Week 4 HW: Protein Design Part I
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
On average 100g of meat 🥩 contains 26g of protein = 26% protein
100:26 ∴ 500:130g
1 amino acid = 100 daltons ≈ 1.66 × 10⁻²²g
130g / 1.66 × 10⁻²²g = 7.83×10²³ amino acids
7.83×10²³ / Avogadro’s number
or 7.83×10²³ /6.02214076×10²³ mol−1= 1.3 mol
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans eat other animals, they break down the foreign animal matter into its constituent parts (i.e. complex proteins become amino acids) in their digestive systems. In this way genetic information is not transferred from the cow or fish to the human, though amino acids, for example, from digestion are used to build the human cells and DNA.
Furthermore, human DNA contains the instructions for human cells only and foreign DNA cannot be read or translated into proteins within human cells.
3. Why are there only 20 natural amino acids?
It is not yet understood why there are only 20 natural amino acids. Francis Crick has proposed the “frozen accident” theory which postulates that the universal genetic code is not optimised, but is rather the result of a random, early and arbitrary arrangement which later became “frozen” 🥶. This would imply that the 20 natural amino acids just so happened to become the foundation for all life. Studies hypothesise that the properties of these 20 natural amino acids, such as polarity/charge, hydrophilicity/hydrophobicity, and size, are evenly distributed throughout the chemical space.
Publication on “What Froze the Genetic Code?” (Pouplana et al. 2017) https://pmc.ncbi.nlm.nih.gov/articles/PMC5492136/
However, for now the reason for there being only 20 natural amino acids is a mystery. Today scientists have created thousands of non-naturally occurring amino acids and continue to do so for medicine, material science, and research purposes.
Article on the applications of “Reprogramming natural proteins using unnatural amino acids” (Adhikari et al. 2021): https://pmc.ncbi.nlm.nih.gov/articles/PMC9044140/#
4. Can you make other non-natural amino acids? Design some new amino acids.
As I touched on briefly in the question above, yes! You can make other non-natural amino acids.
5. Where did amino acids come from before enzymes that make them, and before life started?
“Most models of the origin of life suggest organisms developed from environmentally available organic compounds. A variety of amino acids are easily produced under conditions which were believed to have existed on the primitive Earth or in the early solar nebula.” (Cleaves II 2010:480)
It is believed that before amino acids were manufactured by biotic factors, they were formed through abiotic chemical reactions, with key sources of material finding their way onto Earth through extraterrestrial material such as carbonaceous meteorites.

In the movie 🎥 🍿 I watched with my boyfriend last week, Yi Yi (2000) dir. Edward Yang, there was a scene with lightning 🌩️ ⚡️ ⛈️ and the narrator talked about how scientists posit that it was lightning on Earth 🌍 that helped to create the first amino acids. They said this in passing but it stuck with me and I did some reseach into this and found that yes, research does indicate that the high-temperate shock waves produced by lightning would be a highly efficient energy source for abiotic synthesis of amino acids on prebiotic Earth.
In fact the groundbreaking 1952 Miller-Urey Experiment, conducted by Stanley Miller and supervised by Nobel laureate Harold Urey at the University of Chicago, demonstrated that organic molecules such as amino acids could be synthesised from inorganic precursors under simulated prebiotic Earth conditions. In essence, electrical sparks simulating lightning were passed through a mixture of water, ammonia, methane, and hydrogen. This experiment produced complex organic compounds, thus supporting the theory of abiogenesis.
Miller published a paper detailing the experiment in a 1953 edition of the journal Science: “A production of amino acids under possible primitive earth conditions” (Miller 1953).
https://pubmed.ncbi.nlm.nih.gov/13056598/
Here is a paper reviewing this finding: “The 1953 Stanley L. Miller Experiment: Fifty Years of Prebiotic Organic Chemistry” (Lazcano and Bada 2003).
https://link.springer.com/article/10.1023/A:1024807125069
However, some posit that the results of the Miller-Urey experiment are contentious as the experiment produced a racemix mixture, that is an equal amount of both left and right handed amino acids. This poses a problem as life on Earth requires only left-handed amino acids, and as of yet no natural mechanism has been demonstrated to separate such in early Earth conditions.
Some scholars thus argue that homochirality has extraterrestrial origins: “Chirality and the origin of life” (Bailey 2000).
https://www.sciencedirect.com/science/article/abs/pii/S0094576500000242
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
You would expect left-handedness.
7. Can you discover additional helices in proteins?
Yes! There are not only
- α-helices
there are also
3₁₀-helices
π-helices
8. Why are most molecular helices right-handed?
Most molecular helices such as α-helices in proteins and DNA (in its A and B forms) are right-handed as they are comprised of L-amino acids and D-sugars which favour a right-handed configuration as it maximises hydrogen bonding and minimises steric hindrance.
An exmaple of a molecular helix which is not right-handed is Z-DNA.
9. Why do β-sheets tend to aggregate? - What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate due to the fact that their egdes readily interact with other β-strands, leading to edge-to-edge aggregation. In other words, β-sheets are flatter and tend to stack easily.
Hydrophobicity is the driving force for β-sheet aggregation.
10. Why do many amyloid diseases form β-sheets? - Can you use amyloid β-sheets as materials?
Many amyloid diseases form β-sheets as this structure is highyly stable, low energy, and thermodynamically favourable one which enables the aggregation of misfolded proteins. The β-sheets stack into “cross-β” configurations. This leads to the formation of insoluble fibrils which resist degradation and maximise hydrophobic interations 🚫💧.
This is the essential cause of Alzheimer’s Disease, Type 2 Diabetes, and Spongiform Encephalopathies (i.e. Creutzfeldt-Jakob disease (CJD) 🧠 in humans, and Bovine Spongiform Encephalopathy(BSE)/“Mad Cow Disease” 🦠🐄 in animals).
“Amyloid fibrils” (Rambaran and Serpell 2008) https://pmc.ncbi.nlm.nih.gov/articles/PMC2634529/#:~:text=Introduction,of%20melanosomes%2C%20curli%20and%20hydrophobins.
“Biology of Amyloid: Structure, Function, and Regulation” (Greenwald and Riek 2010) https://www.sciencedirect.com/science/article/pii/S0969212610003084#:~:text=Amyloids%20are%20highly%20ordered%20cross,a%20structural%20framework%20for%20polymorphisms.
And yes! You can use amyloid β-sheets as materials as their stable stable cross-β structure, self-assembly properties, and mechanical robustness can be leveraged upon to create versatile and high-strength eco-friendly 🌱 materials which include hydrogels, nano-wires,and bioplastics. These have the potential for tissue engineering 🫁, biosensors, and drug 💊 delivery 🚚 🚛 📦.
1.Briefly describe the protein you selected and why you selected it.
The protein I have chosen is Sodium-Potassium Pump also written as the Na⁺/K⁺-ATPase enzyme. I chose this protein because I remember learning about it at school and being fascinated by how it works. The Na⁺/K⁺-ATPase enzyme is found in all metazoa (animal) cells, and is located in the plasma membrane of cells.
It essentially maintains an extracellular concentraion of Na⁺ (sodium ions) that is greater than the intracellular concentration, whilst maintaining an intracellular concentration of K⁺ (potassium ions) that is grater than the extracellular concentration.
In other words, it pumps out 3 sodium ions, and takes in 2 potassium ions. It depends on energy in the form of ATP to complete this action. The sodium-potassium pump is essential for understanding how neurons (nerve cells) function, as they depend on this pump to respond to stimuli and transmit impulses.
I just think that it is a masterpiece of biological engineering. Evolution is so crazy!
2.Identify the amino acid sequence of your protein.
I went to NCBI (which was a bloody maze!) and identified the sequence of my protein:
NP_000692.2 sodium/potassium-transporting ATPase subunit alpha-1 [Homo sapiens]
Click to view Na⁺/K⁺-ATPase Enzyme Amino Acid Sequence (CDS)
MGKGVGRDKYEPAAVSEQGDKKGKKGKKDRDMDELKKEVSMDDHKLSLDELHRKYGTDLSRGLTSARAAEILARDGPNALTPPPTTPEWIKFCRQLFGGFSMLLWIGAILCFLAYSIQAATEEEPQNDNLYLGVVLSAVVIITGCFSYYQEAKSSKIMESFKNMVPQQALVIRNGEKMSINAEEVVVGDLVEVKGGDRIPADLRIISANGCKVDNSSLTGESEPQTRSPDFTNENPLETRNIAFFSTNCVEGTARGIVVYTGDRTVMGRIATLASGLEGGQTPIAAEIEHFIHIITGVAVFLGVSFFILSLILEYTWLEAVIFLIGIIVANVPEGLLATVTVCLTLTAKRMARKNCLVKNLEAVETLGSTSTICSDKTGTLTQNRMTVAHMWFDNQIHEADTTENQSGVSFDKTSATWLALSRIAGLCNRAVFQANQENLPILKRAVAGDASESALLKCIELCCGSVKEMRERYAKIVEIPFNSTNKYQLSIHKNPNTSEPQHLLVMKGAPERILDRCSSILLHGKEQPLDEELKDAFQNAYLELGGLGERVLGFCHLFLPDEQFPEGFQFDTDDVNFPIDNLCFVGLISMIDPPRAAVPDAVGKCRSAGIKVIMVTGDHPITAKAIAKGVGIISEGNETVEDIAARLNIPVSQVNPRDAKACVVHGSDLKDMTSEQLDDILKYHTEIVFARTSPQQKLIIVEGCQRQGAIVAVTGDGVNDSPALKKADIGVAMGIAGSDVSKQAADMILLDDNFASIVTGVEEGRLIFDNLKKSIAYTLTSNIPEITPFLIFIIANIPLPLGTVTILCIDLGTDMVPAISLAYEQAESDIMKRQPRNPKTDKLVNERLISMAYGQIGMIQALGGFFTYFVILAENGFLPIHLLGLRVDWDDRWINDVEDSYGQQWTYEQRKIVEFTCHTAFFVSIVVVQWADLVICKTRRNSVFQQGMKNKILIFGLFEETALAAFLSYCPGMGVALRMYPLKPTWWFCAFPYSLLIFVYDEVRKLIIRRRPGGWVEKETYY
https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=476
https://www.ncbi.nlm.nih.gov/protein/NP_000692.2
https://www.ncbi.nlm.nih.gov/protein/NP_000692.2?report=fasta
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is: 1023 amino acids. The most common amino acid is: Leucine, which appears 96 times.
Amino Acid Frequency Analysis
Protein: Na+/K+-ATPase subunit alpha-1
Total Length: 1023 amino acids
| Amino Acid | Count | Frequency (%) |
|---|---|---|
| L (Leucine) | 96 | 9.39% |
| I (Isoleucine) | 83 | 8.12% |
| A (Alanine) | 80 | 7.83% |
| G (Glycine) | 76 | 7.44% |
| V (Valine) | 76 | 7.44% |
| E (Glutamic Acid) | 67 | 6.56% |
| T (Threonine) | 61 | 5.97% |
| D (Aspartic Acid) | 59 | 5.77% |
| K (Lysine) | 56 | 5.48% |
| S (Serine) | 56 | 5.48% |
| R (Arginine) | 45 | 4.40% |
| P (Proline) | 45 | 4.40% |
| F (Phenylalanine) | 44 | 4.31% |
| N (Asparagine) | 42 | 4.11% |
| Q (Glutamine) | 35 | 3.42% |
| M (Methionine) | 26 | 2.54% |
| Y (Tyrosine) | 24 | 2.35% |
| C (Cysteine) | 23 | 2.25% |
| H (Histidine) | 16 | 1.57% |
| W (Tryptophan) | 12 | 1.17% |
- How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
250 found in UniProtKB!
The protein is highly conserved in primates besides humans such as chimpanzees, orangutans, and baboons. This means that the Na⁺/K⁺-ATPase enzyme is so essential it has barely changed over thousands of years of evolution!
https://www.uniprot.org/blast/uniprotkb/ncbiblast-R20260311-160929-0627-30578154-p1m/overview
- Does your protein belong to any protein family?
This protein belongs to the P-type ATPase family ❤️ (Huang et al. 2024).
https://pmc.ncbi.nlm.nih.gov/articles/PMC11168508/
3. Identify the structure page of your protein in RCSB
Using RCBS, I typed in Sodium-Potassium Pump and ended up on the “Crystal structure of the sodium - potassium pump in the E2.2K+.Pi state” for the organism which turned out to be a Squalus acanthias or dogfish. They are so cute. Anyhow I tried again by entering the protein sequence (it helps to be more specific!) and found the entry for homo sapiens “Cryo EM structure of a Na+-bound Na+,K+-ATPase in the E1 state.”
Dogfish https://www.rcsb.org/structure/2ZXE
Human https://www.rcsb.org/structure/7E1Z#entity-1
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
This particular structure was
deposited on the 4th of February 2021
released/solved on the 15th of June 2022
It has a resolution of 3.20 Å - a fair to good quality, which is considerable for a massive complex membrane protein like the Sodium-Potassium Pump.
Are there any other molecules in the solved structure apart from protein?
Yes there are
- Sodium ions (Na)
- a Magnesium ion (Mg)
- Cholesterol Hemisuccinate (Y01)
- Phospholipids (PC1): 1,2-diacyl-sn-glycero-3-phosphocholine
- Sugar (NAG): 2-acetamido-2-deoxy-beta-D-glucopyranose
- Does your protein belong to any structure classification family?
It is a membrane protein (Hydrolase / Transport Protein).
4. Open the structure of your protein in any 3D molecule visualization software:
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
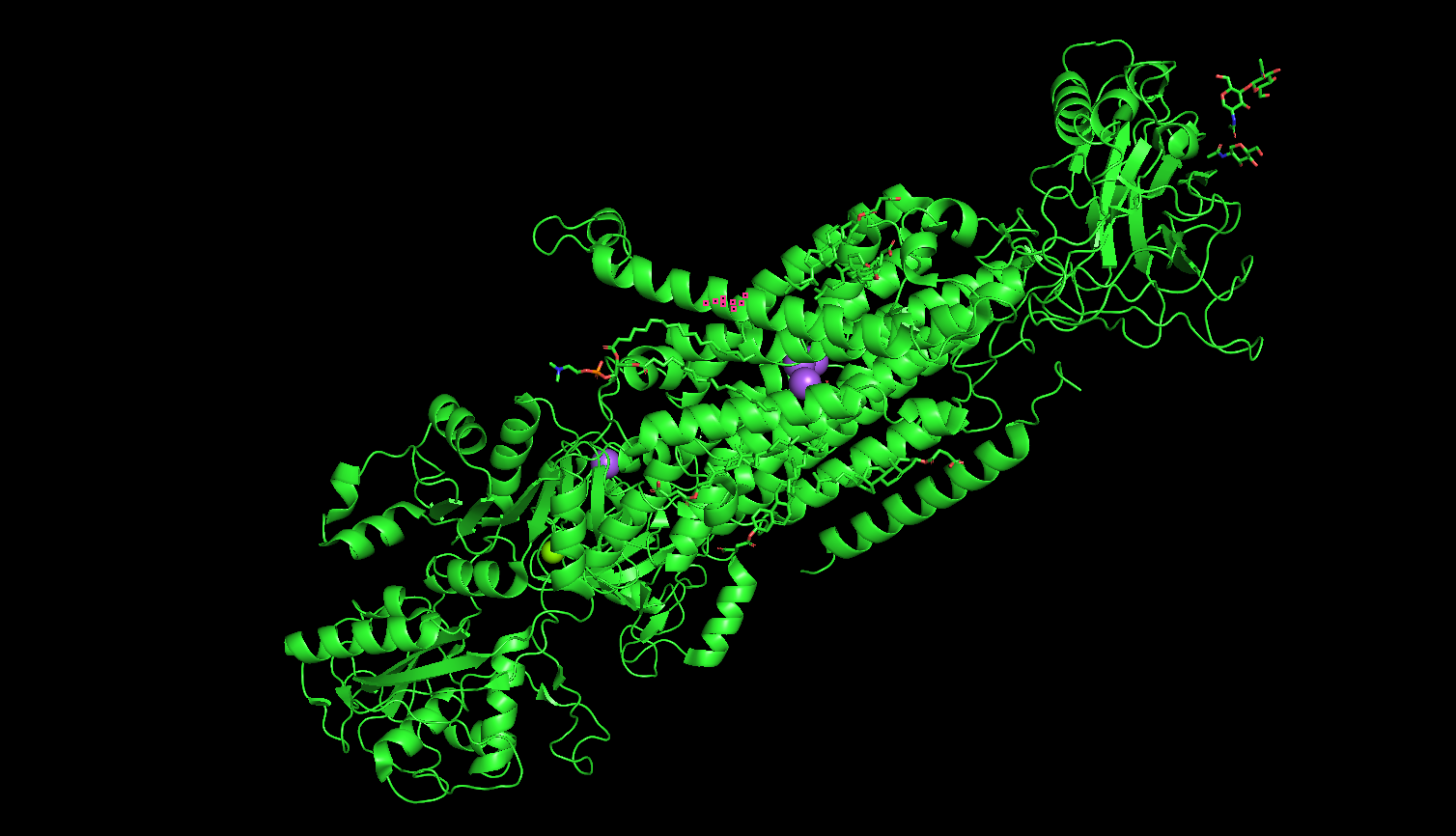
Cartoon
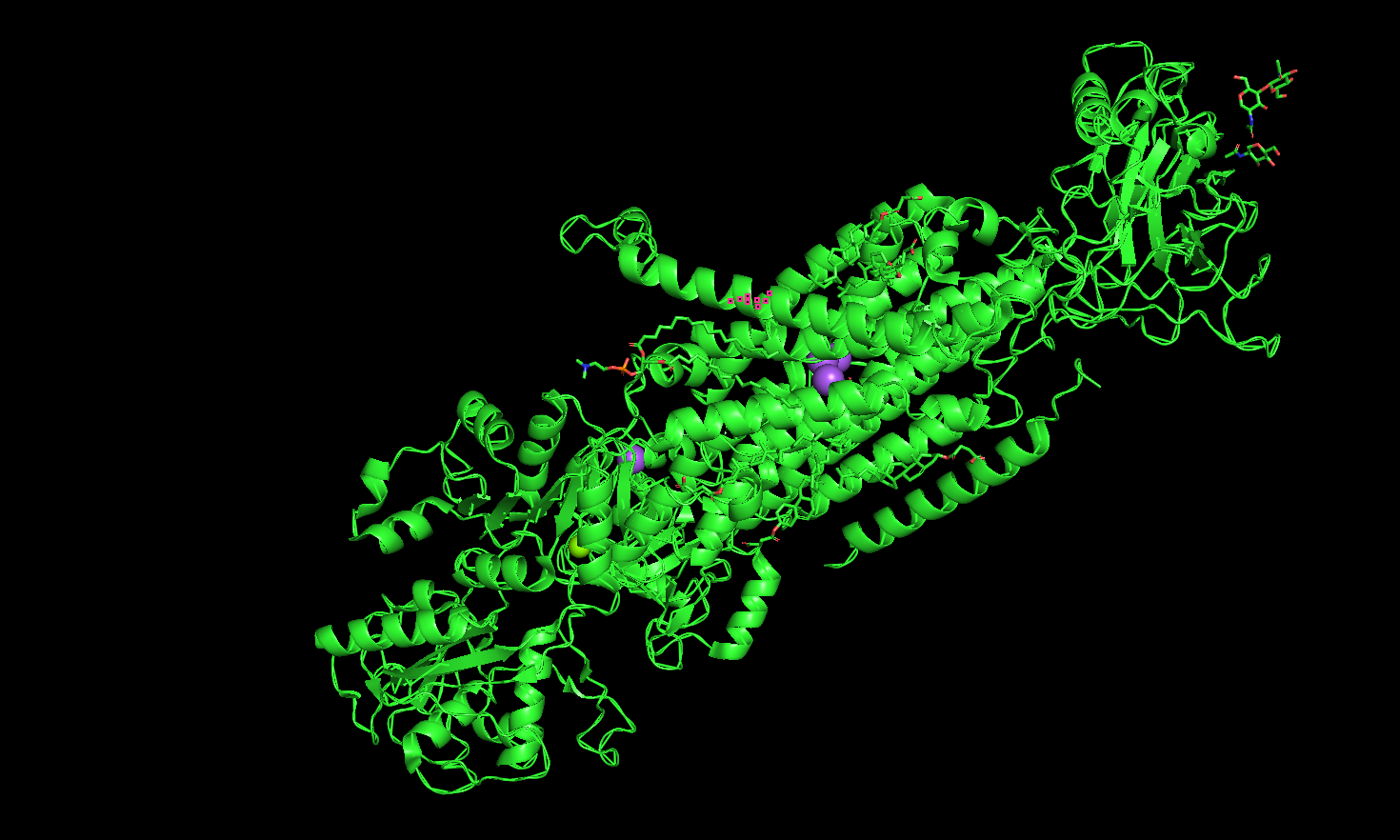
Ribbon
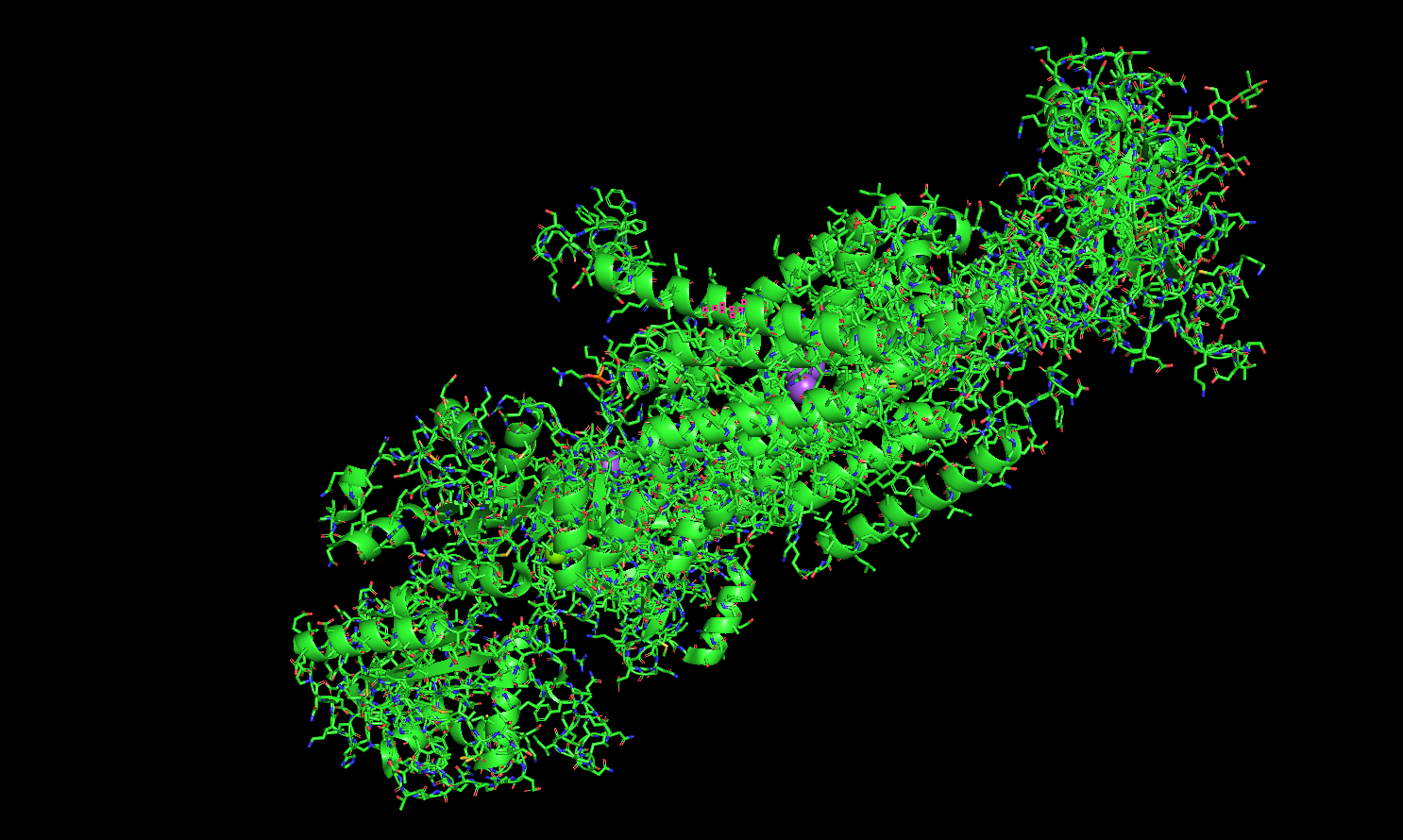
Ball and Stick
Got a bit carried away . . .
Mesh
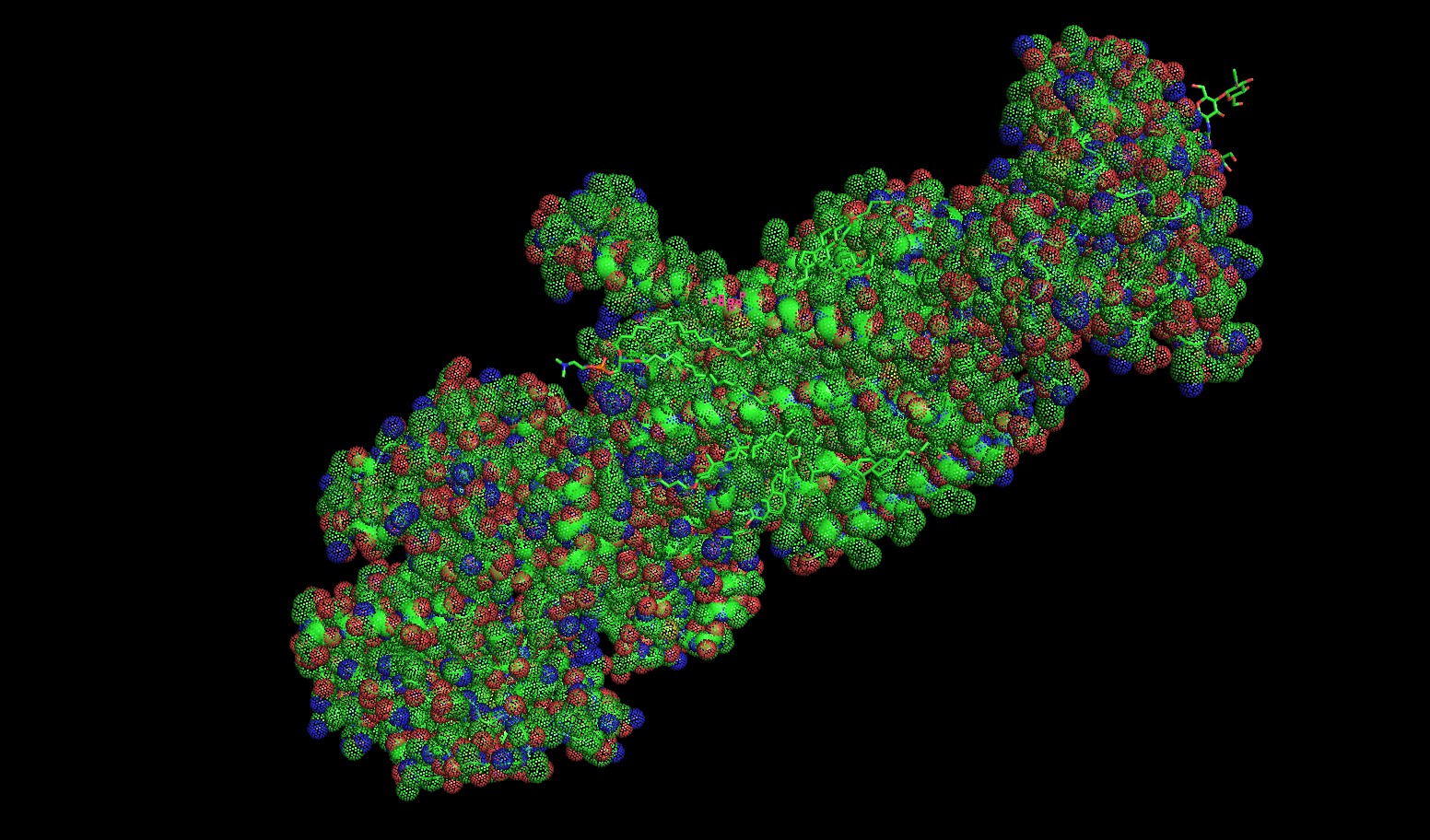
Dots
- Color the protein by secondary structure. Does it have more helices or sheets?
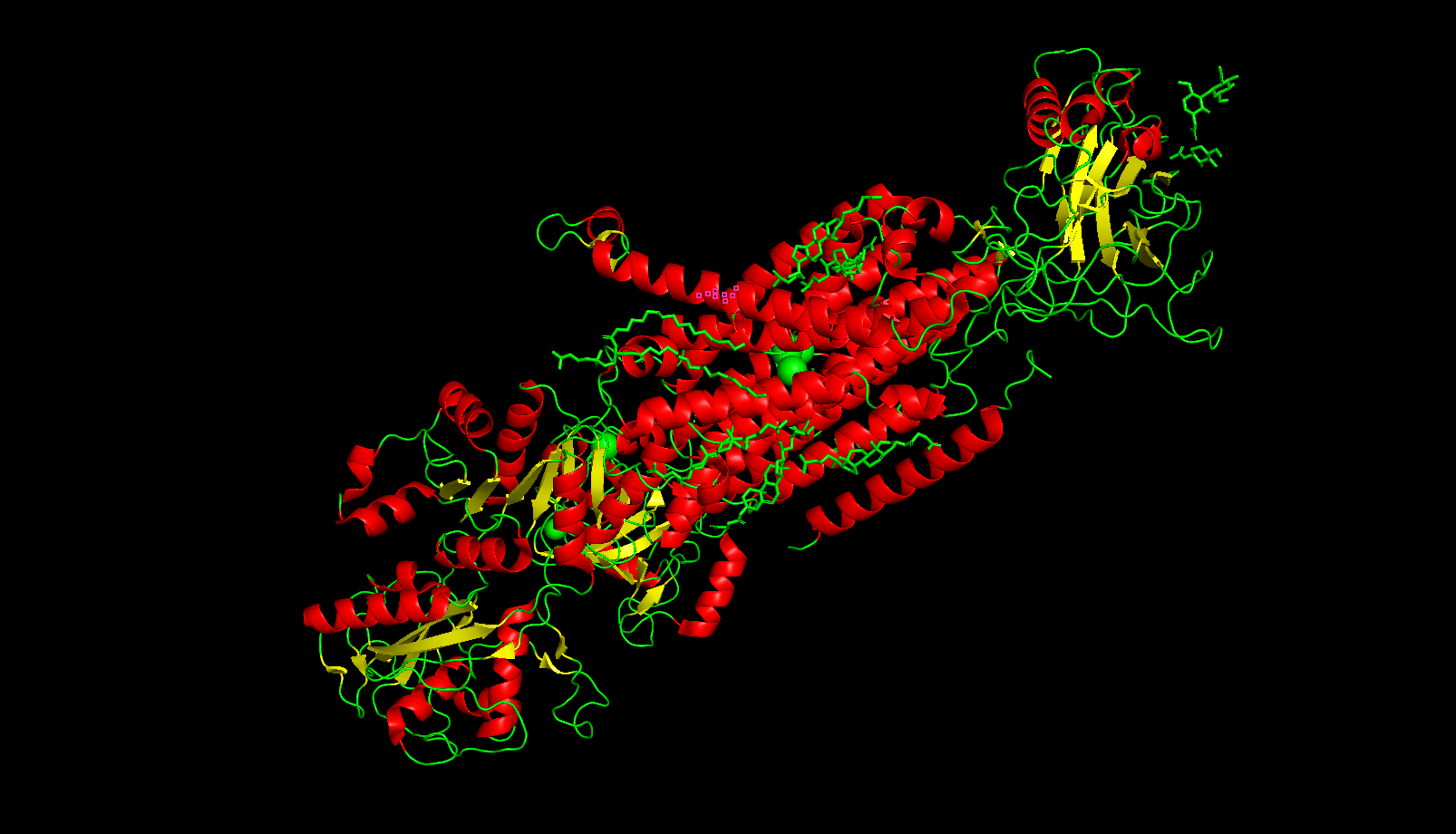
- α-helices = red
- β-sheets = yellow
- loops connecting the two = green
As you can see, there are adundantly more helices. This serves a structural function as the helices act as pillars in the pump.
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
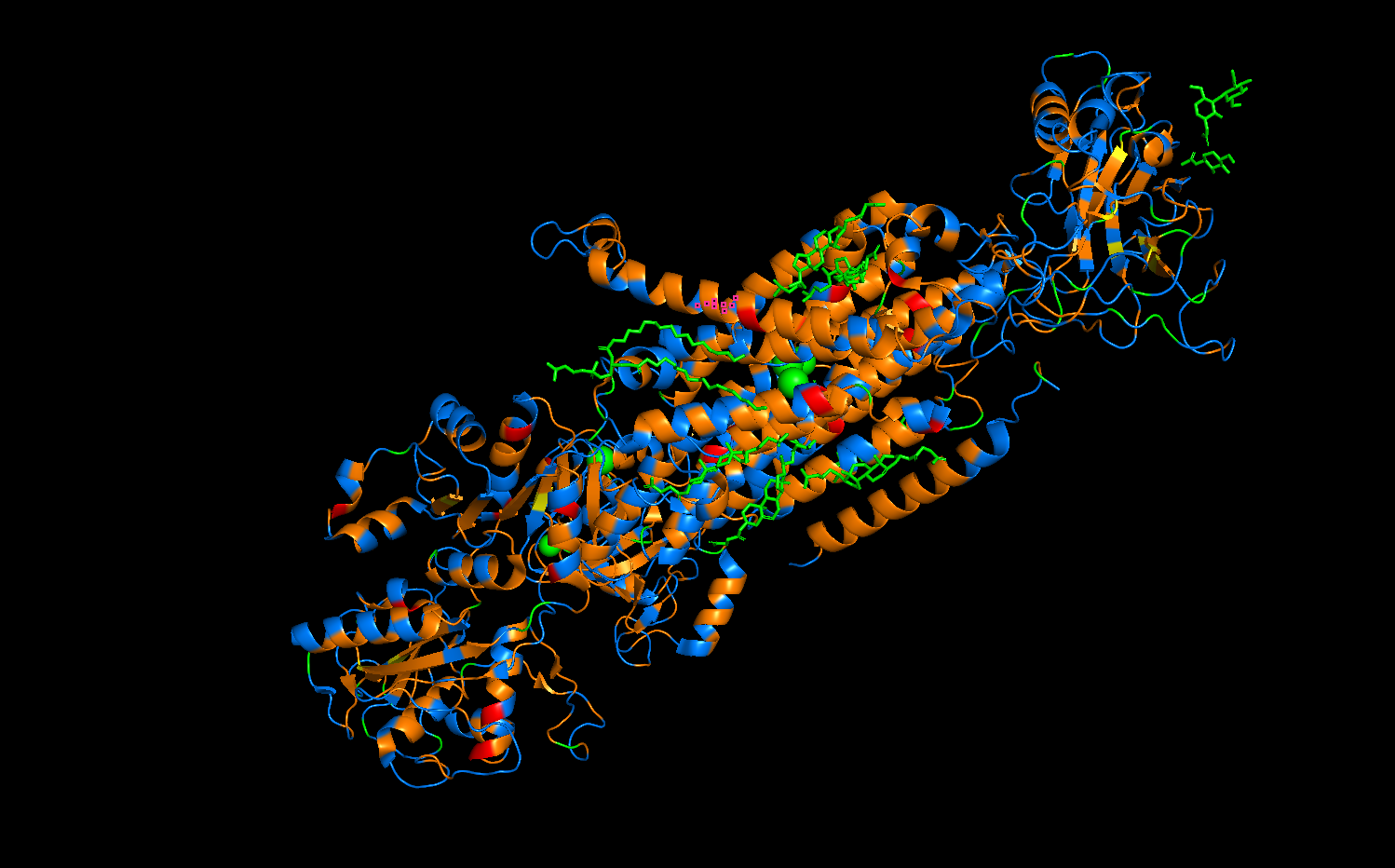
- Orange = hydrophobic
- Blue = hydrophillic
The distribution of protein residues follows a pattern which is much easier to see in the photo below this one. The orange hydrophobic residues are more concentrated the center of the protein, as these form a “transmembrane belt” which interacts with the phospholipid bilayer of a cell. Conversely, the blue hydrophilic residues are located on the exterior surfaces, as these are exposed to the aqeous environments of the cytoplasm and extracellular fluid.
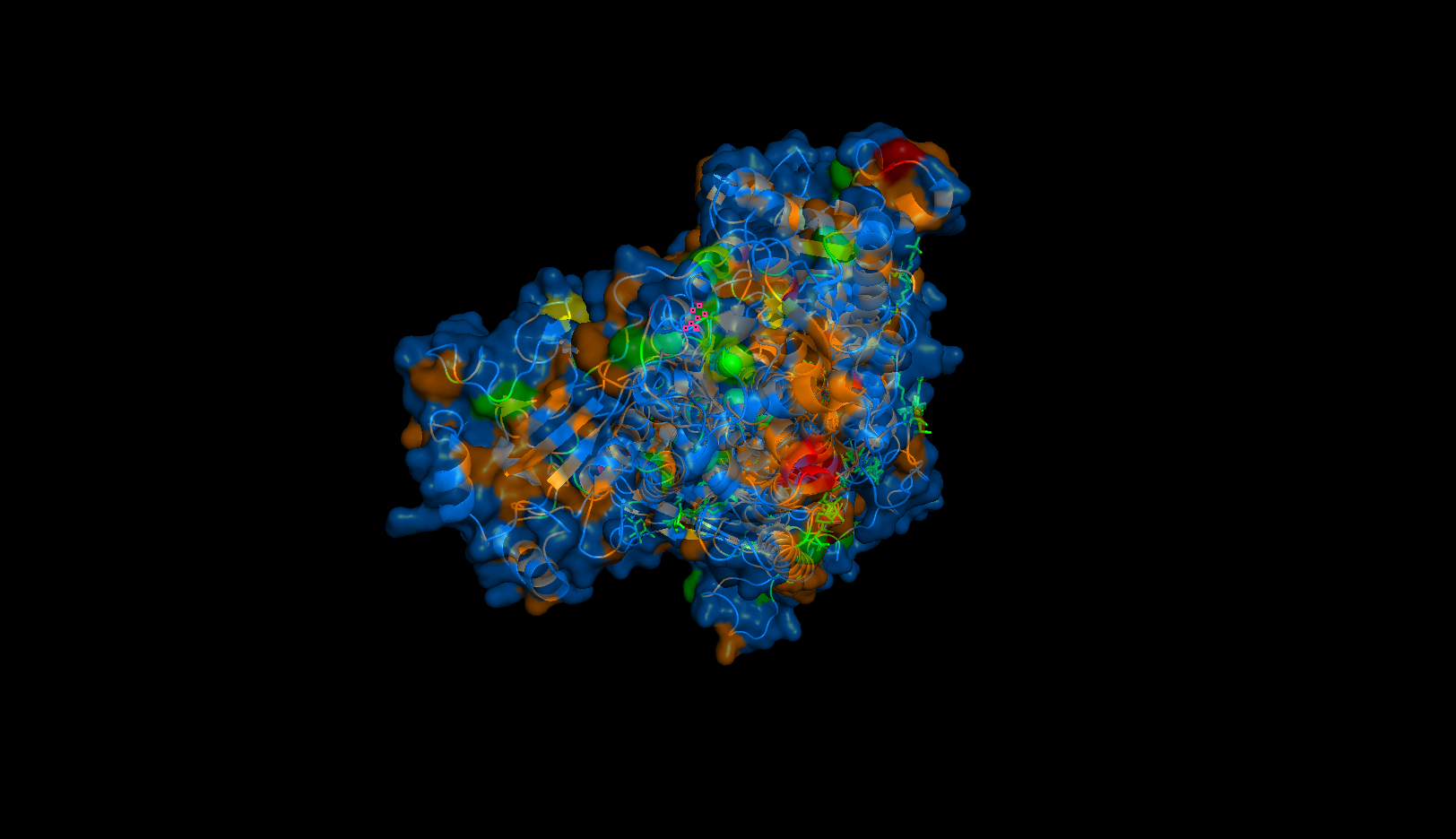
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
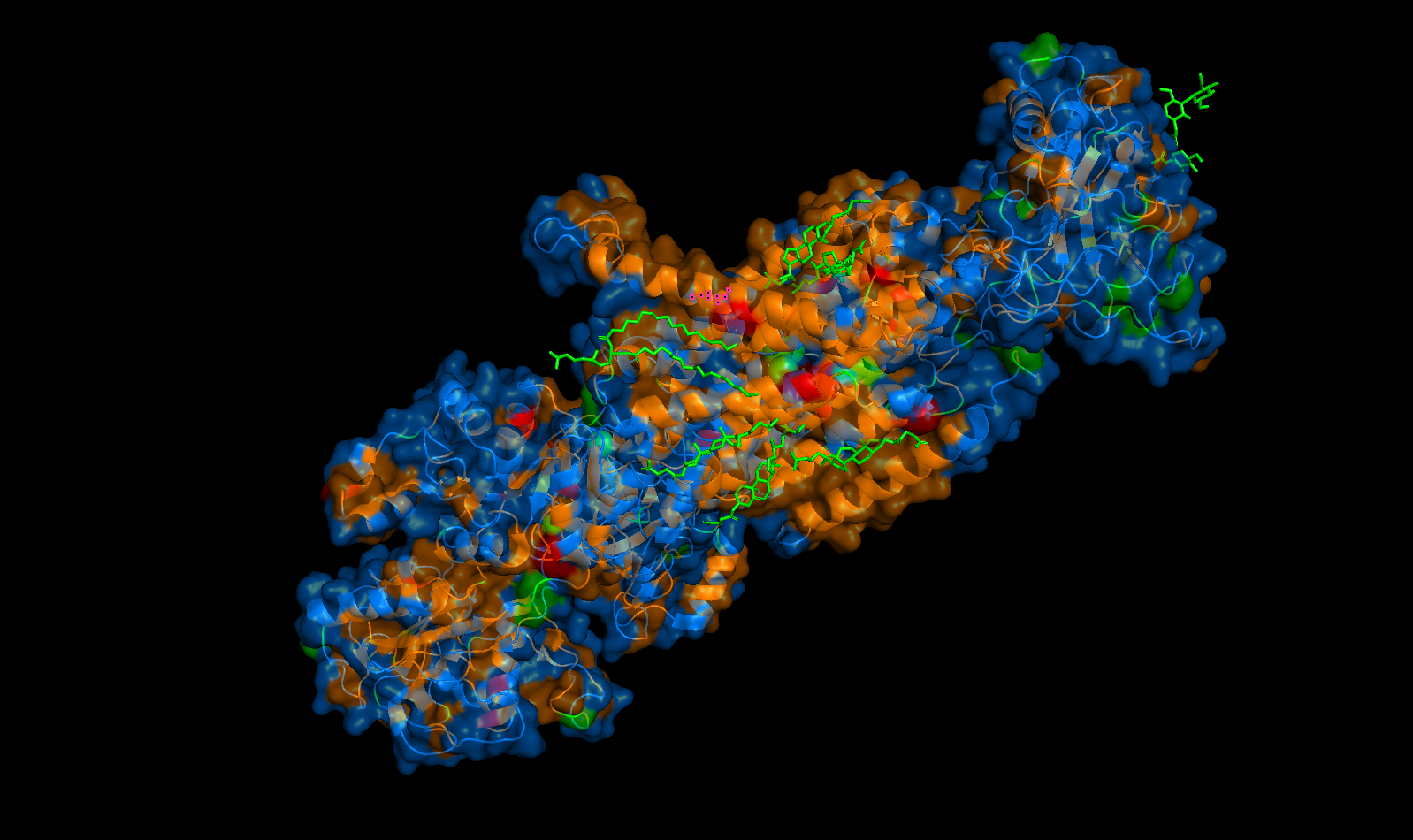
Perspective 1
Perspective 2
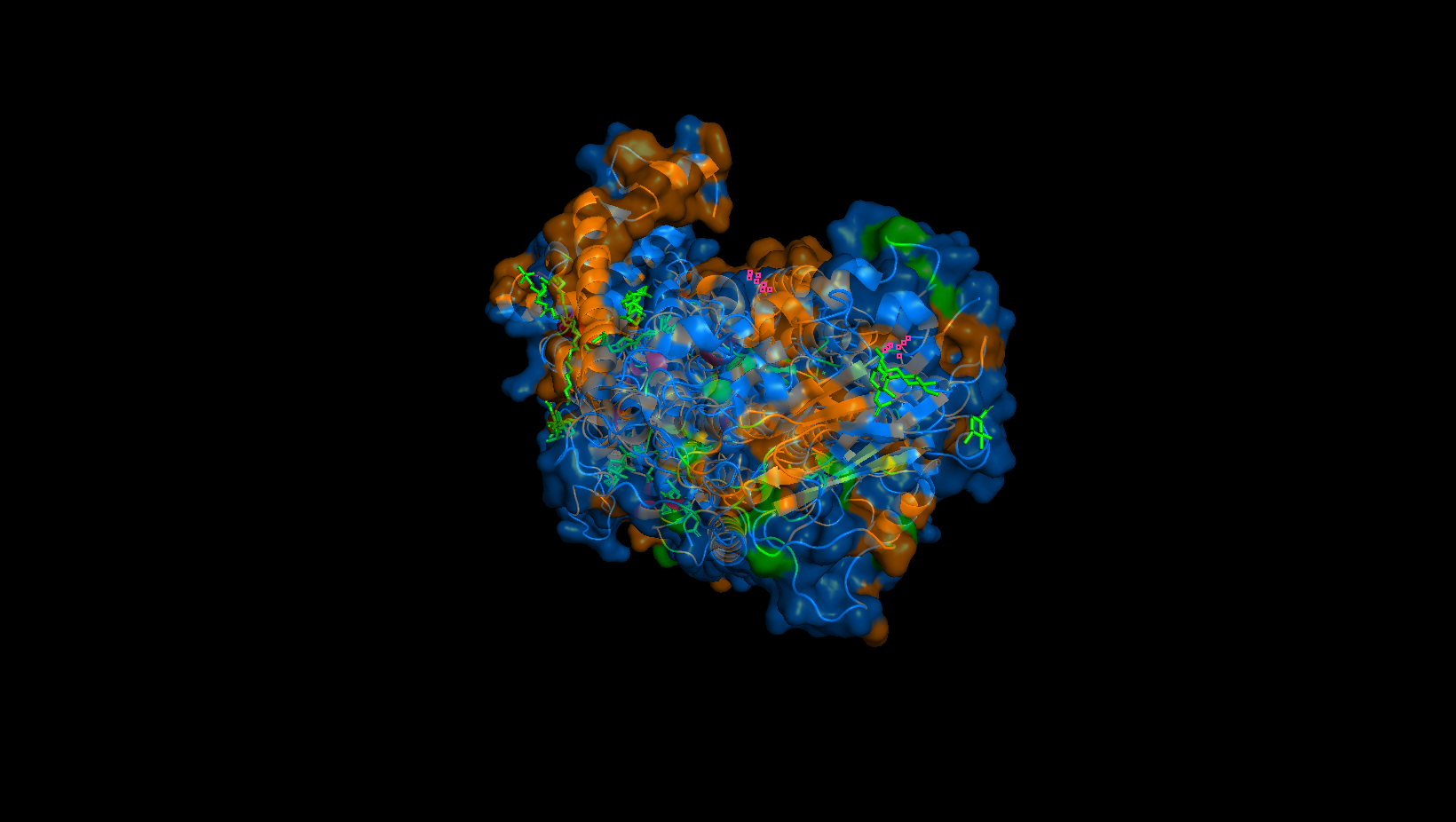
Perspective 3
Yes. It may be hard to tell, but these are largely located in the transmembrane domain where the orange hydrophobic residues are. These “holes” are the binding pockets that hold the sodium ions in the pump.
C1. Protein Language Modeling
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
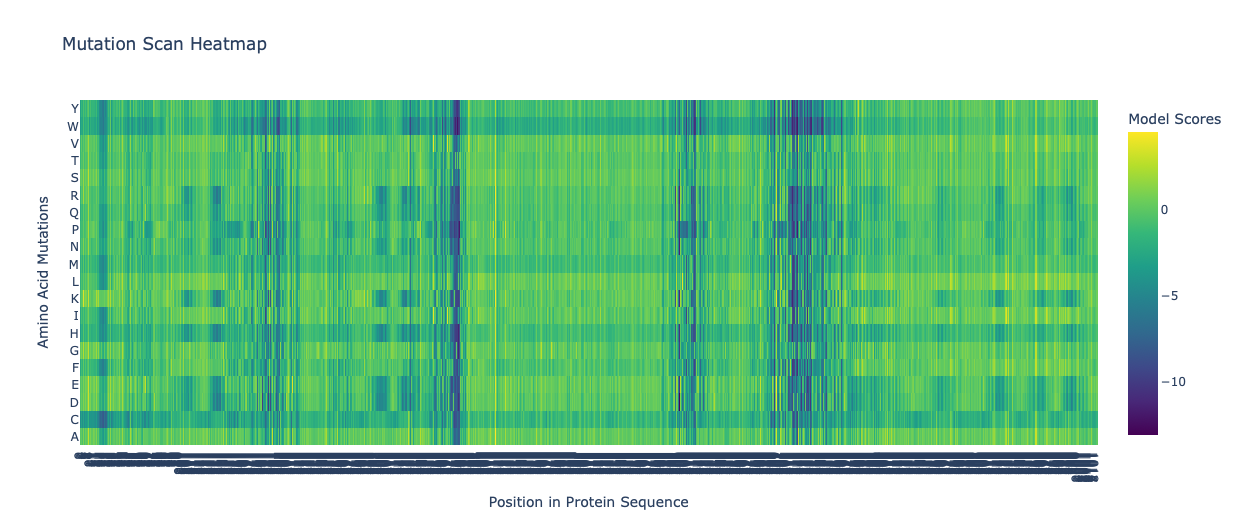
Mutation Scan Heatmap of ATPase
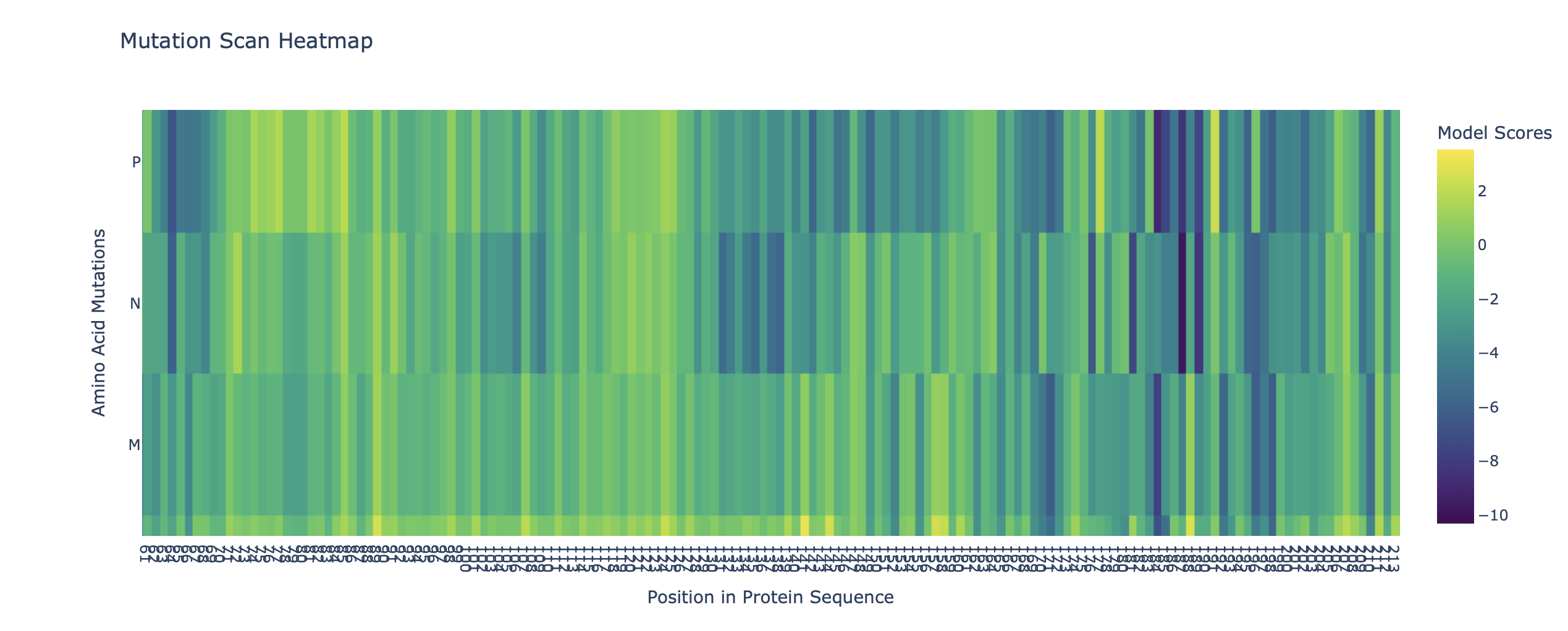
Mutation Scan Heatmap of ATPase (first 400 residues)
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The first image is a beautiful Mutation Scan Heatmap of ATPase which is 1023 residues long! It reminded me of the beautiful tartan skirts of Vivienne Westwood and Chopova Lowena, though the latter I cannot support as she abuses her interns and her garments are bad quality 😶🌫️ I digress . . .
In this image there are a few vertical bands of dark purple. The most prominent is at the rougly residue 370 mark. This likely means that this residue has stayed consistent after thousands or even millions of years of evolution. An alteration of such would probably disrupt the movement of sodium and potassium ions (hence the low score).
However, large swathes of the map are yellow and green, and a signigicant portion is even blue. The lighter areas are probably variable loops which deal with the surface of the protein and not the main sodium and potassium pump mechanism. The protein is able to function even if these residues experience mutation.

Vivienne Westwood Wool Miniskirt! So cute ❤️
The second images is a Mutation Scan Heatmap of the first 400 residues of ATPase. I did this as the protein is very large and its size kept causing CoLab to glitch or warn me that I was running out of memory. Scanning it initially took 10+ minutes and my Mac started to heat up, so I just “trimmed” ATPase to be able to do some modelling.
I chose residue 54 on the heatmap which shows a column with a lot of dark purple (score ≈ 10). This indicates that the position is vital for the ATPase’s function/strucctural integrity. A mutation here likely disrutps the the ion binding mechanism, making the sequence highly improbable in nature. This residue can likely only fit a very specific amino acid.
Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
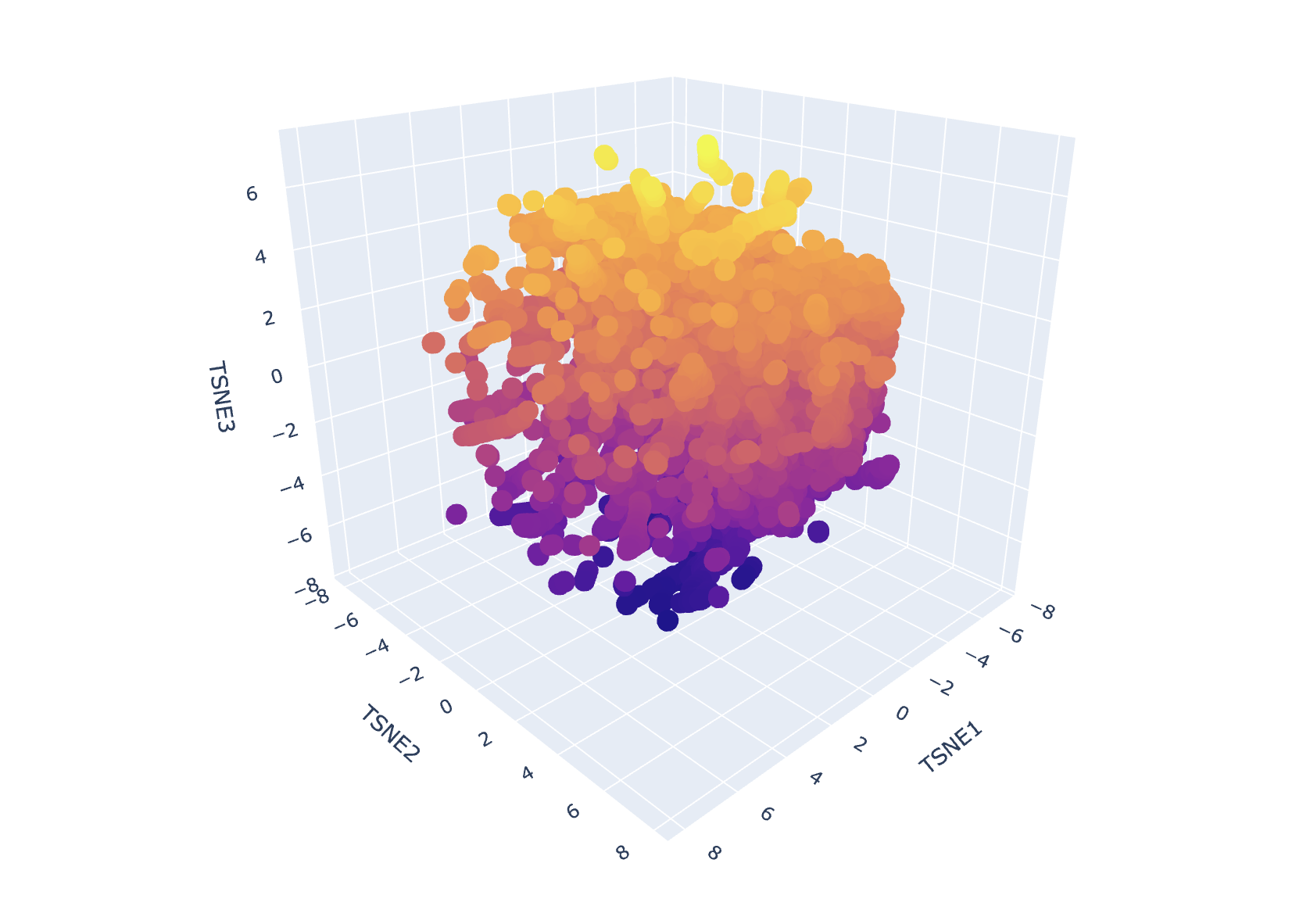
Result of First Run
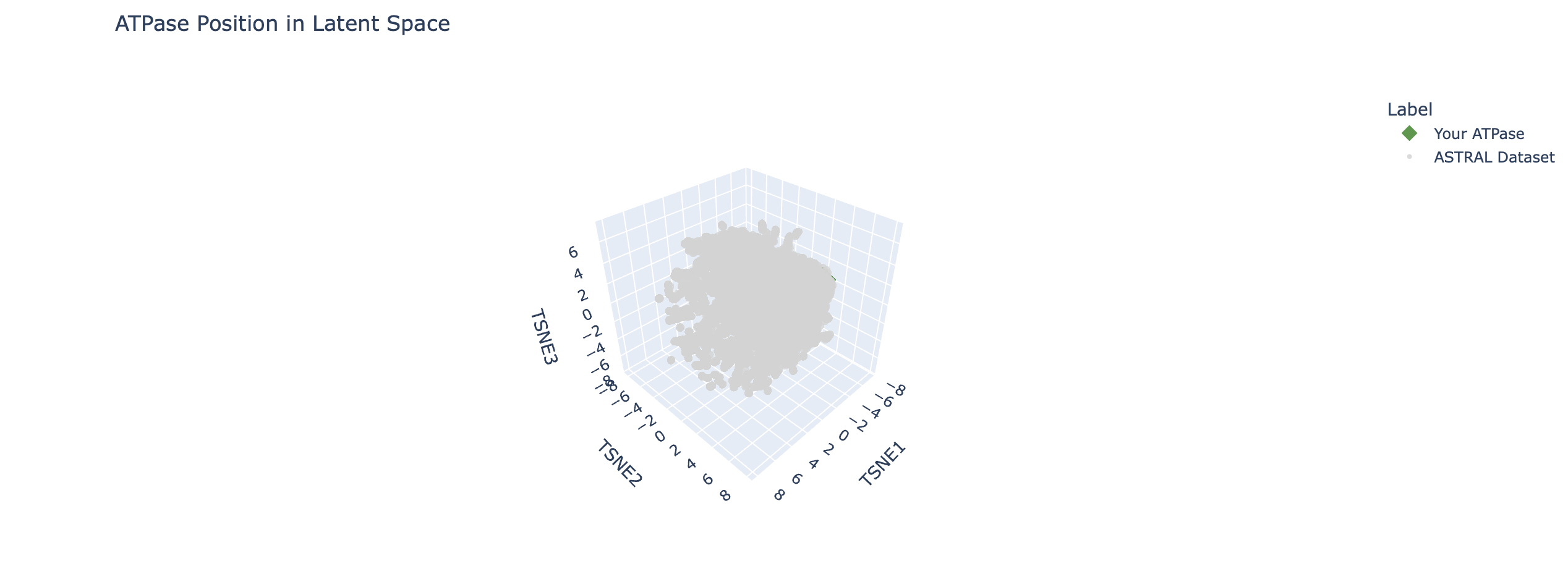
I ran some code to help me isolate ATPase
Let's look closer
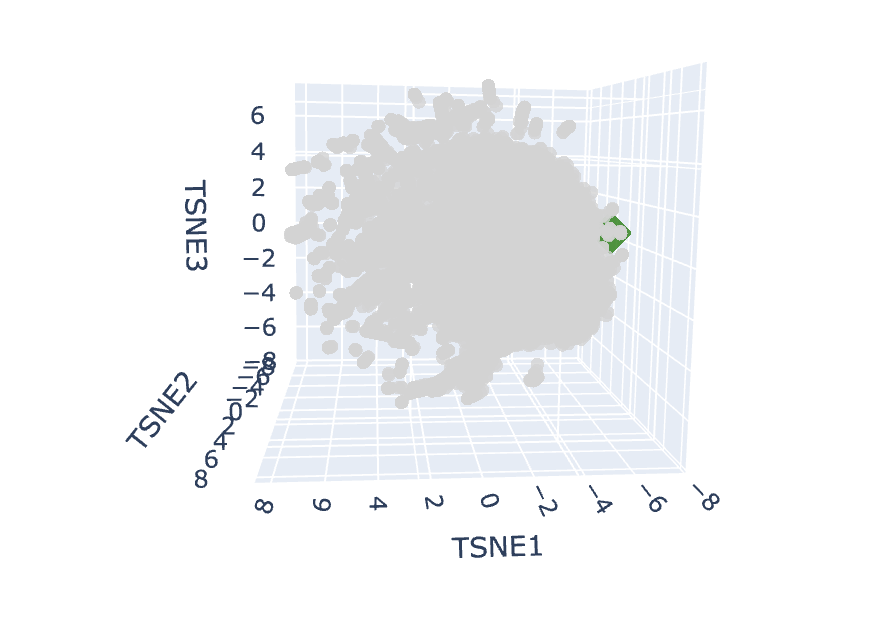
Look at the little green blob! 🟢
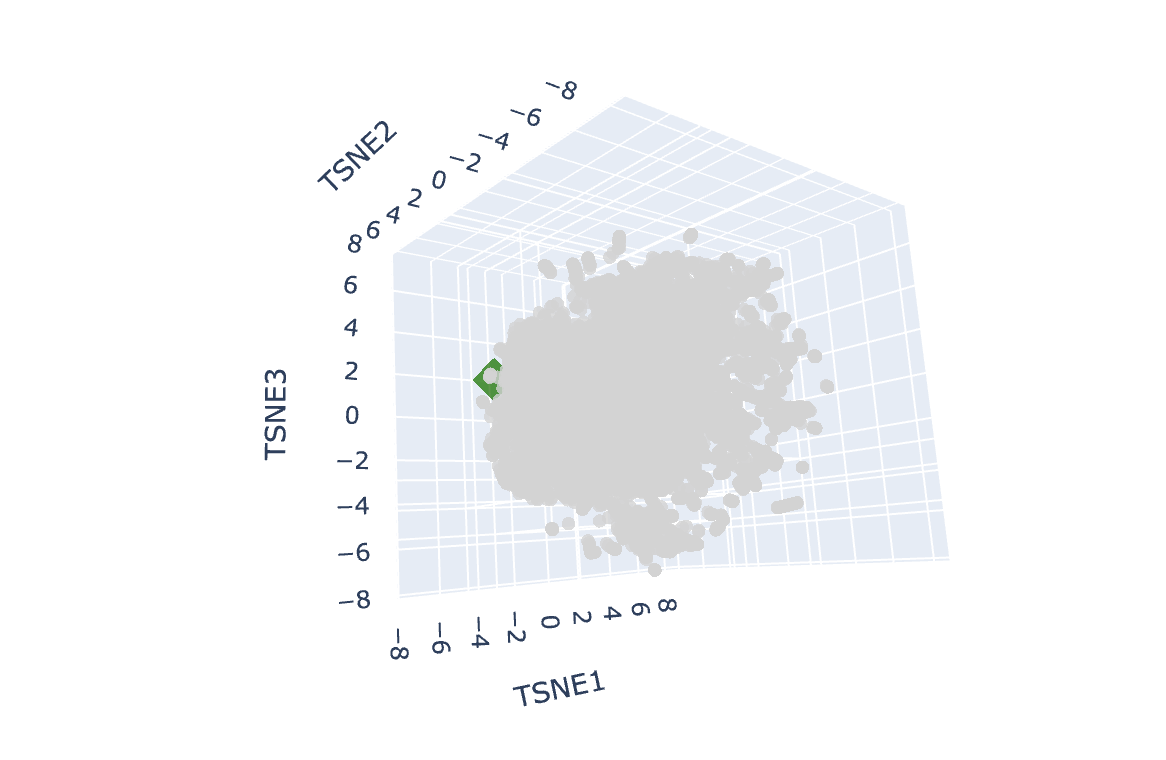
. . . Another perspective

The Wild West out here!

Mistaken identity
So it found the wrong protein. Time to try again.
 Found you maybe
Found you maybe
Another mistaken identity
Could this be it?
 Found you?
Found you?
Found you!
Bingo!
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Above is the ATPase of a fruit fly. Though my ATPase was that of a human, they are both ATPases. This proves that the AI we are using, namely ESM-2 is successfully grouping proteins by their functional neighbourhood rather than simply by which species they come from. So in short yes, the differnet formed neighbourhoods do approximate similar proteins.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
My 400 residue ATPase segment was clustered with other ATPases. This suggests that the model recognises structural elements that are related to ATPase. It was able to situate my protein in the relevant neighbourhood, even if it gave me the ATPase of a different organism.
C2. Protein Folding
Folding a protein
a. Fold your protein with ESMFold.
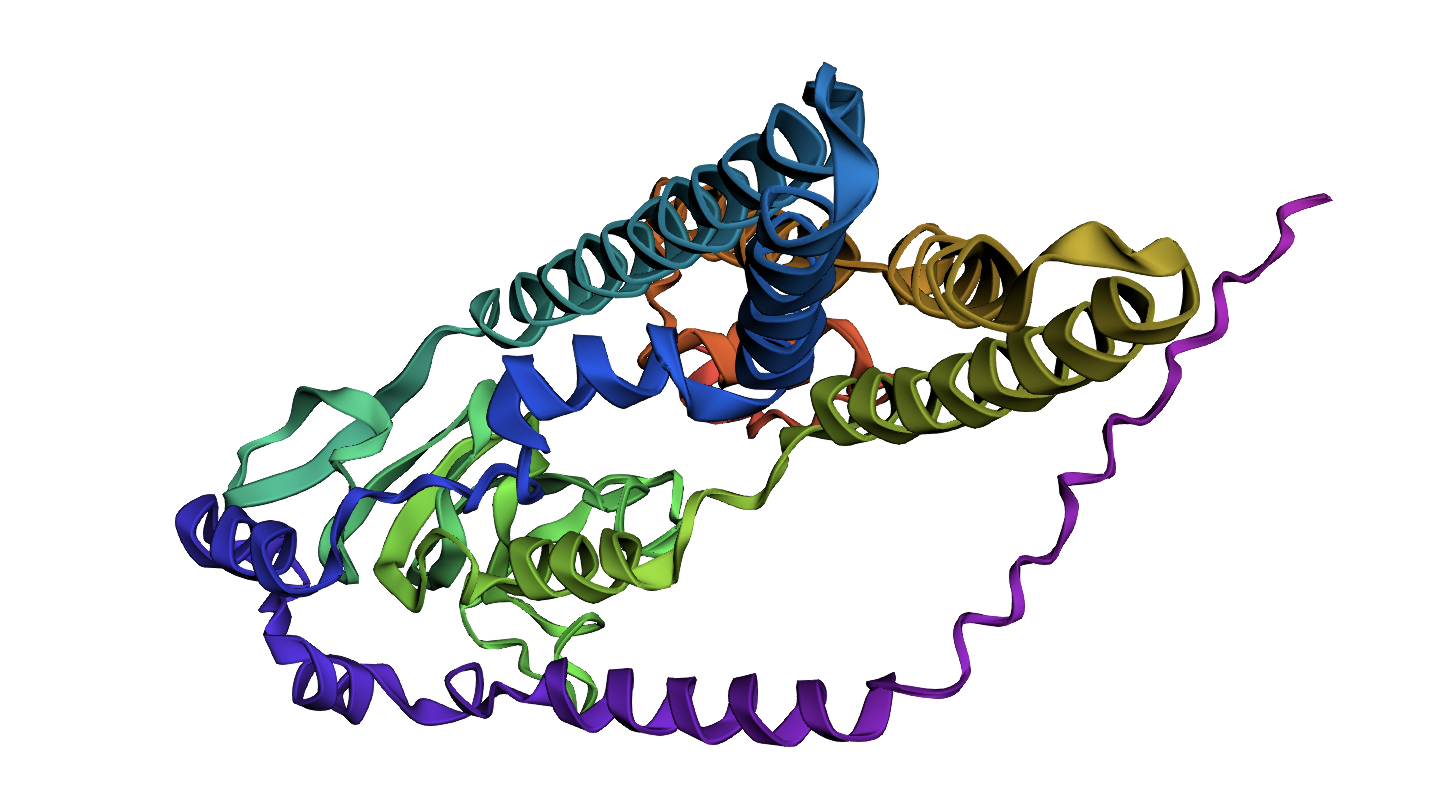
ATPase
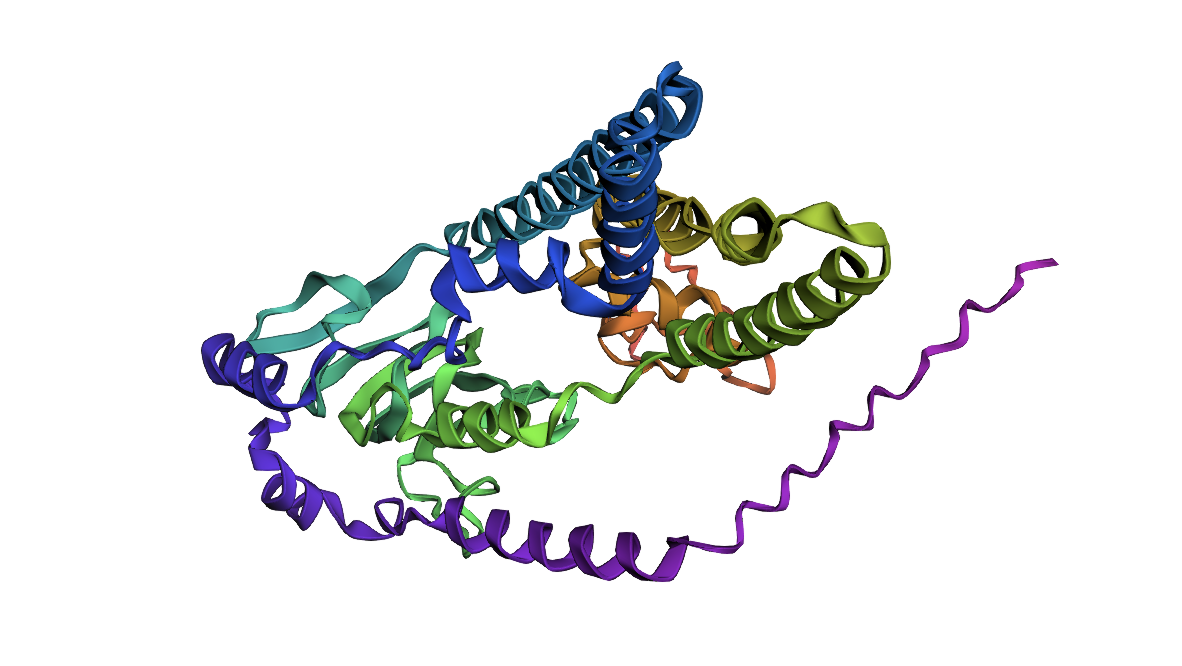
ATPase first 400 residues
b. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
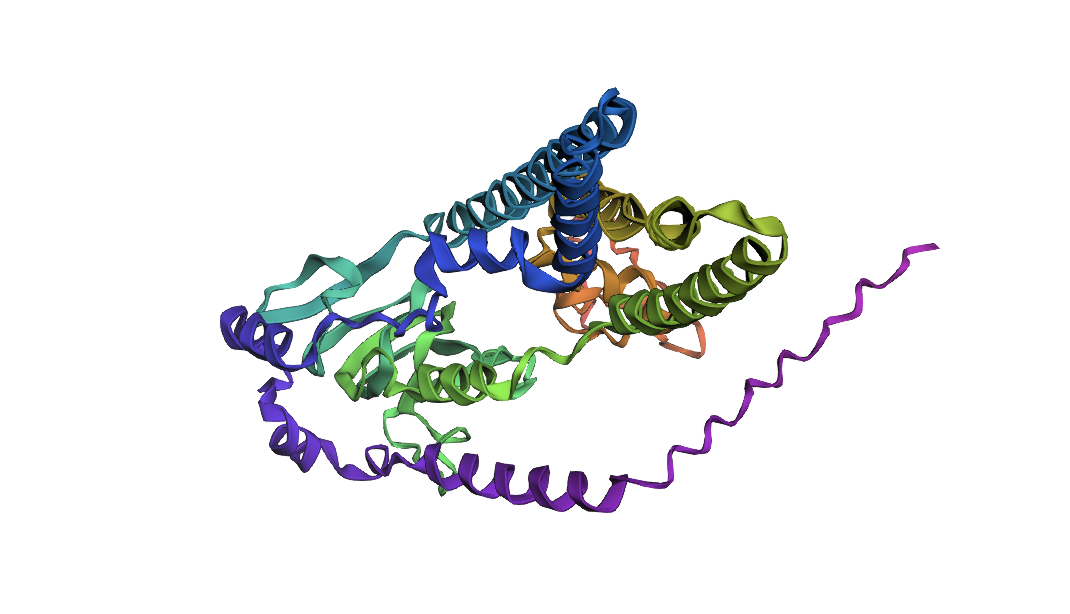 ATPase with 5 mutations
ATPase with 5 mutationsI introduced 5 mutations into the above protein. At the 200 residue mark there is a segment of “LVEVK” which I changed to “EDEVK” to induce a charge reversal to test the protein fold’s stability. As you can see it is pretty stable.
I also deleted a large section of 70 residues and tried to run this but it kept crashing and saying that I had run out of storage on the free tier 😞.
However, from what I have ascertained from the modelling in the above sections, this protein is very stable.