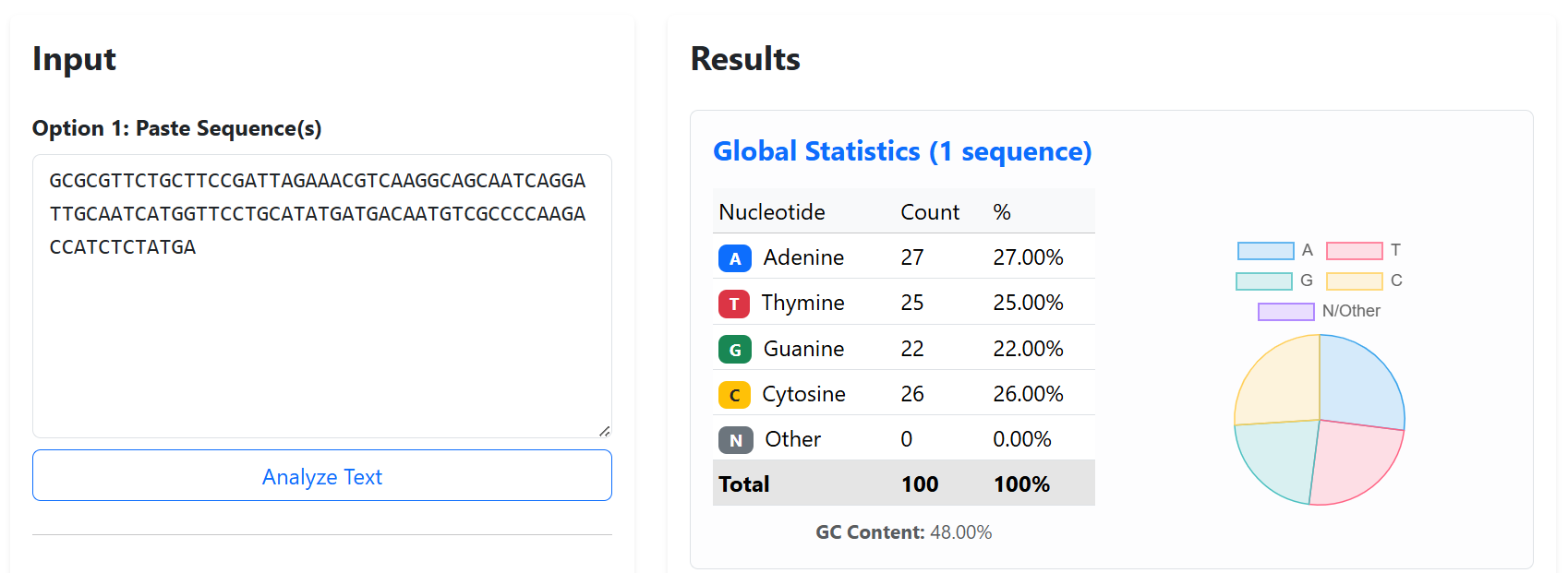
To improve the structural stability and transcriptional insulation of the multi-cassette CODH constructs, neutral spacer sequences were introduced between adjacent expression cassettes. These spacers were designed to reduce promoter and terminator interference, minimize homologous recombination risks, and prevent unwanted interactions between neighboring transcriptional units during cloning and plant expression.
Sources:
Codon Optimization and Sequence Adaptation processes:
1. Start Codon Verification and Correction
As an initial step, all seven CODH genes were carefully inspected to verify the presence of a valid translation initiation codon. A critical adjustment was required for the coxM gene, which was the only gene using an alternative bacterial start codon (GTG) instead of the canonical ATG.
Since plant translation machinery, particularly in Nicotiana tabacum, strictly recognizes ATG as the initiation codon, the native GTG was manually corrected to ATG during the optimization process. This modification ensures proper translation initiation while preserving the original amino acid sequence of the CoxM protein.
2. Codon Optimization Strategy
Codon optimization was performed using the Benchling Codon Optimization Tool, applying the “Match Codon Usage” algorithm. This approach was selected because it reproduces the natural codon distribution of the target organism rather than overusing only the most frequent codons, thereby improving mRNA stability and translation efficiency.
![]()
![]()
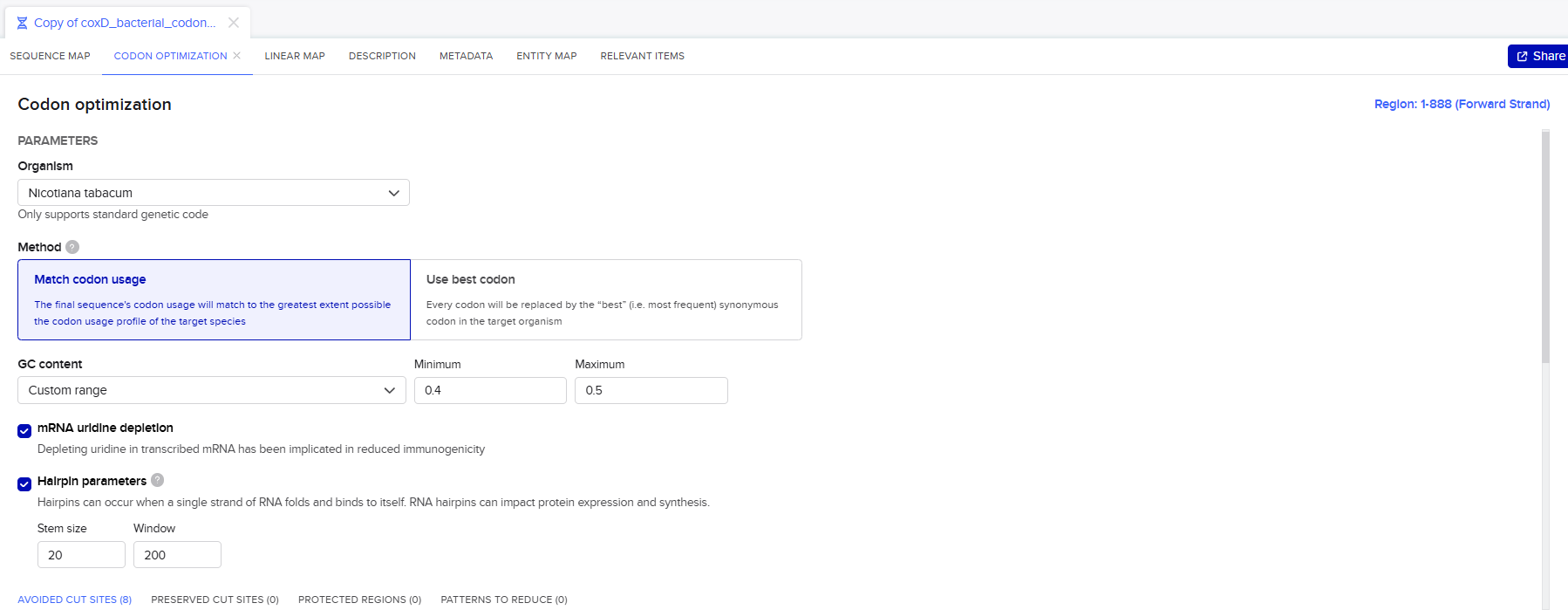
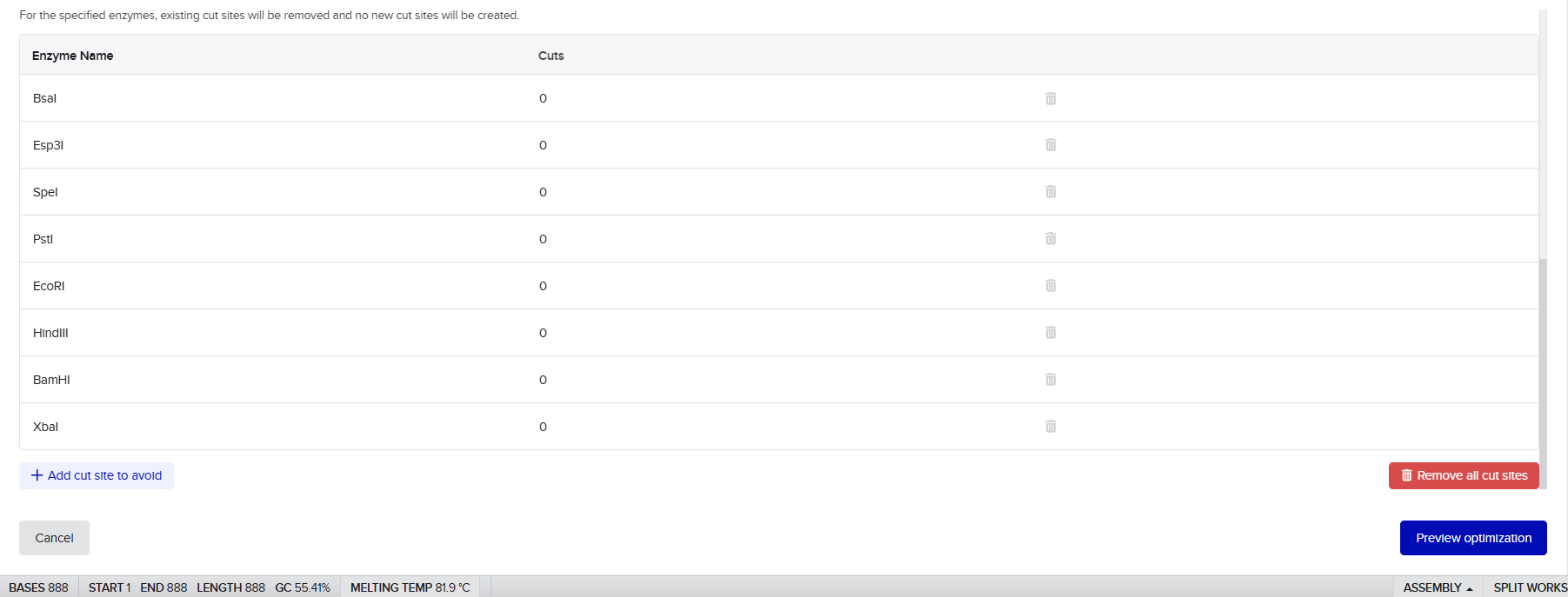
The optimization process was carried out under the following parameters:
3. Results and Validation
Following optimization, all sequences were evaluated using CAIcal to assess codon adaptation and overall sequence quality.
![]()
![]()
The analysis demonstrated consistently strong performance across all seven genes as showed in the following table:
| Gene Name | Length (bp) | CAI Score | Total GC% | GC at 3rd Position | Nc Value | Expression Potential |
|---|---|---|---|---|---|---|
| CoxL | 2430 | 0.773 | 46.3% | 40.0% | 57.0 | Excellent |
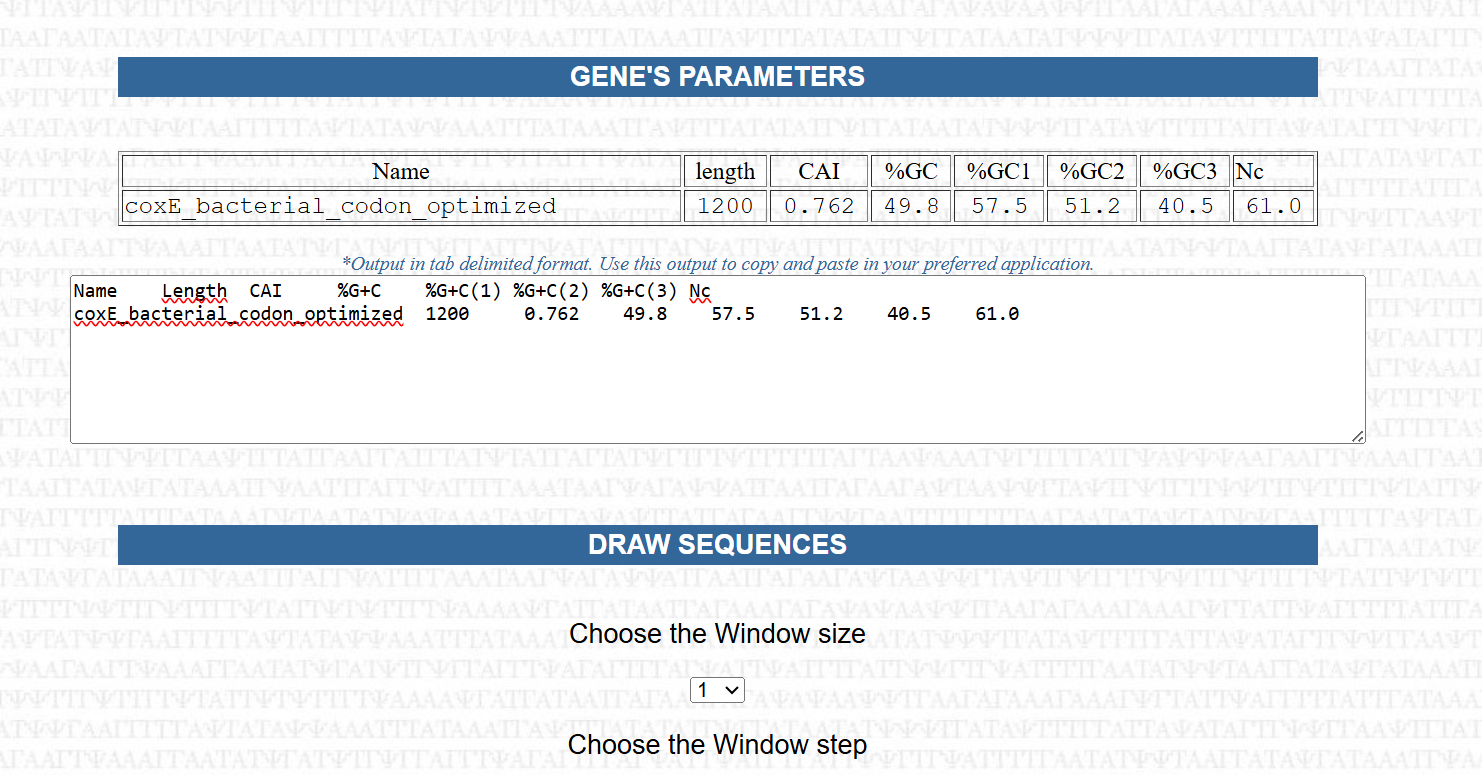
| CoxE | 1200 | 0.762 | 49.8% | 40.5% | 61.0 | Very Good |
| CoxG | 618 | 0.760 | 46.9% | 40.8% | 61.0 | Very Good |
| CoxS | 501 | 0.759 | 47.7% | 43.1% | 61.0 | Very Good |
| CoxD | 888 | 0.756 | 46.8% | 40.5% | 61.0 | Very Good |
| CoxF | 843 | 0.748 | 49.5% | 39.9% | 61.0 | Very Good |
| CoxM | 867 | 0.747 | 49.5% | 39.1% | 61.0 | Very Good |
The Codon Adaptation Index (CAI) values ranged from 0.747 to 0.773, indicating a high level of similarity to codon usage patterns found in highly expressed genes of Nicotiana tabacum. This suggests that the optimized sequences are well-suited for efficient translation in the plant host.
The overall GC content was successfully adjusted to a range of 46.3% to 49.8%, aligning with the typical GC composition of plant genes. This represents a significant improvement compared to the original bacterial sequences and contributes to better transcriptional stability and compatibility with the host genome.
The Effective Number of Codons (Nc) values ranged from 57.0 to 61.0, reflecting a balanced codon usage without excessive repetition. This indicates that the sequences maintain sufficient variability, which is important for avoiding issues such as tRNA depletion or translational bottlenecks.
Additionally, the GC content at the third codon position was maintained at approximately 40%, which is considered optimal for the “wobble” position. This balance supports efficient recognition by plant tRNAs and contributes to overall translation efficiency.
To further validate the integrity of the optimization process, both the raw bacterial sequences and the codon-optimized sequences were translated into their corresponding amino acid sequences.
![]()
![]()
![]()
![]()
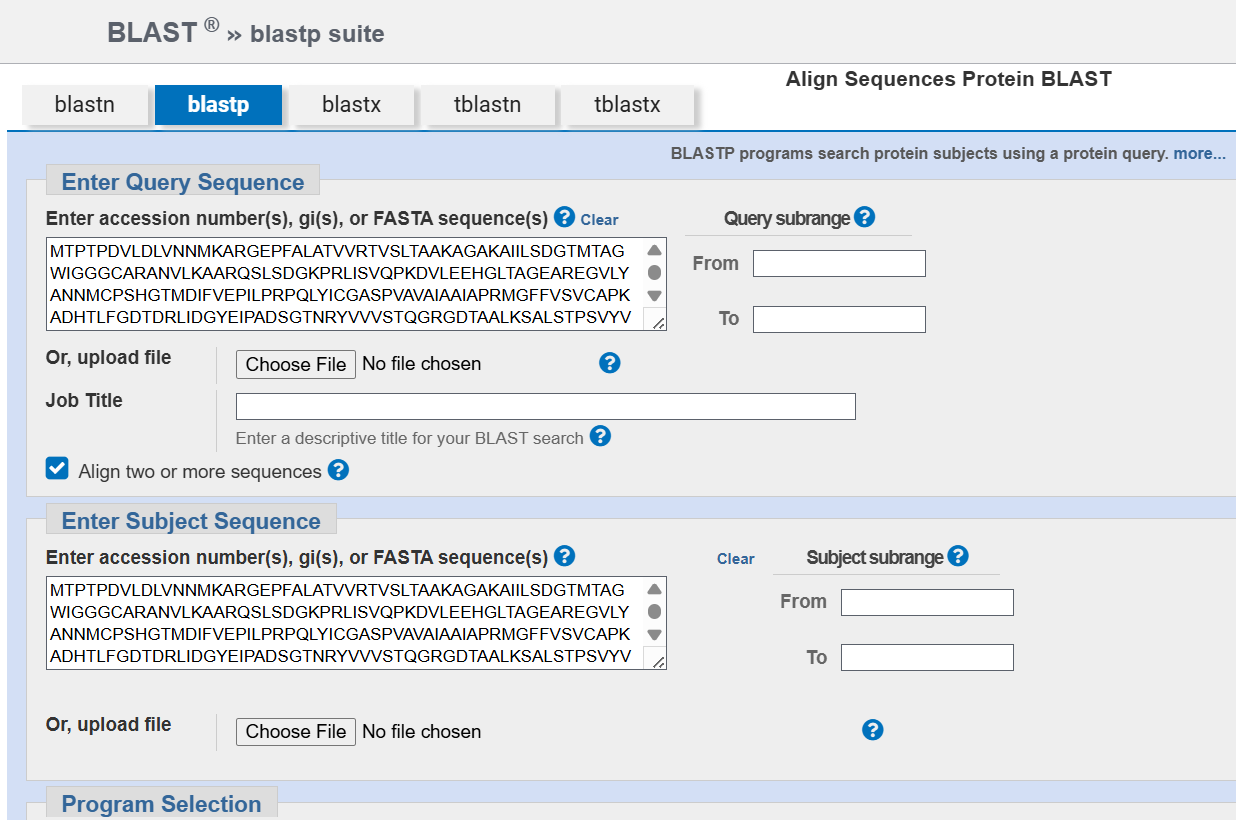
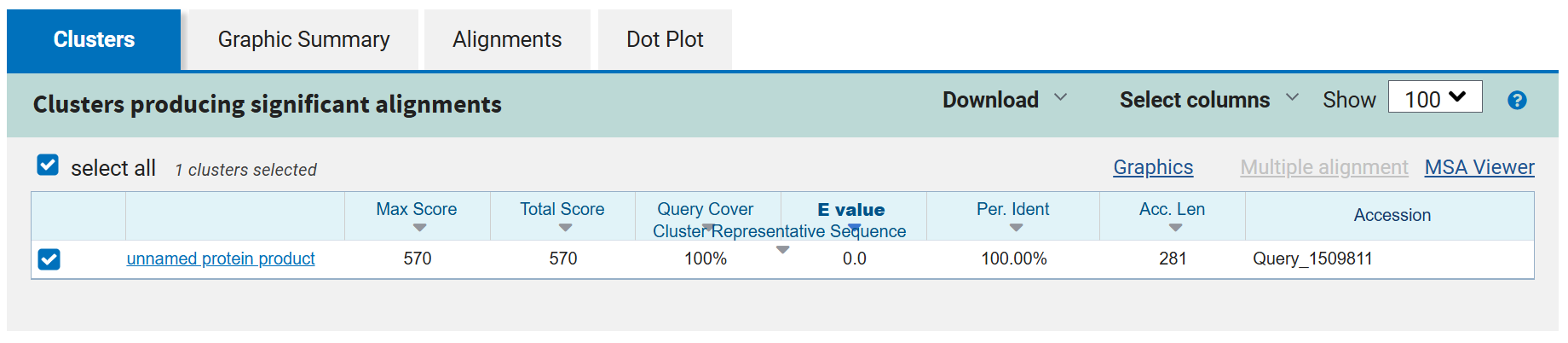
A pairwise comparison was then performed using BLASTp alignment to assess sequence similarity.
 The results confirmed that all optimized proteins are identical to their native counterparts, with no changes in amino acid sequence.
The results confirmed that all optimized proteins are identical to their native counterparts, with no changes in amino acid sequence.
![]()
![]()
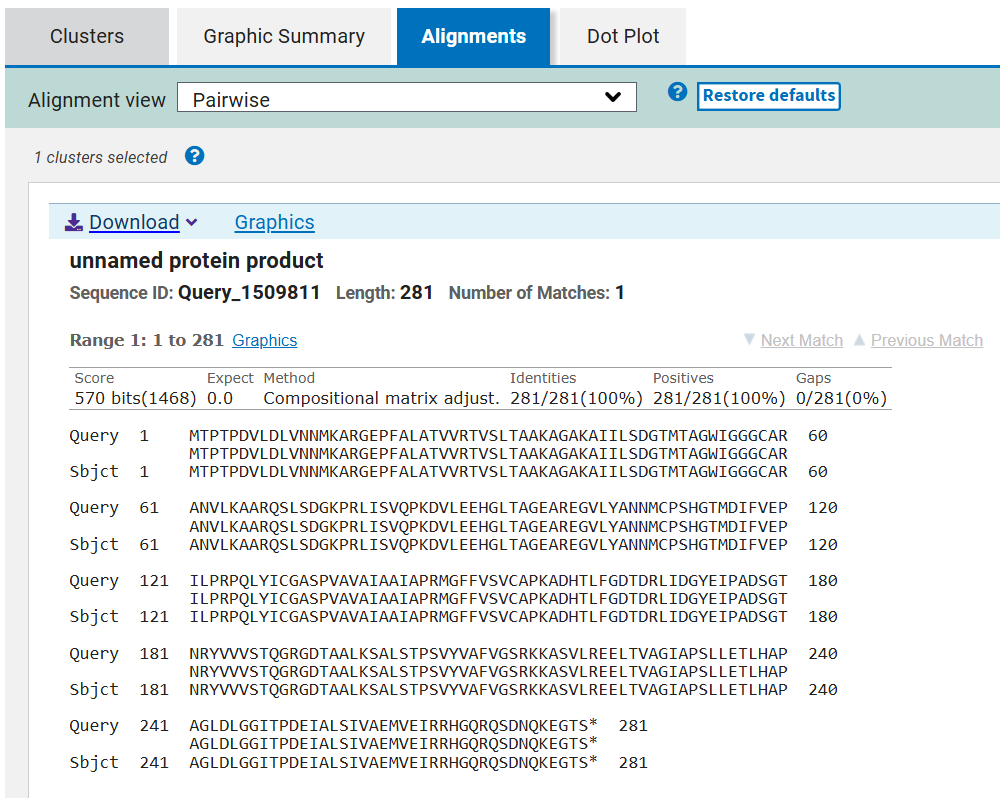 This verification step ensures that codon optimization only affected synonymous codon usage without altering protein structure or function, preserving the biological activity of all seven CODH components.
This verification step ensures that codon optimization only affected synonymous codon usage without altering protein structure or function, preserving the biological activity of all seven CODH components.
The resulting codon-optimized cox genes sequences are as follows:
Back-Translation and Codon Optimization of Engineered CTP Sequences
After designing and validating the engineered chloroplast transit peptides (CTPs) at the amino acid level, the next step was to convert these protein sequences into DNA sequences that are fully compatible with the plant expression system. This process ensures that the “targeting signals” (CTPs) are translated efficiently in Nicotiana tabacum, just like the CODH subunits.
Since these CTPs are fused directly to the N-terminus of the CODH proteins, it is essential that they follow the same genetic design rules as the rest of the system to guarantee consistent expression and proper chloroplast targeting.
Back-Translation Strategy
The engineered CTP amino acid sequences (RbcS, Fer2, and RecA), including the modified junction motifs (VNA–AM, VTA–AM, and TVY–AA), were back-translated into DNA sequences using the Benchling Codon Optimization tool.
This step converted the peptide sequences into nucleotide sequences optimized for expression in Nicotiana tabacum, ensuring compatibility with the plant’s codon usage preferences and translation machinery.
Codon Optimization Consistency
The same optimization framework used for the seven CODH genes was applied to the CTP sequences to maintain full compatibility and expression uniformity across the entire multigene construct. This guarantees that all components of the system follow the same expression logic within the plant cell.
Key Adjustment: Hairpin Structure Control
A specific adjustment was introduced during this step due to the short length of CTP sequences. The standard secondary structure analysis settings were not optimal for short peptide-encoding regions, which can lead to inaccurate prediction of stable RNA hairpins near the translation start site.
To address this, the hairpin analysis window was reduced to 100 to improve sensitivity for short sequences and to ensure that no stable secondary structures form at the 5’ region that could interfere with ribosome binding or early translation.
The following are the final codon-optimized CTP sequences generated in this step:
Objective:
Codon optimization is a fundamental step in synthetic biology when expressing genes across different organisms. Although the genetic code is universal, meaning that most organisms use the same codons to encode the same amino acids, the frequency at which specific codons are used varies between species. This phenomenon is known as codon usage bias.
Each organism has evolved to preferentially use certain codons over others, largely reflecting the abundance of corresponding transfer RNAs (tRNAs). As a result, a gene originating from one organism may be inefficiently translated when introduced into another if its codon usage does not match the host’s preferences.
In this project, the seven genes encoding the Carbon Monoxide Dehydrogenase (CODH) system originate from a bacterium and are being expressed in a plant (Nicotiana tabacum). Without codon optimization, several issues can arise:
Because the CODH system depends on the coordinated expression of multiple subunits and maturation proteins, balanced and efficient expression of each gene is essential. Even a single poorly expressed component could compromise the functionality of the entire enzyme complex.
Therefore, codon optimization is not just a technical adjustment but a critical requirement for functional expression. In this step, each gene sequence is redesigned to match the codon usage preferences of Nicotiana tabacum, while preserving the exact amino acid sequence of the encoded proteins. Additional considerations, such as avoiding mRNA secondary structures, eliminating cryptic splice sites, and maintaining appropriate GC content, are also taken into account.
Sources:
1. Selection of Chloroplast Transit Peptides
To improve targeting efficiency and avoid using repeated sequences, three different plant CTPs were selected:
These CTPs are derived from naturally chloroplast-targeted plant proteins (Arabidopsis thaliana) and are known to efficiently direct proteins into the chloroplast. Instead of using the same CTP for all seven genes, different peptides were intentionally distributed across the CODH subunits.
2. Fusion Design and Junction Engineering
Each CODH protein was fused to a CTP at its N-terminus. To make sure the protein folds correctly after cleavage, the fusion included the first 60 amino acids of each CODH protein, which were obtained using the ExPASy ProtParam tool.
A very important step was designing the junction between the CTP and the CODH protein. This region was carefully modified to include a cleavage motif recognized by the chloroplast enzyme responsible for removing the transit peptide:
(Val/Ile)-X-(Ala/Cys) ↓ Ala
To create this motif, small changes were made at the end of the CTP sequence as showed in the following sequences:
This allowed a smooth transition between the CTP and the CODH protein while keeping both targeting and protein structure intact.
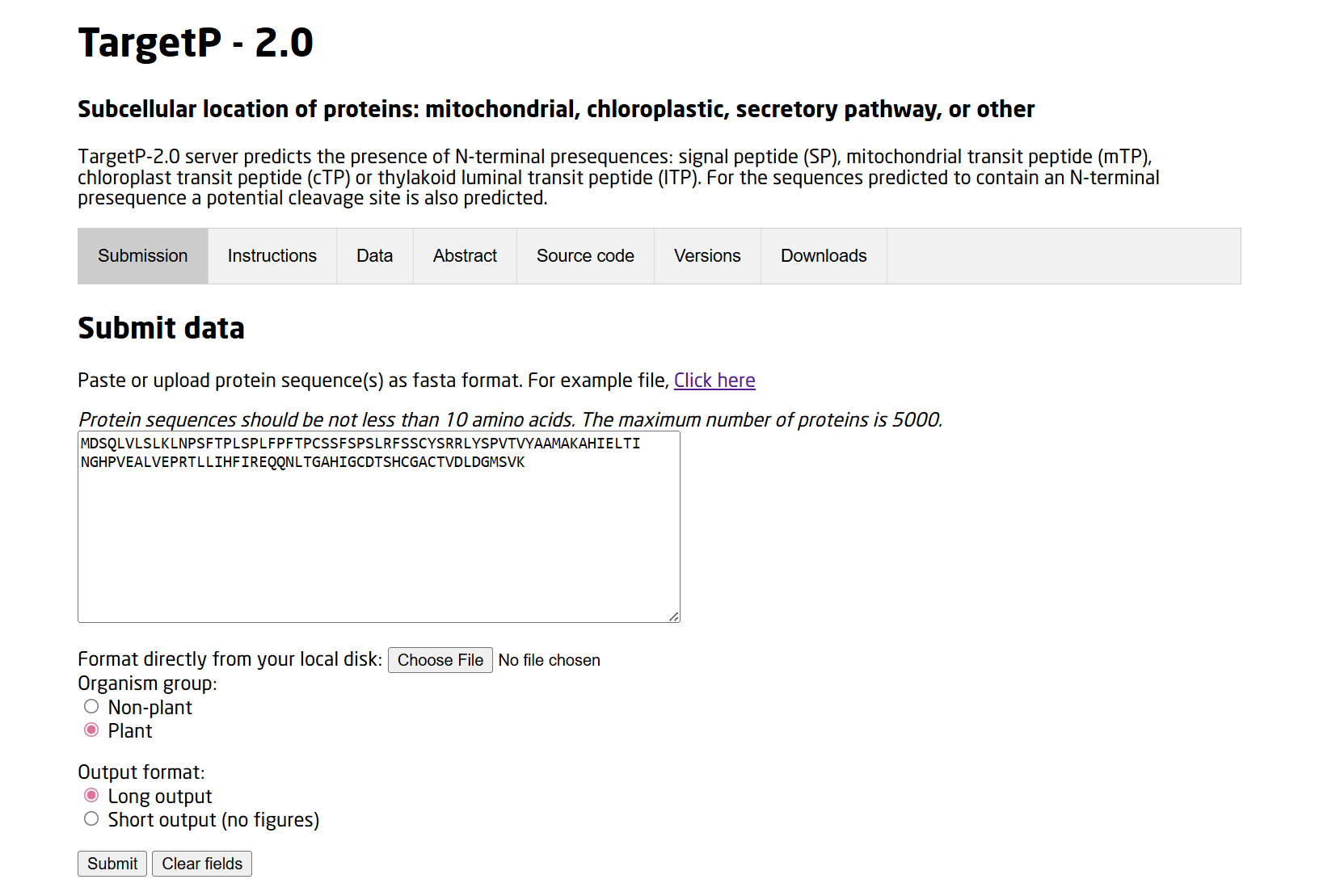
3. In Silico Validation of Targeting and Cleavage
All fusion sequences were analyzed using TargetP 2.0 to check two things:
![]()
![]()
The results showed that all seven proteins are predicted to be targeted to the chloroplast, which confirms that the CTPs are working correctly:
.png)
.png)
.png)
.png)
.png)
.png)
.png)
Summary of the results:
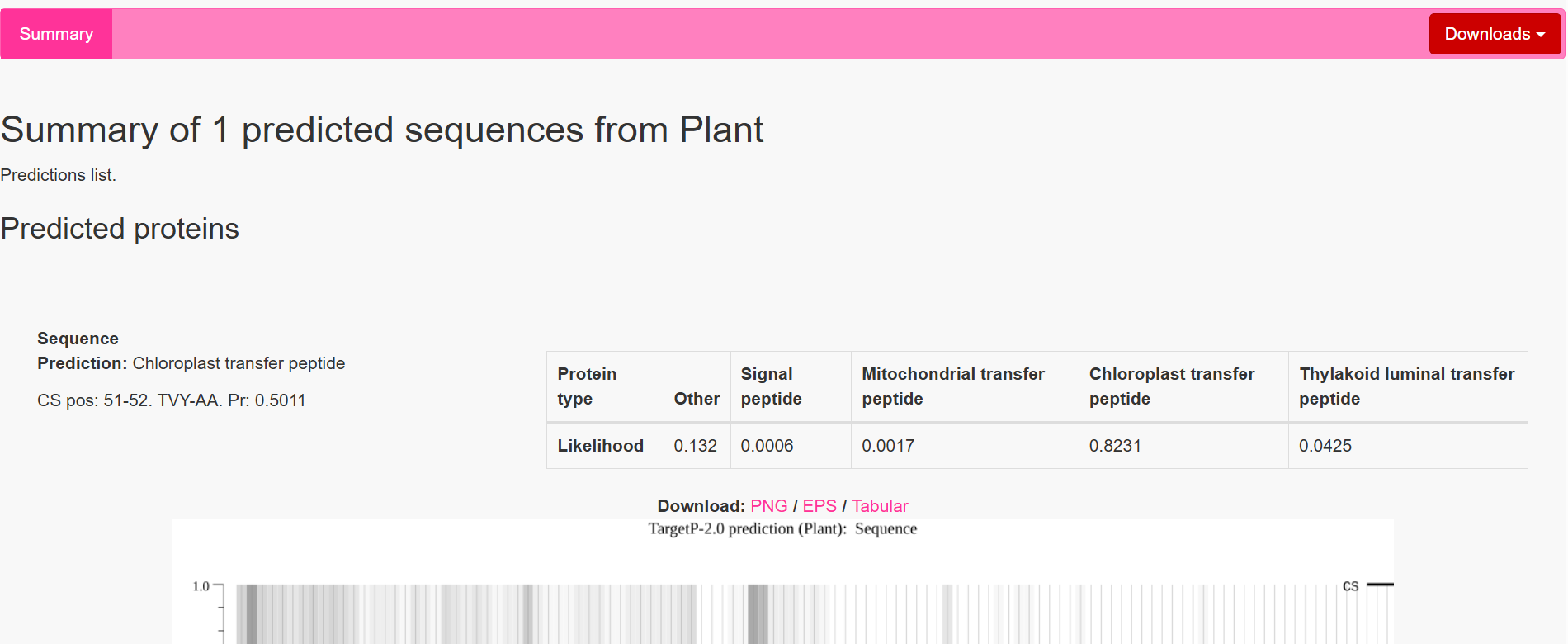
| Gene | CTP Source | Cleavage Site (CS Position) | Junction Motif (CTP → CODH) | Cleavage Probability (Pr) | Prediction |
|---|---|---|---|---|---|
| coxD | RbcS | 55–56 | VNA ↓ AM | 0.5216 | Chloroplast transfer peptide |
| coxE | Fer2 | 51–52 | VTA ↓ AM | 0.3172 | Chloroplast transfer peptide |
| coxF | RecA | 51–52 | TVY ↓ AA | 0.4989 | Chloroplast transfer peptide |
| coxG | RbcS | 55–56 | VNA ↓ AM | 0.5923 | Chloroplast transfer peptide |
| coxL | RbcS | 55–56 | VNA ↓ AM | 0.4842 | Chloroplast transfer peptide |
| coxM | Fer2 | 51–52 | VTA ↓ AM | 0.7188 | Chloroplast transfer peptide |
| coxS | RecA | 51–52 | TVY ↓ AA | 0.5011 | Chloroplast transfer peptide |
Interpretation of Results
Overall, the results indicate successful design of functional targeting signals for all CODH subunits:
All constructs were confidently predicted as chloroplast-targeted proteins, confirming that the added CTPs are functional. The cleavage sites align well with the engineered junction motifs, demonstrating that the proteins are likely to be correctly processed after import.
The coxM fusion showed the highest cleavage probability (Pr = 0.7188), indicating highly efficient targeting and processing. Other subunits showed moderate probabilities (around 0.48–0.59), which are still within acceptable ranges for functional targeting. The coxE fusion presented a lower probability (Pr = 0.3172). Although this suggests potentially less efficient cleavage, the sequence still satisfies the required motif and is expected to remain functional, as variability in cleavage efficiency is common in heterologous systems.
Most constructs showed cleavage occurring exactly at the designed motif, typically between amino acid positions 51–56, depending on the transit peptide used.
However, a notable observation was made for two constructs, coxF and coxS, where the predicted cleavage site occurred slightly upstream of the engineered junction, specifically just before the designed alanine-alanine region rather than directly within it.
This slight variation in cleavage position is consistent with the known behavior of the chloroplast Stromal Processing Peptidase. Rather than recognizing a single fixed sequence, the enzyme identifies a broader structural and sequence context, which allows for some flexibility in the exact cleavage position. As a result, small shifts of one or two amino acids relative to the designed motif are commonly observed in both native and engineered proteins.
In this case, although the cleavage in coxF and coxS occurs marginally earlier than expected, it remains within a functionally acceptable region. The resulting mature proteins retain nearly identical N-terminal sequences and are not expected to lose any essential structural or functional elements. Importantly, the targeting prediction remains strong, confirming that the proteins are still efficiently directed to the chloroplast.
Therefore, this variability does not compromise the overall design. All fusion constructs are considered valid, and no redesign was required. Instead, this observation reflects the inherent flexibility of chloroplast protein processing and further validates the robustness of the engineered system.
Objective
Subcellular targeting is a critical step in synthetic biology when expressing proteins in a new host organism. In plant cells, proteins must be directed to the correct organelle in order to function properly. This is especially important for metabolic pathways that depend on specific cellular environments.
In this project, the seven proteins forming the Carbon Monoxide Dehydrogenase (CODH) system originate from a bacterium. However, in plant cells, these proteins need to function inside the chloroplast, where photosynthesis occurs and where the produced CO₂ can be directly reused.
Bacterial proteins do not naturally contain signals that allow them to enter plant organelles. As a result, if they are expressed without modification, they will remain in the cytosol, where they may not fold correctly, may not interact properly with other subunits, and may fail to form a functional enzyme complex.
To solve this problem, each CODH protein must be fused to a chloroplast transit peptide (CTP). These short sequences are naturally found in plant proteins and act as targeting signals that guide newly synthesized proteins into the chloroplast. Once the protein reaches the chloroplast, the transit peptide is cleaved, releasing the mature protein in its functional form.
Sources:
Objective
After completing sequence collection, codon optimization, chloroplast transit peptide fusion, and cleavage site verification, the next objective was to design the regulatory architecture controlling expression of the seven CODH genes inside Nicotiana tabacum cells.
The CODH pathway is composed of multiple interacting structural and maturation proteins that must function together in a coordinated manner. Because of this, maintaining balanced expression between the genes is critical. Excessive or insufficient expression of specific subunits could negatively affect protein folding, complex assembly, chloroplast burden, and overall enzyme functionality.
Therefore, the main goal of this phase was to design a biologically balanced expression system by selecting suitable promoter–terminator combinations capable of driving efficient and coordinated expression of all seven CODH genes.
The initial plan for this phase was to:
The final promoter–terminator combinations were selected based on relative promoter strengths, functional compatibility between regulatory elements, and expected expression balance across the CODH pathway. Terminator efficiency values were taken from reported comparative plant expression data in Shakhova et al. (2022). The overall performance scores were predicted using an AI-based evaluation (Claude AI) integrating promoter strength, terminator efficiency, and expected transcriptional balance.
| Gene | Promoter | Strength | Terminator | Combined Performance |
|---|---|---|---|---|
| coxL | D100 | 2.2× | tOCS | ★★★★ |
| coxM | SM | 2.1× | tHSP18.2 | ★★★★ |
| coxS | FMV 34S | 2.0× | tATPase | ★★★ |
| coxD | D100 | 2.2× | tOCS | ★★★★ |
| coxE | SM | 2.1× | tHSP18.2 | ★★★★ |
| coxF | S100 | 1.8× | tATPase | ★★★ |
| coxG | FMV 34S | 2.0× | T-35S | ★★★ |
Cassette Architecture Design
Each expression cassette was designed using the same general architecture: Promoter → AMV Enhancer → Chloroplast Transit Peptide (CTP) → CODH Gene → Tag (if applicable) → Terminator
 All seven cassettes were designed individually in Benchling before being assembled into the larger Structural and Maturation multicassette constructs.
All seven cassettes were designed individually in Benchling before being assembled into the larger Structural and Maturation multicassette constructs.
Selection of Regulatory and Functional Elements
The promoter–terminator pairs selected during the previous phase were incorporated into the final cassette designs to drive constitutive expression in tobacco cells. Different promoter strengths were intentionally distributed across the genes to maintain balanced expression between structural and maturation proteins.
Each cassette included the modified AMV RNA4 translational enhancer immediately downstream of the promoter. The endogenous ATG codon was previously removed from the enhancer sequence to ensure that translation initiates only at the intended chloroplast transit peptide start codon.
This enhancer was incorporated to improve ribosome recruitment and increase translational efficiency of the engineered mRNAs.
Because the CODH pathway must function inside chloroplasts, chloroplast transit peptides were fused upstream of each CODH coding sequence. These CTPs act as molecular targeting signals directing the newly synthesized proteins from the cytoplasm into the chloroplast after translation. Different transit peptides were selected based on predicted compatibility and chloroplast import efficiency.
Each codon-optimized CODH gene was fused directly downstream of its corresponding chloroplast transit peptide in order to generate a continuous translational fusion protein.
This design ensures that the targeting peptide is translated first and recognized by the chloroplast import machinery before cleavage by stromal processing peptidase (SPP).
Specific epitope tags were incorporated into selected cassettes to facilitate downstream protein detection, purification, and complex characterization. The following tags were used: FLAG tag for coxL and coxD; His tag for coxS
These tags were included to support future protein purification, Co-IP experiments, PAGE analysis, and enzyme characterization workflows during the experimental validation phase.
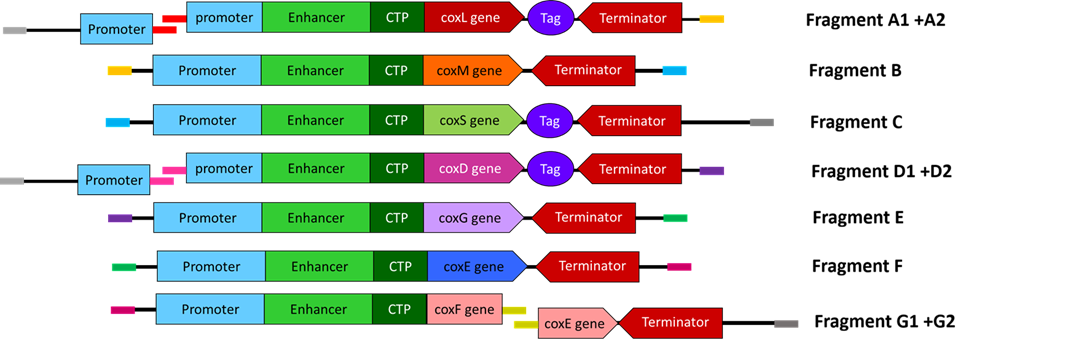
Final Cassette Components
The final regulatory combinations and chloroplast targeting peptides used for each cassette are summarized below.
| Gene | Promoter | CTP Source | Terminator | Tag |
|---|---|---|---|---|
| coxL | D100 | RbcS | tOCS | FLAG |
| coxM | SM | Fer2 | tHSP18.2 | — |
| coxS | FMV 34S | RecA | tATPase | His |
| coxD | D100 | RbcS | tOCS | FLAG |
| coxE | SM | Fer2 | tHSP18.2 | — |
| coxF | S100 | RecA | tATPase | — |
| coxG | FMV 34S | RbcS | T-35S | — |
The objective of this step was to design each cox gene as an independent plant expression cassette containing all the required regulatory elements for efficient expression in Nicotiana tabacum. This included selecting appropriate promoters, terminators, chloroplast transit peptides (CTPs), translational enhancers, purification tags, and spacer sequences, while organizing the multicassette constructs in a modular format compatible with DNA synthesis and Gibson Assembly.
Before assembling the large Structural and Maturation multicassette inserts, the next objective was to identify suitable insertion sites within the pCAMBIA backbones and generate homology arms compatible with Gibson Assembly.
This step was essential to ensure seamless integration of the final multicassette constructs into the plant transformation vectors.
Selection of the Restriction Site
To determine the optimal vector opening site, the multiple cloning site (MCS) maps of both pCAMBIA1300 and pCAMBIA2300 were analyzed in Benchling.
The restriction enzymes previously excluded during gene and cassette design (“Clean List”) were cross-referenced against the vector maps to avoid conflicts with internal restriction sites present in the final constructs.
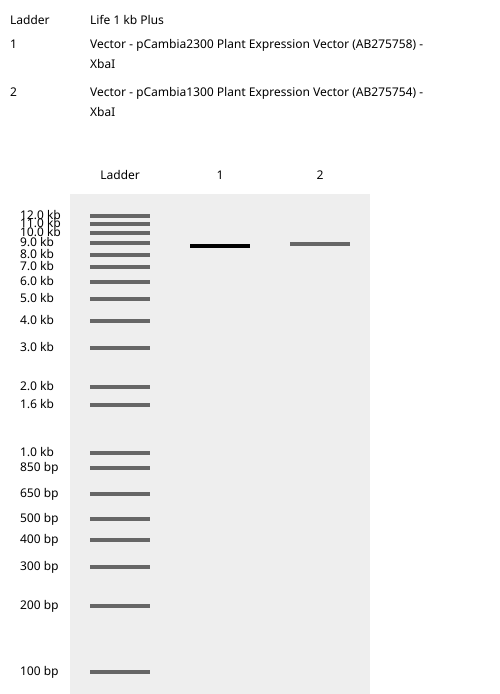
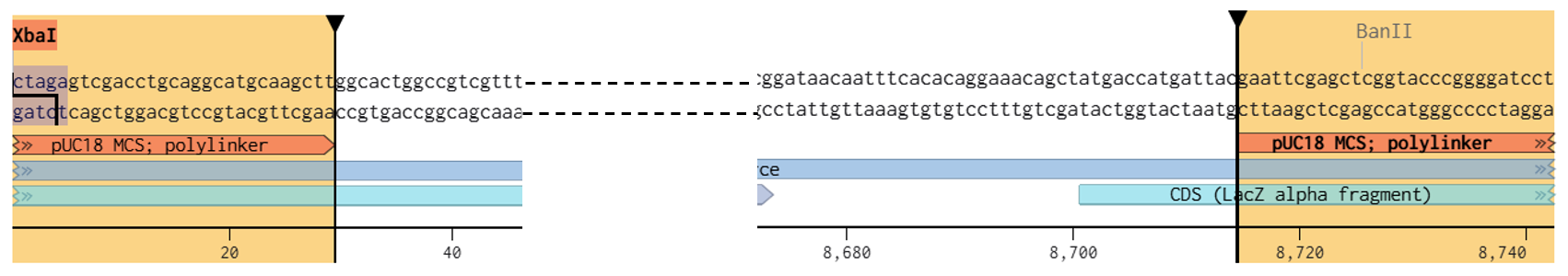
Following this analysis, XbaI was selected as the universal linearization site for both vectors because:
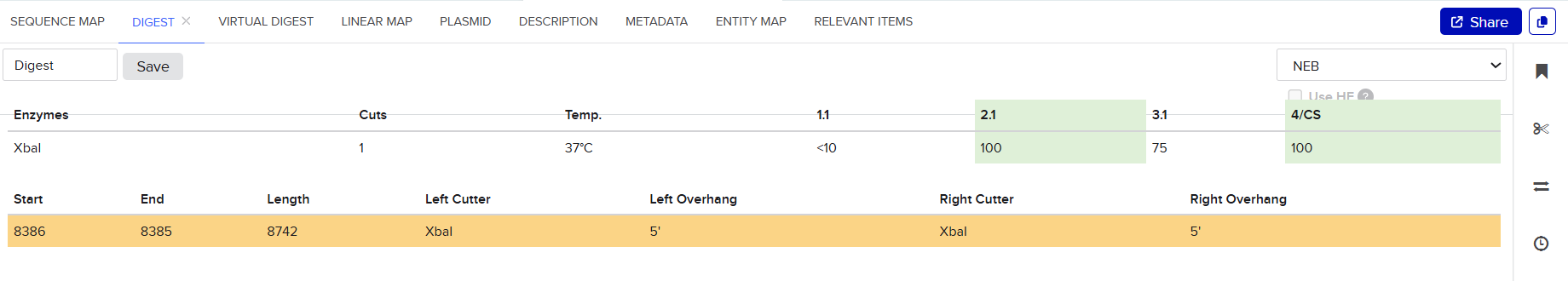
Both pCAMBIA vectors were virtually digested in Benchling using XbaI:

![]()
![]()
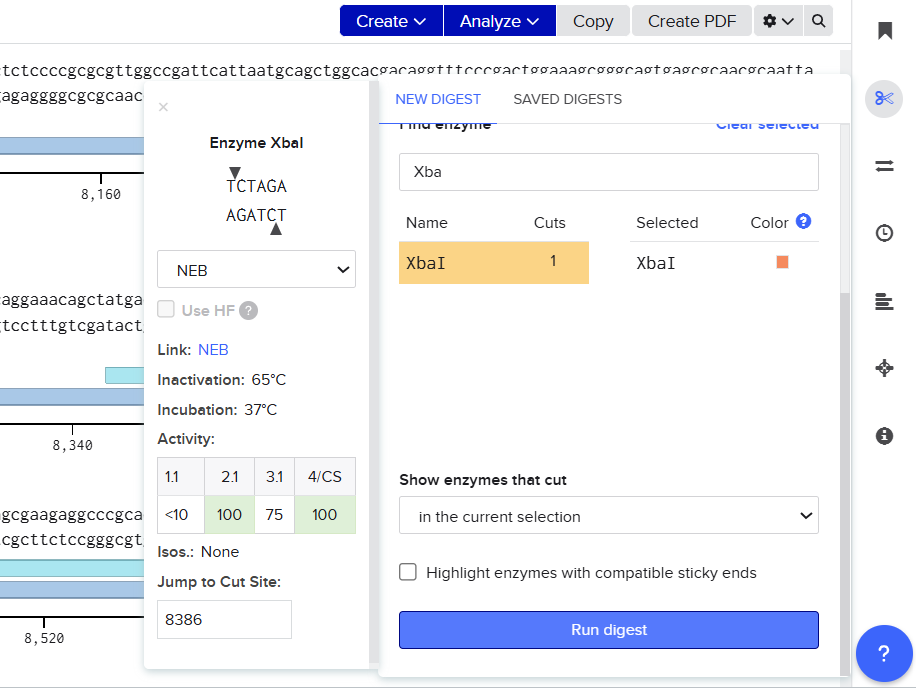
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
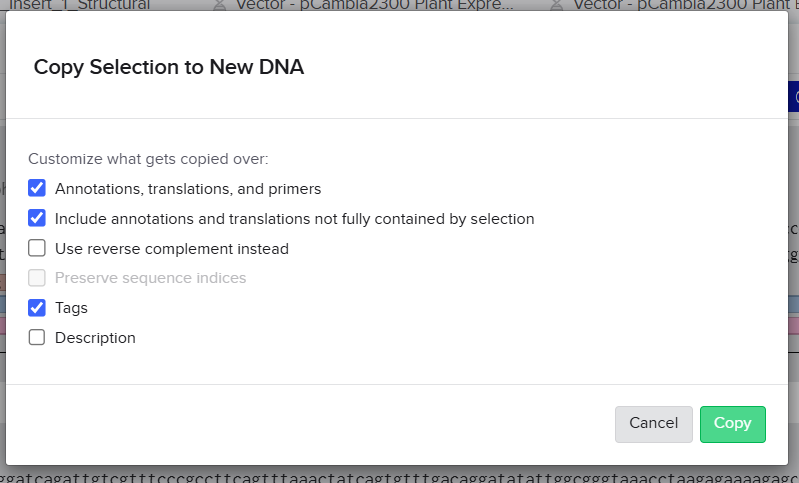
This generated linearized vector backbones with defined left and right insertion junctions.
Homology Arm Design
To enable Gibson Assembly, homology arms were generated directly from the terminal sequences of the XbaI-linearized vectors.
For each construct, 40 bp regions located at the ends of the digested vectors were extracted and incorporated as terminal overlaps (“tails”) on the outer fragments of the multicassette inserts.
These homology arms provide complementary regions between the vector backbone and the insert, allowing seamless enzymatic assembly during Gibson Assembly.
Because both vectors were linearized at the same XbaI site, the resulting homology arms were identical for the two constructs.
Final Homology Arms
Left Homology Arm : gaccatgattacgaattcgagctcggtacccggggatcct
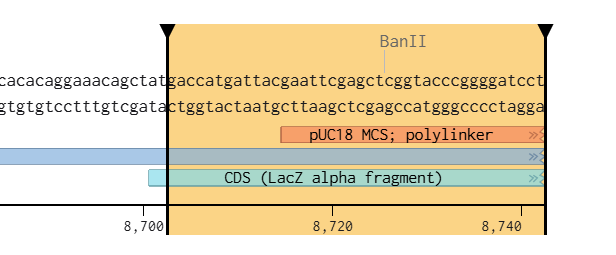
Right Homology Arm: ctagagtcgacctgcaggcatgcaagcttggcactggccg

These sequences were directly extracted from the terminal regions of the XbaI-digested pCAMBIA vectors after virtual linearization in Benchling.
The objective of this step was to prepare the pCAMBIA2300 and pCAMBIA1300 backbones for Gibson Assembly by virtually linearizing the vectors at a selected restriction site and generating homologous overlap regions. These homology arms were designed to guide the precise insertion and seamless assembly of the multicassette fragments into the plasmid backbones.
After finishing the design of all seven expression cassettes in Benchling, I prepared the sequences for synthesis by Twist Bioscience. The objective of this step was to divide the large multicassette constructs into smaller DNA fragments compatible with DNA synthesis and Gibson Assembly.
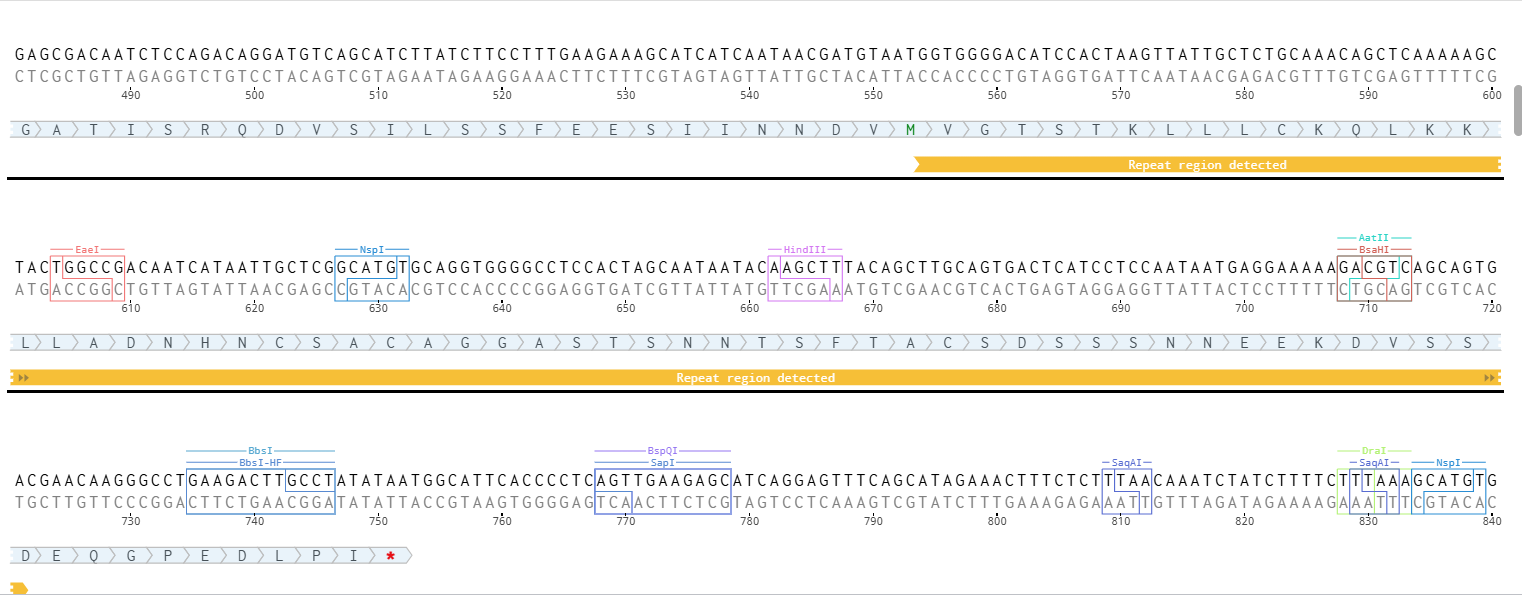
Initially, I tried to submit each complete fragment directly to the Twist synthesis platform. Although several fragments were accepted immediately, others were rejected because the algorithm detected highly repetitive DNA regions.
The major problem came from the synthetic promoters D100 and S100, which contain repeated enhancer motifs. Repetitive DNA is problematic for commercial DNA synthesis because it can:
To solve these issues, I performed several optimization and troubleshooting steps directly in Benchling.
Fragment A
Fragment A was designed for the structural multicassette construct cloned into the pCAMBIA2300 backbone.
This fragment initially contained: [Left Homology Arm] – [Spacer 1] – [coxL Cassette] – [40 bp Spacer 2]
The fragment was rejected by the Twist algorithm because the D100 promoter contained two repeated enhancer regions.

To solve this issue, I first tried to identify the functional transcription factor binding regions inside the promoter sequence. Using the promoter map from the original publication, I localized the consensus sequences (functional boxes) and carefully avoided modifying them.
I then introduced small nucleotide substitutions only in the non-functional repeated regions. The modifications included: A ↔ T, G ↔ C substitutions

I specifically used complementary substitutions in order to maintain approximately the same GC content and preserve promoter stability.
These modifications reduced the number of repeated regions detected by Twist, but the fragment was still rejected.
I also tried to optimize the repeated region located near the end of the coxL cassette. Several synonymous sequence modifications were tested: GGAGAGCAACTTGGACTT→ GGTGAACAGCTGGGTTTG→ GGCGAGCAACTTGGACTT→ GGAGAACAGCTCGGCTTG
However, the Twist algorithm continued detecting problematic repeats.
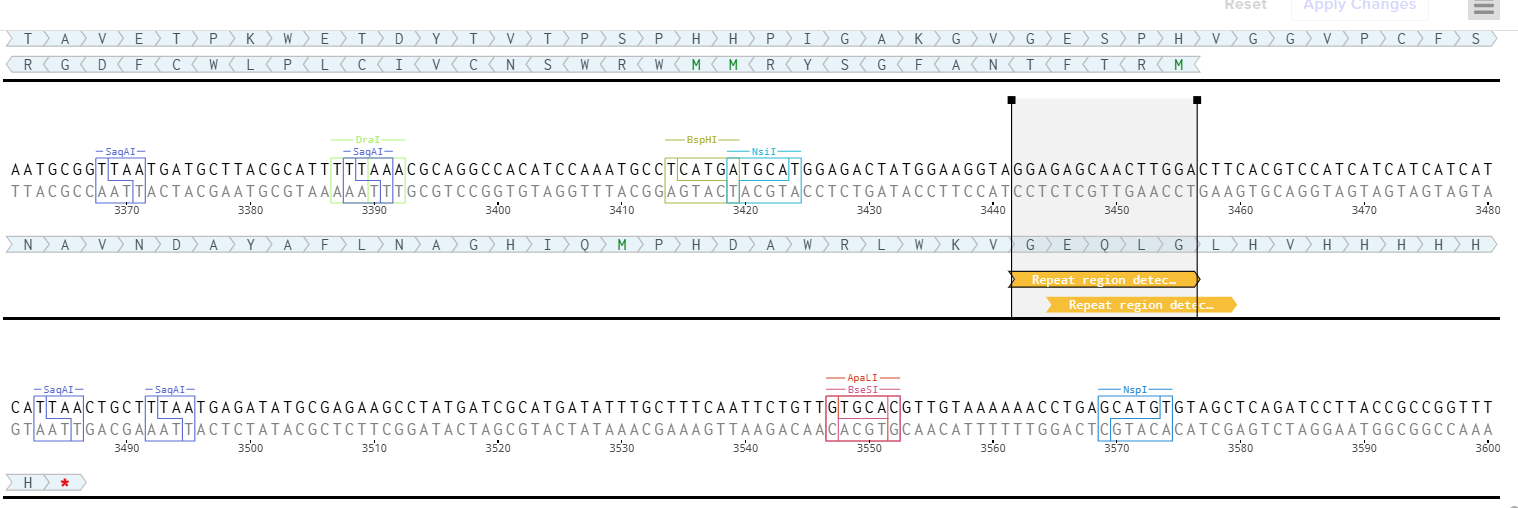
Since sequence optimization alone was insufficient, I decided to split Fragment A into two smaller fragments, A1 and A2.
The objective was to physically separate the repeated enhancer regions of the D100 promoter into different synthesis fragments.
After splitting the construct, both fragments were accepted successfully by Twist Bioscience without additional problems.
Final Fragment Design
Fragment A1 : [Left Homology Arm] – [Spacer 1] – [First Part of D100]
I added a 40 bp overlap between A1 and A2 to allow seamless Gibson Assembly during the final plasmid construction.
Fragment B was accepted directly by the Twist algorithm without requiring any optimization. This fragment contained: [Spacer 2] – [Full coxM Cassette] – [Spacer 3]
The fragment already satisfied all synthesis requirements because it did not contain repetitive regions or problematic GC-rich structures. This overlap design ensured proper assembly continuity during Gibson Assembly.
Fragment C was also accepted directly without major issues. This fragment contained: [Last 40 bp of Spacer 3] – [Full coxS Cassette] – [Right Homology Arm]
The fragment was designed to terminate the structural multicassette assembly inside the pCAMBIA2300 backbone.
This organization ensured proper circularization of the final plasmid during Gibson Assembly.
Fragment D belonged to the maturation multicassette construct cloned into pCAMBIA1300. Initially, the fragment contained: [Left Homology Arm] – [Spacer 1] – [coxD Cassette] – [Spacer 2]
Like Fragment A, this fragment was rejected because the D100 promoter contained repeated enhancer regions detected by the Twist algorithm. Instead of modifying the sequence extensively, I decided to split the fragment into two smaller fragments. The objective was again to physically separate the repeated promoter regions.
Final Fragment Design
Fragment D1: [Left Homology Arm] – [Spacer 1] – [First Part of D100]
I introduced 40 bp overlaps between the fragments to allow Gibson Assembly reconstruction of the complete cassette. After splitting, both fragments were accepted successfully by Twist Bioscience.
Fragment E was accepted directly without requiring optimization. This fragment contained:[Last 40 bp of Spacer 2] – [coxG Cassette] – [Spacer 3]
I designed the overlaps carefully to maintain assembly continuity with the neighboring fragments. The fragment did not contain problematic repeats or synthesis instability regions.
Fragment F contained: [Last 40 bp of Spacer 3] – [coxE Cassette] – [Spacer 4]
Unlike the previous problematic fragments containing the D100 or S100 promoters, this fragment used the SM promoter, which did not contain repetitive enhancer regions.
Therefore, Fragment F was accepted directly by the Twist Bioscience algorithm from the first submission without requiring any optimization, sequence modification, or fragment splitting.
The fragment was synthesized as a complete cassette exactly as originally designed in Benchling.
Fragment G corresponded to the coxF cassette region.
Initially, I designed the complete coxF cassette as a single large fragment containing the S100 promoter. However, the Twist algorithm rejected the sequence because the S100 promoter contained repetitive enhancer regions similar to those previously observed with the D100 promoter.
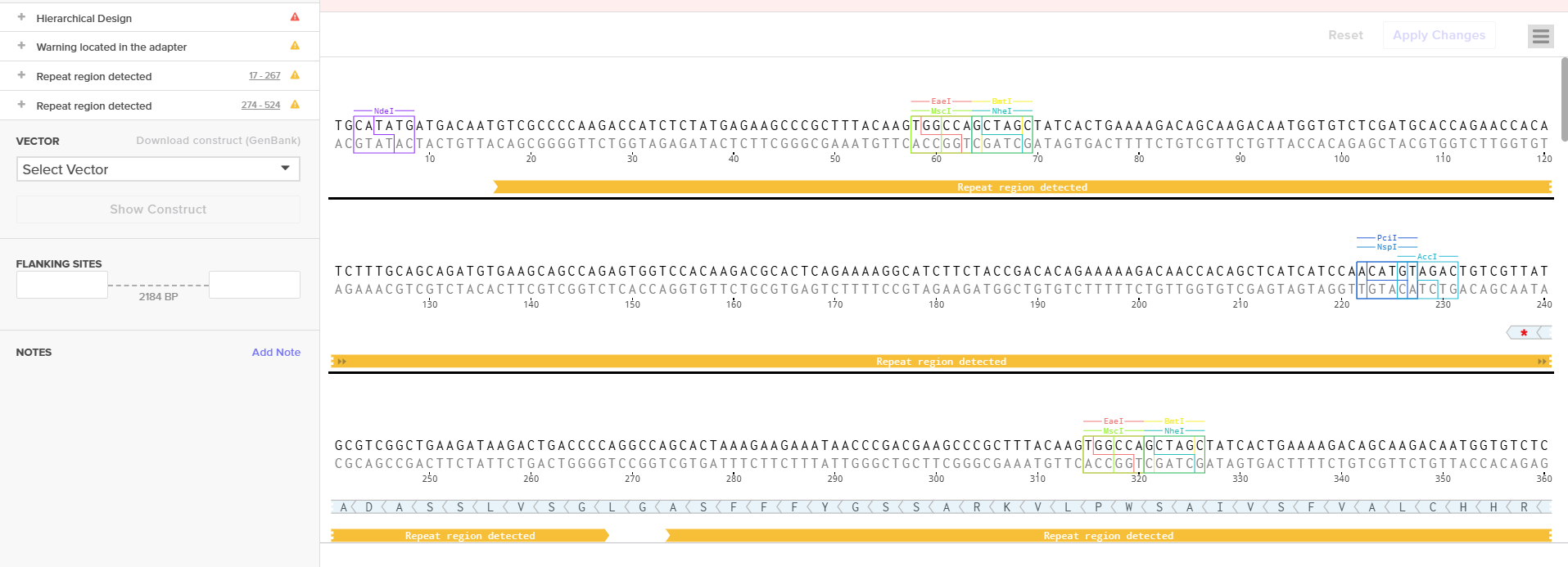 To solve this problem, I divided the large region into multiple smaller fragments. Some fragments were accepted immediately, while the fragment containing the S100 promoter continued to fail.Therefore, I followed the same strategy previously used for the D100 promoter.
To solve this problem, I divided the large region into multiple smaller fragments. Some fragments were accepted immediately, while the fragment containing the S100 promoter continued to fail.Therefore, I followed the same strategy previously used for the D100 promoter.
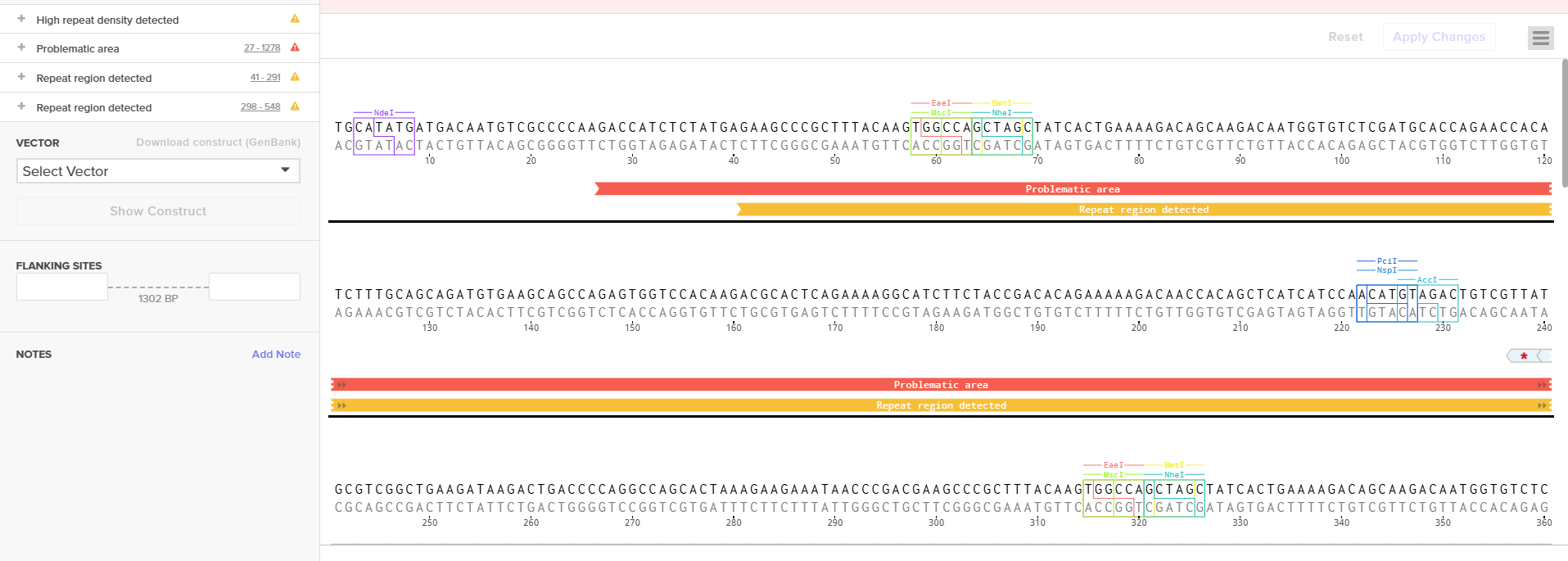 First, I localized the functional consensus regions inside the S100 promoter using the original promoter publication.
First, I localized the functional consensus regions inside the S100 promoter using the original promoter publication.
 Then, I introduced minimal nucleotide substitutions only outside the functional boxes: A ↔ T, G ↔ C substitutions
Then, I introduced minimal nucleotide substitutions only outside the functional boxes: A ↔ T, G ↔ C substitutions
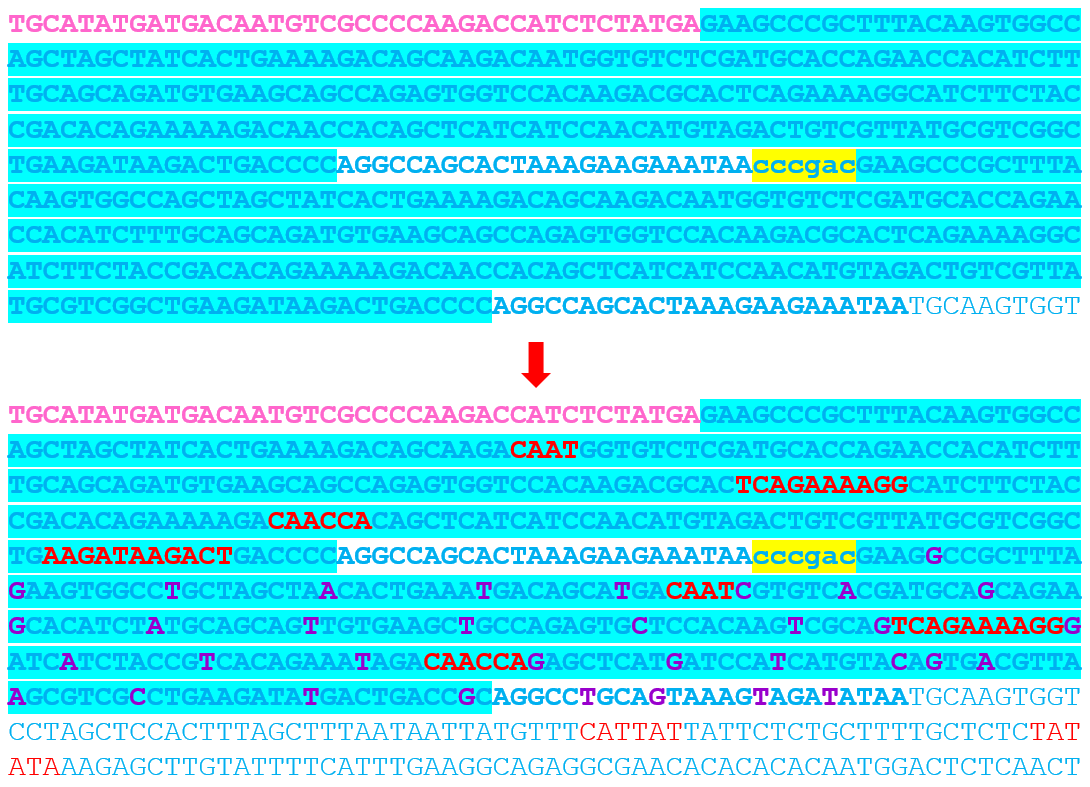 These modifications preserved: GC content balance, Promoter architecture, Functional regulatory motifs
These modifications preserved: GC content balance, Promoter architecture, Functional regulatory motifs
After these adjustments, the fragment was finally accepted by the Twist algorithm. Fragment G corresponded to the coxF cassette region. Initially, I designed the fragment as a single large sequence, but the S100 promoter repeats again caused synthesis rejection.
To solve this issue, I split the region into two smaller fragments:
The 40 bp overlap allowed seamless Gibson Assembly between both fragments. After splitting and promoter optimization, both fragments were accepted successfully.
Final twist validated fragments:
| Fragment | Construct | Main Components | Special Notes |
|---|---|---|---|
| A1 | Structural construct (pCAMBIA2300) | Left homology arm + Spacer 1 + First part of D100 promoter | Fragment created after splitting Fragment A to separate repeated enhancer regions |
| A2 | Structural construct (pCAMBIA2300) | 40 bp overlap from A1 + Remaining D100 promoter + Complete coxL cassette + First 40 bp of Spacer 2 | Accepted after promoter splitting |
| B | Structural construct (pCAMBIA2300) | Spacer 2 + Complete coxM cassette + Spacer 3 | Accepted directly without optimization |
| C | Structural construct (pCAMBIA2300) | Last 40 bp of Spacer 3 + Complete coxS cassette + Right homology arm | Accepted directly without optimization |
| D1 | Maturation construct (pCAMBIA1300) | Left homology arm + Spacer 1 + First part of D100 promoter | Generated after splitting Fragment D to separate repeated promoter regions |
| D2 | Maturation construct (pCAMBIA1300) | 40 bp overlap from D1 + Remaining D100 promoter + Complete coxD cassette + Spacer 2 | Accepted after fragment splitting |
| E | Maturation construct (pCAMBIA1300) | Last 40 bp of Spacer 2 + Complete coxG cassette + Spacer 3 | Accepted directly without optimization |
| F | Maturation construct (pCAMBIA1300) | Last 40 bp of Spacer 3 + Complete coxE cassette + Spacer 4 | Contains SM promoter; accepted directly from the first submission |
| G1 | Maturation construct (pCAMBIA1300) | First 40 bp of Spacer 4 + First part of coxF cassette | Fragment generated after splitting the coxF region because of S100 promoter repeats |
| G2 | Maturation construct (pCAMBIA1300) | 40 bp overlap from G1 + Remaining coxF cassette + Right homology arm | Accepted after S100 promoter optimization and fragment splitting |
The objective of this step was to adapt the designed multicassette constructs to the synthesis requirements of Twist Bioscience by identifying and resolving problematic repetitive regions, optimizing synthesis compatibility, and ensuring that all final fragments could be successfully synthesized and assembled through Gibson Assembly.
After completing the design and optimization of all fragments in Benchling
, I exported the finalized sequences in FASTA format. Each fragment corresponded to a specific part of the structural or maturation multicassette constructs and already contained the required overlaps for Gibson Assembly.
I then uploaded the FASTA files into the Twist Bioscience Platform
using the gene synthesis workflow. The platform automatically analyzed each sequence to evaluate synthesis compatibility, including repetitive regions, GC balance, and sequence complexity.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Fragments that failed the screening due to repetitive promoter regions were optimized during the previous phase either by introducing minimal nucleotide modifications in non-functional regions or by splitting the constructs into smaller fragments. After re-uploading the corrected sequences, all fragments were successfully accepted by the Twist algorithm.
This final simulation confirmed that the complete multicassette constructs were synthesis-compatible and ready for downstream Gibson Assembly and cloning experiments.
The objective of this phase was to simulate the commercial DNA synthesis workflow by exporting the finalized multicassette fragments from Benchling in FASTA format and evaluating their compatibility with the synthesis requirements of Twist Bioscience. This step aimed to verify sequence manufacturability, detect potential synthesis issues such as repetitive regions or sequence complexity, and confirm that all fragments were fully ready for commercial synthesis and downstream Gibson Assembly.
After preparing and validating all the synthesis fragments, I moved to the in silico assembly step in Benchling to digitally reconstruct the complete Structural and Maturation multicassettes before any physical cloning work. For this step, I used the native Gibson Assembly tool available in Benchling because all fragments were already designed with 40 bp overlaps to enable seamless assembly.
First, I opened Benchling and clicked on the Create (+) button from the left sidebar. Then, from the Assembly options, I selected “Assemble DNA sequences by cloning.” This opened the Gibson Assembly workflow interface where I configured all the assembly parameters.
 For the assembly settings, I selected the destination project folder dedicated to the multicassette constructs. Since I wanted to generate standalone insert sequences rather than circular plasmids at this stage, I set the construct topology to Linear. Then, I selected Gibson Assembly as the cloning method. For fragment joining, I chose the option “Find existing overlaps” because all overlaps had already been engineered during the previous fragment preparation phase.
For the assembly settings, I selected the destination project folder dedicated to the multicassette constructs. Since I wanted to generate standalone insert sequences rather than circular plasmids at this stage, I set the construct topology to Linear. Then, I selected Gibson Assembly as the cloning method. For fragment joining, I chose the option “Find existing overlaps” because all overlaps had already been engineered during the previous fragment preparation phase.
Next, I adjusted the homology parameters to match the overlaps used in my fragment design. I fixed both the minimum and maximum overlap length to 40 bp, corresponding to the overlaps added between all neighboring fragments. I also kept the minimum melting temperature around 39°C to ensure proper overlap recognition during the digital assembly process.
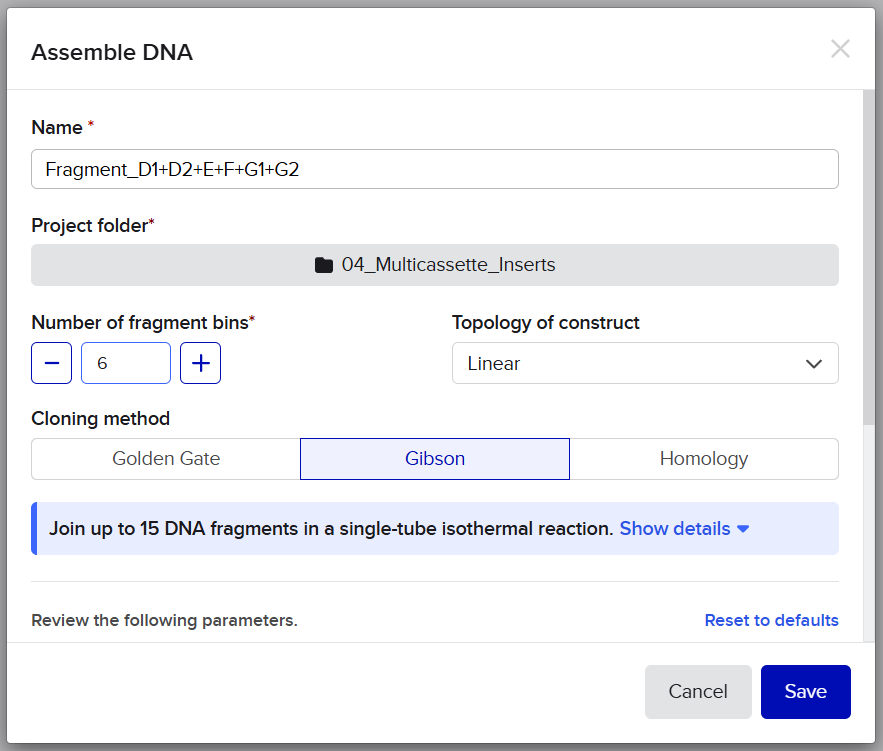
![]()
![]()
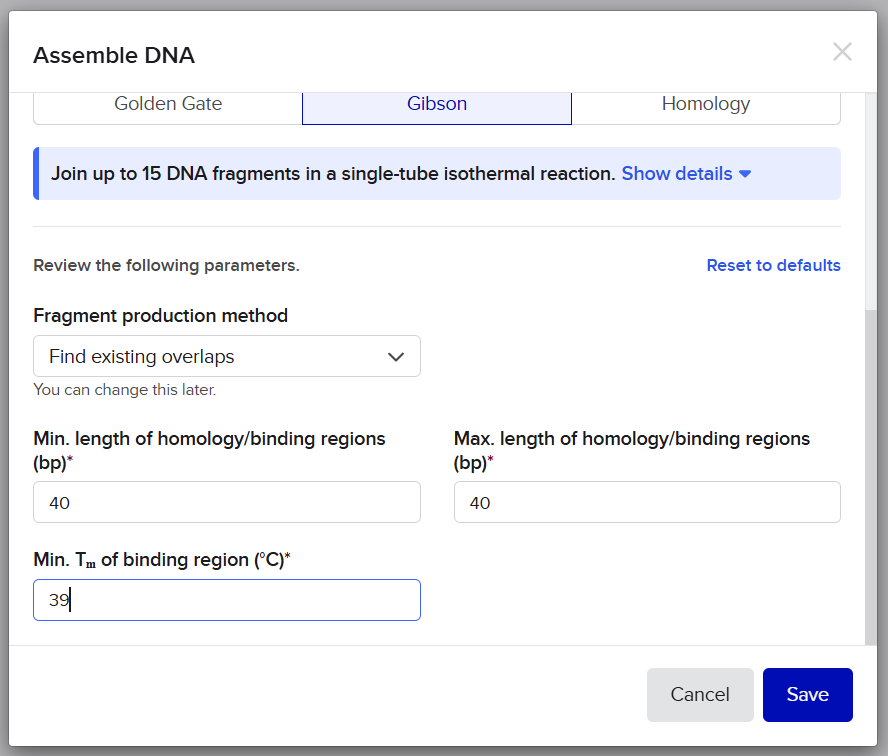 After configuring the assembly settings, Benchling generated a linear assembly lane containing several fragment bins. I then imported all fragments sequentially from left to right according to their assembly order.
After configuring the assembly settings, Benchling generated a linear assembly lane containing several fragment bins. I then imported all fragments sequentially from left to right according to their assembly order.
 For the Structural multicassette, I imported the fragments in the following order:
For the Structural multicassette, I imported the fragments in the following order:
For the Maturation multicassette, I imported:
Inside each bin, I used the “Search for sequences” option to retrieve the fragments directly from my Benchling project files. I also ensured that every junction remained configured on “Find existing overlaps” so Benchling could automatically detect and validate the engineered Gibson homology regions between adjacent fragments.
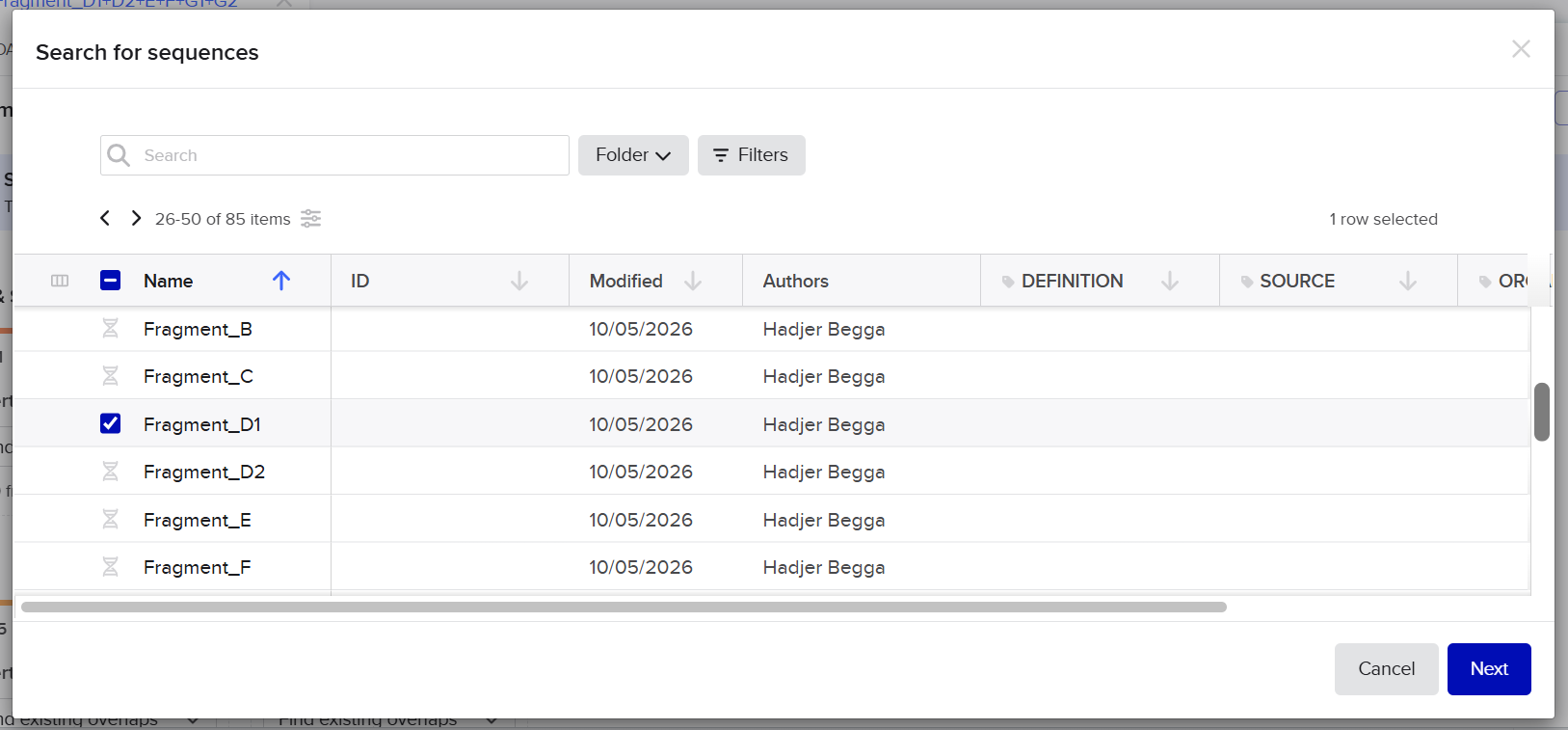
![]()
![]()
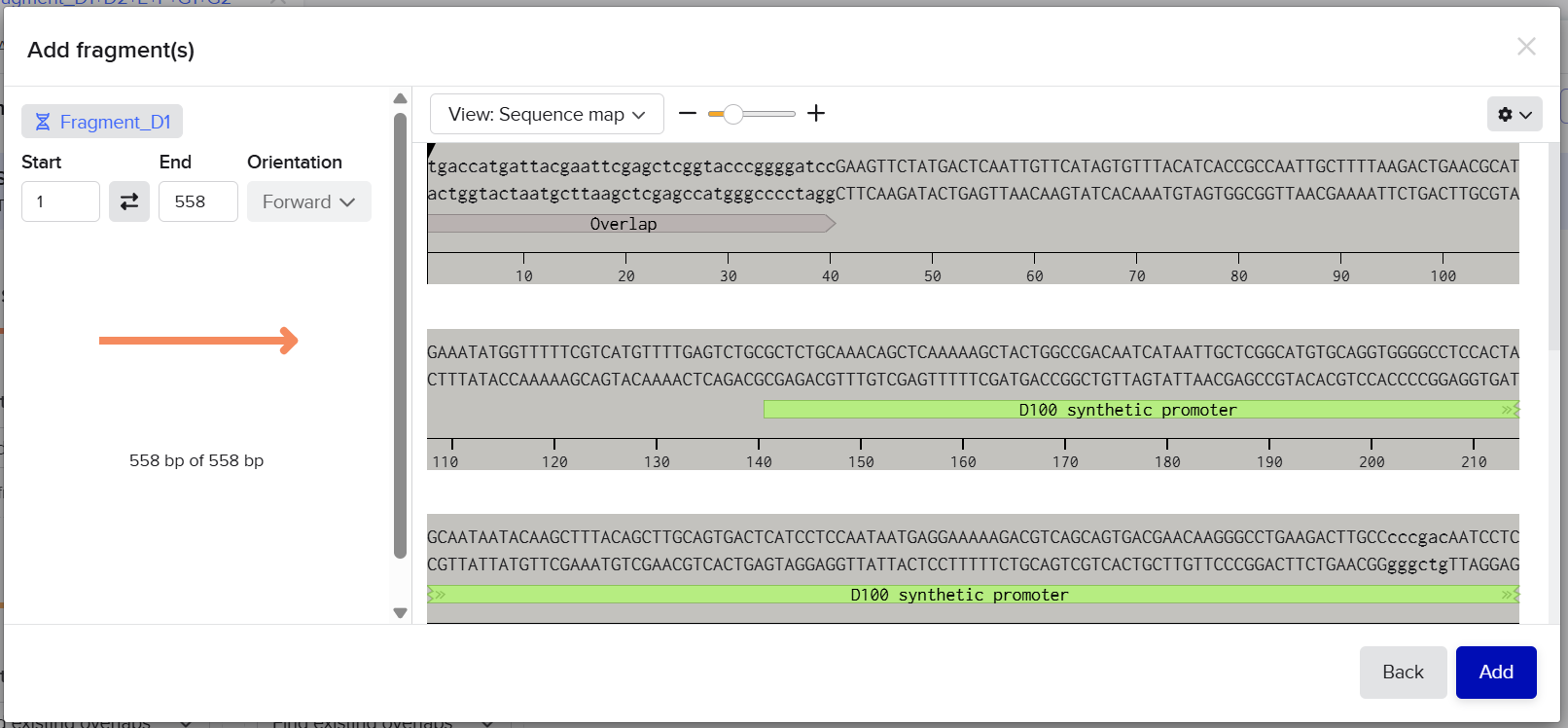 Once all fragments were added, Benchling automatically analyzed the overlaps between neighboring fragments.
Once all fragments were added, Benchling automatically analyzed the overlaps between neighboring fragments.
![]()
![]()
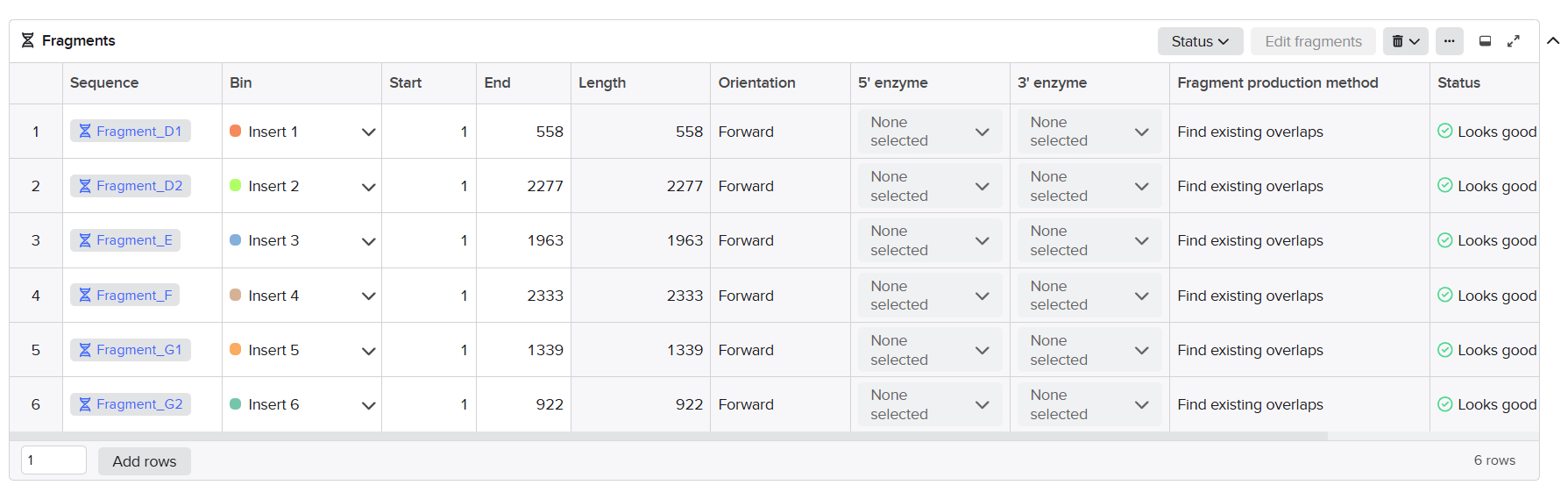
![]()
![]()
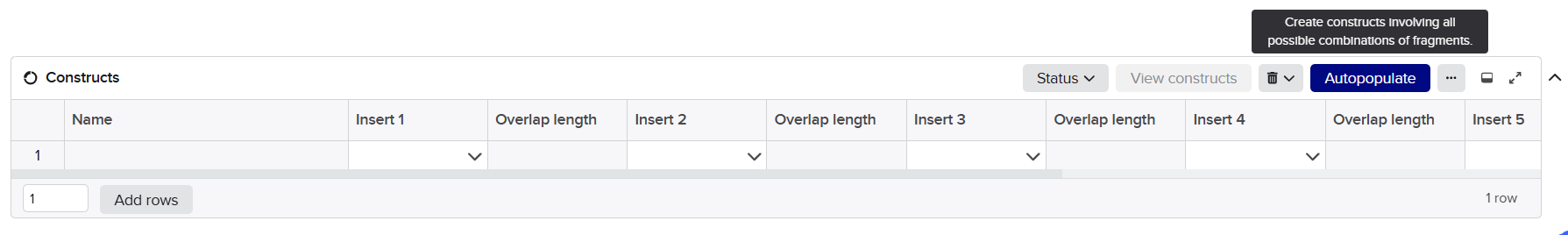
![]()
![]()
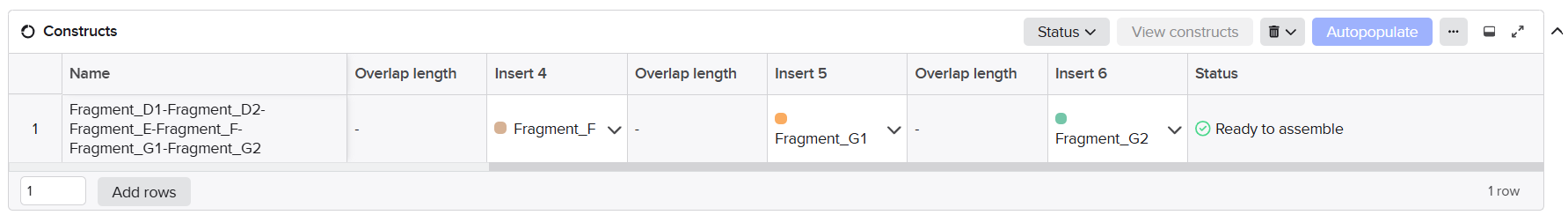 When all homology regions matched correctly, the assembly status changed from 0 constructs to 1 construct, confirming that the fragments were compatible and could assemble successfully into a continuous sequence. I then clicked the “Assemble” button to generate the final multicassette sequences. Benchling created new linear DNA constructs corresponding to the complete Structural and Maturation inserts assembled from all validated sub-fragments.
When all homology regions matched correctly, the assembly status changed from 0 constructs to 1 construct, confirming that the fragments were compatible and could assemble successfully into a continuous sequence. I then clicked the “Assemble” button to generate the final multicassette sequences. Benchling created new linear DNA constructs corresponding to the complete Structural and Maturation inserts assembled from all validated sub-fragments.
![]()
![]()
![]()
![]()
![]()
![]()
 Finally, I opened the resulting linear maps to verify the integrity of both assembled structural and maturation multicassettes. I carefully checked that all annotations were preserved correctly across the final constructs, including promoters, AMV enhancers, chloroplast transit peptides (CTPs), coding sequences, purification tags, spacers, and terminators. I also verified that all junctions were seamless and that no gaps, inversions, or frame disruptions appeared between adjacent fragments.
Finally, I opened the resulting linear maps to verify the integrity of both assembled structural and maturation multicassettes. I carefully checked that all annotations were preserved correctly across the final constructs, including promoters, AMV enhancers, chloroplast transit peptides (CTPs), coding sequences, purification tags, spacers, and terminators. I also verified that all junctions were seamless and that no gaps, inversions, or frame disruptions appeared between adjacent fragments.
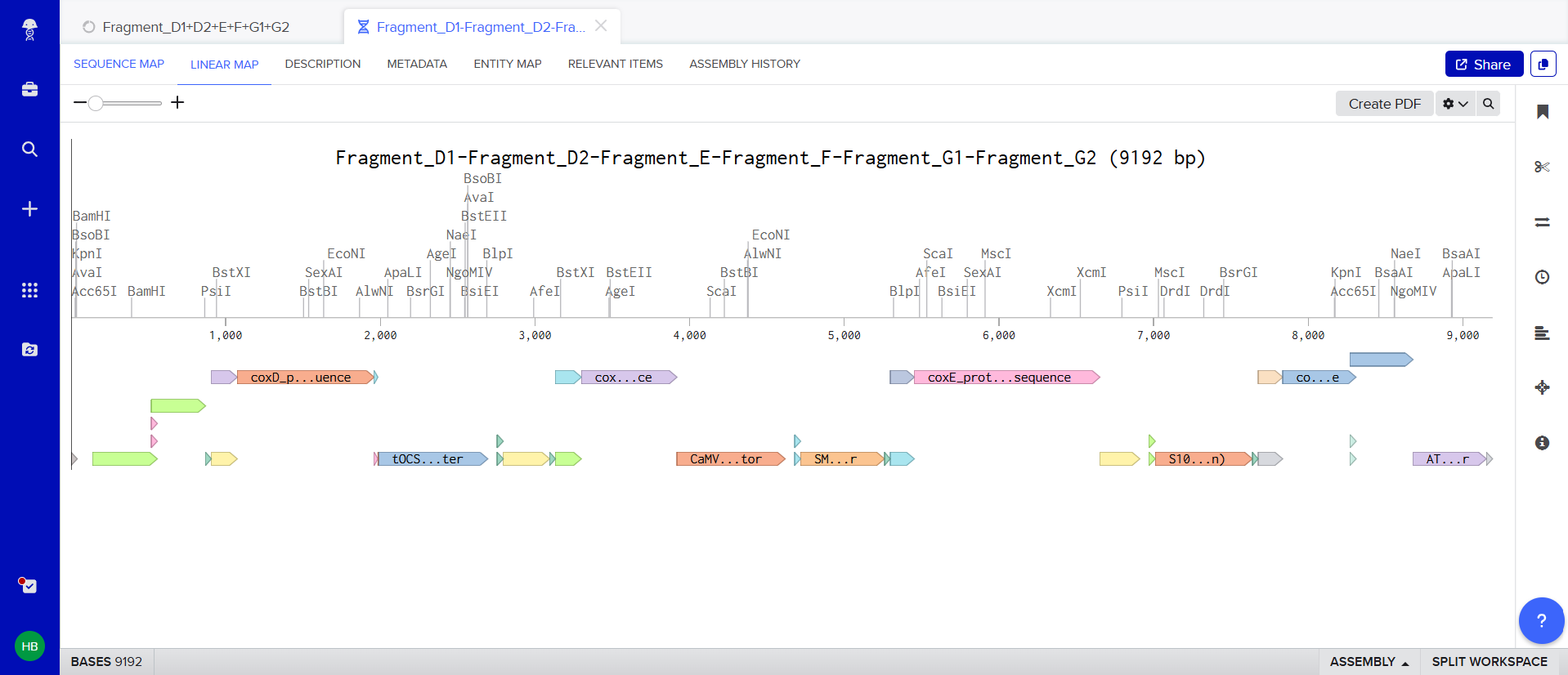
![]()
![]()
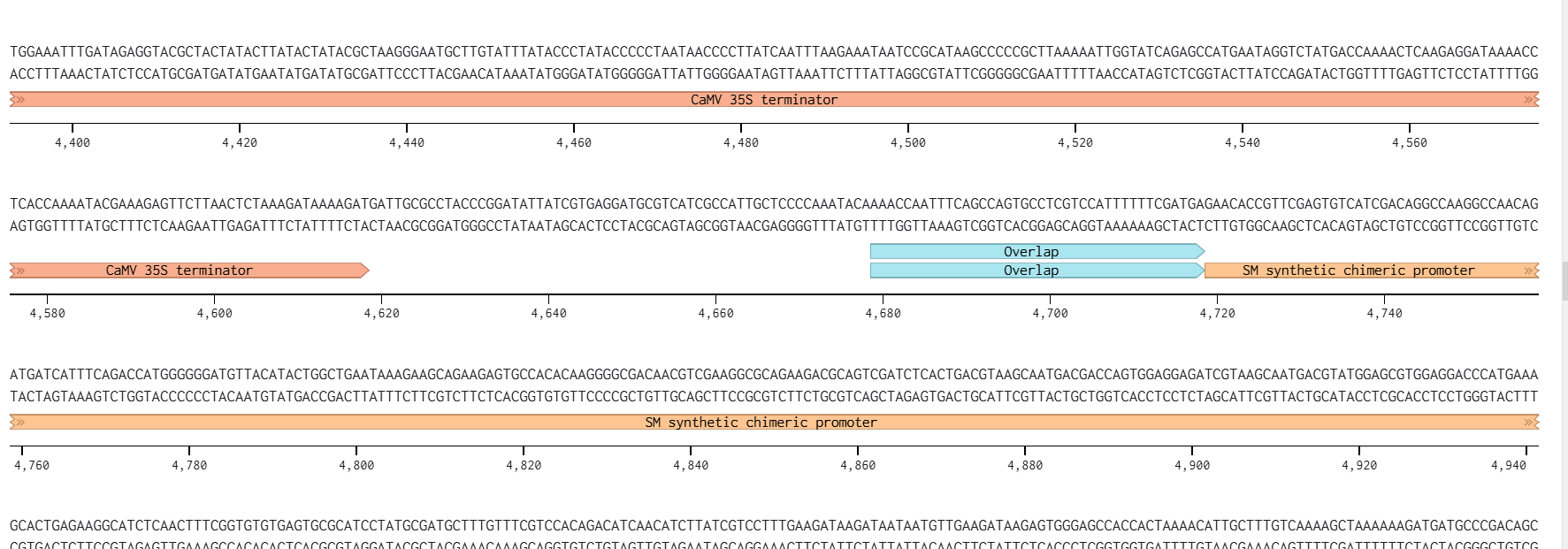 This final in silico Gibson Assembly simulation confirmed that both multicassette inserts were correctly designed and fully ready for downstream cloning and plasmid integration steps.
This final in silico Gibson Assembly simulation confirmed that both multicassette inserts were correctly designed and fully ready for downstream cloning and plasmid integration steps.
The objective of this phase was to digitally assemble all synthesized DNA fragments into the final structural and maturation multicassette constructs using Gibson Assembly simulation in Benchling . This step allowed me to verify fragment compatibility, overlap integrity, correct orientation, and successful reconstruction of the complete plasmids before experimental cloning.
After successfully assembling the Structural and Maturation multicassette inserts in Phase 7, I moved to the final cloning step where I inserted each complete multicassette block into its corresponding binary vector backbone using in silico Gibson Assembly in Benchling.
For this phase, I followed the same general Gibson Assembly workflow previously used for multicassette reconstruction. However, instead of assembling several independent fragments together, I assembled only two major components: the linearized pCAMBIA vector backbone and the complete multicassette insert.
I first opened the Benchling Assembly tool by selecting Create (+) → Assemble DNA sequences by cloning. Then, I configured the assembly parameters similarly to the previous phase. The cloning method was set to Gibson Assembly, and the overlap detection mode remained configured on “Find existing overlaps.”
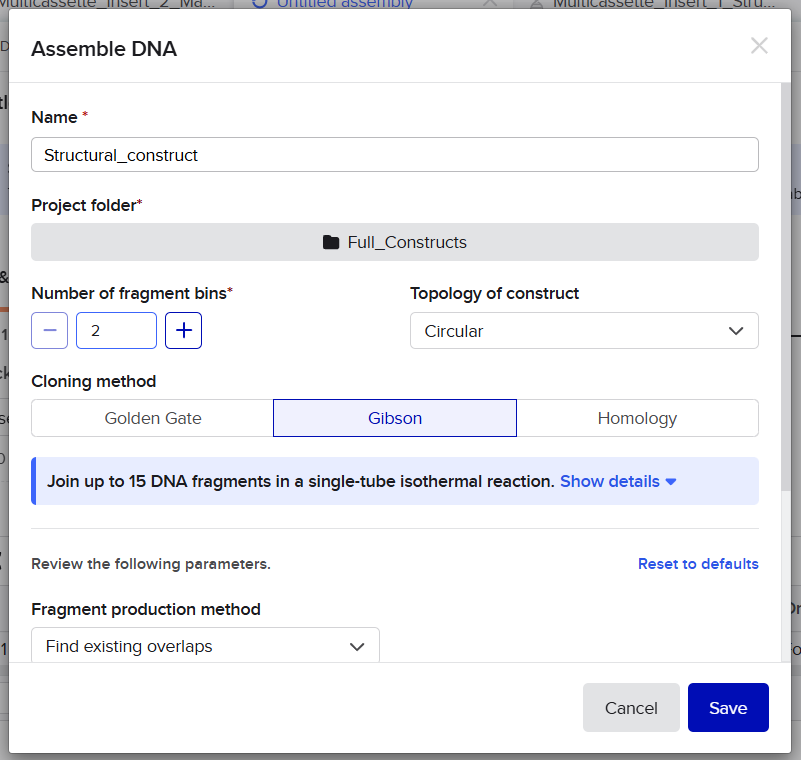 Unlike Phase 7, where the constructs were generated as linear inserts, I configured the topology of the final constructs as Circular because the insert and vector backbone needed to re-circularize to form complete binary plasmids.
Unlike Phase 7, where the constructs were generated as linear inserts, I configured the topology of the final constructs as Circular because the insert and vector backbone needed to re-circularize to form complete binary plasmids.
For the Structural construct, I imported:
For the Maturation construct, I imported:
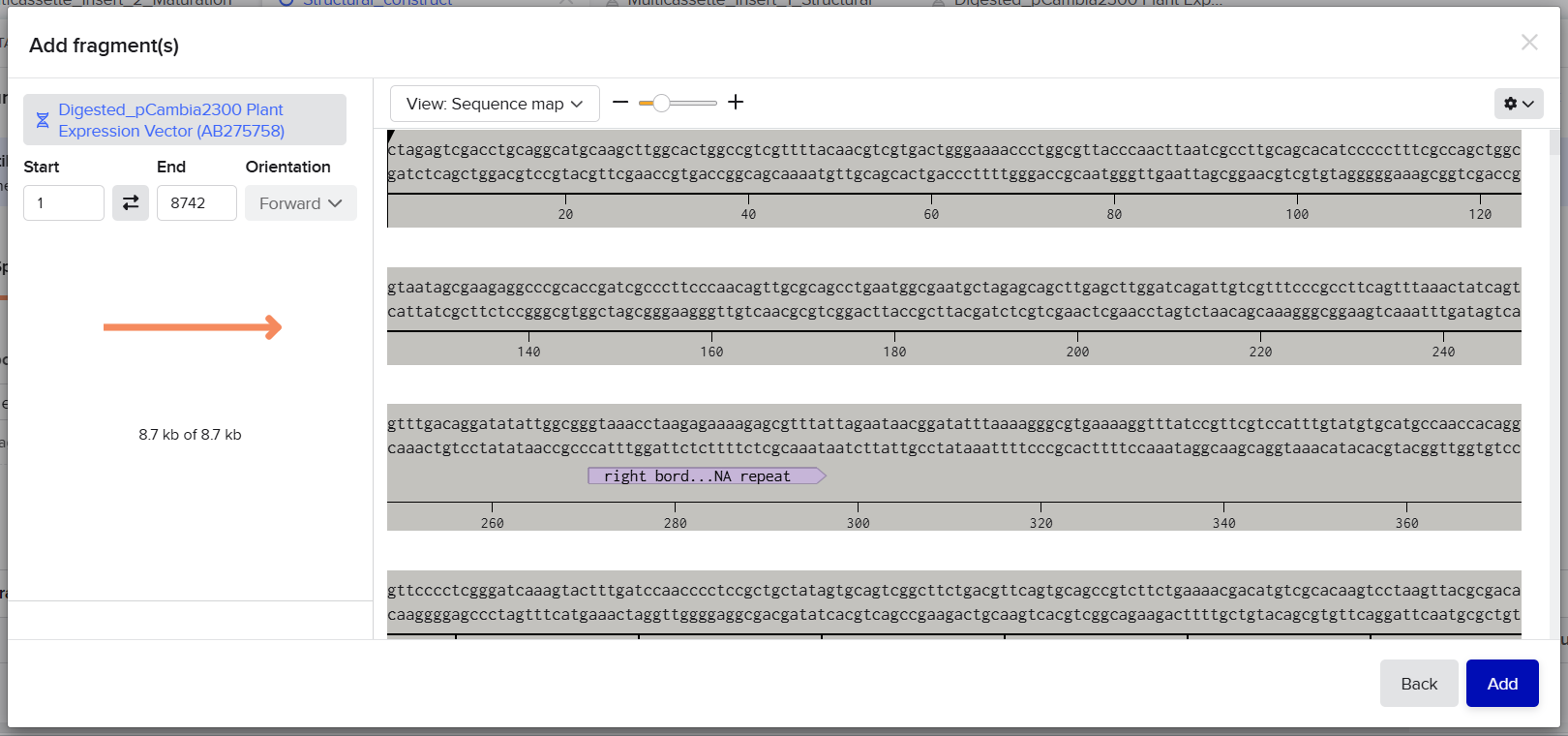
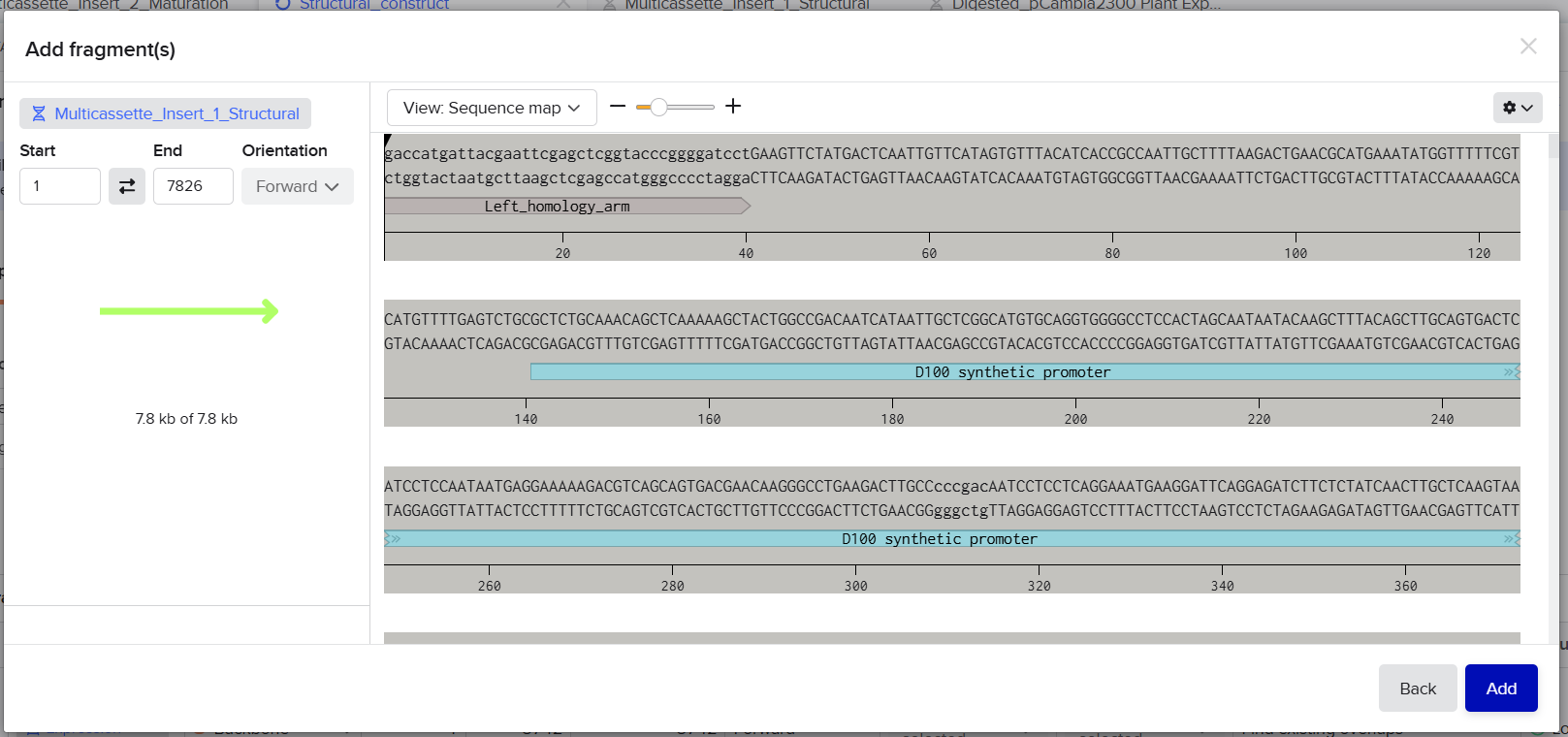 Benchling automatically analyzed the homology regions between the insert ends and the vector backbone extremities to validate correct assembly compatibility.
Benchling automatically analyzed the homology regions between the insert ends and the vector backbone extremities to validate correct assembly compatibility.Once both components were loaded into the assembly bins, Benchling successfully detected the overlap regions and generated one valid construct for each assembly. I then clicked the “Assemble” button to create the final circular plant expression plasmids.
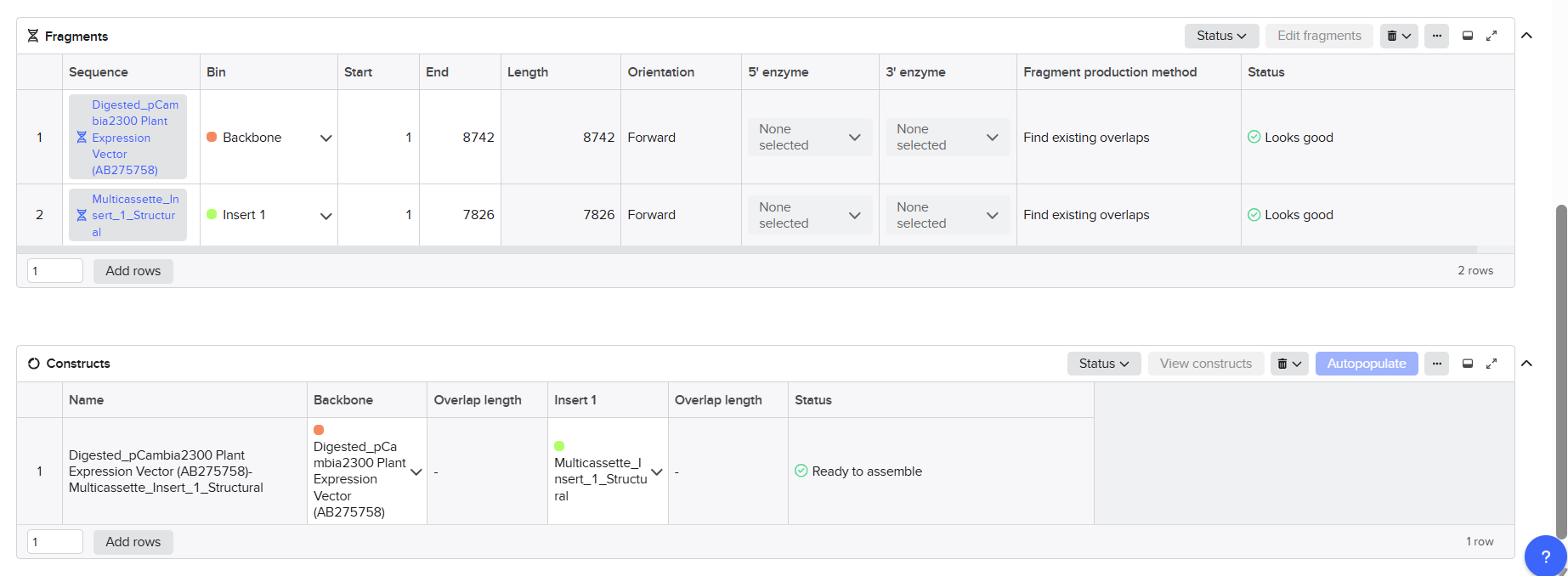
![]()
![]()
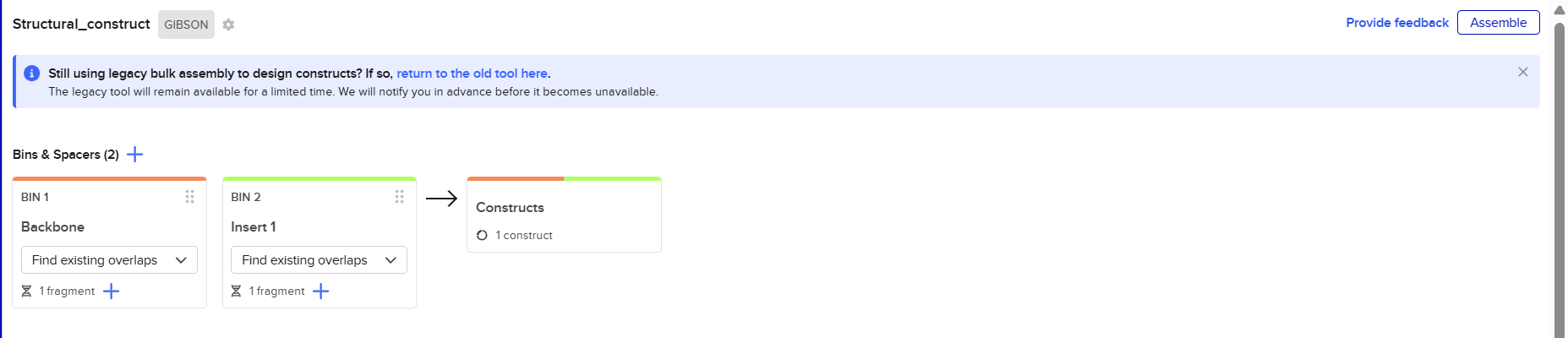 The resulting constructs were then analyzed using both the Plasmid Map and Linear Map visualization modes in Benchling. This final verification step allowed me to confirm that the multicassette inserts were correctly integrated into the vectors without inversions, sequence interruptions, or junction mismatches.
The resulting constructs were then analyzed using both the Plasmid Map and Linear Map visualization modes in Benchling. This final verification step allowed me to confirm that the multicassette inserts were correctly integrated into the vectors without inversions, sequence interruptions, or junction mismatches.
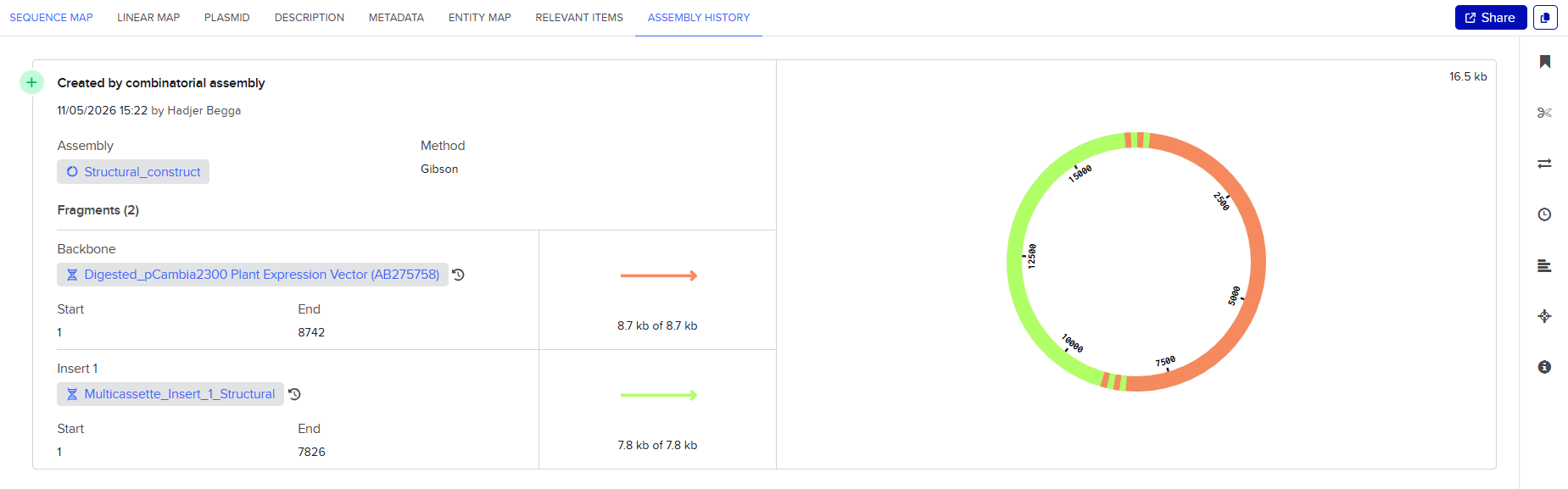 The final Structural construct generated a circular plasmid of approximately 16,488 bp, while the final Maturation construct generated a circular plasmid of approximately 18,070 bp.
The final Structural construct generated a circular plasmid of approximately 16,488 bp, while the final Maturation construct generated a circular plasmid of approximately 18,070 bp.
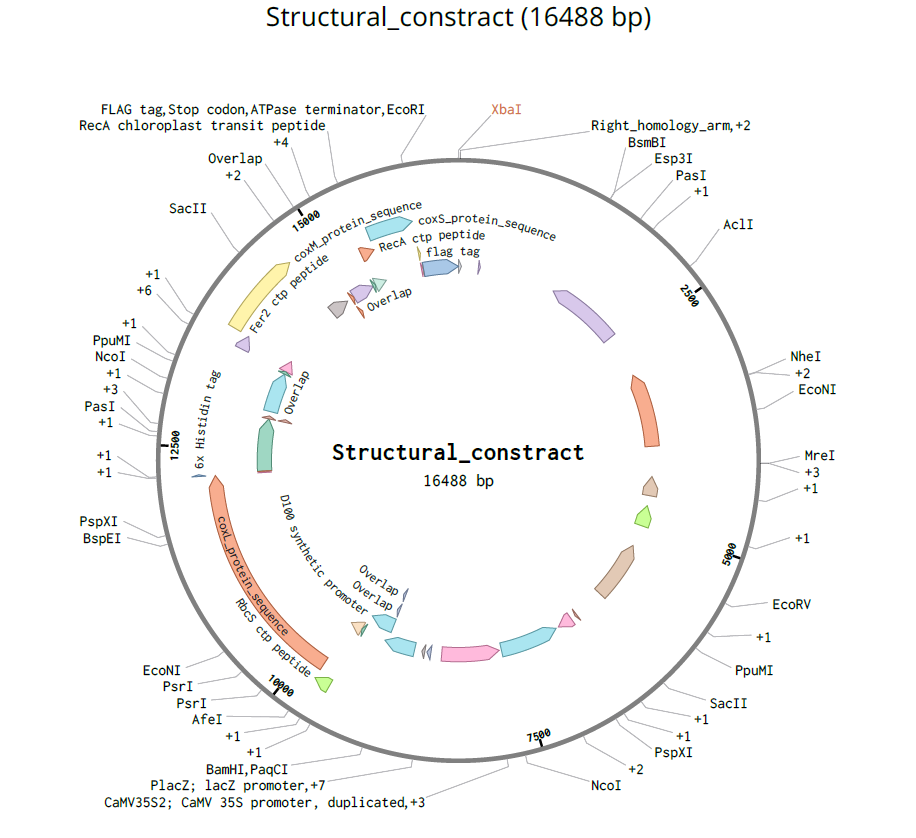
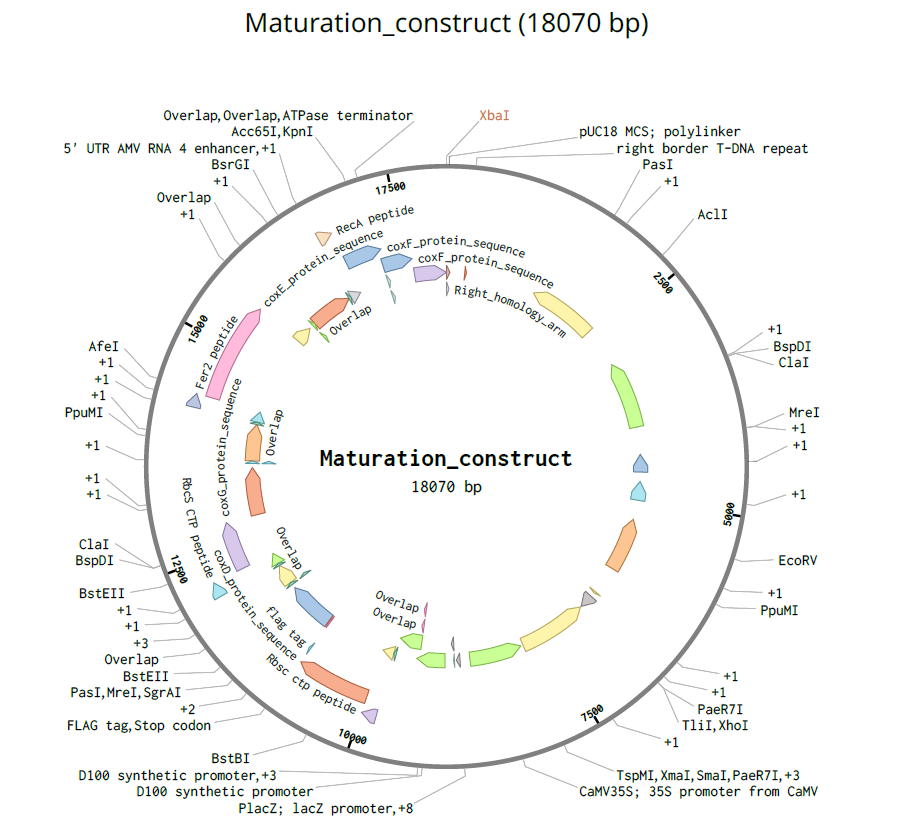
During the final quality-control inspection, I verified that the entire multicassette payload was correctly positioned between the Left Border (LB) and Right Border (RB) T-DNA sequences, ensuring compatibility with future Agrobacterium-mediated plant transformation.
I also confirmed that all original backbone features remained intact after assembly. In pCAMBIA2300, the nptII kanamycin resistance cassette used for plant selection was preserved correctly. Similarly, the hygromycin resistance cassette of pCAMBIA1300 remained unaffected.
Finally, I checked the integrity of the essential bacterial backbone elements outside the T-DNA region, including the pVS1 replication/stability regions, the pBR322 origin of replication, and the bacterial antibiotic resistance marker. All these elements remained fully conserved after circularization of the final plasmids.
The objective of this phase was to digitally assemble the fully reconstructed Structural and Maturation multicassette inserts into their corresponding binary plant expression vectors, pCAMBIA2300 and pCAMBIA1300, using in silico Gibson Assembly in Benchling. This step aimed to generate complete circular plant transformation plasmids, verify the integrity of all assembly junctions and vector backbone elements, and confirm that the final constructs were fully compatible with downstream cloning, bacterial propagation, and Agrobacterium-mediated plant transformation applications.
Verification 1 — Monomer Architecture, Confidence Profiles (pLDDT), & Tag Exposure Analysis
Objective & Methods
To comprehensively evaluate the structural integrity, predictive confidence, and purification tag behavior of my engineered plant-targeted constructs, I performed an integrated macro-scale monomer analysis using AlphaFold 3. For this first verification step, I analyzed each engineered fusion protein separately as an individual monomeric prediction. This allowed me to specifically evaluate the local structural effects of the added chloroplast transit peptides (CTPs) and purification tags on each protein independently before studying higher-order assembly behavior in later verification steps.
For each fusion protein, I systematically cross-examined four key design parameters within a single diagnostic profile:
Structural Subunits Analysis
.gif)
–> Design Verdict: PASSED ✅.
.gif)
–> Design Verdict: PASSED ✅. The Fer2 transit peptide preserves necessary terminal flexibility despite containing a brief, non-interfering junctional alpha-helix.
.gif)
–> Design Verdict: PASSED ✅.
Maturation Component Analysis
.gif)
–> Design Verdict: PASSED ✅.
.gif)
o The N-Terminal Domain Block: Aligning native residues 1 – 85 against engineered residues 53 – 136 confirmed a highly preserved structural match (RMSD = 0.4 Å and a TM-score = 0.98).
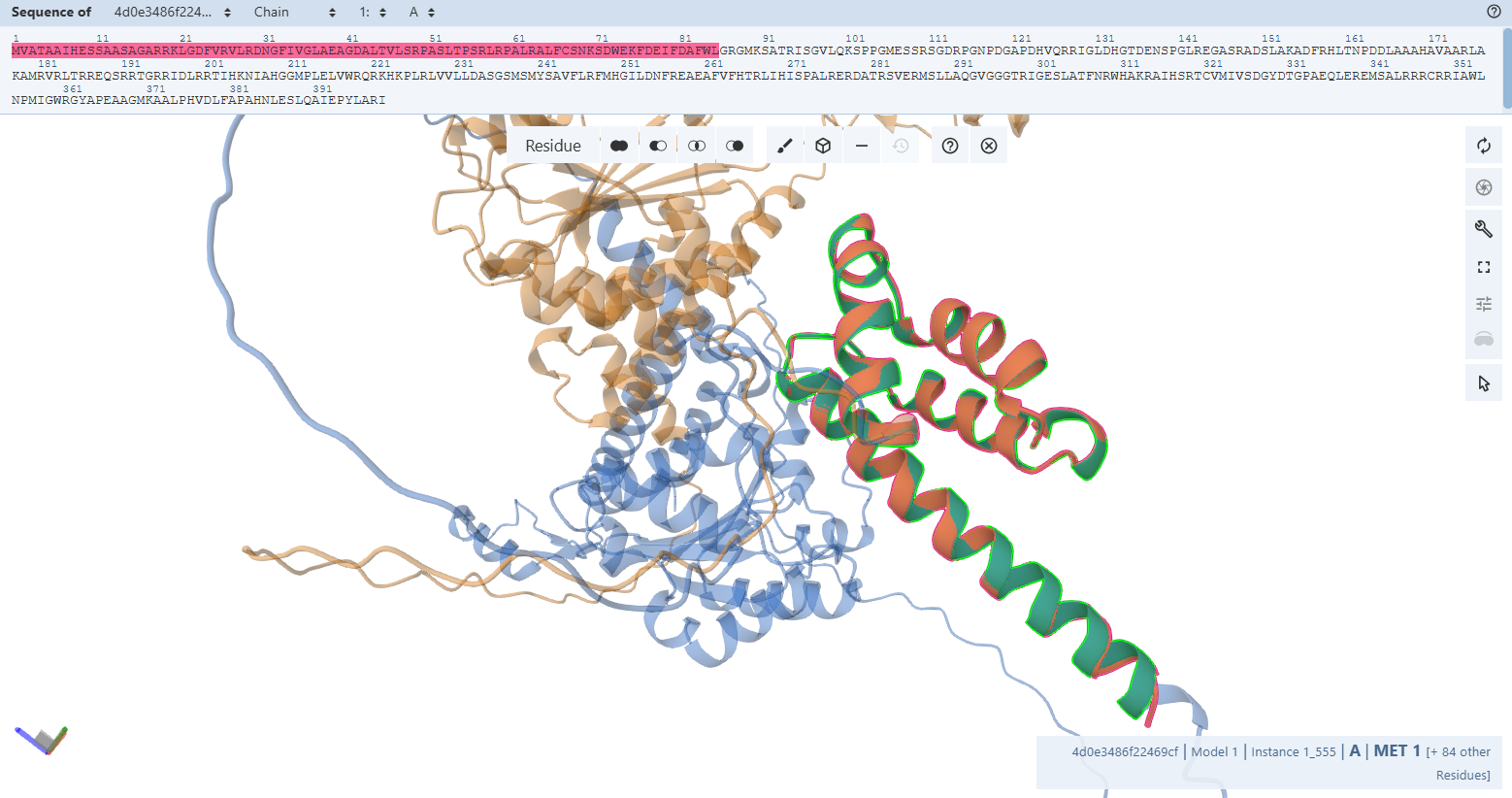 o The C-Terminal Domain Block: Aligning native residues 138 – 399 against engineered residues 190 – 451 yielded an identical, unwarped topology (RMSD = 0.95 Å and a TM-score = 1.48).
o The C-Terminal Domain Block: Aligning native residues 138 – 399 against engineered residues 190 – 451 yielded an identical, unwarped topology (RMSD = 0.95 Å and a TM-score = 1.48).
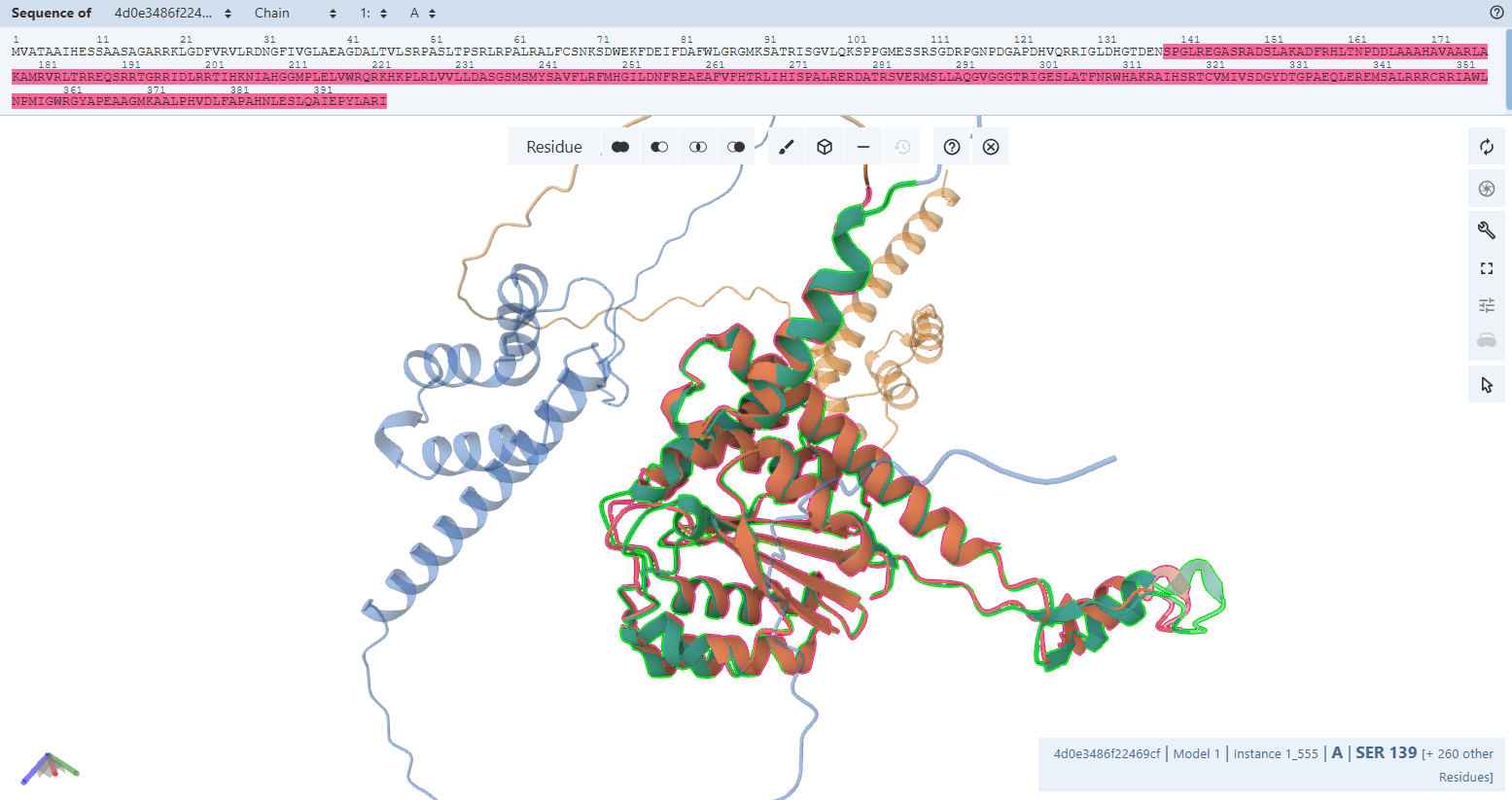
–> Design Verdict: PASSED ✅. Segmented domain matching confirms the rigid blocks are structurally pristine, and the global variation is mathematically proven to be a harmless reflection of native hinge flexibility.
.gif)
–> Design Verdict: PASSED ✅.
.gif)
Justification of Internal Core Flexibility: The minor coordinate displacement (RMSD = 1.6 Å) and unaligned residue window capture a native functional mechanism. As an accessory maturation chaperone, CoxG natively utilizes localized, flexible loop segments to bind and process its target enzyme partners. The orange patch you see inside the core is an intrinsically flexible docking loop. AlphaFold models this loop in alternative sweeping orientations when accommodating the added N-terminal RbcS CTP, confirming that the structural framework remains completely uncompromised.
–> Design Verdict: PASSED ✅. Control alignments mathematically validate that the core fold is conserved (TM-score = 0.76), and the internal core orange region is verified as a native, flexible chaperone loop.
Verification 2 — Is the Core Enzyme Domain Fold Preserved?
Objective & Methods
To verify that my engineered, codon-optimized plant-targeted fusions folded into their native, active bacterial conformations, I performed a high-resolution pairwise structural alignment. I compared each predicted monomer structure against the corresponding chain from the Oligotropha carboxidovorans gold-standard crystal structure (PDB: 1N5W) using the RCSB PDB alignment server.
To achieve this, I isolated the core catalytic domains of my models to bypass the unaligned, highly flexible synthetic additions (specifically the N-terminal chloroplast transit peptides (CTPs) and C-terminal purification tags) allowing the algorithm to evaluate the true functional enzyme scaffolds.
![]()
![]()
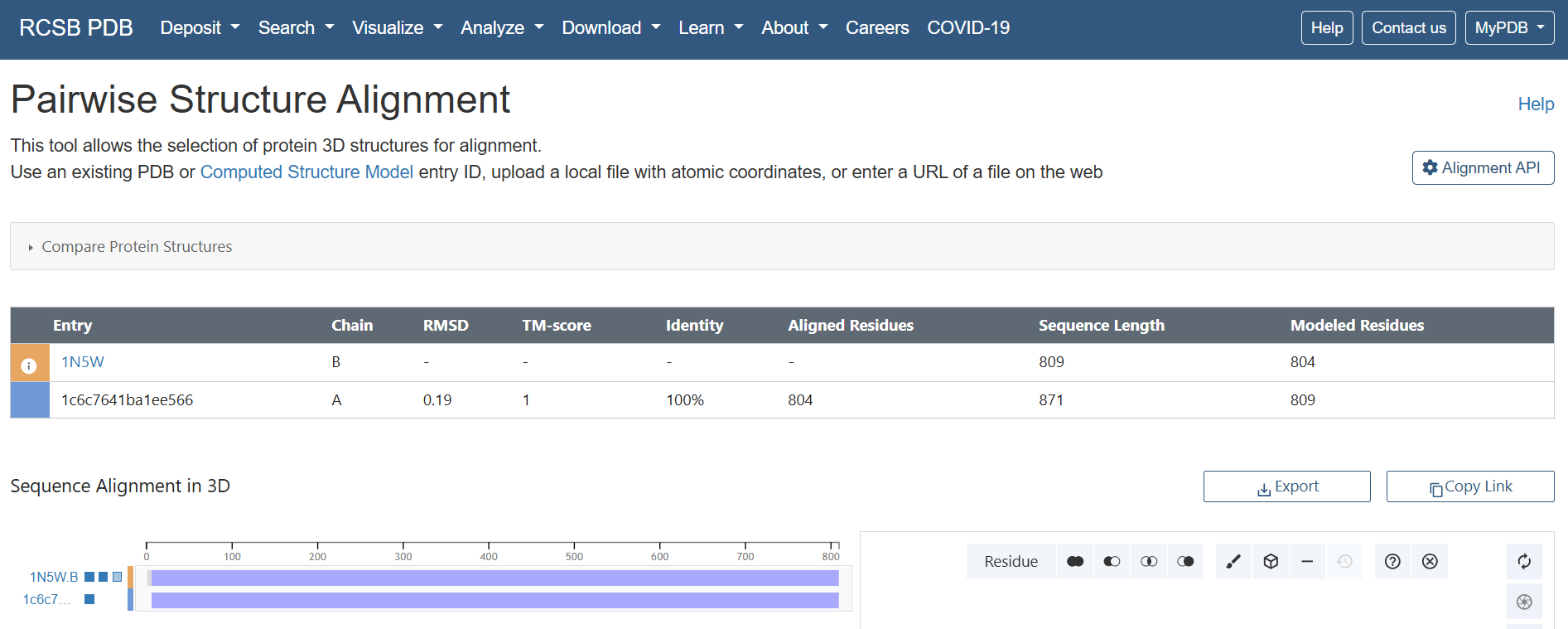 Results & Quantitative Metrics
Results & Quantitative Metrics
The alignment yielded exceptionally strong quantitative validation metrics across all three structural blocks:
| Target Subunit | Reference Chain (1N5W) | Sequence Identity | Aligned / Native Residues | Backbone RMSD (Å) | Global TM-score | Design Validation Status |
|---|---|---|---|---|---|---|
| CoxL Fusion | Chain B | 100 % | 804 / 809 | 0.19 Å | 1.00 | ✅ PASSED: Flawless active core preservation. |
| CoxM Fusion | Chain C | 100 % | 287 / 288 | 0.17 Å | 1.00 | ✅ PASSED: Pristine backbone trace topology. |
| CoxS Fusion | Chain A | 99 % | 159 / 166 | 0.87 Å | 0.98 | ✅ PASSED: Core stable; score captures flexible terminal loops. |
Structural Interpretation
.gif?width=500px)
.gif?width=500px)
.gif?width=500px)
Verification 3 — Is the Active Site Geometry of CoxL Preserved?
Objective & Methods
While global backbone alignments (Verification 2) verify macroscopic folding, true enzymatic function strictly depends on the micro-spatial positioning of active site side-chains. To prove that my plant-targeted, codon-optimized fusions preserve these crucial chemical environments, I executed a high-resolution, atom-by-atom visual audit using the Mol* molecular viewer. For each subunit, I applied a two-tiered inspection method:
Note on Sequence Numbering: Due to the engineered addition of N-terminal chloroplast transit peptides (CTPs) required for organelle targeting, the amino acid coordinates in my custom fusions are shifted forward relative to the historical bacterial literature numbering:
Literature Context & Key Residues
According to foundational structural data (Schübel et al., 1995; Dobbek et al., 1999):
L Subunit (Molybdoprotein Subunit)
The massive CoxL subunit forms the catalytic heart of the carbon monoxide dehydrogenase complex. It coordinates the unique bimetallic molybdenum-copper [CuSMoO_2] cluster and a molybdopterin cytosine dinucleotide (MCD) cofactor:
M Subunit (Flavoprotein Subunit)
The CoxM flavoprotein subunit binds a flavin adenine dinucleotide (FAD) cofactor to facilitate electron transport from the molybdenum center to downstream cellular acceptors:
S Subunit (Iron-Sulfur Subunit)
The small CoxS subunit acts as an electronic wire, channeling electrons from the molybdenum active site in CoxL to the FAD cofactor in CoxM via two distinct iron-sulfur ([2Fe-2S]) clusters. Literature establishes that CoxS is split into two rigid functional domains:
CoxL Subunit Molybdoprotein Active Site Validation
The Catalytic Core Anchor (Cys-444L & Ala-441L)
VAYRC388LSFR Loop sequence forms the active-site loop, which is unique to CO dehydrogenases and may be involved in substrate binding, it includes two critical amino acides : Cys388L (S-selanylcysteine) and Ala385L:
Molybdenum Sphere Stabilization (Gln-296L)
The Catalytic Base Proxy (Glu-819L)
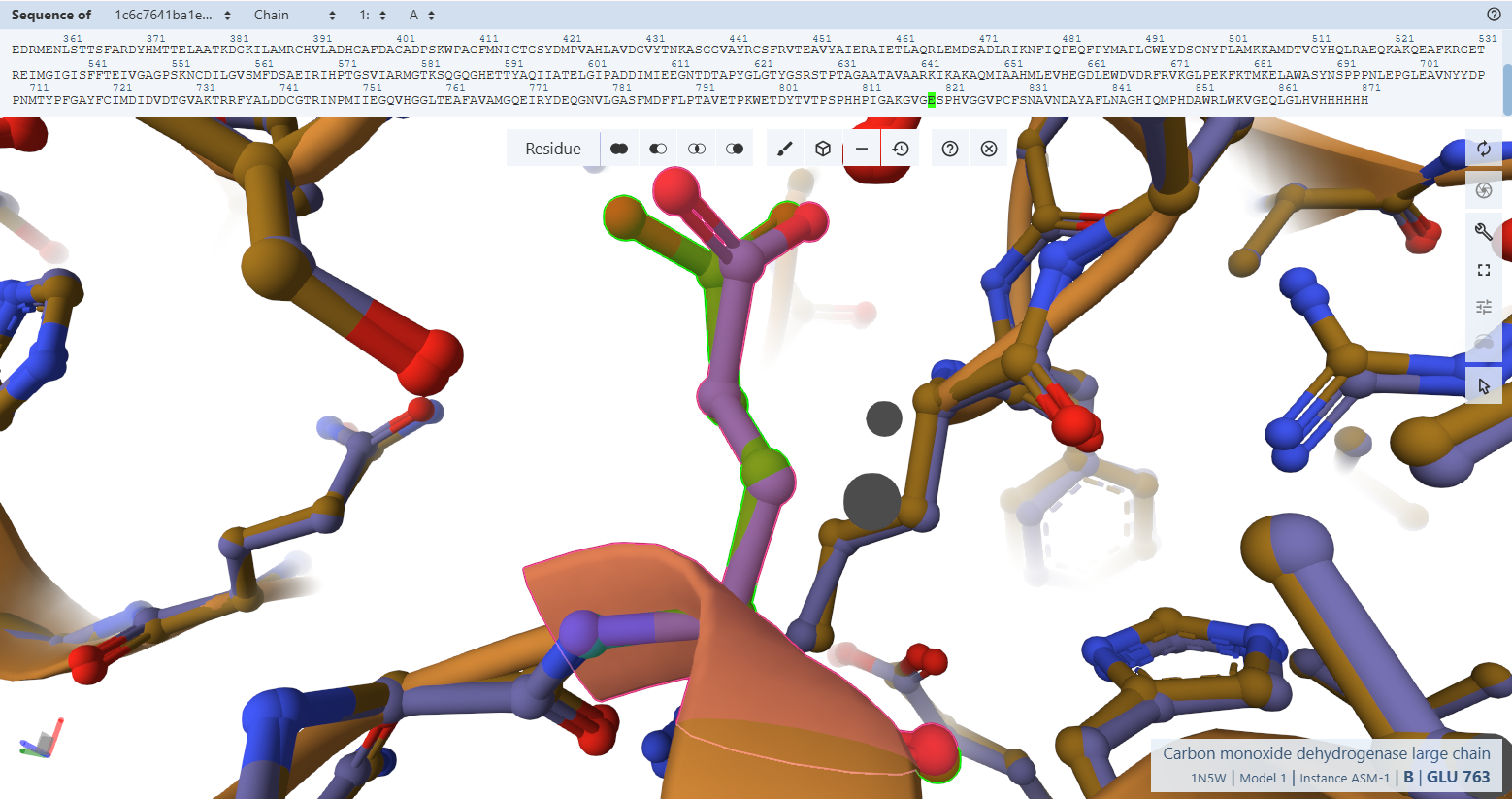 The plant-targeted, codon-optimized CoxL subunit is an exact spatial duplicate of the native Oligotropha carboxidovorans enzyme. The structural preservation proven macroscopically in Verification 2 holds true all the way down to individual chemical atoms in Verification 3. The addition of the N-terminal RbcS CTP and the C-terminal HA-tag induces no structural tension or side-chain displacement inside the catalytic core, ensuring that the engineered enzyme is fully capable of binding its cofactors and conducting chemical carbon monoxide oxidation.
The plant-targeted, codon-optimized CoxL subunit is an exact spatial duplicate of the native Oligotropha carboxidovorans enzyme. The structural preservation proven macroscopically in Verification 2 holds true all the way down to individual chemical atoms in Verification 3. The addition of the N-terminal RbcS CTP and the C-terminal HA-tag induces no structural tension or side-chain displacement inside the catalytic core, ensuring that the engineered enzyme is fully capable of binding its cofactors and conducting chemical carbon monoxide oxidation.CoxM Flavoprotein Subunit & FAD-Binding Pocket Validation
In Verification 2, the global alignment of the CoxM flavoprotein subunit achieved a backbone trace matching down to a 0.17 Å RMSD. To verify that this structural preservation translates to biochemical functionality, we must confirm that the micro-spatial positioning of the FAD cofactor cage is maintained.
The Solvent-Shielding Gatekeeper (Tyr-193M)
Residues Verified: Native Tyr-193M –> Engineered Tyr-245M (193 + 52).
Ball-and-Stick Analysis: Tyr-245M plays a crucial gatekeeping role by shielding the reactive isoalloxazine ring of FAD from unwanted solvent interactions. The phenolic ring of this tyrosine shows excellent spatial overlay with no steric conflicts, preserving its native capacity to swing out slightly during hydride transfer pathways.
 By switching to a ball-and-stick rendering, the engineered variant’s residues (rendered in light green) were compared directly to the native bacterial template (rendered in pink).
The Pyrophosphate-Binding Motif (AGGHS loop):
By switching to a ball-and-stick rendering, the engineered variant’s residues (rendered in light green) were compared directly to the native bacterial template (rendered in pink).
The Pyrophosphate-Binding Motif (AGGHS loop):
Residues Verified: Native 32MAGGHS36 on the M subunit Engineered 84MAGGHS88 (32 + 52).
Ball-and-Stick Analysis: This loop contains a highly conserved double-glycine fingerprint. Because glycine lacks a bulky side-chain, its backbone is highly flexible, allowing it to wrap closely around the charged pyrophosphate arm of the FAD molecule. The atomic overlay demonstrates a very similar match, ensuring that the main anchoring loop for the FAD center remains unwarped.
The Adenosine-Binding Motif (TIGG loop):
The FAD Stabilization Hydrogen-Bonding Network
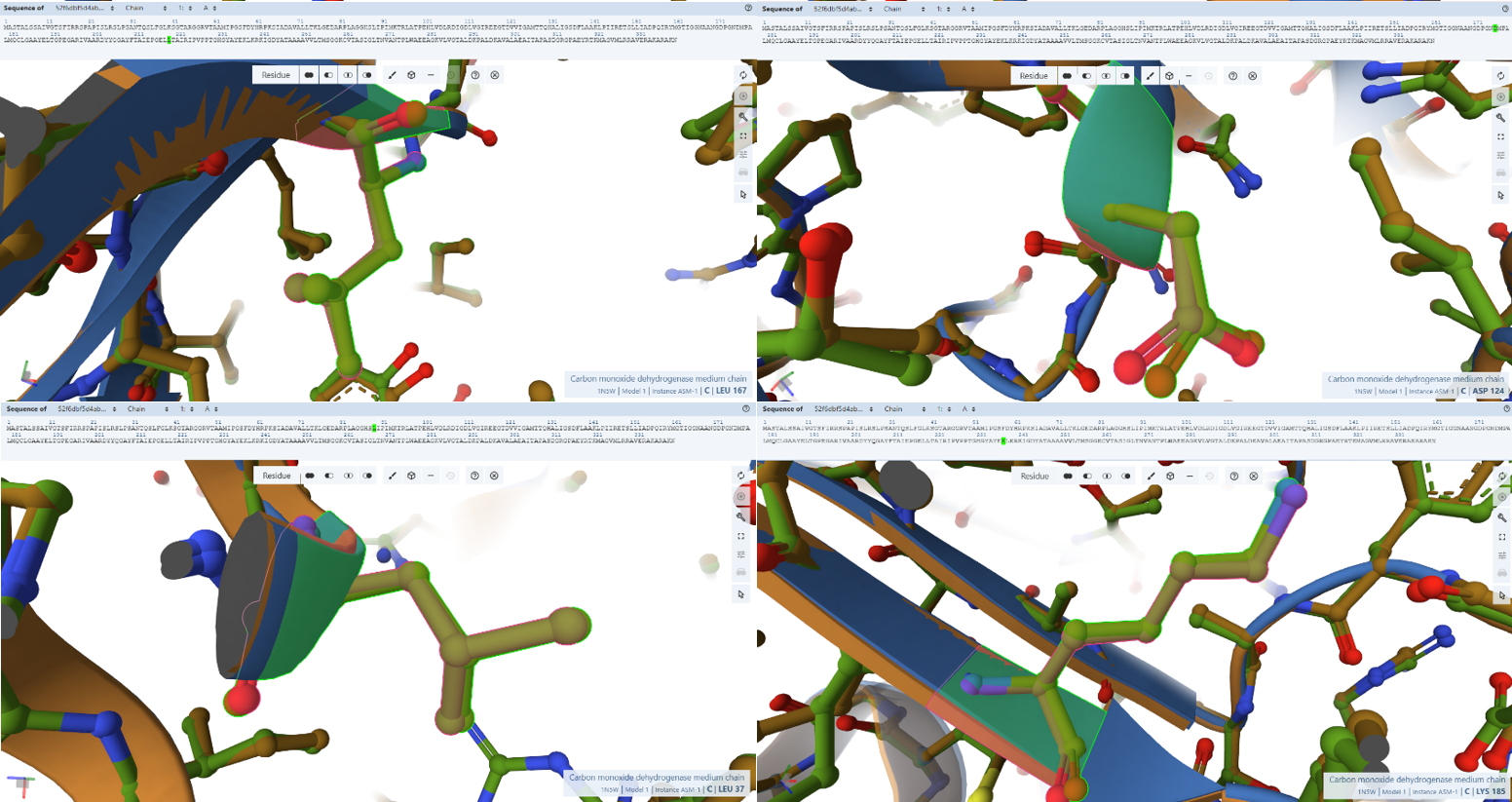
The NAD+ Electron-Exit Docking Gateway
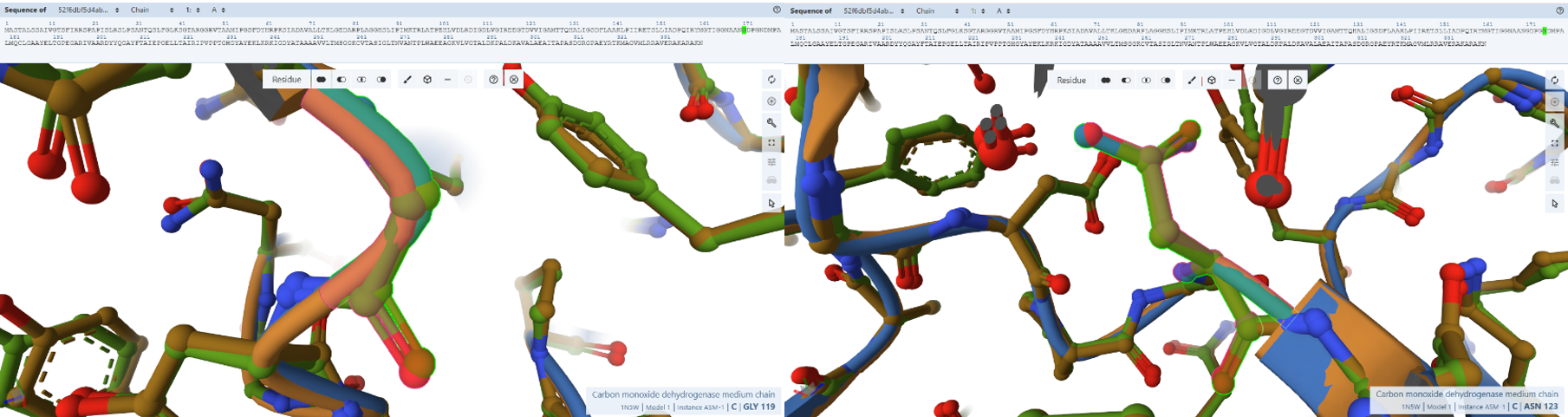
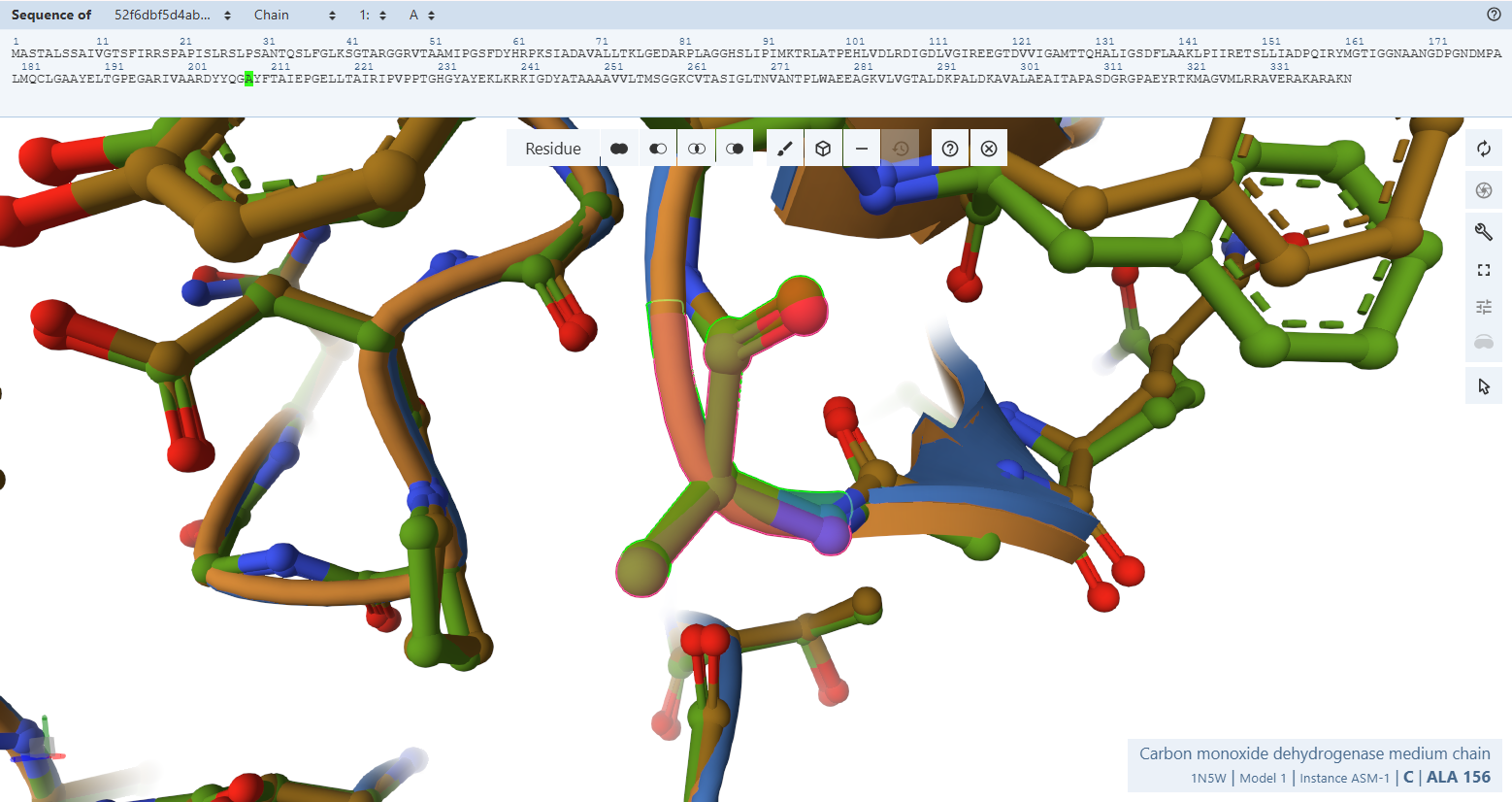
Subunit S Iron-Sulfur Subunit validation
Globally, when looking at the cartoon representations, the engineered variant’s ribbon layout (light green) matches the native bacterial template beautifully. Both the N-terminal domain (FeS II) and the C-terminal domain (FeS I) fold into their correct secondary structure orientations. This macroscopic overlay proves that the general physical envelope required to cradle the two vital [2Fe-2S] clusters is fully preserved.
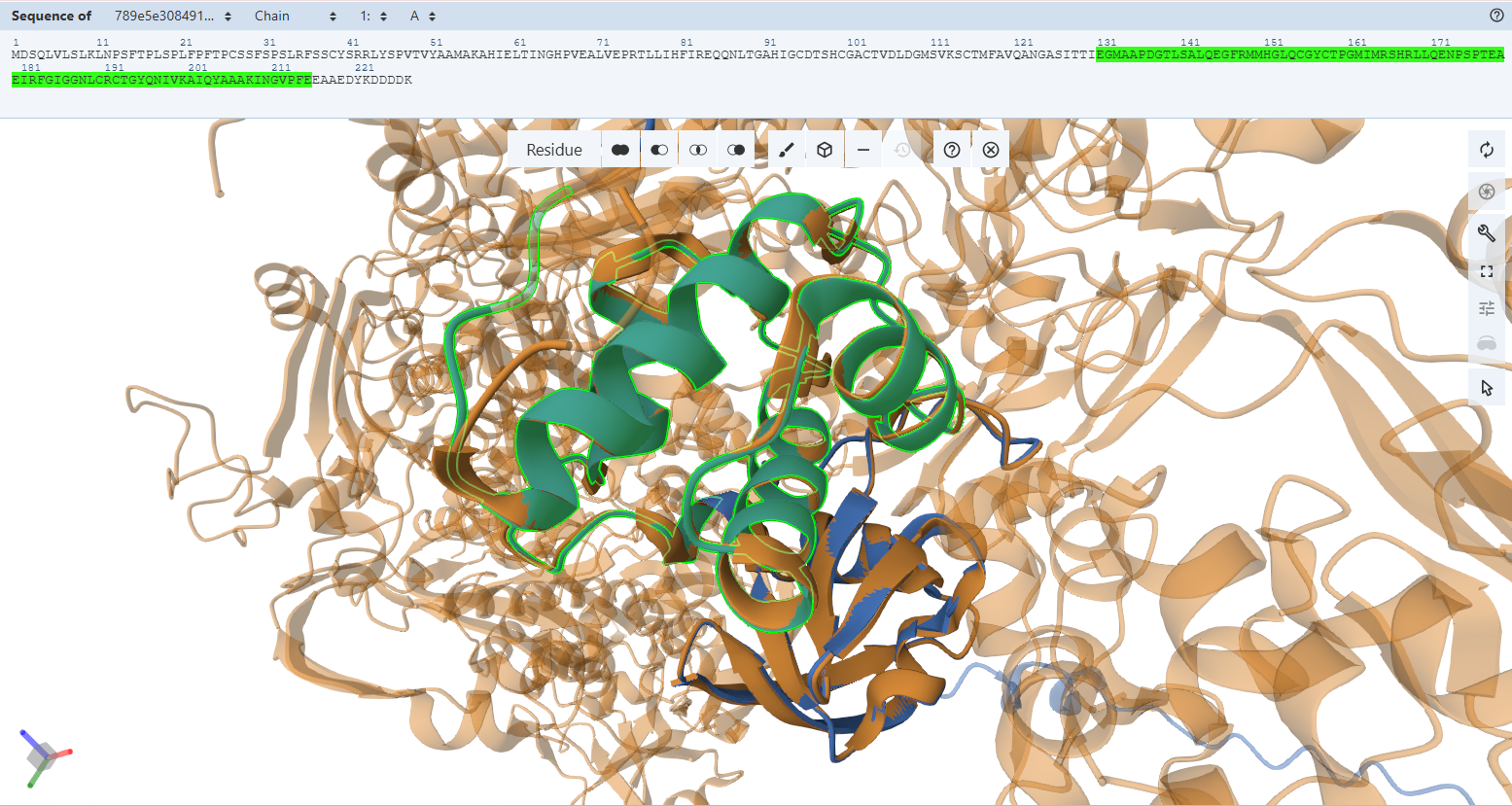
However, when we zoom in to inspect the explicit amino acid trajectories using stick representations, we can find clear structural divergences in some specific amino acids at both the absolute N-terminus and C-terminus boundaries.
 These local mismatches are predictable computational phenomena that do not compromise enzymatic function. The absolute terminal ends of CoxS directly border the engineered modifications: the RecA transit peptide junction at the N-terminus and the 9-amino-acid FLAG epitope tag at the C-terminus. Terminal tails are inherently highly dynamic, flexible “flapping tails” that lack fixed secondary structure constraints in monomeric predictions. While they adopt alternative loop paths in a relaxed fluid simulation, the core alpha-helices and beta-sheets holding the iron-sulfur clusters remain stable and unwarped.
These local mismatches are predictable computational phenomena that do not compromise enzymatic function. The absolute terminal ends of CoxS directly border the engineered modifications: the RecA transit peptide junction at the N-terminus and the 9-amino-acid FLAG epitope tag at the C-terminus. Terminal tails are inherently highly dynamic, flexible “flapping tails” that lack fixed secondary structure constraints in monomeric predictions. While they adopt alternative loop paths in a relaxed fluid simulation, the core alpha-helices and beta-sheets holding the iron-sulfur clusters remain stable and unwarped.
Verification 4 — Complex Assembly and Interface Accessibility Analysis
Objective & method
To verify whether the engineered system correctly assembles into its expected functional macromolecular complex, I performed a full structural validation of the predicted heterohexameric enzyme. The goal was to confirm that all engineered subunits properly assemble without disrupting native-like interaction networks, and that chloroplast targeting sequences and fusion modifications do not interfere with oligomerization.
Instead of analyzing isolated subunits, the full biological assembly was evaluated as a complete six-chain heterohexameric complex predicted by AlphaFold Multimer.
System Architecture (Hexameric Model)
The modeled system corresponds to a functional symmetric heterohexamer composed of two trimeric units:
This defines a complete (LMS)₂ assembly, representing two identical trimeric functional units forming a higher-order oligomer.
Global Structural Validation (AlphaFold Multimer)
The full six-chain complex was first evaluated using AlphaFold Multimer prediction.
 The model shows: A stable and symmetric heterohexameric assembly with proper organization of all six subunits. The model displayed well-defined packing between the functional chains, indicating that the proteins assemble correctly into the expected complex. The Predicted Aligned Error (PAE) analysis revealed low-error values at the different interfaces, supporting a high level of confidence in the inter-chain interactions and overall oligomeric arrangement. No signs of chain dissociation, structural deformation, or collapse were observed in the predicted structure. In addition, the chloroplast transit peptides were oriented outward toward solvent-exposed regions and remained separated from the structural core, indicating that the introduced targeting sequences do not interfere with protein folding or complex assembly.
The model shows: A stable and symmetric heterohexameric assembly with proper organization of all six subunits. The model displayed well-defined packing between the functional chains, indicating that the proteins assemble correctly into the expected complex. The Predicted Aligned Error (PAE) analysis revealed low-error values at the different interfaces, supporting a high level of confidence in the inter-chain interactions and overall oligomeric arrangement. No signs of chain dissociation, structural deformation, or collapse were observed in the predicted structure. In addition, the chloroplast transit peptides were oriented outward toward solvent-exposed regions and remained separated from the structural core, indicating that the introduced targeting sequences do not interfere with protein folding or complex assembly.
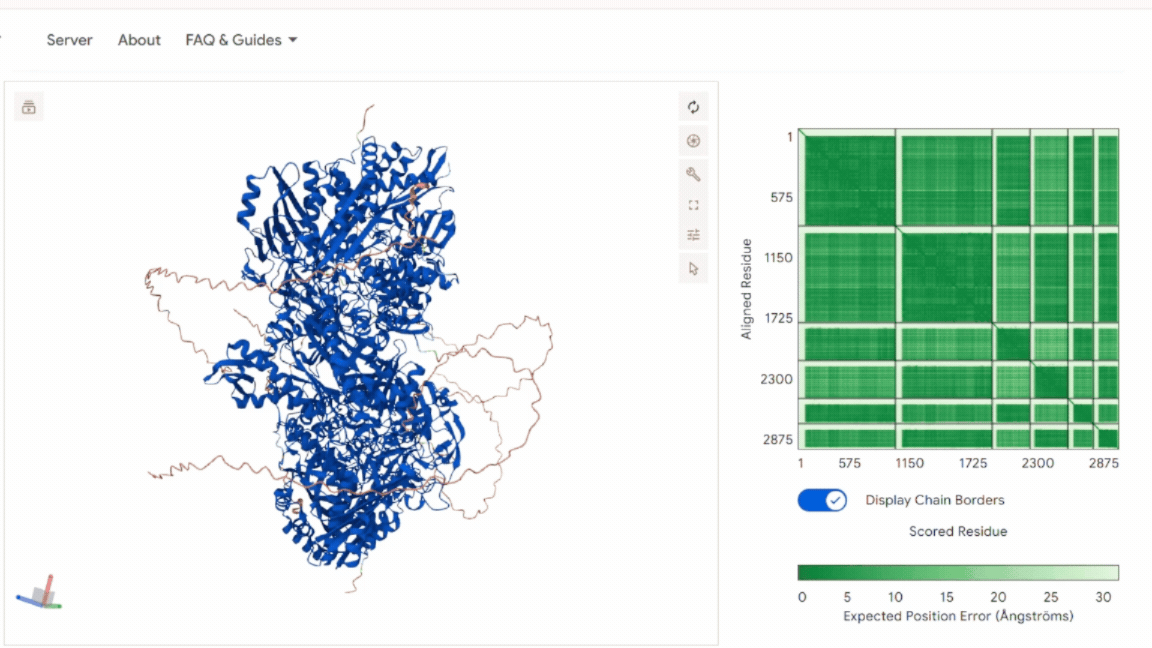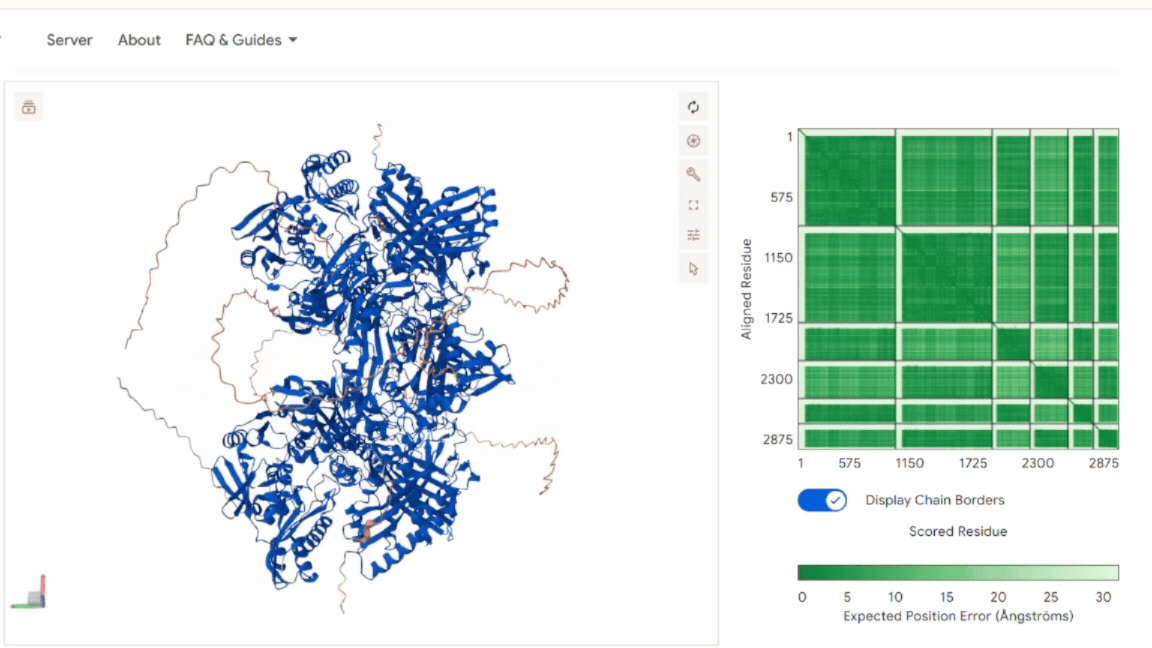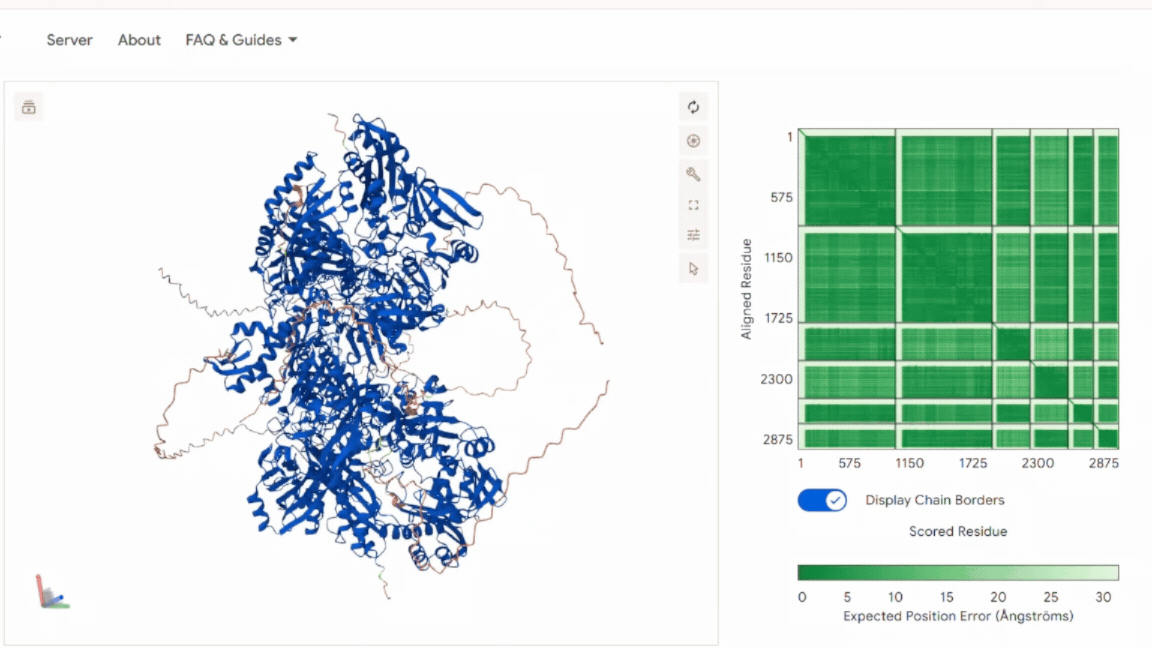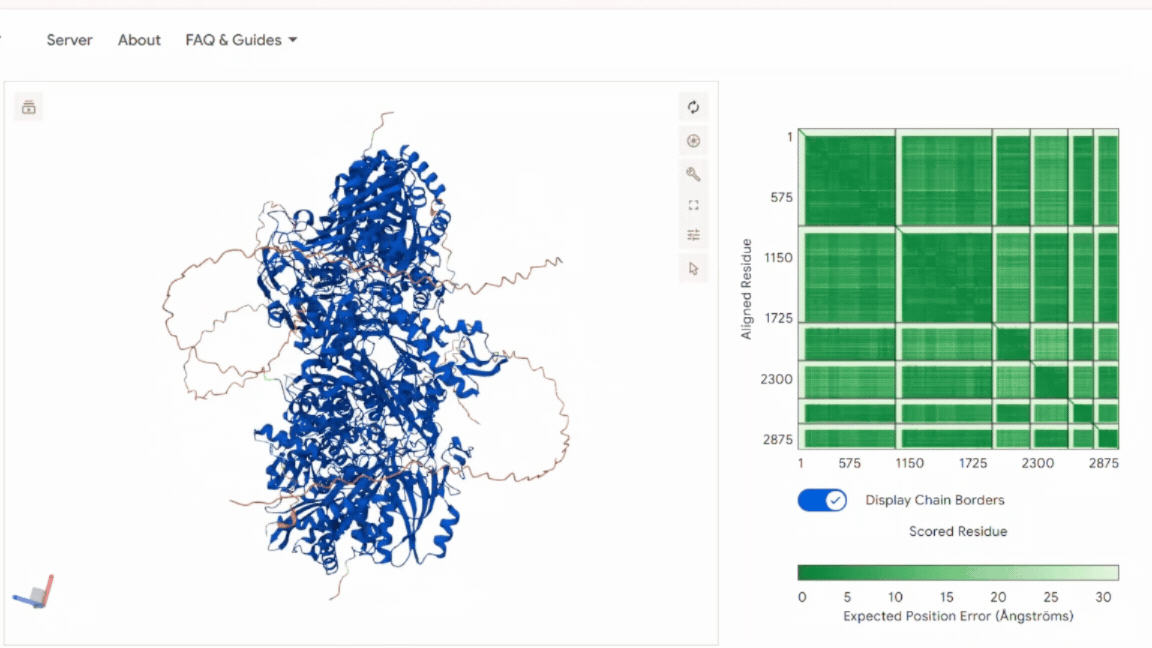
These results confirm that the global architecture is structurally consistent with a functional oligomeric enzyme.
Unbiased Interaction Mapping Strategy (PyMOL Analysis)
To identify all possible atomic interactions without bias toward predefined interfaces, I used a fully unrestricted contact-scan approach in PyMOL.
 Instead of selecting specific interfaces manually, the script:
Instead of selecting specific interfaces manually, the script:
Although all possible chain combinations were allowed in the script (A–B–C–D–E–F), the analysis naturally converged into only four physically meaningful interaction networks, indicating that only specific interfaces are structurally stable and biologically relevant:
A–C Interface (CoxL ↔ CoxM core interaction)
CoxL (Chain A) and CoxM (Chain C) form a strong central interface where both proteins are tightly packed together and build the structural core of the trimer.
This interface is stabilized by different types of interactions, including salt bridges, hydrogen bonds, and hydrophobic contacts. The residues listed below are examples taken from the full interaction set identified in PyMOL (not the complete list):
These interactions show that CoxL and CoxM are strongly connected through a combination of electrostatic attraction and hydrophobic packing, which stabilizes the core structure of each trimer unit.
A–E Interface (CoxL ↔ CoxS interaction)
CoxS (Chain E), the smaller functional subunit, interacts with CoxL on the external surface of the complex. This interface ensures that CoxS is properly anchored and positioned for its functional role.
The residues shown below are representative examples from the full set of interactions detected in PyMOL (not exhaustive):
These interactions confirm that CoxS is firmly attached to the main complex and is not loosely associated or freely moving.
C–E Interface (CoxM ↔ CoxS outer stabilization interface)
CoxM (Chain C) and CoxS (Chain E) form additional stabilizing interactions that reinforce the outer structure of each trimer unit.
The interactions below are examples selected from the complete interaction network detected by PyMOL (not the full list):
These interactions indicate that the outer surface of the trimer is stabilized by multiple weak and strong forces working together.
A–B Interface (CoxL ↔ CoxL dimerization axis)
This interface represents the central dimerization boundary where two trimeric units assemble into the full hexameric structure. The interaction is highly symmetric and indicates a strong and specific docking interface.
The residues shown below are examples from the full symmetric interaction network identified in PyMOL (not exhaustive):
These interactions are further stabilized by nearby charged residues, including ASP605 and ASP606, which contribute to the electrostatic stability of the interface. This confirms that the two trimer halves assemble in a highly specific and symmetric manner, forming a stable functional hexamer.
I used the Gemini AI tool to interpret the structural results and to predict how these specific CTP modifications and AA junctions influence protein folding, stability, and chloroplast targeting efficiency. while ChatGPT was employed for technical editing, ensuring the documentation was clear, concise, and grammatically precise.
Objective:
The objective of this verification step was to evaluate whether the engineered fusion subunits retained their ability to correctly assemble into the complete functional enzyme complex after the addition of chloroplast transit peptides (CTPs) and purification tags. Instead of predicting only the CoxL–CoxM–CoxS trimer, I modeled the entire (LMS)2 heterohexameric complex using AlphaFold 3 in order to perform a more realistic structural validation of the final engineered system.
This analysis aimed to verify that all modified subunits still formed stable inter-chain interactions comparable to the native enzyme architecture, while also confirming that the added CTP regions remained solvent-exposed and spatially separated from the subunit–subunit interaction interfaces. In addition, this step was used to assess whether the native assembly surfaces between CoxL, CoxM, and CoxS remained structurally accessible and unaffected by the engineered modifications, ensuring that the final enzyme complex could theoretically self-assemble correctly inside the chloroplast environment.
Sources:
Golden Gate Assembly Design Strategy
To enable modular Golden Gate Assembly, all fragments were flanked with BsaI recognition sites and custom-designed overhangs. These overhangs were selected to guide the ordered assembly of the fragments into the pCAMBIA1300 backbone after digestion.
The same assembly architecture was used for all three constructs, with the only difference being the CTP sequence.
The assembly order was:
Vector → FMV promoter → AMV enhancer → CTP → eGFP → tE9 terminator
Design of Junction Overhangs
The pCAMBIA1300 vector was linearized using XbaI digestion. The last four nucleotides remaining from the digested vector (“TCCT”) were directly incorporated as the assembly scar between the vector backbone and the FMV promoter fragment.
 This strategy avoided unnecessary sequence modifications and maintained compatibility with the Golden Gate assembly design.
This strategy avoided unnecessary sequence modifications and maintained compatibility with the Golden Gate assembly design.
A custom “TACT” overhang was designed between the FMV promoter and the AMV RNA4 enhancer.
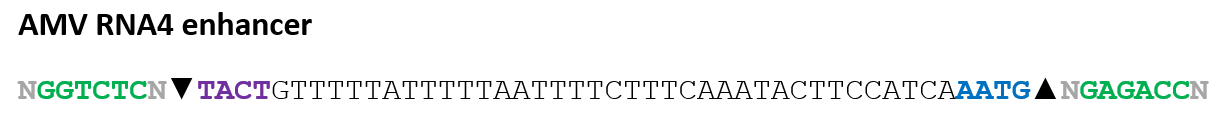 This sequence functioned as a neutral assembly scar that allowed directional ligation while preserving the integrity of both regulatory elements.
This sequence functioned as a neutral assembly scar that allowed directional ligation while preserving the integrity of both regulatory elements.
The “AATG” overhang was designed between the AMV enhancer and the chloroplast transit peptide (CTP) sequence.
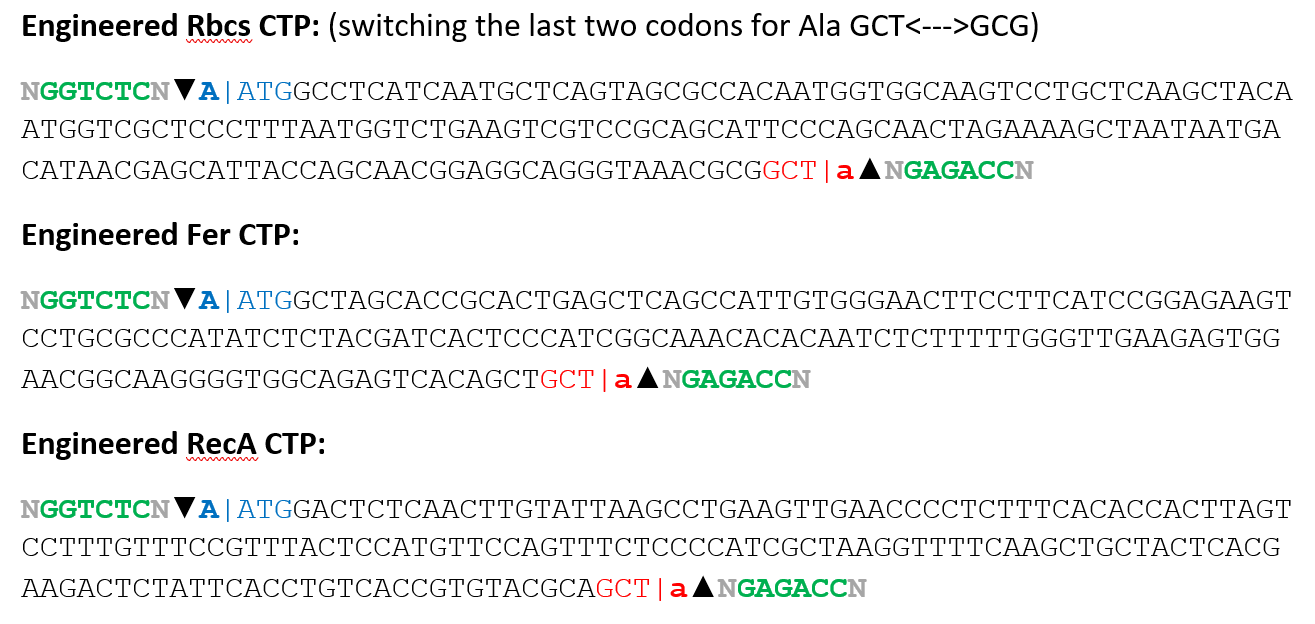 This overhang was selected because it contains the ATG start codon required for translation initiation. The design therefore allowed the translational start site to be incorporated directly into the assembly junction while preserving the correct reading frame.
This overhang was selected because it contains the ATG start codon required for translation initiation. The design therefore allowed the translational start site to be incorporated directly into the assembly junction while preserving the correct reading frame.
The junction between the CTP coding sequence and eGFP was designed carefully to preserve the open reading frame and avoid frameshift mutations.
Because this junction connected two coding sequences, the overhang was designed using:
Together, these nucleotides formed the “GCTA” overhang.

For the RbcS chloroplast transit peptide construct, the last two alanine codons were simply rearranged by switching: GCT↔GCG
Because both codons encode alanine, this modification did not alter the amino acid sequence of the transit peptide. The change was performed only to expose the required “GCT” sequence needed for the Golden Gate Assembly overhang at the CTP–eGFP junction while preserving the correct reading frame and maintaining the native peptide composition.
This strategy allowed the translational reading frame to remain continuous across the fusion protein while minimizing unnecessary amino acid changes.
The reading frame continuity was verified during sequence design, as represented by the “|” positions in the coding sequences.
For the junction between eGFP and the tE9 terminator, the “CGCT” overhang was designed.
In this case:
 This overhang was added at the end of the eGFP fragment after the stop codon, ensuring proper assembly without affecting the translated protein sequence.
This overhang was added at the end of the eGFP fragment after the stop codon, ensuring proper assembly without affecting the translated protein sequence.Golden Gate Assembly of Reporter Constructs in Benchling
First, I opened Benchling and clicked on the Create (+) button from the left sidebar. From the cloning options, I selected “Assemble DNA sequences by cloning”, then chose the Golden Gate Assembly workflow. This opened the assembly interface where all cloning parameters were configured.
For the assembly settings, I selected the destination project folder dedicated to the reporter constructs. Since the final products were designed as plasmids, I set the construct topology to Circular. I then selected Golden Gate Assembly as the cloning method and specified BsaI as the Type IIS restriction enzyme used for assembly.
![]()
![]()
 Next, I imported all DNA fragments in their correct assembly order. The fragments included:
Next, I imported all DNA fragments in their correct assembly order. The fragments included:
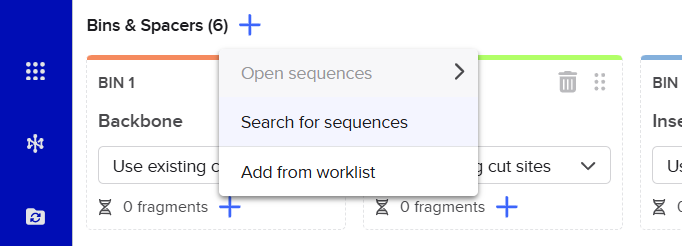
![]()
![]()
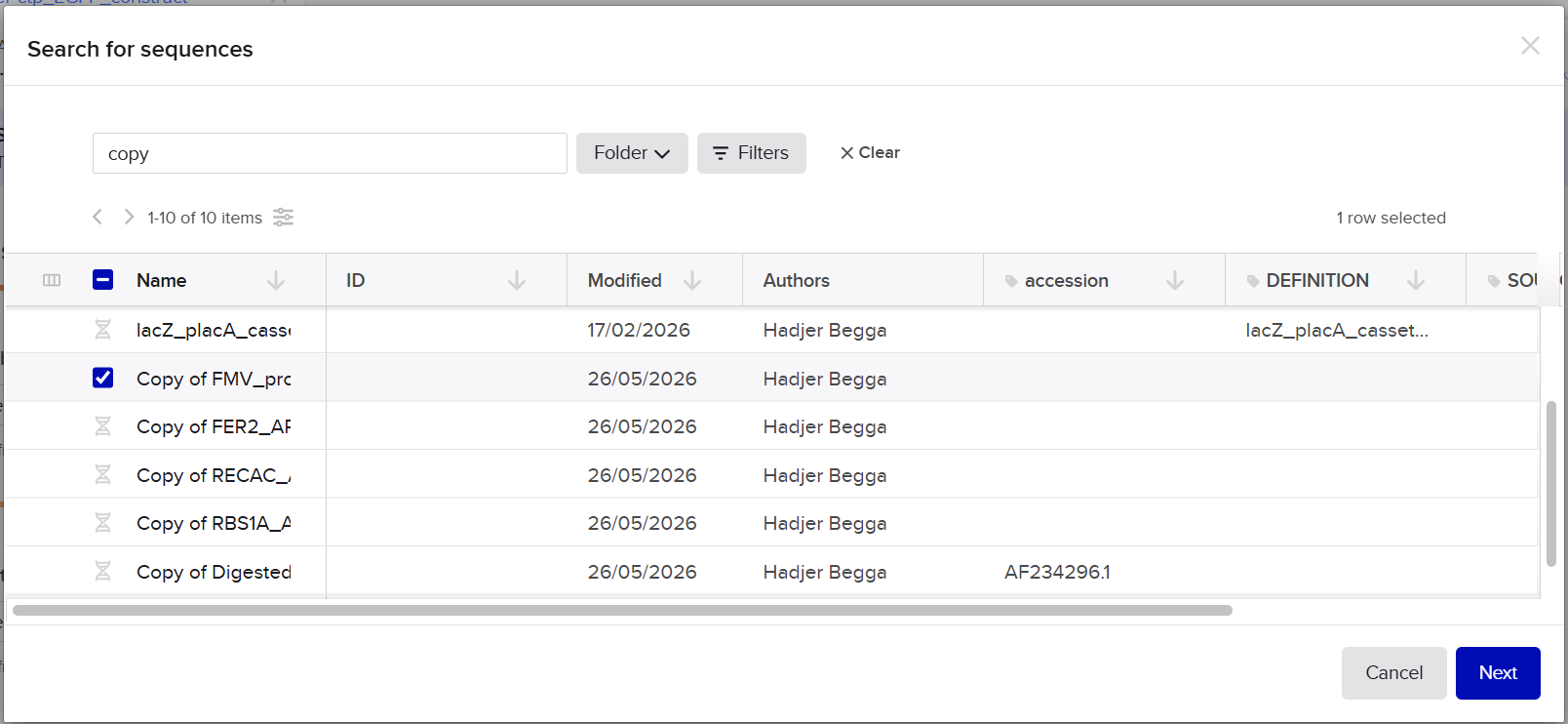
![]()
![]()
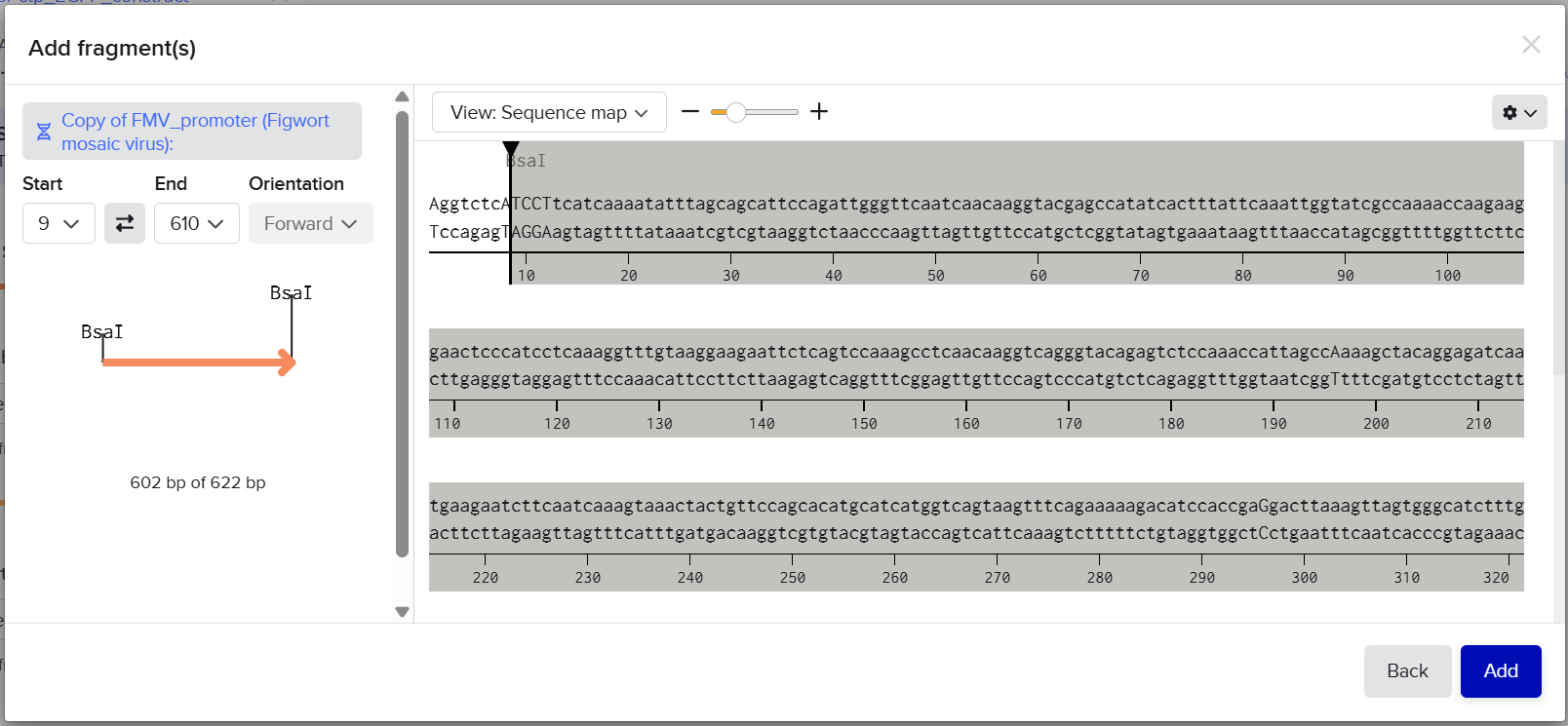
![]()
![]()
After importing all fragments, Benchling automatically analyzed the BsaI digestion products and checked the compatibility of all adjacent overhangs. When all overhangs matched correctly and fragment orientation was valid, Benchling generated a complete circular assembly product corresponding to the final reporter plasmid.
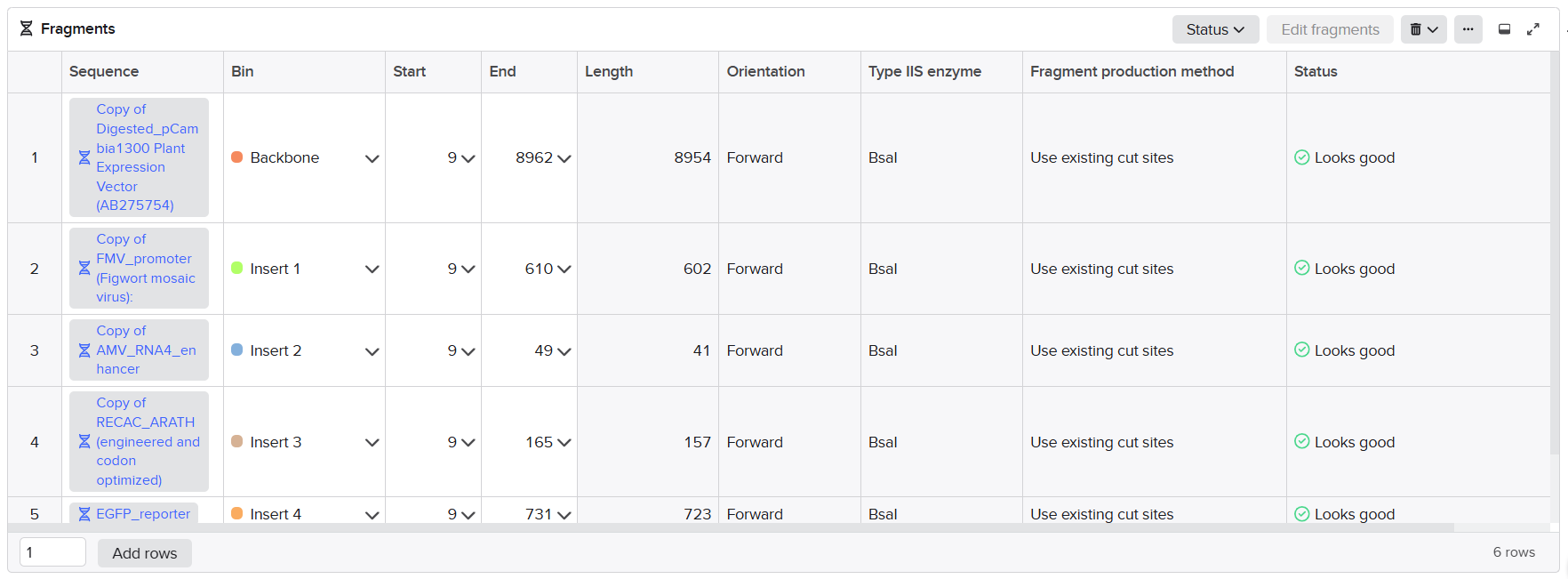
![]()
![]()
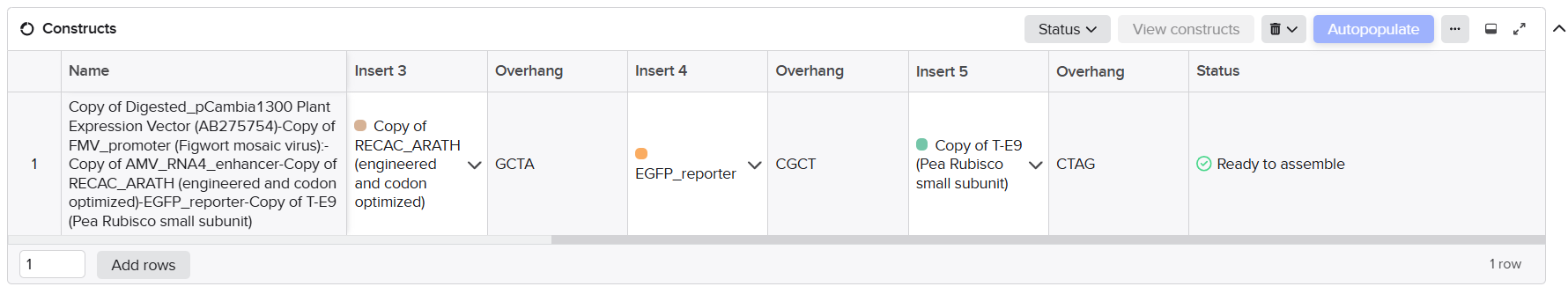 I then clicked the “Assemble” button to generate the final constructs.
I then clicked the “Assemble” button to generate the final constructs.
![]()
![]()
 Benchling created three independent circular plasmid sequences corresponding to:
Benchling created three independent circular plasmid sequences corresponding to:
Rbcs-CTP_EGFP_Benchling_Design: FMV promoter → AMV enhancer → RbcS CTP → eGFP → tE9
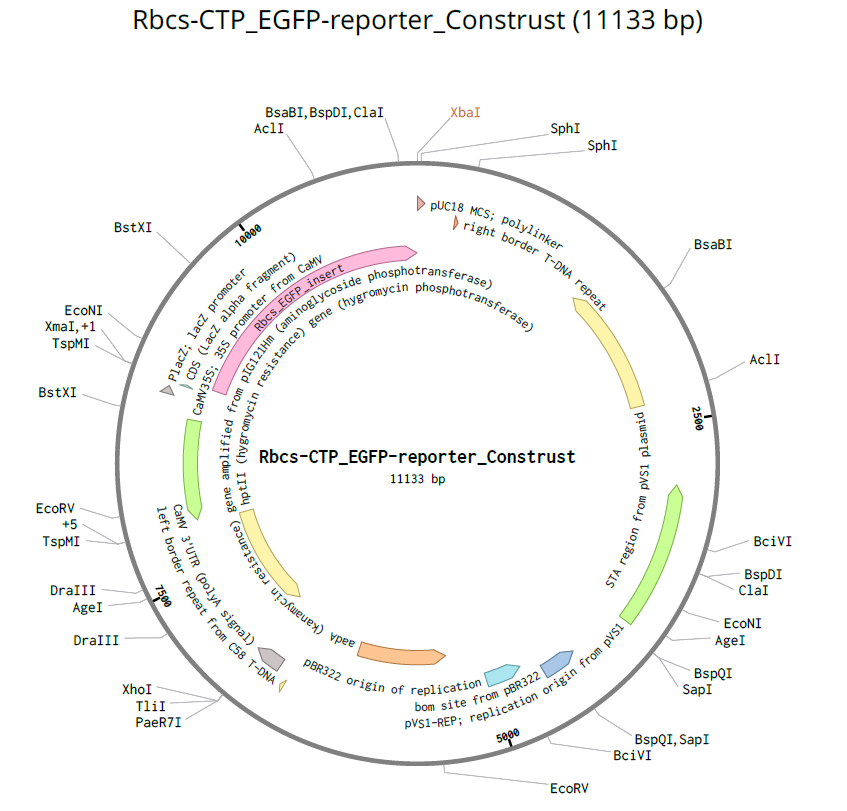 Fer-CTP_EGFP_Benchling_Design: FMV promoter → AMV enhancer → Fer2(M→A) CTP → eGFP → tE9
Fer-CTP_EGFP_Benchling_Design: FMV promoter → AMV enhancer → Fer2(M→A) CTP → eGFP → tE9
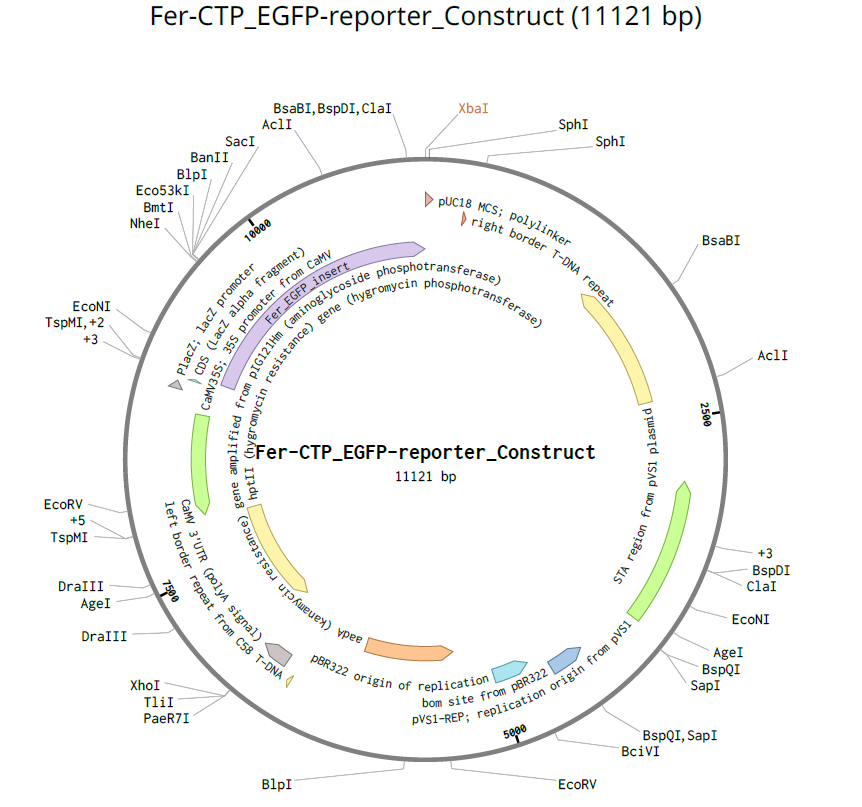 RecA-CTP_EGFP_Benchling_Design: FMV promoter → AMV enhancer → RecA CTP → eGFP → tE9
RecA-CTP_EGFP_Benchling_Design: FMV promoter → AMV enhancer → RecA CTP → eGFP → tE9
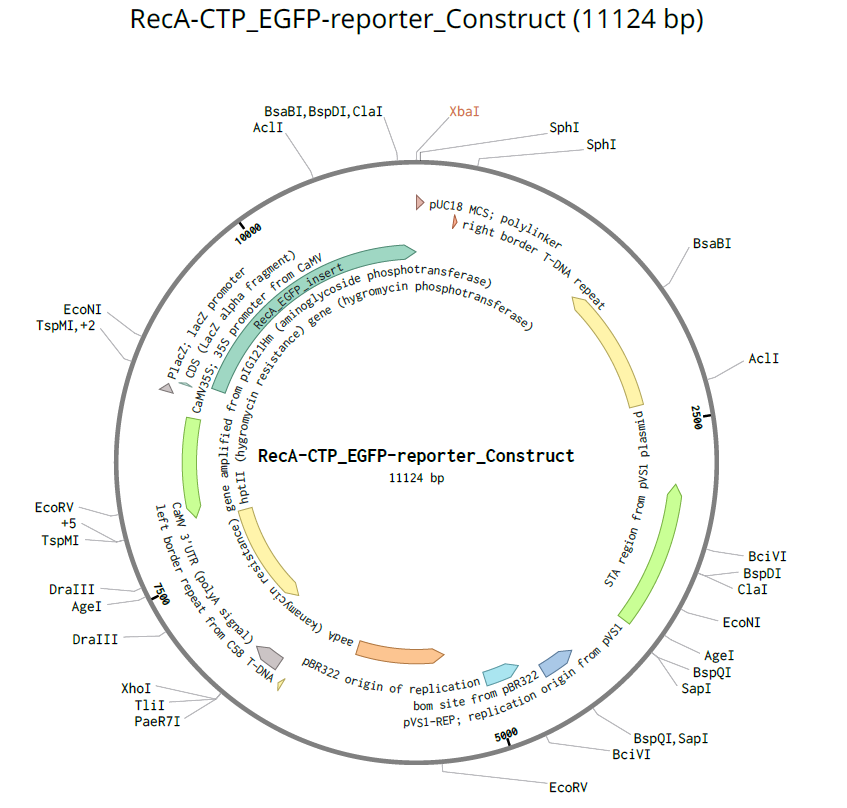
Finally, I opened the resulting plasmid maps to verify construct integrity. I carefully checked that all annotations were preserved correctly across the assembled plasmids, including promoters, enhancers, transit peptides, coding sequences, and terminators. I also verified that all junctions were seamless, that the reading frame remained continuous across fusion regions, and that no inversions, missing fragments, or unintended mutations were introduced during the assembly simulation.
Objective
The objective of this step was to design three plant expression constructs that could be efficiently assembled into circular plasmids using the Golden Gate Assembly (GGA) method. All three constructs were designed with the same regulatory and reporter elements, while only the chloroplast transit peptide (CTP) sequence was changed in order assess the correct localization of the three engineered ctp sequences using a GFP reporter and confocal microscopy. The final constructs were designed as follows:
Each construct was assembled using BsaI-mediated Golden Gate cloning with specifically designed 4 bp overhangs to ensure correct orientation and seamless ligation between adjacent fragments.