Governance 1. Bioengineering Tool Phage satellites are a diverse class of mobile genetic elements that parasitize a phage. Extracellular Prophage-Inducing Particles (EPIPs), are a novel class of phage satellite discovered by the Saha Lab that induces the extremely stable prophage, HerbertWM, in Mycolicibacterium aichiense. Due to their novelty, much of their mechanisms of action are unknown, but it is hypothesized that they contain antirepressors or partial antirepressors due to annotated genes that bear resemblance to BRO domains which have been noted to have some influence over transcription (Zemskov et al., 2000).
Because of the observed ability of EPIPs to induce a stable prophage, developing a lytic-lysogenic switch from their putative antirepressors would be valuable for many applications including medical and environmental applications. This switch could ensure that a temperate phage lyses bacterial pathogens in phage therapy or control the transcription of engineered constructs in the soil.
For properly formatted equations, read the version on GitHub.
Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
I would measure both fluorescence and OD600. Fluorescence will tell me how well the antirepressors are being expressed, and OD600 will tell me how much the antirepressors affecting bacterial growth.
The 1,536 Pixel Artwork Canvas | Collective Artwork I contributed a blue pixel to the bottom right corner. I liked being able to see the project evolve over time. One thing I’d improve for next year is to either make more pixels so people aren’t writing over previously drawn pixels or to limit the number of pixels someone can add and see how a collective artwork emerges that way.
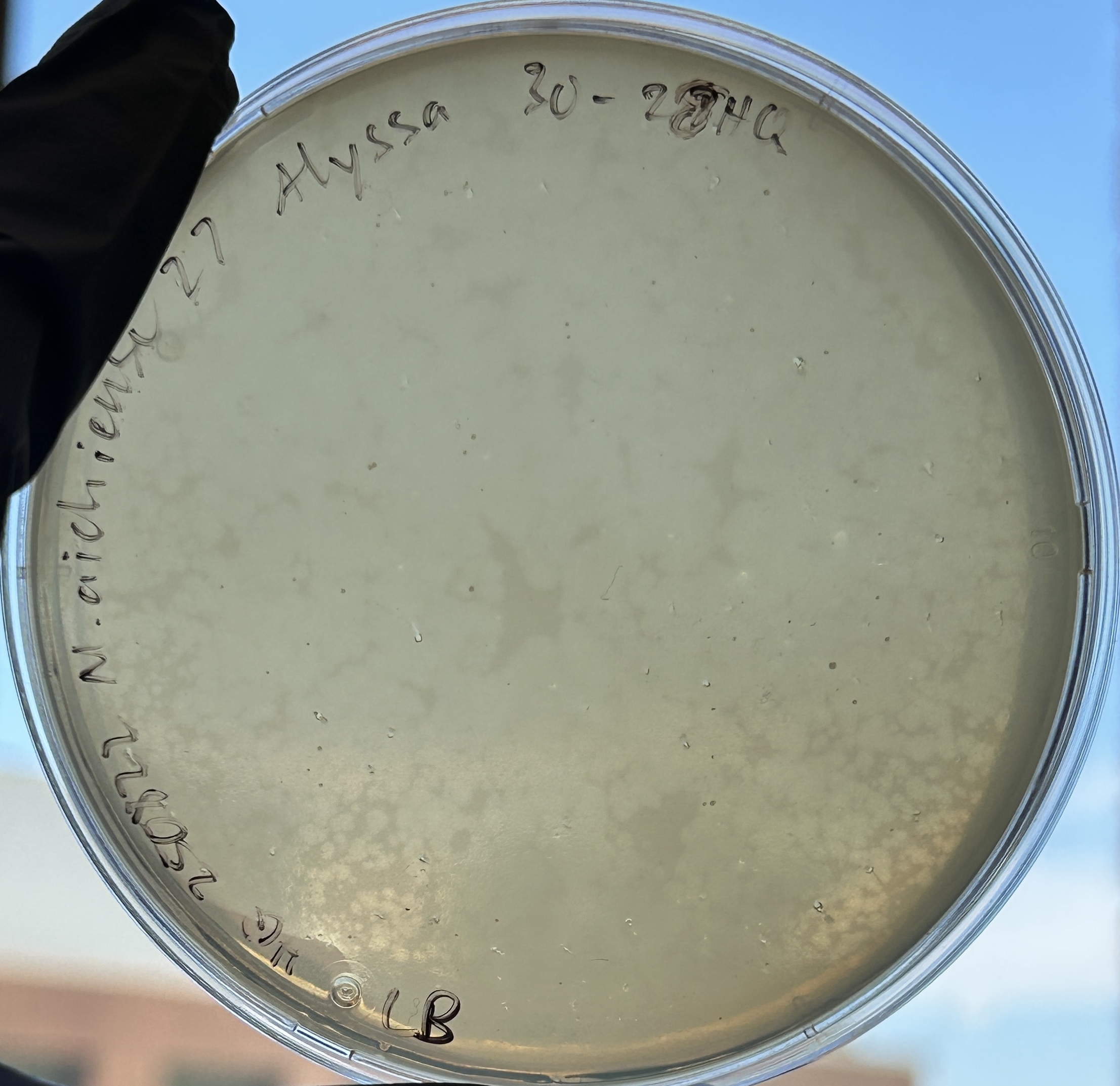
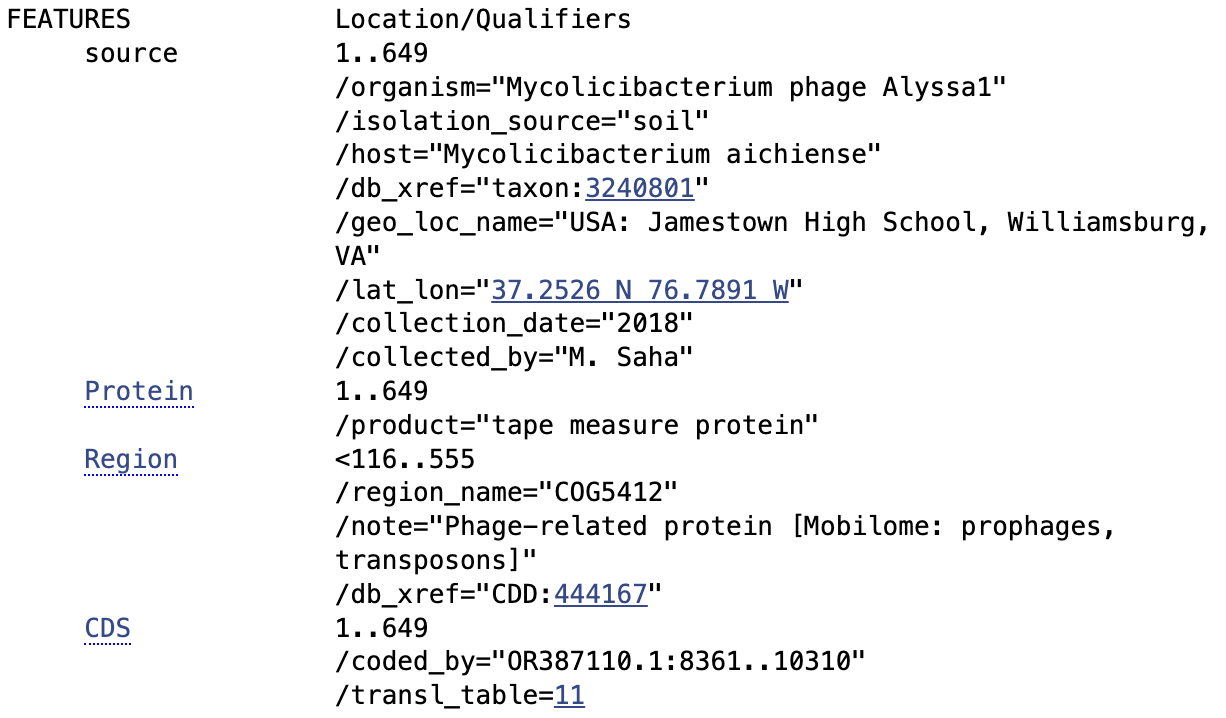
DNA Design Challenge Protein chosen: tape measure protein of Alyssa1, a Mycolicibacterium phage satellite called an “Extracellular Prophage-Inducing Particle” (EPIP). Tape measure proteins are rare in phage satellites, and TEM imaging has shown that EPIPs have uniquely long tails compared to the helper phage, the phage they parasitize, suggesting the tape measure protein may contribute to a unique mechanism of parasitism.
Paper Discussion Paper: AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots
This paper reports using the Opentrons robot to conduct PCR, Golden Gate assembly, and Gibson assembly. It appears that the robot was able to determine specific parameters for experiments such as annealing temperature. The paper did not go into much detail about how this occurred, but after looking at some of the supplementary materials, the script likely parses .csv files that include primer and fragement sequences.
Conceptual Questions Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans eat, the macromolecules the beef are made of are broken down during digestion into monomers. These monomers are common to all life, and humans use them to build human-specific macromolecules.
Why are there only 20 natural amino acids?
These 20 amino acids are what evolution happened to select for. These 20 amino acids happen to be enough to build all the proteins that are necessary for life that has evolved on Earth. Theoretically, there could be more, but in our “system” of life, these 20 are enough.
SOD1 Binder Peptide Design (From Pranam) Generate Binders with PepMLM PepMLM Colab File
SOD1 sequence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Sequence with A4V mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binders, perplexity score:
WRVPPAALRHKE, 22.653588 HRSPPVAAEHWK, 19.512332 WRYYPVAAAWKK, 11.081843 WRYYVAALRHGK, 15.691672 known SOD1 binder: FLYRWLPSRRGG, 20.635231 Evaluate Binders with AlphaFold3 Binder ipTM Score Binding Location WRVPPAALRHKE 0.39 near β-barrel and globular part HRSPPVAAEHWK 0.26 near β-barrel, across the seam and onto globular part WRYYPVAAAWKK 0.28 near β-barrel, across the seam WRYYVAALRHGK 0.30 near β-barrel, across the seam known: FLYRWLPSRRGG 20.635231 0.31 The ipTM score is highest for the first binder, but none of the binders bind in a similar location compared to the known binder. All of the predicted binders bound a similar part of the protein that was very different from where the known binder is predicted to bind.
DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
DNA polymerase - amplifies DNA by adding nucleotides
dNTPs - bases added by DNA polymerase
Buffer - maintains pH and contains cofactors the DNA polymerase requires
What are some factors that determine primer annealing temperature during PCR?
Primer length and GC content.
Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are able to process multiple inputs and gradients, unlike Boolean functions that largely process “black or white” options. This allows them to make more nuanced “decisions.”"
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
General Homework Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
In cell-free protein synthesis, variables such as pH, ion concentration, components, etc. are more adjustable. Additionally, researchers can add or remove components at any time. Two cases where cell-free expression is more beneficial than cell production are when the protein is toxic or when large amounts of protein are needed quickly.
Subsections of Homework
Week 1 HW: Principles and Practices
Governance
1. Bioengineering Tool
Phage satellites are a diverse class of mobile genetic elements that parasitize a phage. Extracellular Prophage-Inducing Particles (EPIPs), are a novel class of phage satellite discovered by the Saha Lab that induces the extremely stable prophage, HerbertWM, in Mycolicibacterium aichiense. Due to their novelty, much of their mechanisms of action are unknown, but it is hypothesized that they contain antirepressors or partial antirepressors due to annotated genes that bear resemblance to BRO domains which have been noted to have some influence over transcription (Zemskov et al., 2000). Because of the observed ability of EPIPs to induce a stable prophage, developing a lytic-lysogenic switch from their putative antirepressors would be valuable for many applications including medical and environmental applications. This switch could ensure that a temperate phage lyses bacterial pathogens in phage therapy or control the transcription of engineered constructs in the soil.
2. Governance/Policy Goals
Enhancebiosecurity: making sure this tool won’t activate genes or viruses that could harm people
Preventing incidents
Helping respond
Foster lab safety: while M. aichiense is BSL-1, safety precautions should still be used
Preventing incidents
Helping respond
Protect the environment: monitor how using this tool in the environment effects ecology
Preventing incidents
Helping respond
Other considerations
Minimizing costs/burdens to stakeholders
Feasibility
Not impede research
Promote constructive applications
3. Actions
Option 1: Make grant funding easier to obtain if the researcher is working toward using this tool in a beneficial application
Purpose: encourage beneficence
Design: funding sources such as the NIH or NSF must agree to this. These sources also must have enough money allocated by the government to give out, so the government must also agree to this.
Assumptions: researchers want money; it is already required of researchers to discuss potential applications of their work when applying for funding
Risks of failure and “success”: this could accidentally discourage basic science research—research for the sake of knowledge or a better understanding of the world that could benefit more people in the long run.
Option 2: Ensure individuals are well-trained to prevent contamination by more harmful microbes
Purpose: policies regarding BSL-1 waste disposal are already in place; however, in the past, M. aichiense cultures have become contaminated with bacteria that could have been a higher BSL level (but thankfully were not)
Design: more senior researchers should give newer researchers more hands-on focused training
Assumptions: senior researchers have the time to give newer researchers this training
Risks of failure and “success”: this could detract from the time spent researching, making the lab less productive
Option 3: Researchers must study long-term effects of using this tool in the environment
Purpose: ensure the application the tool is used for does not harm the environment
Design: researchers must have the means to track environmental changes over time. Some regulatory agency must enforce this standard.
Assumptions: researchers will do this to some extent anyway to see how durable/effective their work is in the environment
Risks of failure and “success”: researchers may not have the resources or time to allocate to study this. They may feel that their resources are better spent elsewhere
4. Scoring
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
2
1
1
• By preventing incidents
1
1
2
• By helping respond
3
2
1
Foster Lab Safety
n/a
1
n/a
• By preventing incidents
n/a
1
n/a
• By helping respond
n/a
1
n/a
Protect the environment
2
n/a
1
• By preventing incidents
2
n/a
2
• By helping respond
3
n/a
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
2
3
• Feasibility?
2
1
3
• Not impede research
1
2
2
• Promote constructive applications
1
3
2
5. Prioritization
Options 1 and 2 should be prioritized because they are more easily enforced and are based on pre-existing regulations and practices, making them fairly feasible. Option 3 is difficult to enforce and offers researchers no incentive to comply, whereas option 1 provides incentive and works through positive reinforcement. Additionally, both options 1 and 2 focus on preventing incidents by offering incentives toward beneficence (option 1) or providing better safety training (option 2). Meanwhile, option 3 prioritizes reacting to possible incidents by tracking environmental changes, noticing damage to the environment, and responding. Lastly, option 3 most obviously hinders research by dictating what resaerchers must investigate, while option 1 does not hinder research and option 2 will help research in the long run, even if it initially takes time away from research to teach good practices.
Pre-Lecture Work
Professor Jacobson
DNA polymerase’s error rate is 1:106. The human genome is ~3 billion bp, so on average, there would be 3000 mistakes made per round of DNA replication. Biology deals with this by destroying misfolded proteins and/or killing cells with too many mutations.
Due to third-base wobble, there are many ways to code for a protein of interest. In real life, many of these don’t work because certain organisms have a bias toward particular tRNAs, so mRNA codons must match the tRNA anticodons.
Dr. LeProust
The most common method of oligo synthesis uses phosphoramidite.
Chemical errors accumulate, so making fragments longer than 200 bp is difficult with direct synthesis.
Errors have accumulated so much that there are basically no fragments with the correct sequence.
George Church
10 essential amino acids: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine. The “lysine contingnecy” suggests that humans (and other animals) must eat plants because they are our only source of lysine, but they are also the only source of all of these amino acids, not just lysine.
I would use slide 4, the NA to AA conversion to know what nucleotides to use that would result in amino acids that would chemically interact.
BoSS: It would be great to find a way to store medications/treatments at room temperature; however, this could lead to unintended consequences such as vulnerability to contamination. As someone who works in a lab with bacteria, although the bacteria I work with could survive at room temperature, we store them in a fridge to help prevent contamination. I would also be concerned about using protective molecules from organisms that have adapted to survive at extreme temperatures to store medications/treatments, as these protective molecules could harm humans if they contaminate the treatment.
HTGAA Website
I added information about me, my email, and a cover image to the home page of the website. I have also learned the basics of markdown (including how to embed links and add images) during that process and have added this homework assignment to the website as well.
Week 10 HW: Imaging and Measurement
For properly formatted equations, read the version on GitHub.
Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
I would measure both fluorescence and OD600. Fluorescence will tell me how well the antirepressors are being expressed, and OD600 will tell me how much the antirepressors affecting bacterial growth.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Fluorescence: measured using a plate reader. OD600: measured using a spectrophotometer.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Plate reader and spectrophotometer.
Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/.
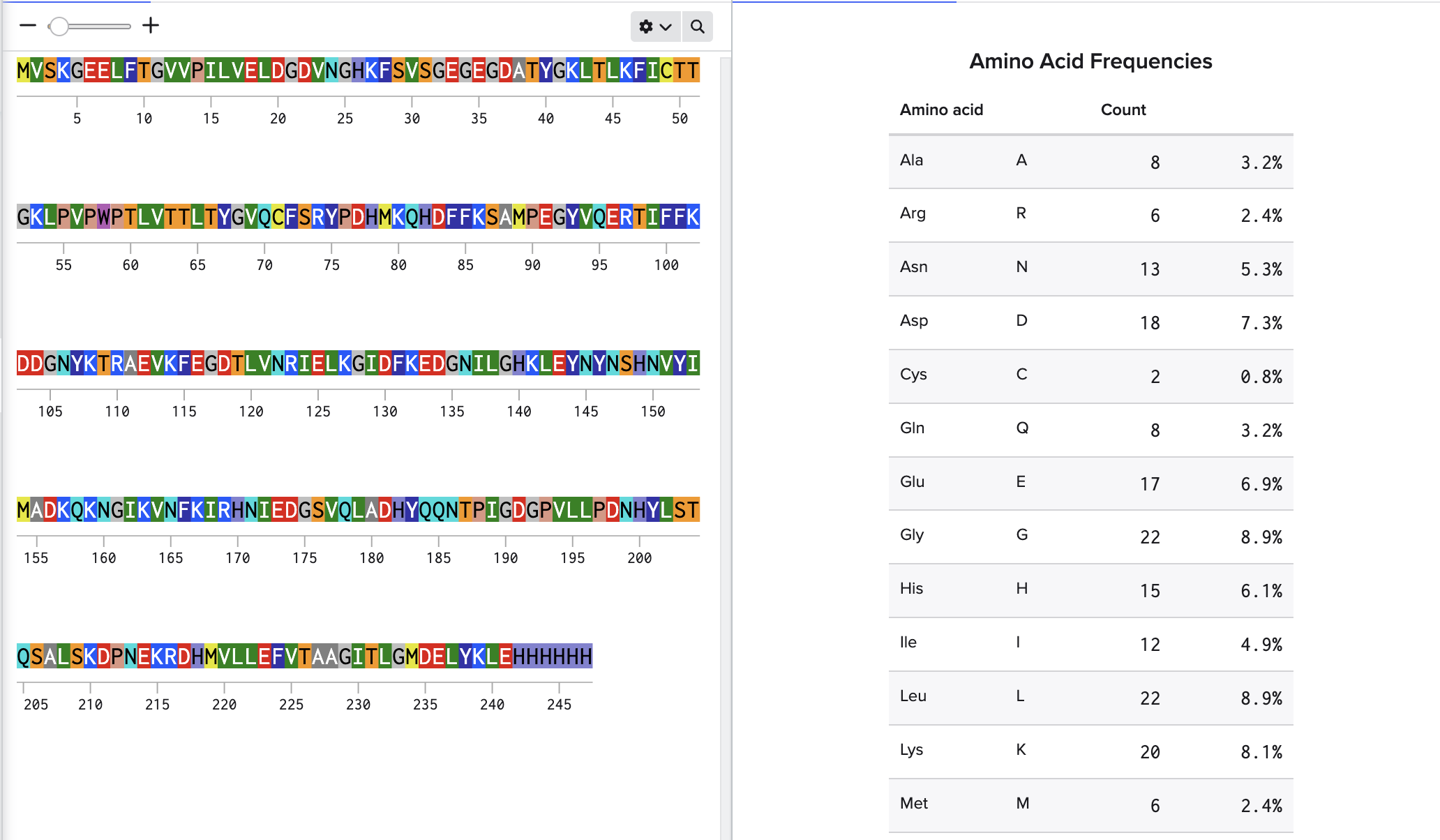
eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Mw = 28006.60 Da
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
1. Determine $z$ for each adjacent pair of peaks $(n, n + 1)$ using:
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No, because there are many peaks closely clustered together that aren’t fully resolved, so you cannot observe the charge state of the intact eGFP.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
19 peptides
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
~18 peaks that are >10% relative abundnace.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
It almost matches the number of predicted peptides. It is one fewer.
Identify the mass-to-charge ($\frac{m}{z}$) of the peptide shown in Figure 5b. What is the charge ($z$) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ($[M + H]^+$) based on its $\frac{m}{z}$ and $z$.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
What is the percentage of the sequence that is confirmed by peptide mapping?
90.7%
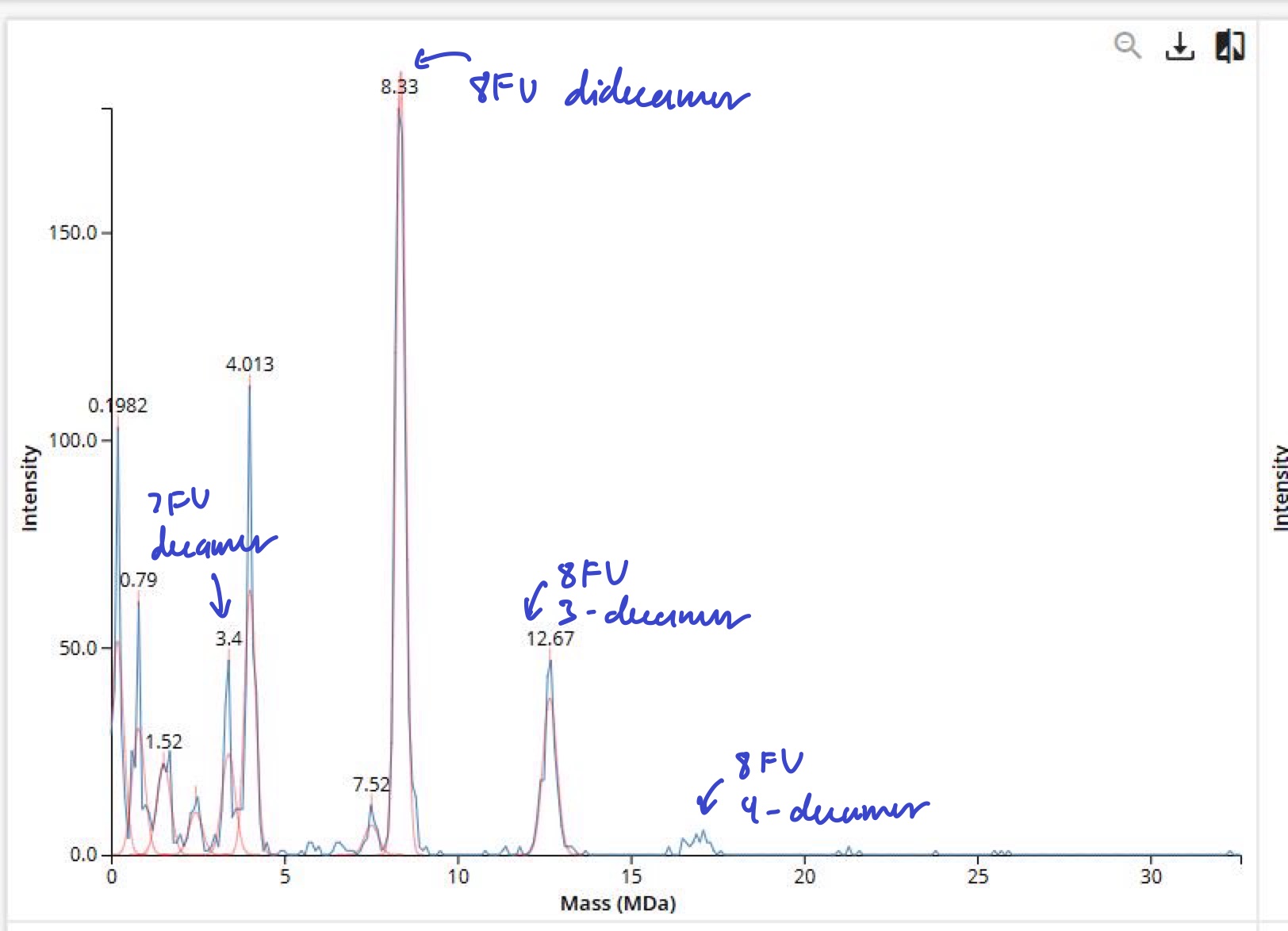
Waters Part IV — Oligomers
Waters Part V — Did I make GFP?
From screenshot in lab document (LEHHHHHH peptide):
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
1.083498
1.083492
5.26
Week 12 HW: Bioproduction and Cloud Labs
The 1,536 Pixel Artwork Canvas | Collective Artwork
I contributed a blue pixel to the bottom right corner. I liked being able to see the project evolve over time. One thing I’d improve for next year is to either make more pixels so people aren’t writing over previously drawn pixels or to limit the number of pixels someone can add and see how a collective artwork emerges that way.
Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): contains all the cellular machinery to complete transcription and translation (polymerase, ribosome, tRNAs, etc.)
Salts/Buffer
Potassium Glutamate: stabilizes protein folding, facilitates ribosome function
HEPES-KOH pH 7.5: maintains pH
Magnesium Glutamate: ribosome structure (and RNA folding), tRNA charging, etc.
Guanine: used for extra GTP required by translation
Translation Mix (Amino Acids)
17 Amino Acid Mix: necessary for making up the protein
Tyrosine: necessary for making up the protein, separate because of solubility issues
Cysteine: necessary for making up the protein, separate because reactive (oxidizes)
Additives
Nicotinamide: precursor to NAD+/NADP+
Backfill
Nuclease Free Water: ensure everything reaches the correct concentration
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour master mix lacks some pH stabilizing salts, and uses a different carbon source. It also uses different additives. Also, the nucleotides are already tri-phosphorylated, rather than just having one phosphate.
Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: matures quickly, allowing for quick fluorescence after synthesis
mRFP1: matures slowly, so it may not fluoresce as brightly, but is not very sensitive to acid, making it resistant to pH changes
mKO2: somewhat sensitive to acid changes, potentially causing it to not fluoresce as brightly in suboptimal conditions
mTurquoise2: matures quickly and is not sensitive to acid, allowing it to fluoresce quickly and brightly in many varying pH
mScarlet_I: somewhat sensitive to acid changes, potentially causing it to not fluoresce as brightly in suboptimal conditions
Electra2: oxygen dependent, so it may not function properly in poorly-oxygenated conditions
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
For mRFP1, perhaps adding chaperone proteins to help it fold could stabilize its fold, and ensuring that the pH does not change much during the incubation using a buffer would help.
Week 2 HW: DNA Read, Write, and Edit
DNA Design Challenge
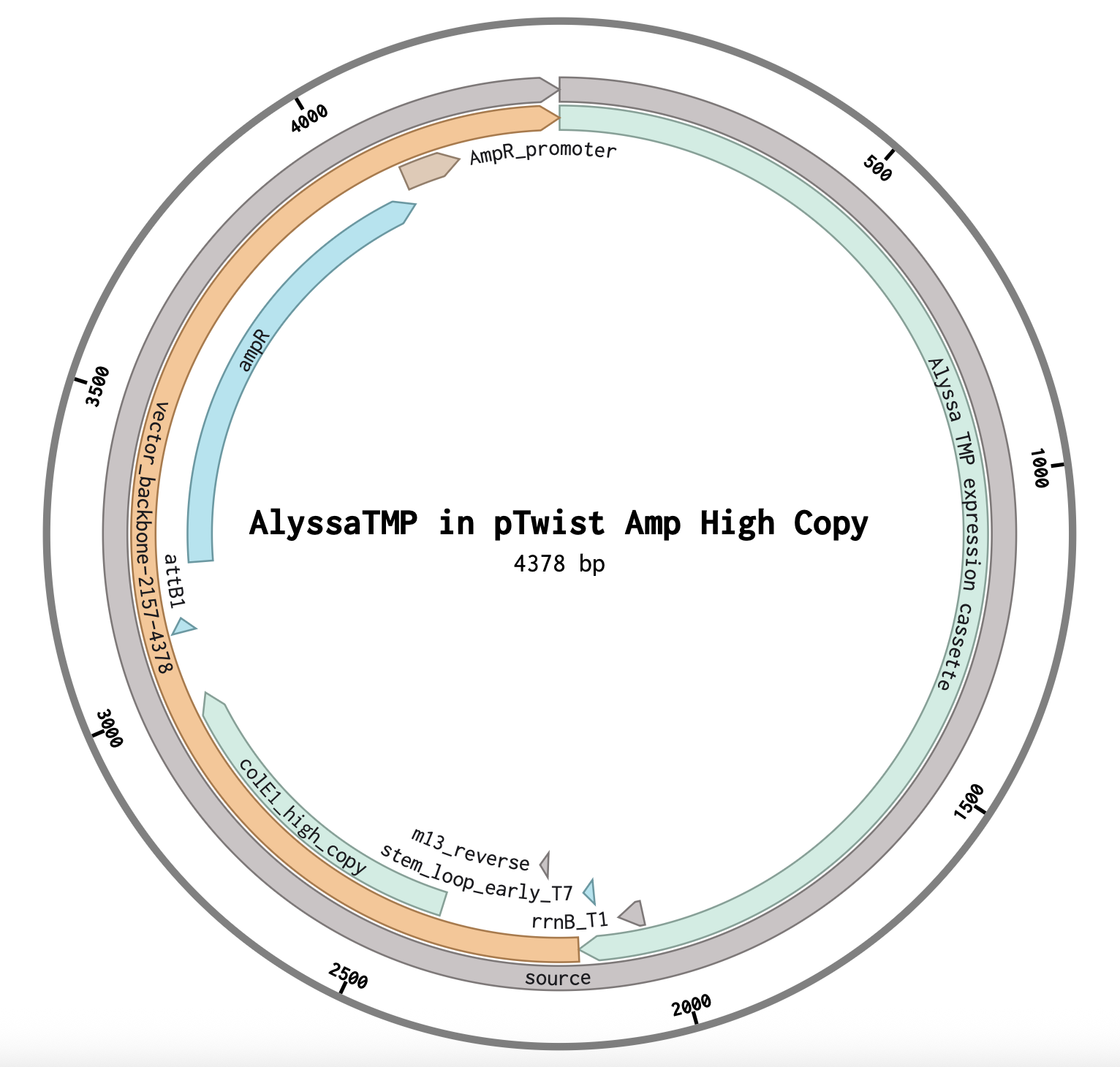
Protein chosen: tape measure protein of Alyssa1, a Mycolicibacterium phage satellite called an “Extracellular Prophage-Inducing Particle” (EPIP). Tape measure proteins are rare in phage satellites, and TEM imaging has shown that EPIPs have uniquely long tails compared to the helper phage, the phage they parasitize, suggesting the tape measure protein may contribute to a unique mechanism of parasitism.
This sequence has been optimized for expression in E. coli. In this instance, it would be helpful to do this as these phage satellites were discovered in Mycolicibacterium aichiense, a non-model organism, so expressing this protein in E. coli would make it easier to work with. In general, nucleotide sequences are optimized so that the protein can be more efficiently expressed in a given organism, so that working with it is easier (such as by removing restriction enzyme cut sites), or so that synthesis is easier (such as by lowering the GC content).
Choosing a Vector in Twist: I chose pTwist Amp High Copy because the Saha Lab works with ampicillin and if I want to express this protein in E. coli, I assume I’d want a lot of protein and for every E. coli descendent to have the plasmid, so a high copy plasmid would work best.
What DNA would you want to sequence (e.g., read) and why? I would like to sequence my gut microbiome. I’ve read about how the microbiome, specifically the gut microbiome can influence a person’s behaviors and send signals to the brain. This raises some philosophical questions of whether humans are truly governed by their own thoughts or if their thoughts are being subtly influenced by bacteria in their gut. I think it would be interesting to see what species dominate my microbiome.
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Nanopore sequencing it can generate long reads, so it is easier to identify unique features of each species. This makes it easier to assemble genomes.
Write
What DNA would you want to synthesize (e.g., write) and why? I have always wondered if it would one day be possible to create an organism with “no evolutionary history.” Many of the synthetic biology products/parts now are from preexisting organisms, but is it possible to create an entirely new sequence of DNA that produces proteins with no orthologs? Theoretically, this may be possible, similar to the “monkey at a type-writer” idea, where by chance a random sequence of nucleotides could not only produce a novel protein, but a novel organism. I would like to do this if only to see if it is possible.
What technology or technologies would you use to perform this DNA synthesis and why? To design this sequence, I would need some kind of machine learning model because biology is extremely complex and cannot be modeled with traditional methods. To synthesize this DNA, I could use the most common method of DNA synthesis, phosphoramidite chemistry, to synthesize pieces of this DNA, then use an assembly technique like Gibson assembly or Golden Gate assembly to construct the genome. These methods are most accessible and are commonly used.
Essential steps: deprotection, add next nucleotide, cap to stop strands that didn’t get the new nucleotide added from growing, and oxidation. Limitations: this method can’t make fragments longer than ~200 bp due to errors that accumulate.
Edit
What DNA would you want to edit and why?
I would like to edit my dog’s DNA to make him age more slowly. My dog makes me very happy but I don’t get to see him much because I’m at college and he lives with my parents. I would love it if he could stick around long enough that I could take care of him once I am out of school.
What technology or technologies would you use to perform these DNA edits and why? I would use CRISPR to knock in or out genes that I’d need to change. Editing an organism to change its life span would require many edits to the genome, and those edits would likely have to be precise. Because of that, CRISPR would be the best option.
Essential steps: guide RNA recognizes DNA sequence, and the Cas 9 enzyme makes a double-stranded break in the DNA. Preparation and input: prepare the guide RNA, so I’d need to know the sequence. Inputs include cells, some sort of vector to get the CRISPR-Cas9 system into the cells, and the CRISPR-Cas9 system (enzyme and guide RNA) Limitations: cuts in off target places, requirement of a PAM sequence, delivery mechanism (finding a vector)
This paper reports using the Opentrons robot to conduct PCR, Golden Gate assembly, and Gibson assembly. It appears that the robot was able to determine specific parameters for experiments such as annealing temperature. The paper did not go into much detail about how this occurred, but after looking at some of the supplementary materials, the script likely parses .csv files that include primer and fragement sequences.
Automation in Final Project
Since my final project will likely involve assembly techniques as described above, I could use automation by writing a Python script that automatically calculates experiment parameters such as temperatures and concentrations based on information that I put in such as DNA sequences and concentrations. This part of experiment planning is tedious and often involves multiple tools/pages (such as NEB’s Tm calculator, and various protocols for each method of assembly), so having one script that can do this automatically would be helpful. Additionally, it would be interesting to see how the Opentrons robot can run this script, though I predict that that would require some amount of troubleshooting that I may not have time for.
Why do humans eat beef but do not become a cow, eat fish but do not become fish? When humans eat, the macromolecules the beef are made of are broken down during digestion into monomers. These monomers are common to all life, and humans use them to build human-specific macromolecules.
Why are there only 20 natural amino acids? These 20 amino acids are what evolution happened to select for. These 20 amino acids happen to be enough to build all the proteins that are necessary for life that has evolved on Earth. Theoretically, there could be more, but in our “system” of life, these 20 are enough.
Can you make other non-natural amino acids? Design some new amino acids. Theoretically, yes. The R group can be anything, but only some will be functional with the 20 natural amino acids. Below are designs of new amino acids; I based them off of preexisting ones and added/removed atoms
Where did amino acids come from before enzymes that make them, and before life started? Amino acids formed under natural conditions on Earth. One example is demonstrated by the Miller-Urey experiments where they demonstrated that atmospheric gases could form amino acids spontaneously when energy is put in in the form of lightning.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? Left-handed because natural amino acids are L and create right-handed α-helixes. This means D-amino acids mirror L-amino acids, so the α-helix would also be mirrored.
Can you discover additional helices in proteins? Yes, you can discover new α-helices that already exist in proteins using tools/techniques like AlphaFold and X-ray crystallography. Discovering novel types of helices would require changing the chemistry of the amino acids, so discovering pre-existing ones would be unlikely. Creating new types of helices would be interesting, though.
Why are most molecular helices right-handed? This is because most biological molecules are D-enantiomers (right-handed), so they also create right-handed helices.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? β-sheets are formed using hydrogen bonds across backbones. β-sheet backbones “want” to form more hydrogen bonds with another peptide backbone, and can do that by stacking with other β-sheets.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Many amyloid diseases form β-sheets because hydrogen bonding in β-sheets is extremely favorable, so if the protein is able to “misfold” into β-sheets, it will do that. β-sheets can be used as materials. In fact, silk is rich in β-sheets, which is why it has so much tensile strength.
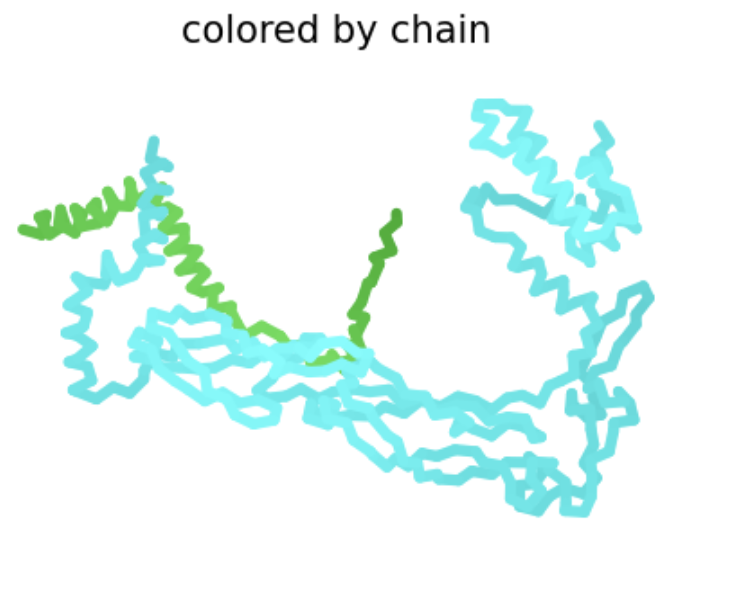
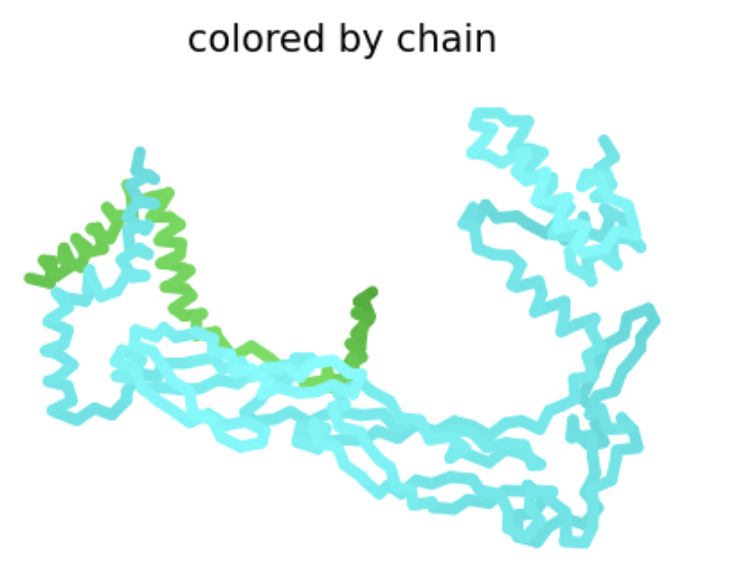
Protein Analysis and Visualization
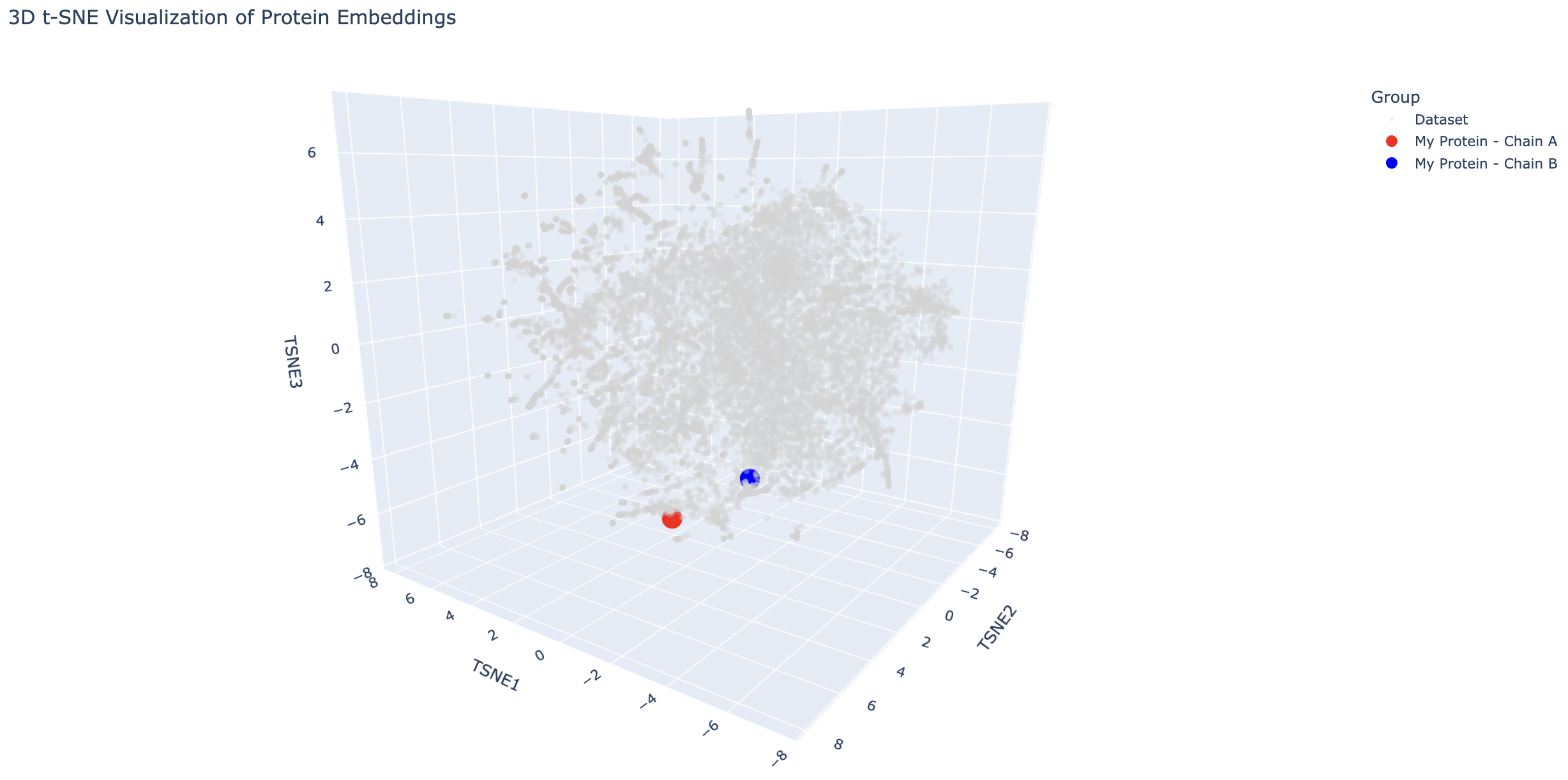
Briefly describe the protein you selected and why you selected it. I selected the antirepressor protein in phage Phi106 bound to a transcriptional activator in Bacillus. In the Saha Lab, we are interested in how Extracellular Prophage-Inducing Particles (EPIPs) are able to induce a prophage, HerberWM, that has a repressor. We hypothesize that the EPIPs may have an antirepressor, but the antirepressors are not in RCSB.
Identify the amino acid sequence of your protein.
Amino acid sequence of transcriptional activator: MSNNTGVKLKKLRKSKKLTLRDLADKLGVTHSYLSKIERGVTNPSLKMINSLAEFFDVDQSYFFTDEKNLDNFTDEELELTFERDLSIENLREKYNLTLGGKEVSDDEIKVMLEVLKAYRESKGGSRSD
Amino acid sequence of antirepressor: MKKEISLDEYLEKLKQLLENESVGTRAAL
How long is it? What is the most frequent amino acid?
Transcriptional activator: 129 amino acids, most frequent is leucine.
Antirepressor: 29 amino acids, most frequent is leucine.
How many protein sequence homologs are there for your protein?
Transcriptional activator: 250 found in UniProt
Antirepressor: 247 found in UniProt
Does your protein belong to any protein family? Yes, transcriptional activator and antirepressor.
Identify the structure page of your protein in RCSB
PDB ID: pdb_00009sf2
When was the structure solved? Is it a good quality structure? Deposited 8/19/2025, released 1/14/2025. The resolution is 1.70 Å, which is small, so it’s a good quality structure.
Are there any other molecules in the solved structure apart from protein? No.
Does your protein belong to any structure classification family? No results showed up when I searched the PDB ID in SCOP, so no.
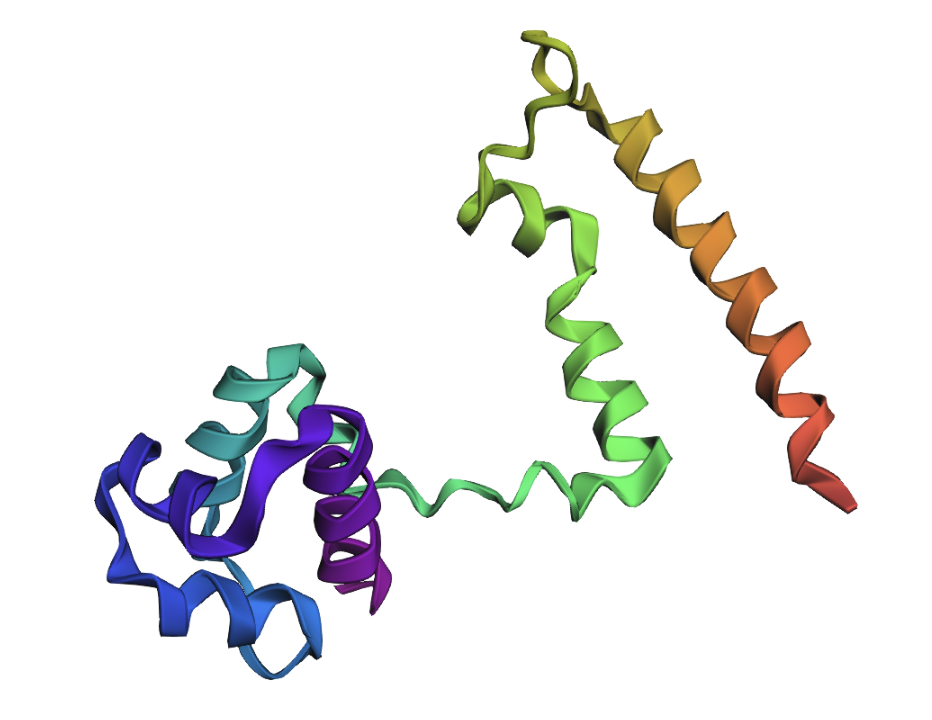
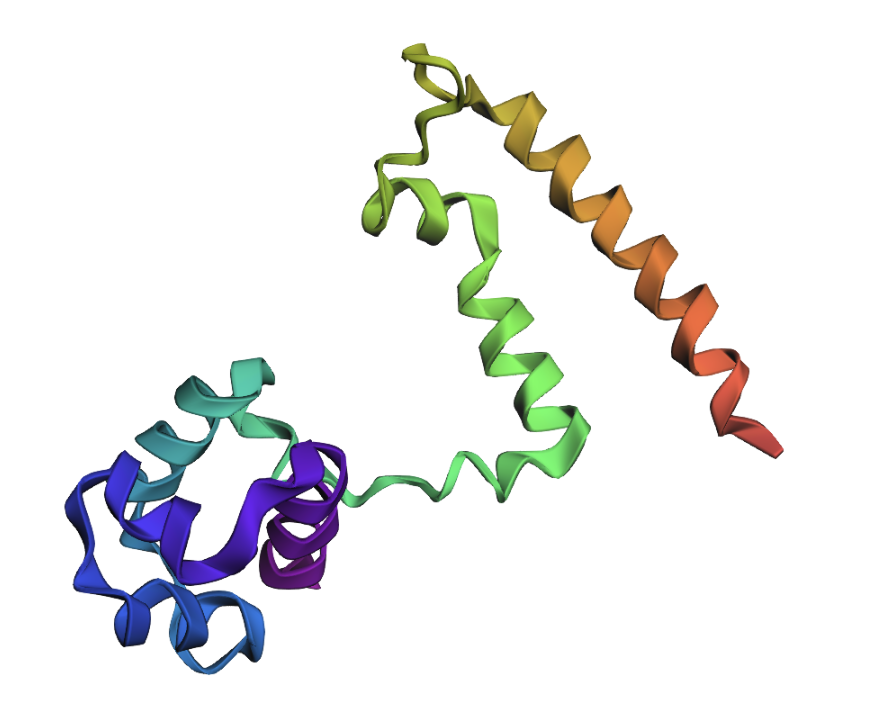
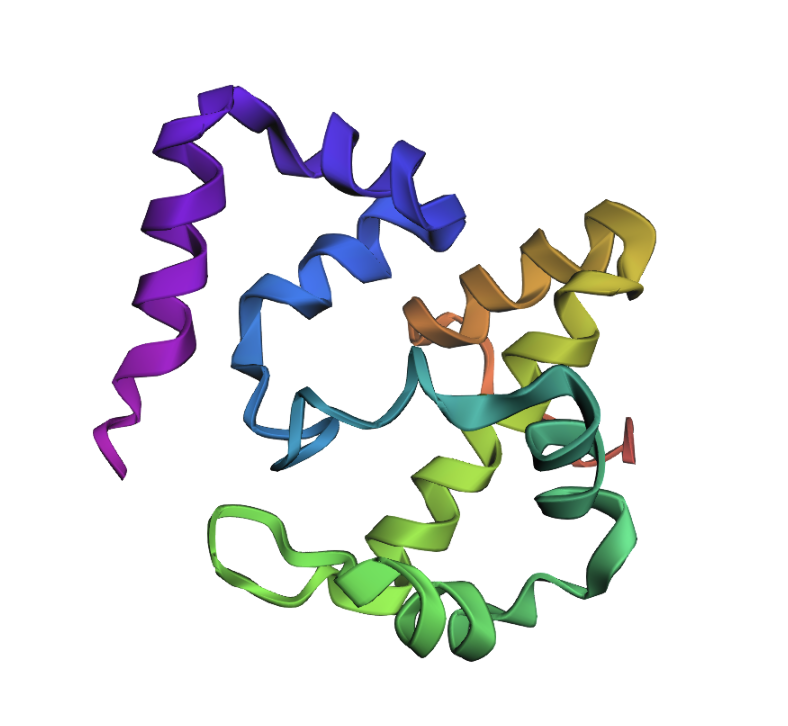
Open the structure of your protein in any 3D molecule visualization software
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
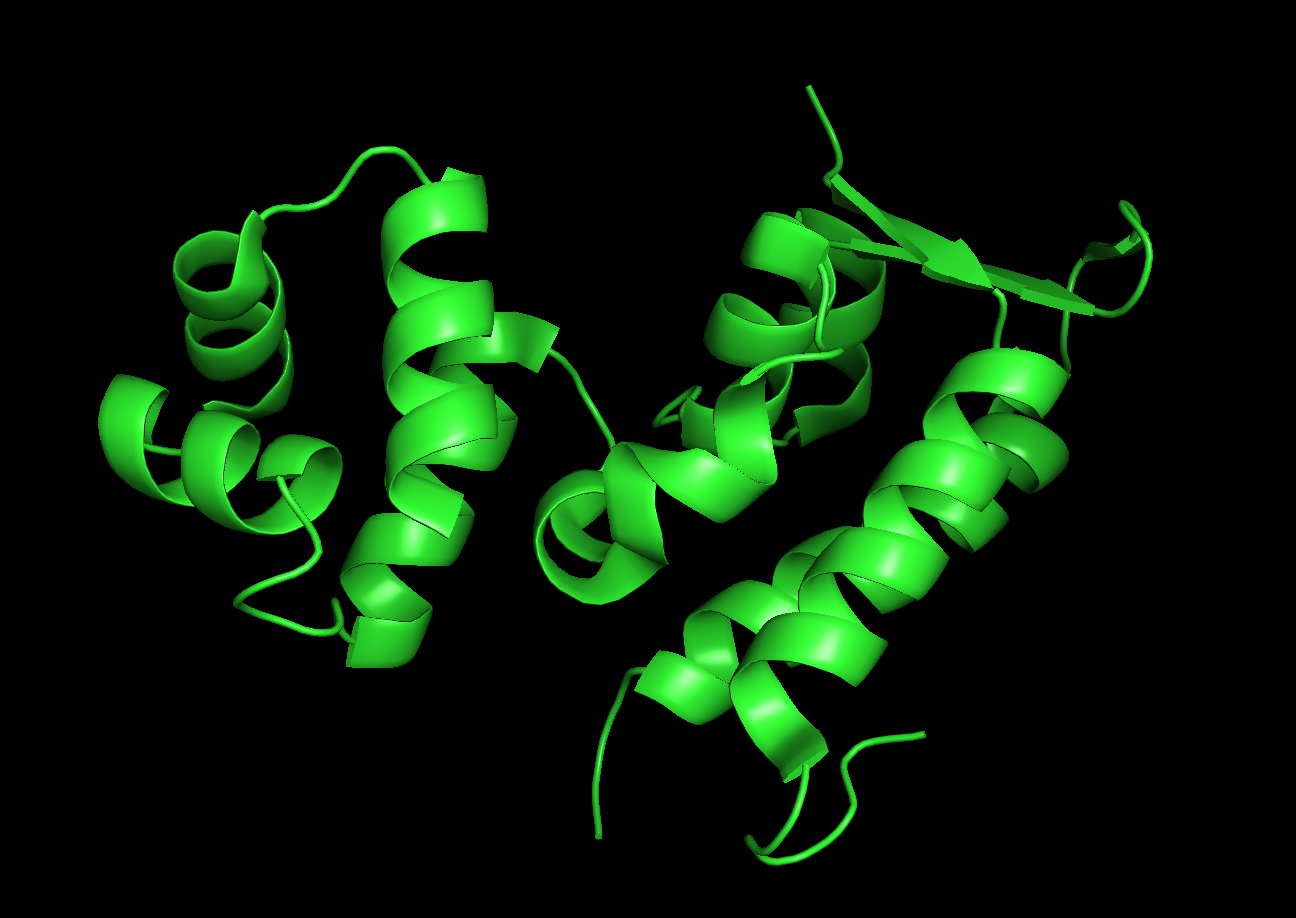
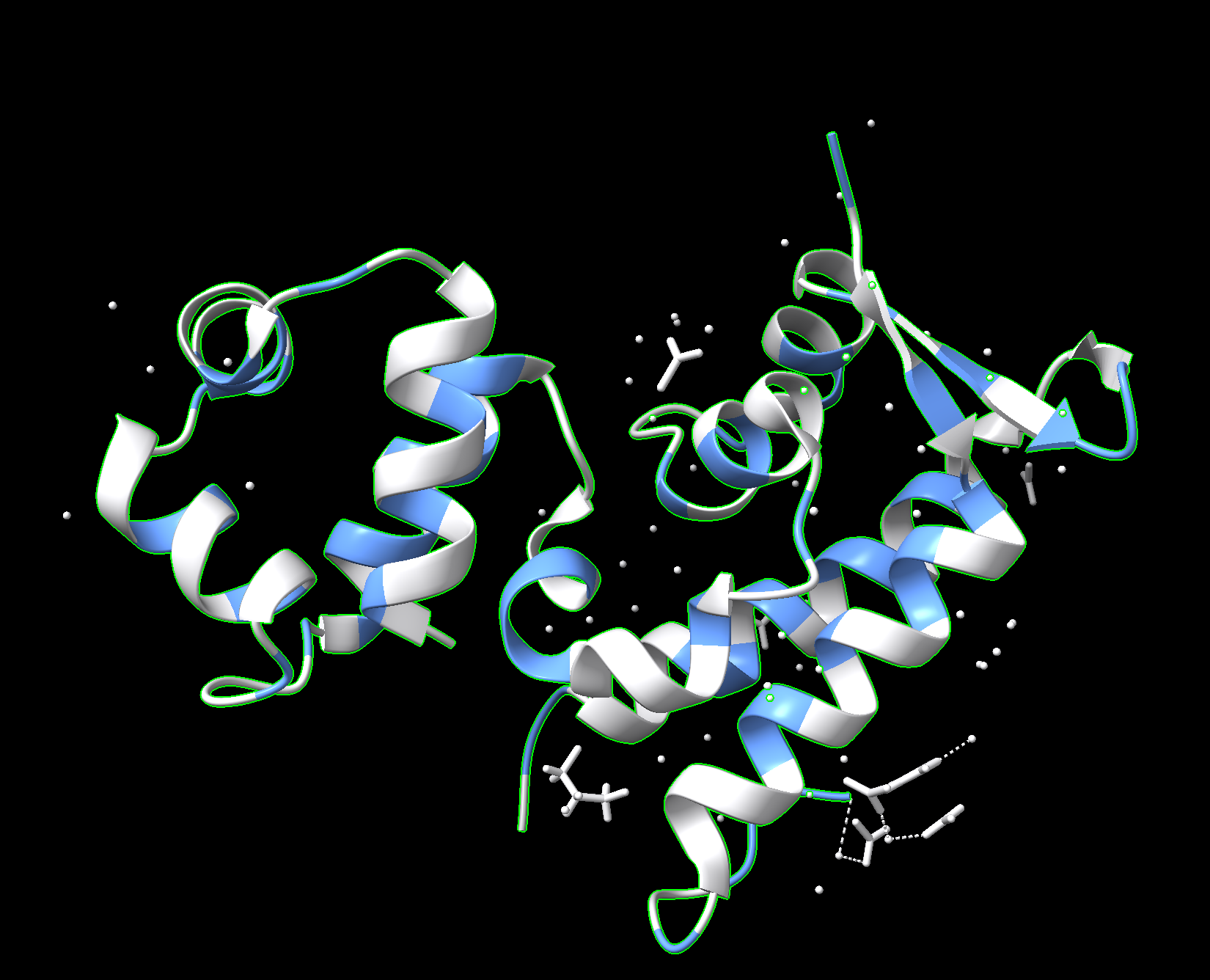
Cartoon:
Ribbon:
Ball and stick:
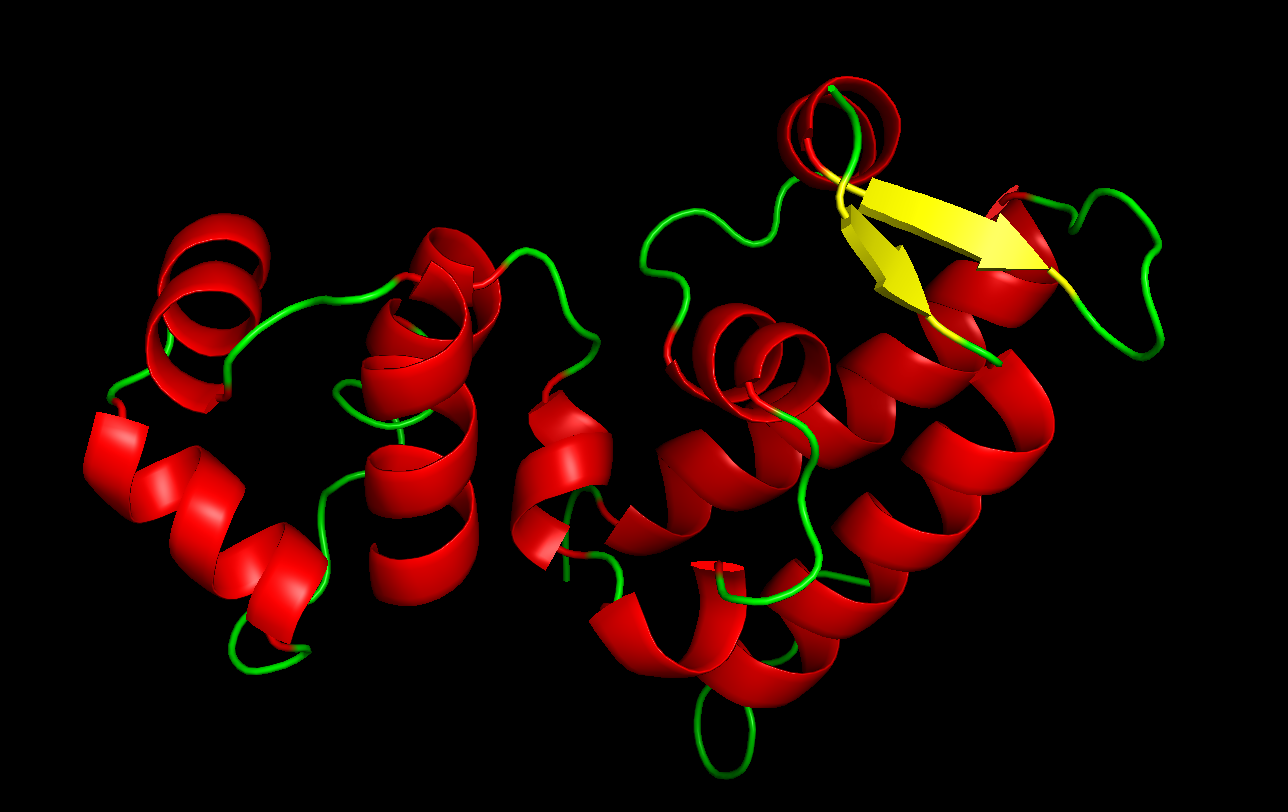
Color the protein by secondary structure. Does it have more helices or sheets?
Red: helices Yellow: sheets
The protein has more helices.
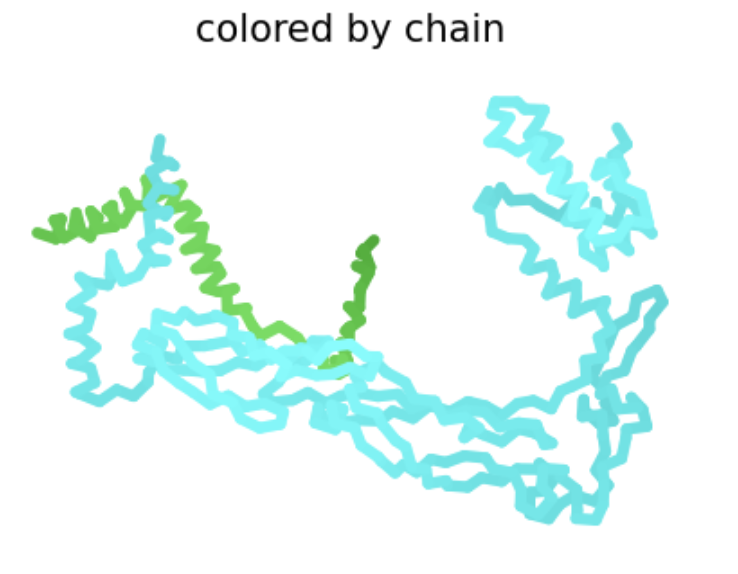
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Used ChimeraX because pyMOL was difficult to work with.
Blue: hydrophobic White: hydrophilic
The proteins mostly have hydrophobic residues on the inside and hydrophobic residues on the outside.
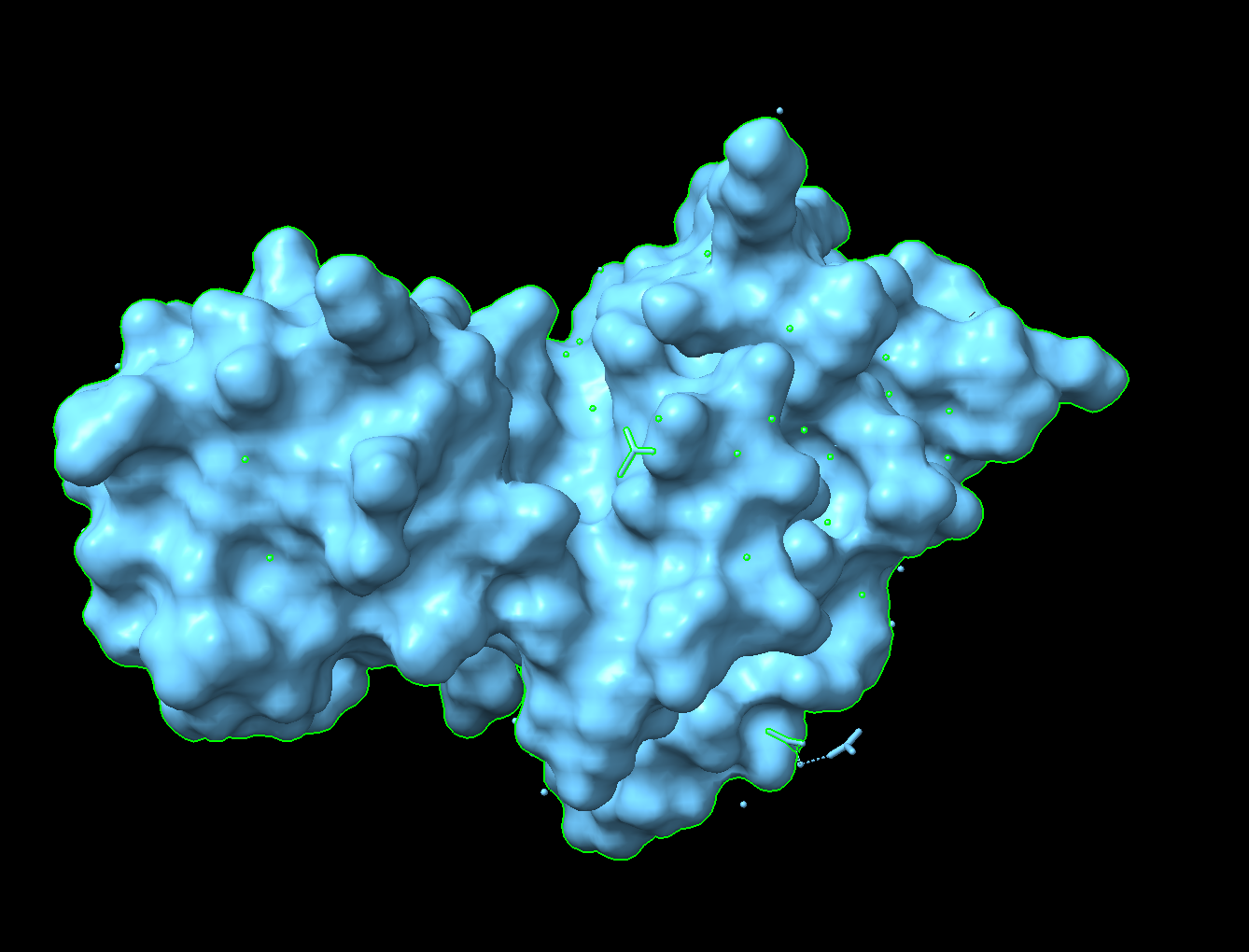
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
There is a “hole” that could be a binding pocket in the middle of this image.
Using ML-Based Protein Design Tools
Chose to use the same proteins as above (antirepressor in phage Phi106) for the same reasons.
Protein Language Modeling
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? Amino acids at the beginning of the two sequences appear to be more conserved. This could be because it is a binding site either for a ligand or for the other protein. Also, cystine and tryptophan are less likely to be substituted into the protein throughout the chains. This could be that cystine is unique as it is able to form disulfide bonds, which could disrupt the structure of the proteins. Tryptophan is large and bulky, so substituting it in could also disrupt the proteins’ structure.
Latent Space Analysis
Analyze the different formed neighborhoods: do they approximate similar proteins? Yes, I have found clusters of proteins of similar function (eg. dehydrogenases, phosphatases).
Place your protein in the resulting map and explain its position and similarity to its neighbors.
The transcriptional activator is close to proteins such as a transcriptional activator and a hypothetical protein with a DNA-binding domain, which makes sense because these are all DNA-binding proteins. The antirepressor is also near DNA-binding proteins such as transcription factors and a helicase. This is interesting because it suggests the antirepressor may also bind to DNA.
Protein Folding
Only did the transcriptional activator for simplicity and because it has a more interesting fold.
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
This predicted structure matches the original structure well.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Transcriptional activator G6P substitution:
Protein appears more or less the same as the native protein, so this substitution did not have an impact.
Transcriptional activator - moved middle chunk of sequence to end:
Protein appears different compared to native protein. It appears more scrunched, so translocating a segment of sequence had a large impact.
Protein Generation
I used the example sequence for this because I couldn’t figure out how to upload the PDF file and get the output for both chains for the protein I had been looking at.
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The score for the predicted sequence is much lower than the score for the original one - the predicted sequence matches the fold better. This could be due to evolutionary constraints on the original one. Also, the sequence recovery was 0.5137, meaning ~51% of the amino acids from the original sequence were correctly recovered.
Input this sequence into ESMFold and compare the predicted structure to your original.
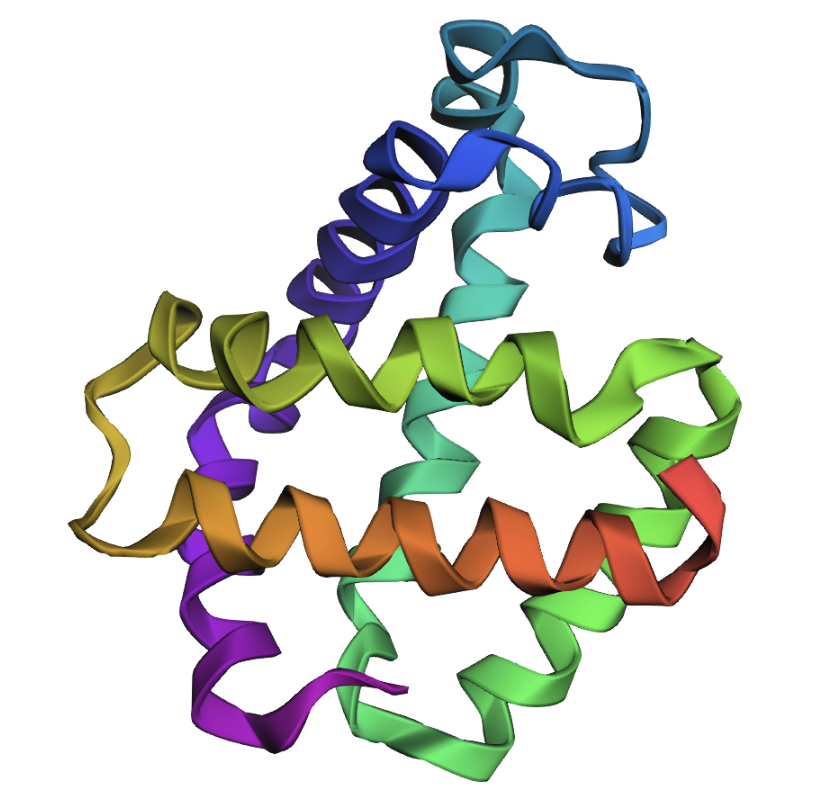
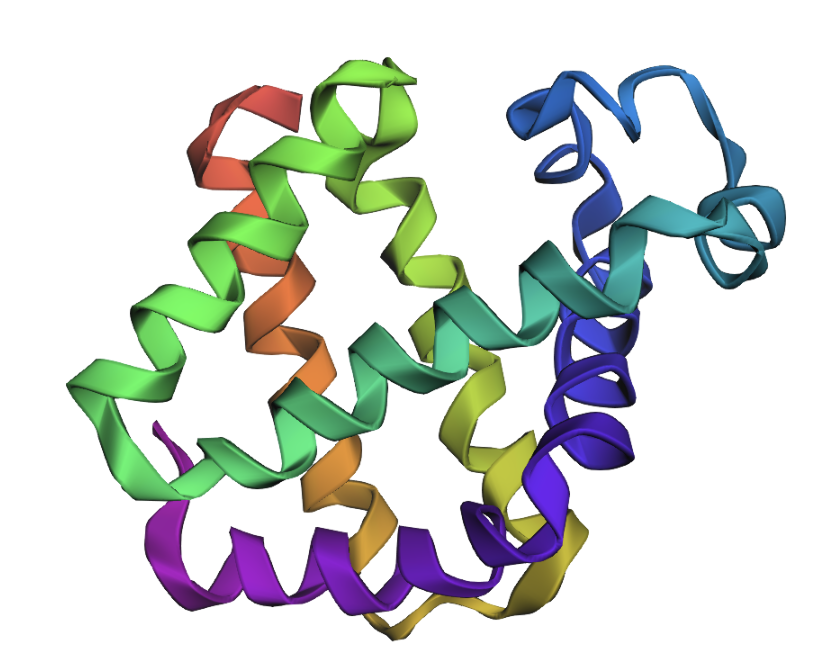
Original (backbone in PDB file):
Predicted (based on input sequence and PDB file backbone):
The folds appear very similar. Inverse folding appears to have worked well.
Group Brainstorm on Bacteriophage Engineering
I worked with Grace Hussey for this.
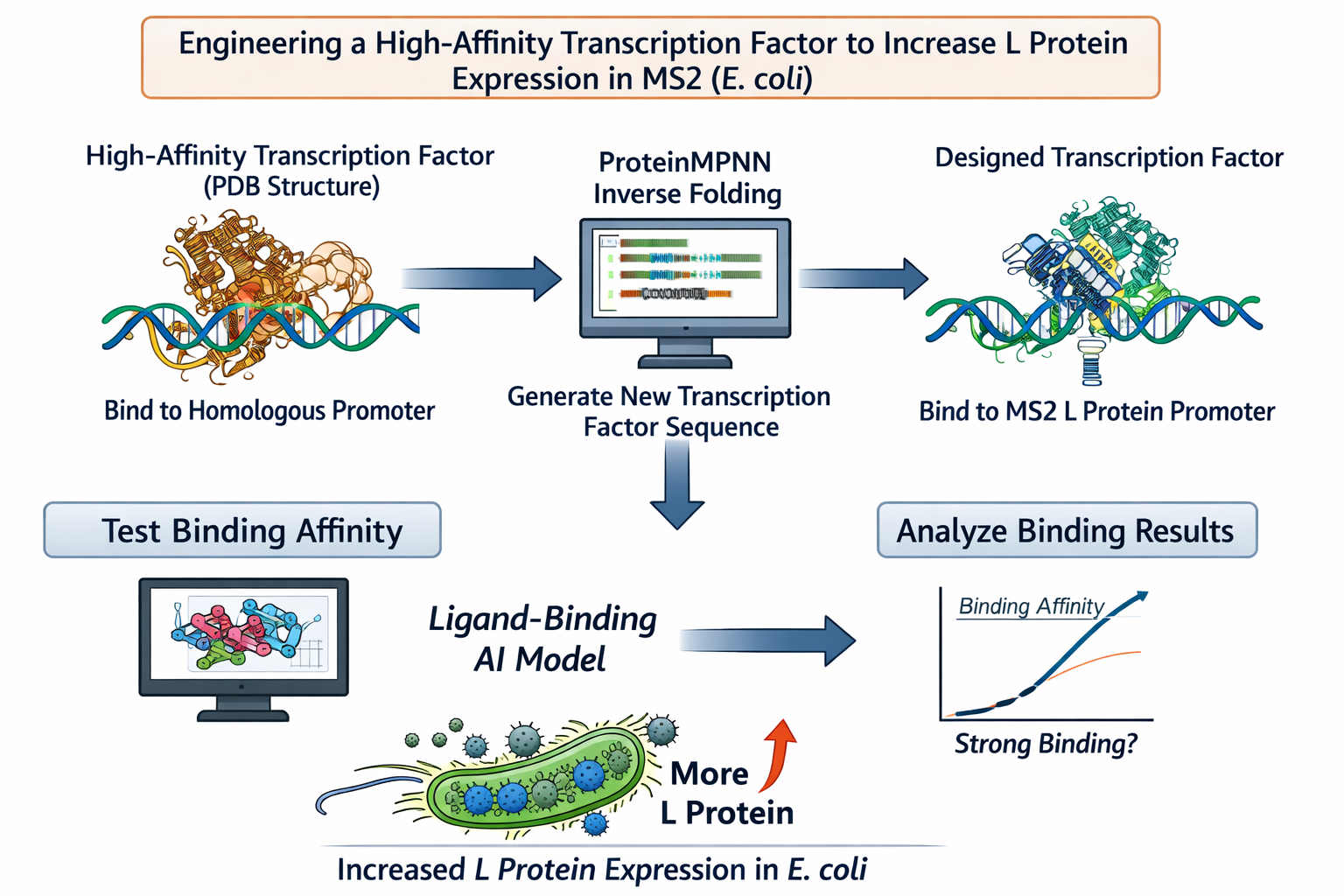
Computational Goal: We will attempt to increase the titer of the L protein expressed by MS2 in the E. coli host.
Overall solution: create a new transcription factor that binds very tightly to the promoter, increasing expression of the L protein
Inverse protein folding using ProteinMPNN
Use structure of a transcription factor (a PDB file) that binds with very high affinity to a promoter that is highly homologous to the L protein promoter
Input the structure of the aforementioned transcription factor into ProteinMPNN along with the amino acid sequence of a native transcription factor that binds to the L protein promoter
This will theoretically generate the sequence of a protein that is structurally similar to a transcription factor with high DNA-binding affinity but is specific to the L protein promoter
Confirm the binding affinity between our designed transcription factor and the MS2 L protein promoter with a ligand-binding AI model
Why will these tools accomplish our computational goal?
Protein MPNN is an inverse-folding algorithm. The sequence of the L protein promoter and other transcription factors that bind to this promoter are known. However, to generate a transcription factor with higher affinity for this promoter sequence than the native L protein transcription factors, we will model our inverse-folding after an existing transcription factor that behaves in the manner we envision (binds with high affinity) for our engineered transcription factor. As discussed in this week’s lecture, existing AI algorithms are good at designing structures similar to previously-characterized structures but less good at designing different (novel) structures. As such, our inverse-folding approach hinges on the existence of another high affinity transcription factor for a homologous promoter
Using a ligand-binding AI model will provide a computational indication of the success of our engineering without the need to use an in vitro or in vivo model
Possible pitfalls:
We are unable to find a transcription factor that binds to a highly homologous promoter sequence with high affinity :(
Sequence with A4V mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binders, perplexity score:
WRVPPAALRHKE, 22.653588
HRSPPVAAEHWK, 19.512332
WRYYPVAAAWKK, 11.081843
WRYYVAALRHGK, 15.691672
known SOD1 binder: FLYRWLPSRRGG, 20.635231
Evaluate Binders with AlphaFold3
Binder
ipTM Score
Binding Location
WRVPPAALRHKE
0.39
near β-barrel and globular part
HRSPPVAAEHWK
0.26
near β-barrel, across the seam and onto globular part
WRYYPVAAAWKK
0.28
near β-barrel, across the seam
WRYYVAALRHGK
0.30
near β-barrel, across the seam
known: FLYRWLPSRRGG
20.635231
0.31
The ipTM score is highest for the first binder, but none of the binders bind in a similar location compared to the known binder. All of the predicted binders bound a similar part of the protein that was very different from where the known binder is predicted to bind.
Evaluate Properties of Generated Peptides in the PeptiVerse
Binder
Binding Affinity (pKd/pKi)
Solubility Probability
Hemolysis Probability
Net Charge
Molecular Weight (Da)
WRVPPAALRHKE
Weak, 5.114
Soluble, 1
Non-hemolytic, 0.020
1.85
1450.7
HRSPPVAAEHWK
Weak, 4.576
Soluble, 1
Non-hemolytic, 0.014
0.94
1414.6
WRYYPVAAAWKK
Weak, 6.124
Soluble, 1
Non-hemolytic, 0.021
2.76
1538.8
WRYYVAALRHGK
Weak, 6.474
Soluble, 1
Non-hemolytic, 0.023
2.84
1519.8
known: FLYRWLPSRRGG
Weak, 5.968
Soluble, 1
Non-hemolytic, 0.047
2.76
1507.7
Binding affinity is weak for all peptides. There does not seem to be a strong correlation between ipTM score and binding affinity. From PeptiVerse results, it appears that all peptides are soluble and non-hemolytic. The ones with stronger charges have a higher binding affinity.
I’m choosing to continue with the 4th generated peptide, WRYYVAALRHGK, because it has the highest binding affinity from PeptiVerse and it has a ipTM score similar to the known peptide from AlphaFold.
Generate Optimized Peptides with moPPIt
Motif positions used: 95-100, 85-90
Peptide generated: PKHCLQRLLSKH
ipTM score: 0.45
Binding location: end of the β-barrel that is closer to the termini
Binding affinity (pKd/pKi): weak, 6.408
Solubility probability: soluble, 1
Hemolysis probability: non-hemolytic, 0.057
Net charge: 3.12
Molecular weight (Da): 1459.8
This peptide has one of the strongest binding affinities according to PeptiVerse along with the highest ipTM score from AlphaFold. Like the other peptides, it is soluble and non-hemolytic. Consistent with previous observations, it has the strongest charge, which could be why it binds mutated SOD1 relatively strongly.
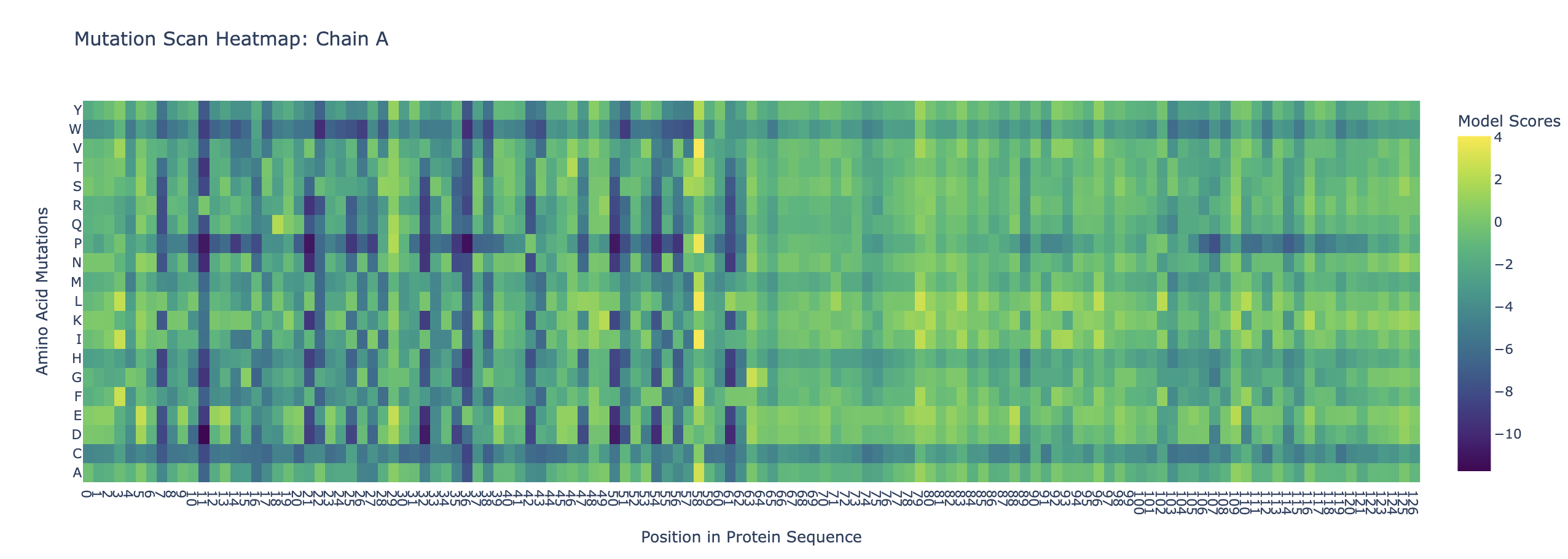
Final Project: L-Protein Mutants
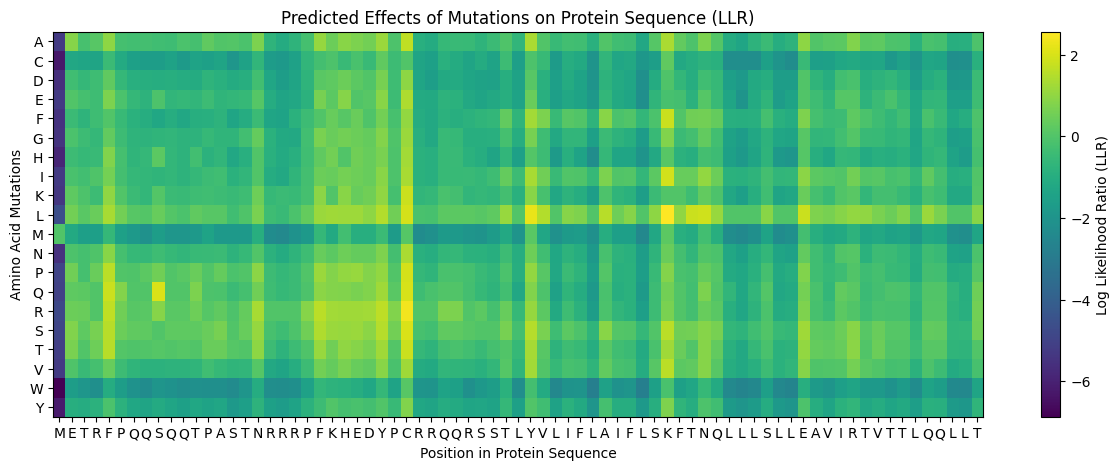
L-protein sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT Last 35 residues are the transmembrane domain
For the most part, experiments where lysis still occured correlates to a yellowish green point on the heatmap, suggesting that the heatmap is somewhat reliable.
Proposed mutations:
Mutation
Domain
Reasoning
L-protein Multimer
R19S
Soluble
This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.14, which is low, suggesting low confidence in this assembly of a multimer.
R31I
Soluble
This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.16, which is low, suggesting low confidence in this assembly of a multimer.
H24L
Soluble
This amino acid is shown to change in BLASTp results, and the heatmap shows a very positive score for this mutation. There is no experimental data, so it is possible this mutation would result in a functional protein, even though other amino acid substitutions at this position have not, especially since histidine and lysine are both positively charged amino acids.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.15, which is low, suggesting low confidence in this assembly of a multimer.
T69S
Transmembrane
This amino acid is shown to change in BLASTp results, and the heatmap shows a score near 0, so it’s not strongly negative. However, experimental data suggests this protein does not result in lysis. This makes sense because if the transmembrane domain is mutated, the L-protein will not be able to oligomerize in the membrane and create pores.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.15, which is low, suggesting low confidence in this assembly of a multimer.
N53T
Transmembrane
This amino acid is shown to change in BLASTp results, and the heatmap shows a very positive score for this mutation. There is no experimental data, so it is possible this mutation would result in a functional protein, even though other amino acid substitutions at this position have not. Also, this mutation is the only polar-to-polar mutation that was not experimentally tested.
Multimer forms but appears asymmetrial. ipTM score is 0.16, which is low, suggesting low confidence in this assembly of a multimer.
Mutagenesis using Af2-Multimer
Proposed mutations:
Mutation
Domain
Reasoning
L-protein-DnaJ Multimer
R19S
Soluble
This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative.
The L-protein and DnaJ appear to make contact. ipTM is 0.17, which is low, suggesting low confidence in this interaction.
R31I
Soluble
This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative.
The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction.
H24L
Soluble
This amino acid is shown to change in BLASTp results, and the heatmap shows a very positive score for this mutation. There is no experimental data, so it is possible this mutation would result in a functional protein, even though other amino acid substitutions at this position have not, especially since histidine and lysine are both positively charged amino acids.
The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction.
F5S
Soluble
This particular substitution exists in BLASTp results, and the heatmap shows a very positive score. There is no experimental data, so it is possible this mutation would result in a functional protein.
The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction.
A14S
Soluble
This amino acid is shown to change in BLASTp results, and the heat map shows a positive score. There is no experimental data, so it is possible this mutation would result in a functional protein.
The L-protein and DnaJ appear to make contact ipTM is 0.16, which is low, suggesting low confidence in this interaction.
Week 6 HW: Genetic Circuits Part I
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? DNA polymerase - amplifies DNA by adding nucleotides dNTPs - bases added by DNA polymerase Buffer - maintains pH and contains cofactors the DNA polymerase requires
What are some factors that determine primer annealing temperature during PCR? Primer length and GC content.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. PCR uses primers to specify which regions of DNA are targeted through amplification. Restriction enzyme digests do not amplify DNA but use restriction enzymes to cut DNA in specific places, potentially cutting out a region of interest. PCR is more flexible in where the primers can bind, so it is easier to extract a region of interest. PCR would also be used when a lot of the region of interest is needed, since it amplifies DNA. Restriction digests are useful for linearizing a plasmid, for example, or creating sticky ends.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The ends of the fragments must be identical. This can be done by positioning PCR primers appropriately.
How does the plasmid DNA enter the E. coli cells during transformation? Heat-shocking the E. coli will create pores in the membrane so that DNA can enter.
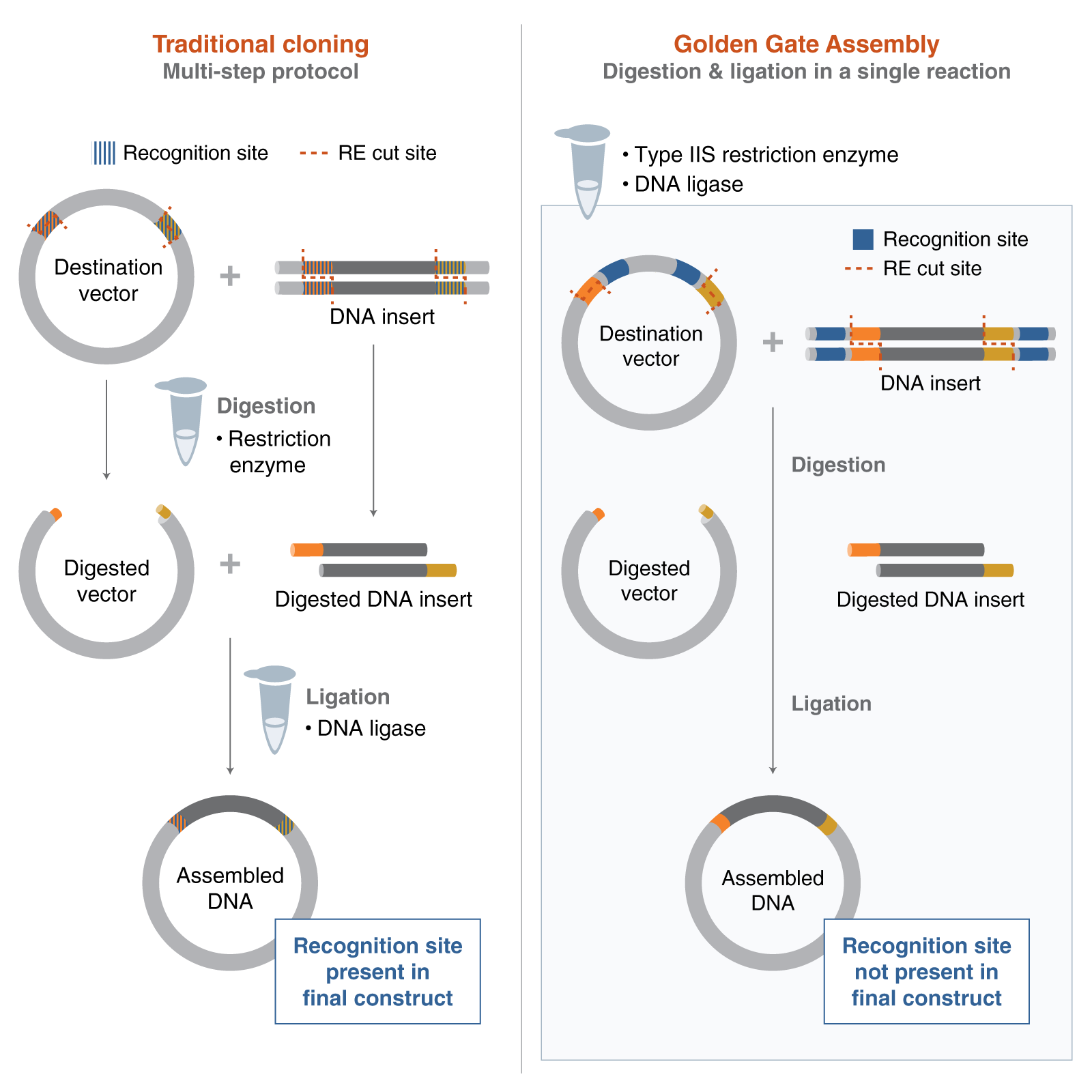
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online). Golden Gate assembly uses a restriction enzyme that cuts outside of the enzyme binding site to create sticky ends without scarring. Primers are designed to amplify each fragment used in assembly and to add on the enzyme binding and cut site. The fragments are amplified, then a restriction digest is run to create the sticky ends before assembly. Usually, the fragments are first cloned into a plasmid for stability before being used for assembly.
Model this assembly method with Benchling or Asimov Kernel!
Asimov Kernel
Copied from my notebook in Asimov:
Repressilator
Recreated Repressilator
The graphs appear the same for the repressilator in the Bacterial Parts Demo repository and the one that I’ve created, which makes sense because the same DNA parts and simulation conditions were used.
Construct 1
This construct behaves how I expect it to because there is only one promoter and RBS, so a singular on/off switch. Since there is nothing in this system that would inhibit the expression of LacI, the expression remains constant throughout the time period, which is reflected in the two graphs on the right.
Construct 2
I would expect TetR and LitR levels to start high then decrease as they repress themselves.
The graphs somewhat match my predictions. The RNA graph does have a high concentration at the beginning then rapidly decrease, though I’m not sure why the concentration then stays constant. The protein graph shows less of a change in concentration after the initial spike and also stays constant after dropping. I also don’t know why there would be more expression of TetR than LitR. Maybe the pTetR promoter is stronger?
Construct 3
I would expect this construct to function similarly to the repressilator where expression oscillates over time.
This did not match my prediction, and I’m not sure why. Expression levels mostly rise then stay level throughout time, though some genes are highly expressed initially, then decrease to a stable level.
Week 7 HW: Genetic Circuits Part II
Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are able to process multiple inputs and gradients, unlike Boolean functions that largely process “black or white” options. This allows them to make more nuanced “decisions.”"
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
An IANN could be used to predict a cell’s response to a combination of environmental signals/ligands at various combinations of concentrations. Since ligands can elicit multiple responses by triggering multiple pathways and pathways that impact other pathways, an IANN could be a good choice to deal with this complexity. Some limitations that an IANN might face is that there are so many parts involved in this situation that molecules could interact with the IANN, and a large IANN would place a large metabolic burden on the cell it’s in.
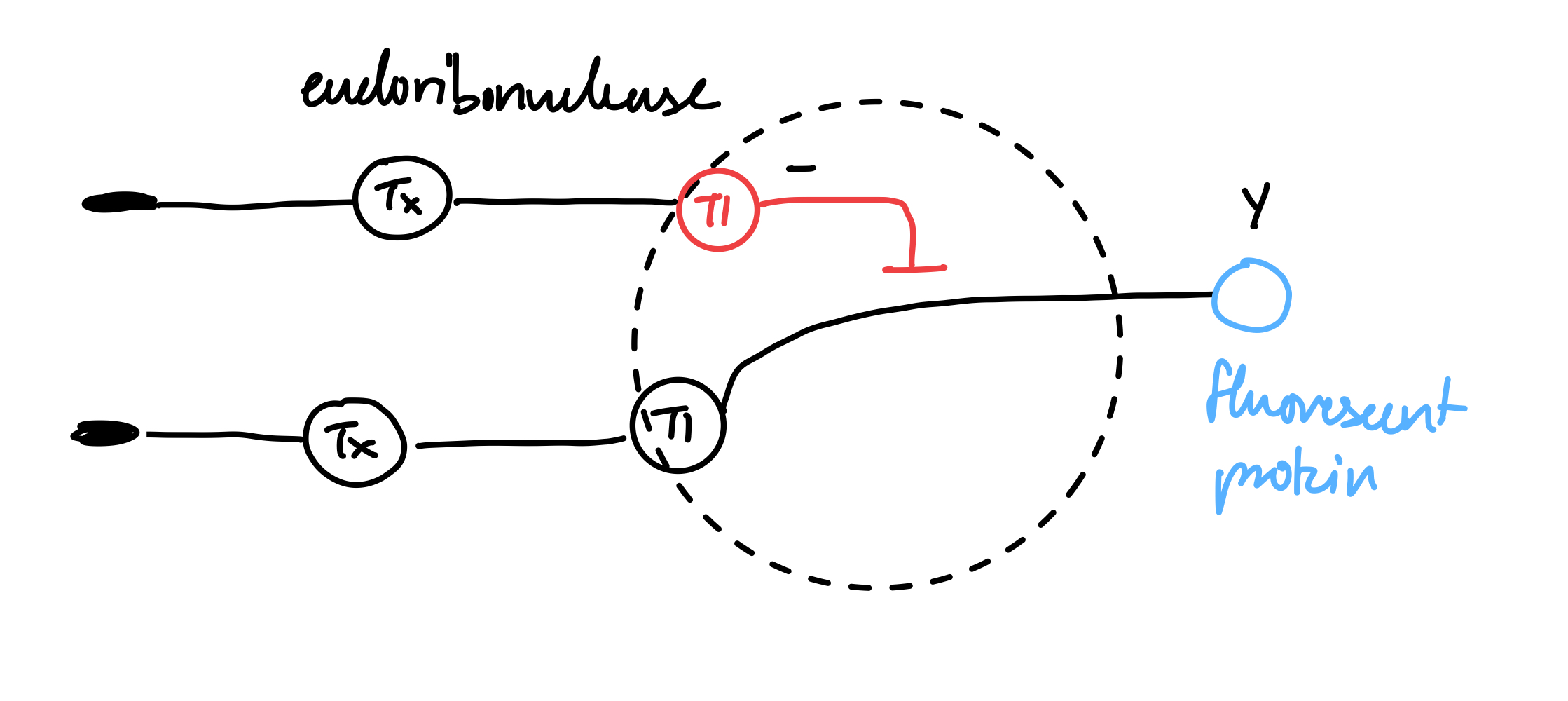
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials have been used as biomedical scaffolds Antinori et al., 2021. An advantage is that this material is cheap, regenerates itself, and is sustainable. A disadvantage is that in general, fungal materials are not well-studied, so more investigation must be done before this is used widely.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
If I were to engineer fungi, I would choose to make it produce something that would help humans, so something like a pharmaceutical. One advantage of doing synthetic biology in fungi instead of bacteria are that fungi are eukaryotic, so proteins that would be found in humans may be able to be produced in fungi. Many human proteins are post-translationally modified in ways that aren’t done in bacteria such as glycosylation. This allows synthetic biologists to produce proteins that might be more directly applicable to humans.
Week 9 HW: Cell Free Systems
General Homework Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
In cell-free protein synthesis, variables such as pH, ion concentration, components, etc. are more adjustable. Additionally, researchers can add or remove components at any time. Two cases where cell-free expression is more beneficial than cell production are when the protein is toxic or when large amounts of protein are needed quickly.
Describe the main components of a cell-free expression system and explain the role of each component.
ATP (energy source), DNA (template for transcription and translation), nucleotides (for mRNA synthesis), ribosomes and tRNAs with amino acids (for translation), RNA polymerase (for translation).
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision regeneration is critical because ATP is required both for RNA synthesis and for protein synthesis (formation of phosphodiester and peptide bonds). One method that could be used is to add phosphoenol pyruvate (the precursor to pyruvate in glycolysis) and pyruvate kinase to produce ATP as the cell-free system runs.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Prokaryotic cell-free expression systems provide very high yield and quick reactions, but eukaryotic cell-free expression system allow for proper folding of more complex proteins. Eukaryotic cell-free systems may also be able to add post-translational modifications to proteins or insert proteins into membranes (microsomes, which are essentially vesicles).
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Poor/weak promotor or RBS. Troubleshoot by changing the promoter/RBS to something that is known to be stronger.
Running out of energy/resources. Troubleshoot by adding more ATP, PEP, and pyruvate kinase or more nucleotides and amino acids.
Protein misfolding. Troubleshoot by lowering the temperature to slow the reaction or add chaperone proteins.
Homework Question from Kate Adamala
1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
My synthetic cell would assemble a phage. The input would be a phage’s genome, and the output would be phage proteins that would (hopefully) assemble into a working phage. Perhaps I would also need to add some bacterial chaperone proteins that I determine are necessary for phage assembly.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, this could be realized by cell-free Tx/Tl alone because no phage proteins are membrane-bound.
c. Could this function be realized by genetically modified natural cell?
Yes, that is essentially phage rebooting, where the phage’s genome is electroporated into its host, and it replicates as usual.
d. Describe the desired outcome of your synthetic cell operation.
A working phage, so essentially a lysate that can be used to infect the host and create more phages.
2. Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
It doesn’t really matter since phage proteins don’t depend on a membrane to fold.
b. What would you encapsulate inside? Enzymes, small molecules.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial is enough because phages infect bacteria and use their machinery.
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
I’m not sure that my synthetic cell would have to communicate with the environment. Perhaps it would still need channels to import macromolecules/nutrients, but everything to make a phage would be contained within the cell.
4. Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Genes: capsid proteins, tail proteins (tape measure, tail tube, tail fibers), terminase, head-to-tail adaptors/stoppers, etc. Lipids: as far as I know, nothing specific is necessary, so phospholipids are enough.
b. How will you measure the function of your system?
I would conduct plaque assays to calculate the titer of the lysate made by the synthetic cells.
Homework Question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
Cloth that doesn’t let oil stains set by releasing an emulsifier when run under water.
How will the idea work, in more detail? Write 3-4 sentences or more.
One problem I envision encountering is that an emulsifier (which detergents are) kill cells, so how could a cell produce an emulsifier? Perhaps it would be possible for a cell to secrete the parts of an emulsifier—the hydrophobic part and the hydrophilic part—separately. Then, due to the extracellular conditions outside the cell being different from those inside the cell, the hydrophobic and hydrophilic part could self-assemble outside of the cell, so it doesn’t kill the cell as it’s being made. This emulsifier would not spread very far in the fabric, so cells from the surrounding area would eventually grow back over the cells that were killed when the emulsifer was released.
What societal challenge or market need will this address?
Clothing sustainability. It is very difficult to get oil/grease stains out of clothing if the stain is not washed immediately, so stained clothing gets thrown out. If it is possible to make it so that the stain is easier to wash out, more clothes would last longer.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
I’ve already addressed this a bit in the second question, but another limitation is making sure the cells have enough resources to continue living and making the emulsifier.
Homework Question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/.
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
During space travel, there are limited resources, as additional weight can inhibit take off of a rocket. Additionally, while in outerspace, astronauts’ immune systems are dampened. As such, they are more susceptible to infections. This means that it would be helpful to have a way to produce medicines, such as common antibiotics, on demand.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Amoxicillin. This is a commonly used antibioitic used to treat ear, sinus, throat, and pneumonia infections.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Ear, sinus, and throat infections along with pneumonia are common infections that may happen in space. It is possible that there are no medications to eradicate the bacterial pathogen causing these infections, so having a way to produce it on demand in space would be helpful.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
This project aims to produce amoxicillin in vitro using a cell-free expression system. I hypothesize that it may be possible to produce the enzymes involed in the synthesis pathway using cell-free expression, then use those enzymes under the right conditions with the right starting materials to synthesize amoxicillin in space.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would use separate cell-free expression systems to synthesize each of the enzymes in the amoxicillin synthesis pathway. I can determine that the enzymes have been synthesized correctly by running something like a western blot using an antibody if it exists. Then, I would combine the reagents necessary for amoxicillin synthesis with the sythesized enzymes. It is possible that some enzymes cannot be in the same tube together, in which case a separation technique would be necessary to isolate the amoxicillin intermediate and transfer it to the tube with the next enzyme. I would work to limit the number of times I’d have to do this, as that would decrease the yield of amoxicillin each time. I would assess whether amoxicillin had been correctly synthesized by the end of the procedure using a chemistry technique such as high-performance liquid chromatography (de Marco et al., 2017).