Week 5 HW: Protein Design Part II
Human SOD1 Sequence from UniProt (154 amino acids): https://www.uniprot.org/uniprotkb/P00441/entry https://www.uniprot.org/uniprotkb/P00441/entry#sequences
Click to view Human SOD1 Sequence
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Click to view Human SOD1 Sequence with A4V mutation which causes ALS
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I had to manually reset the code as I was not able to change the parameters with the sliding scales. I was stuck with generating a single 15 peptide long binder at a time. Thankfully I was able to change this by editing the back end of the form, but also by forcing the code to make 4 binders that were 12 peptides long.
Here is a table with the binders ranked and compared against a known binder:
| Rank | Peptide Source | Sequence | Pseudo Perplexity |
|---|---|---|---|
| 1 | Reference (Experimental) | FLYRWLPSRRGG | 2.2833 |
| 2 | PepMLM (Candidate 0) | KLVPAVVLAHKX | 7.4714 |
| 3 | PepMLM (Candidate 1) | KRSYPTALRHWX | 10.1367 |
| 4 | PepMLM (Candidate 2) | WRYPVAABHGK | 11.0383 |
| 5 | PepMLM (Candidate 3) | WHVYVVGLRHKE | 25.8914 |
The perplexity metric measures how perplexed or “surprised” as it were, a model is by a sequence. Hence a lower score represents higher model confidence or predicted affinity. Here, the known binder FLYRWLPSRRGG acts as a benchmark, scoring 2.28 on the pseudo perplexity rating, which is significantly lower than the newly generated designs. As you can see, I have ranked the binders in order of their respective perplexity ratings.
| Rank | Job Name | ipTM | pTM | Primary Binding Location | Target Engagement |
|---|---|---|---|---|---|
| 1 | SOD1 and KLVPAVVLAHK | 0.58 | 0.82 | N-terminus Groove | High (Pocket) |
| 2 | SOD1 and WHVYVVGLRHKE | 0.49 | 0.81 | Upper β-barrel Ridge | Moderate (Surface) |
| 3 | SOD1 and KRSYPTALRHW | 0.44 | 0.90 | β-barrel Loops | Moderate (Surface) |
| 4 | SOD1 and WRYPVAABHGK | 0.39 | 0.83 | Lower Dimer Interface | Low/Mod (Surface) |
| 5 | SOD1 and FLYRWLPSRRGG (Ref) | 0.26 | 0.81 | Surface Loops | Low (Transient) |
Key
| Confidence Level | pLDDT Range | Emoji | Corresponding Color |
|---|---|---|---|
| Very High | pLDDT > 90 | 💙 | Dark Blue |
| Confident | 90 > pLDDT > 70 | 🩵 | Light Blue (Cyan) |
| Low | 70 > pLDDT > 50 | 💛 | Yellow |
| Very Low | pLDDT < 50 | 🧡 | Orange |
Protein-peptide complex Models using AlphaFold3 and Residue Alignment Charts (Green)
They are ordered according to their ipTM score, with the first (KLVPAVVLAHK) having the greatest score (0.58) etc Please refer to the relevant table above if necessary
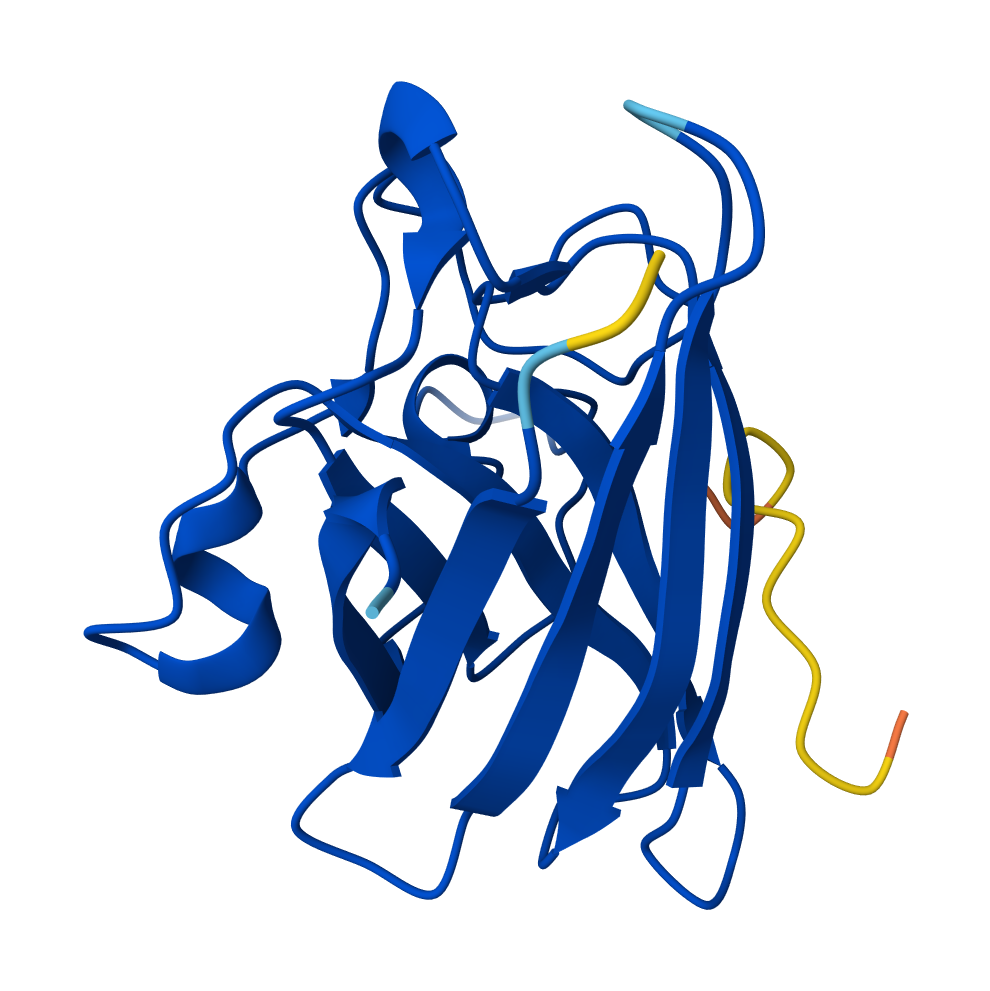
SOD1 & KLVPAVVLAHK

SOD1 & WHVYVVGLRHKE
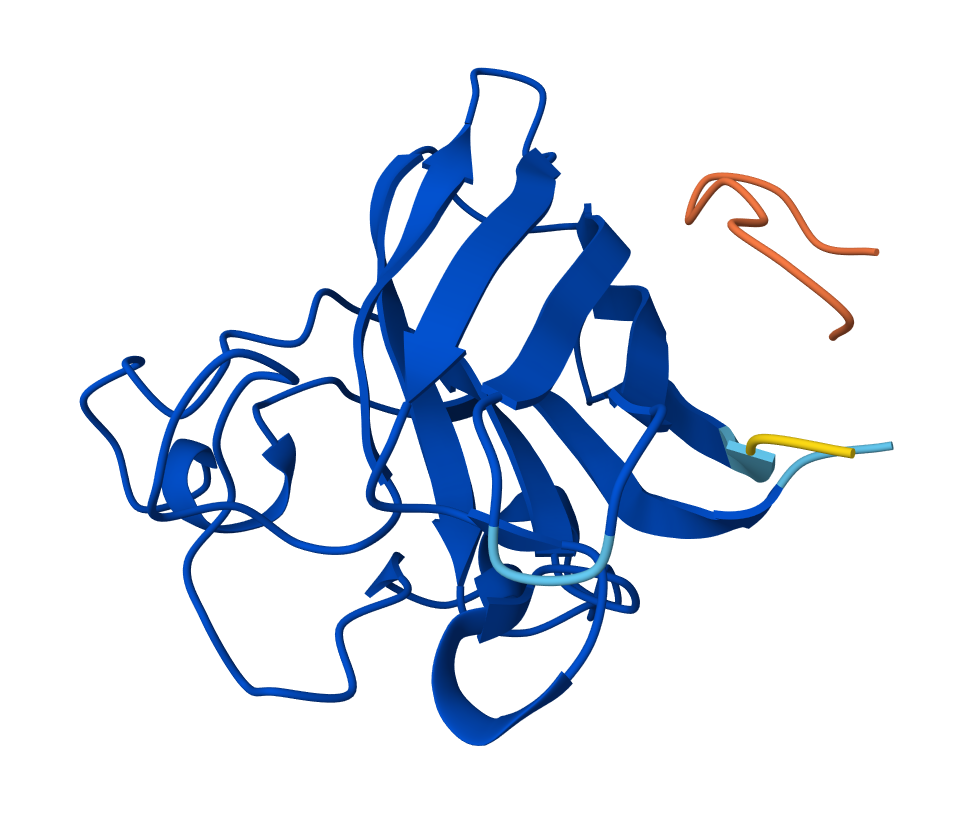
SOD1 & KRSYPTALRHW
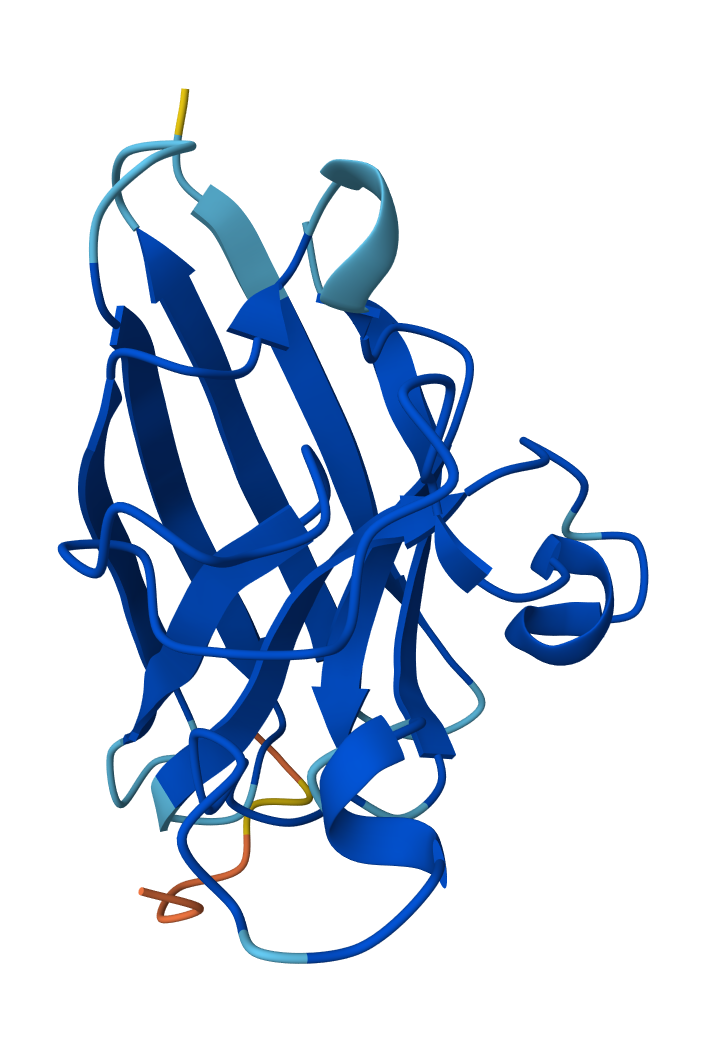
SOD1 & WRYPVAABHGK
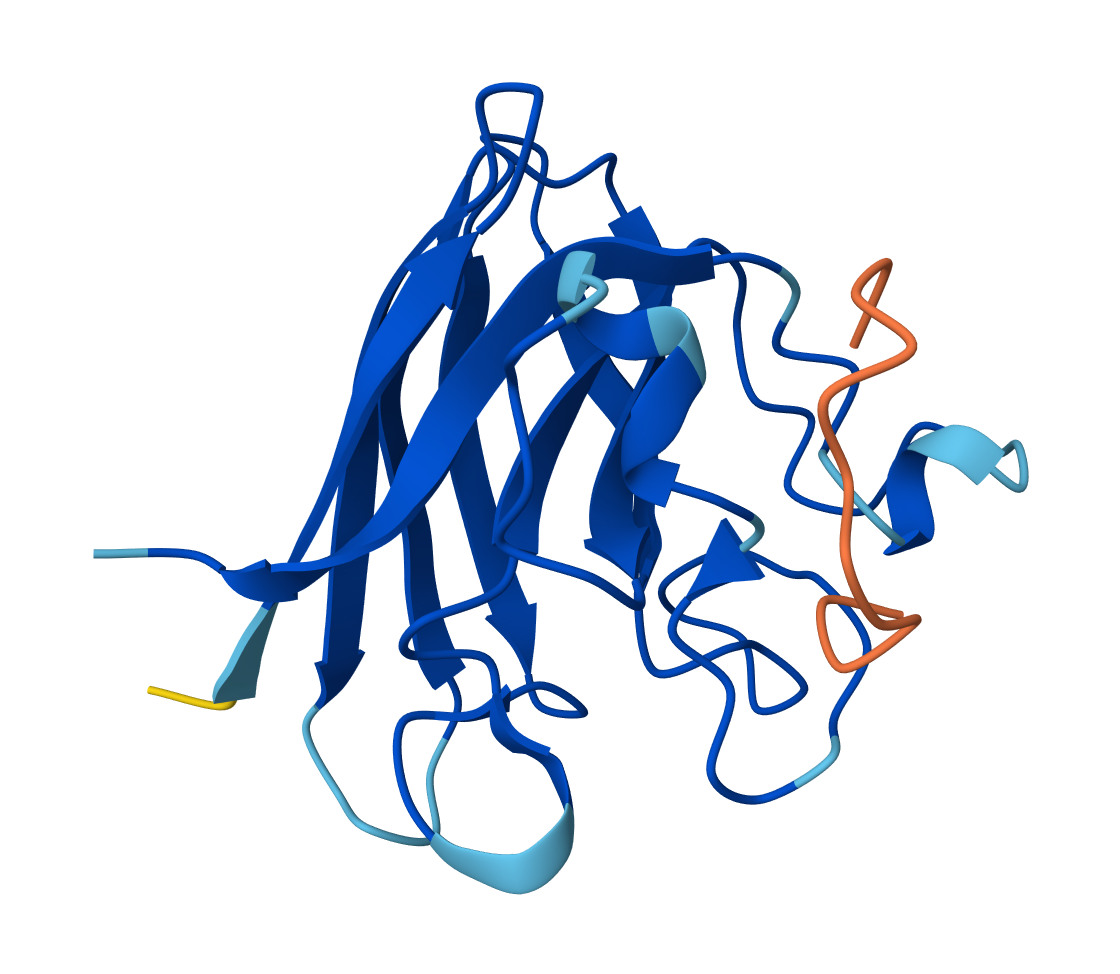
SOD1 & FLYRWLPSRRGG - Reference
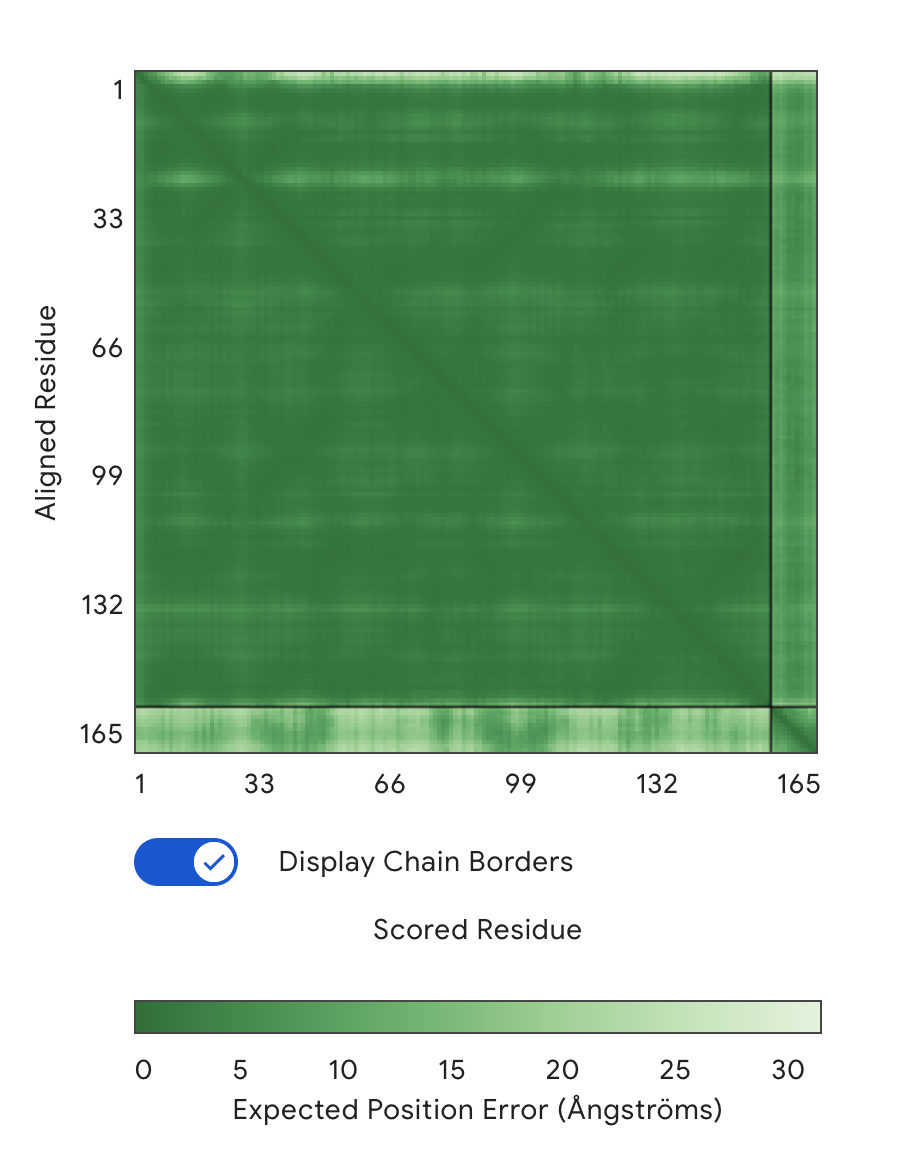
Alignment: SOD1 & KLVPAVVLAHKX
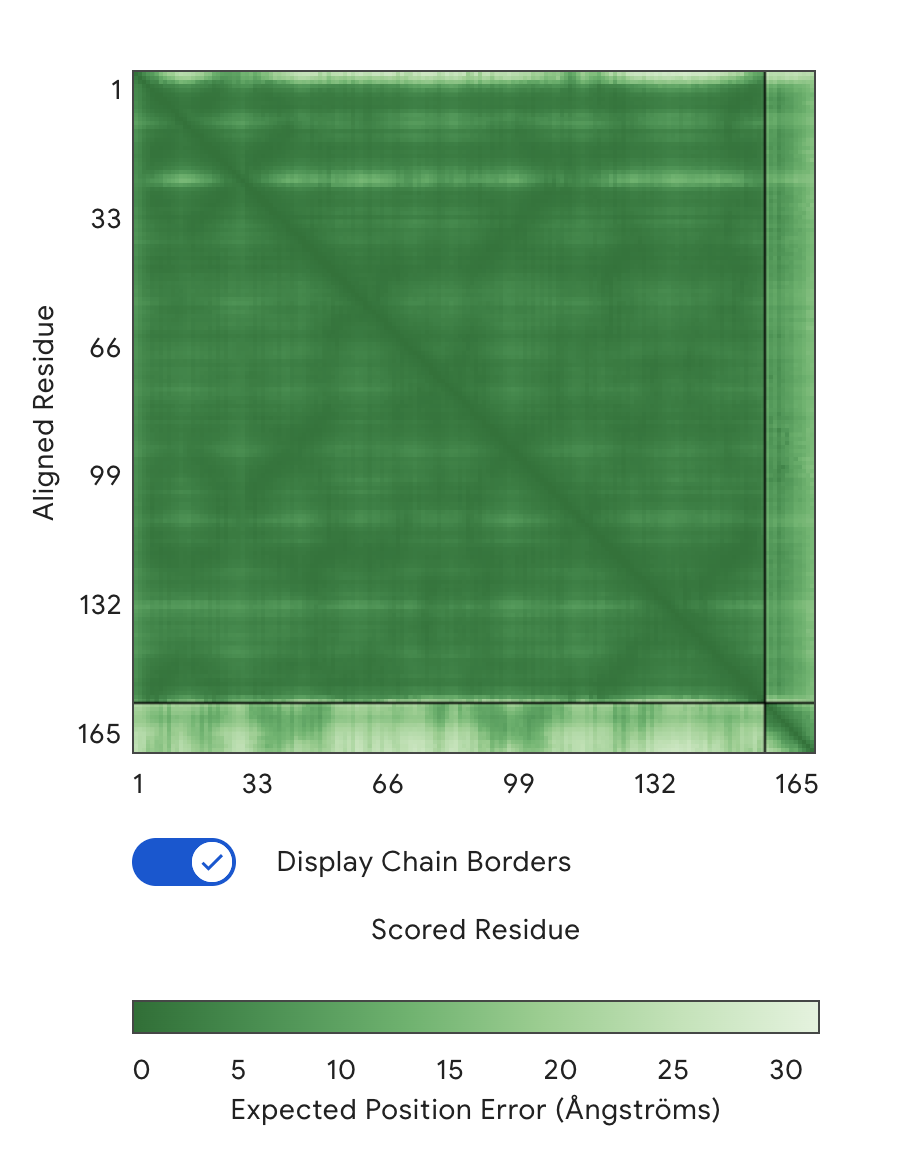
Alignment: SOD1 & WHVYVVGLRHKE
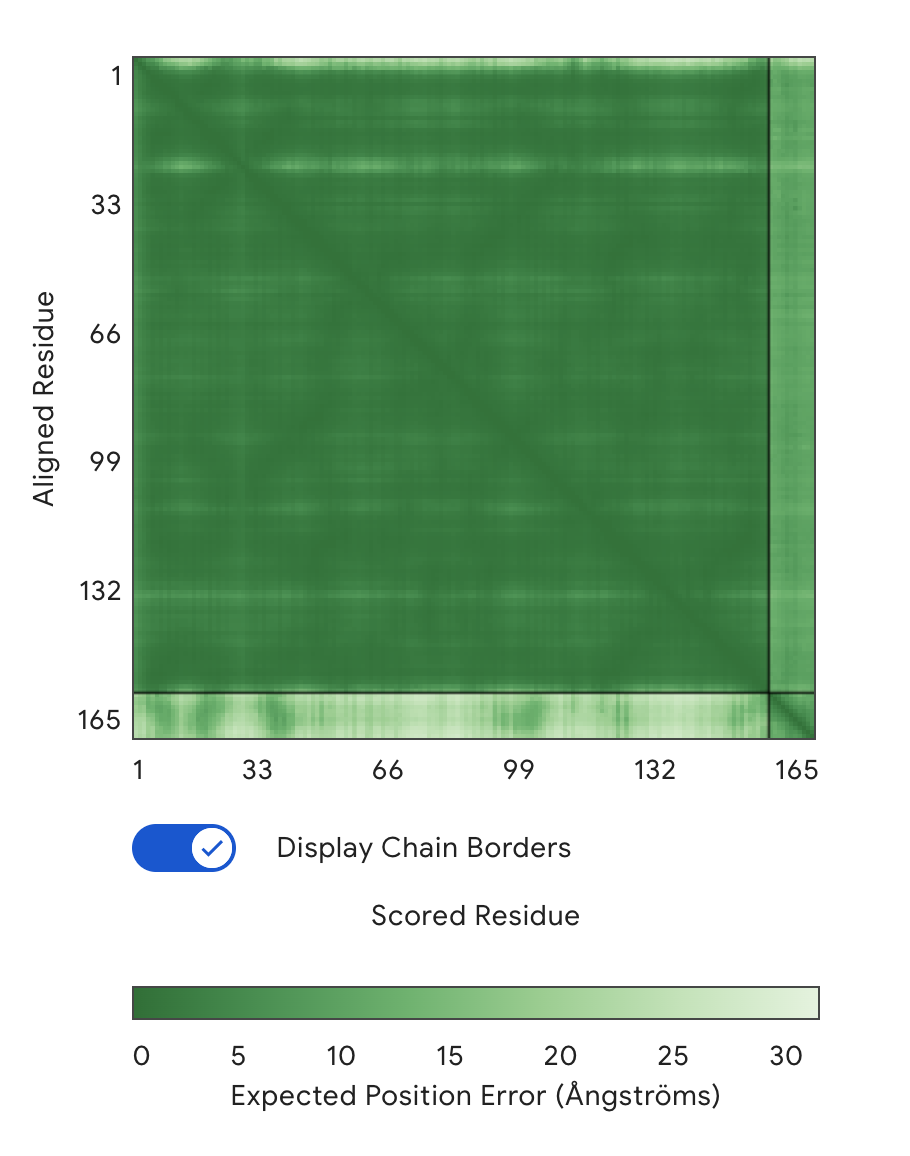
Alignment: SOD1 & KRSYPTALRHW
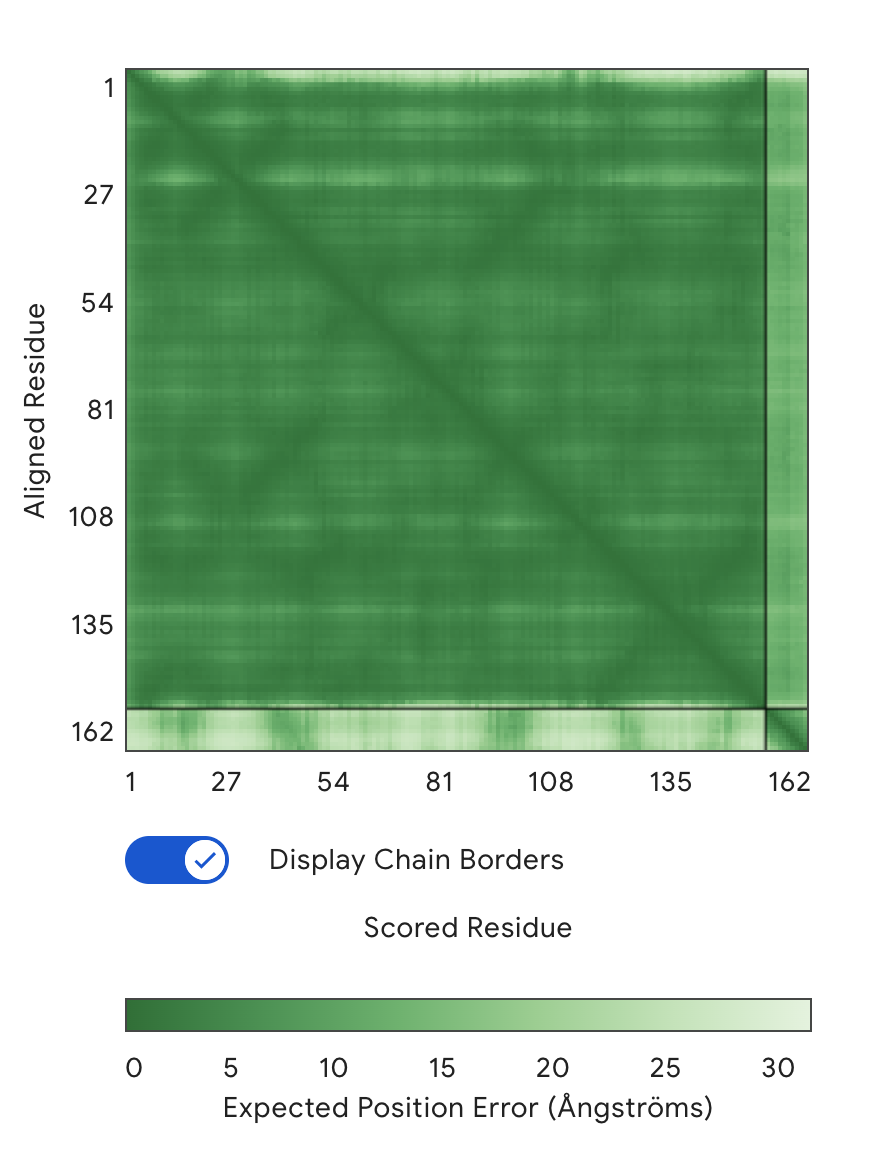
Alignment: SOD1 & WRYPVAABHGK
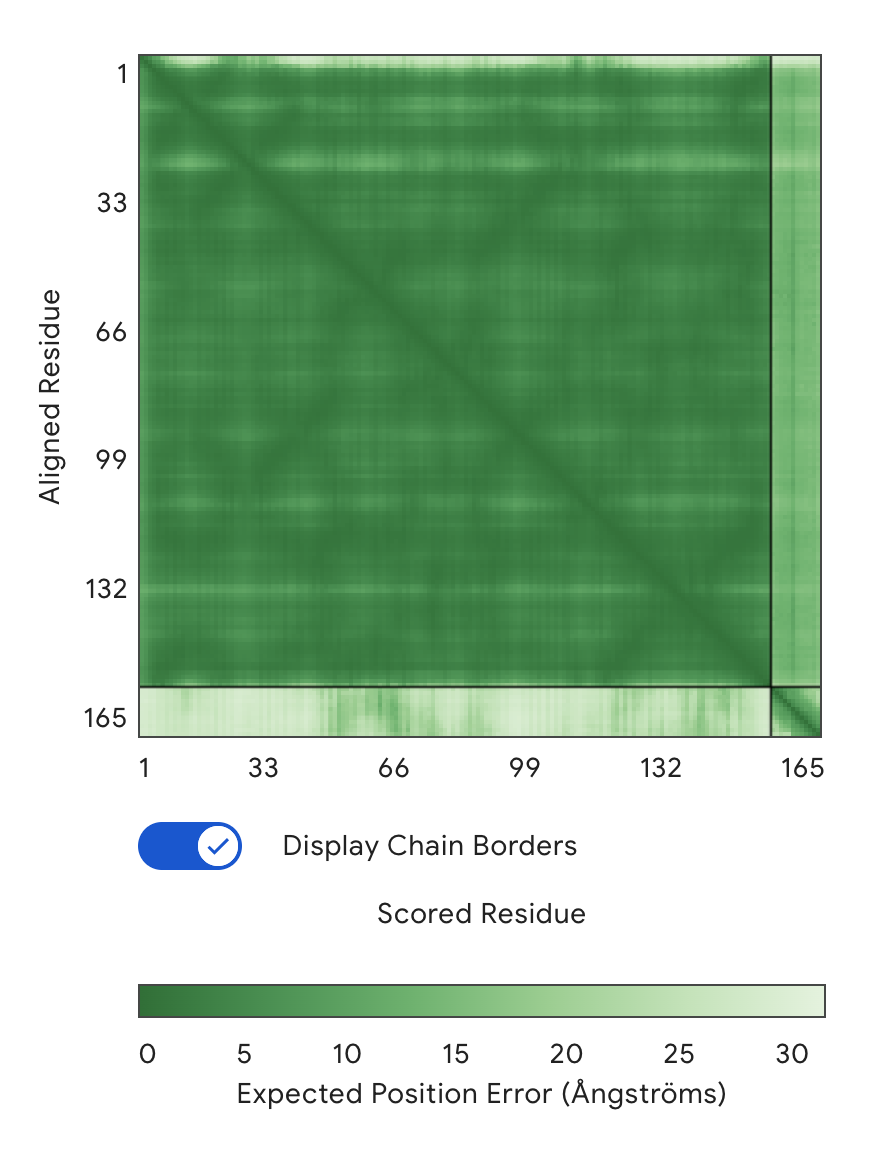
Alignment: SOD1 & FLYRWLPSRRGG - Reference
So the AlphaFold 3 modelling confirmed the potential of the peptides I designed using PepMLM. Interestingly all four peptides that I generated using the model outperformed FLYRWLPSRRGG - the known experimental reference, in terms of ipTM (interface confidence). This was despite it having the lowest pseudo perplexity score. Candidate 0 (KLVPAVVLAHK) emerged as having the greatest ipTM score with 0.58; it also had the lowest perplexity score of 7.4714. It’s high ipTM score demonstrates its unique ability to dock deeply within the N-terminus groove where the A4V (ALS causing) mutation of SOD1 sits. The other peptides showed varying affinities for the β-barrel and dimer interface.
The second highest performing binder was Candidate 3 (WHVYVVGLRHKE) - which had an ipTM score of 0.49, though it had the highest pseudo perplexity score of 25.8914. This means that it binds quite well to the mutated SOD1 though it is less likely to be found in nature than the other peptides.
The first thing I noticed was that the PeptiVerse Website Interface is so cute and easy to use. Wow! Pranam and his team really popped off with this one 👏🏼 so hats 🎩 🧢 👒 off to them!

So kawaii!
In the search for a peptide that can stabilize the SOD1 protein—a primary target in ALS research we move from structural modeling in AlphaFold 3 to therapeutic profiling in PeptiVerse. While a 3D model shows us how a peptide looks, these 11 metrics provided by PeptiVerse tell us how it will behave in a human body. Here is the result of testing the four PepMLM-designed candidates against a known reference binder (FLYRWLPSRRGG) in order of ipTM score (highest to lowest):
| Metric / Property | KLVPAVVLAHK | WHVYVVGLRHKE | KRSYPTALRHW | WRYPVAABHGK | FLYRWLPSRRGG (Ref) |
|---|---|---|---|---|---|
| 🐊 ipTM (Structural) | 0.58 | 0.49 | 0.44 | 0.39 | 0.26 |
| 💧 Solubility | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| 🔬 Permeability | 0.242 | 0.143 | 0.849 | 0.359 | 0.862 |
| 🩸 Hemolysis | 0.032 | 0.052 | 0.022 | 0.010 | 0.047 |
| 👯 Non-Fouling | 0.285 | 0.297 | 0.549 | 0.480 | 0.666 |
| ⏱️ Half-Life (hrs) | 0.438 | 0.412 | 0.342 | 0.339 | 0.310 |
| 🔗 Binding (pKd) | 5.528 | 5.919 | 5.965 | 5.300 | 5.968 |
| 📏 Length (aa) | 11 | 12 | 11 | 11 | 12 |
| ⚖️ Mol. Weight (Da) | 1174.5 | 1522.8 | 1414.6 | 1166.5 | 1507.7 |
| ⚡ Net Charge (pH 7) | +1.59 | +0.94 | +2.85 | +1.85 | +2.76 |
| 🎯 Isoelectric Point | 10.00 | 8.60 | 11.00 | 9.99 | 11.71 |
| 💦 GRAVY (Hydrophobicity) | 1.02 | -0.38 | -1.44 | -0.73 | -0.71 |
The results revealed a fascinating trade-off between structural integrity and medicinal viability!
As we know Candidate 0 (KLVPAVVLAHK) had the greatest ipTM score of 0.58, meaning that AlphaFold3 has great confidence in its structural fit to the mutated SOD1. However, based on PeptiVerse’s therapeutic metrics, Candidate 1 (KRSYPTALRHW) comes out on top though it had a lower ipTM score of 0.44.
Why is this?
Candidate 1 (KRSYPTALRHW) had the second lowest pseudo perplexity score of the generated peptides with 7.4714, meaning that it is reasonable to assume that it could occur in nature. Regarding the generated peptides it achieved the highest predicted affinity score with 5.965 pKd, as well as the best permeability score of 0.849, suggesting that it could actually reach the mutated SOD1 proteins inside cells. It also has the highest positive net charge of all the peptides (including the reference), with a score of +2.85 which makes it uniquely suited to crossing the blood-brain barrier and binding to the negatively charged aggregates of mutant SOD1.
Therefore I would advance Candidate 1 (KRSYPTALRHW).
| Run | Sequence | Affinity (pKd) | Solubility | Specificity | Motif Score | Hemolysis |
|---|---|---|---|---|---|---|
| #1 | RFKCIVKVMVRR | 8.881 | 0.500 | 0.615 | 0.553 | 0.944 |
| #2 | KRLQLYRKKCAE | 7.193 | 0.750 | 0.737 | 0.634 | 0.964 |
| #3 | QRACDYFRDDED | 7.783 | 0.833 | 0.679 | 0.059 | 0.895 |
| #4 | KEKEGPCWESEK | 7.360 | 0.833 | 0.871 | 0.002 | 0.962 |
Lysis Protein Sequence from UniProt (75 amino acids): https://www.uniprot.org/uniprotkb/P03609/entry
Click to view Lysis Protein Sequence
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DnaJ Protein Sequence from UniProt (376 amino acids): https://www.uniprot.org/uniprotkb/P08622/entry
Click to view Lysis Protein Sequence
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR