Week 4 HW: Protein Design Part I
Conceptual Questions
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans eat, the macromolecules the beef are made of are broken down during digestion into monomers. These monomers are common to all life, and humans use them to build human-specific macromolecules.
Why are there only 20 natural amino acids?
These 20 amino acids are what evolution happened to select for. These 20 amino acids happen to be enough to build all the proteins that are necessary for life that has evolved on Earth. Theoretically, there could be more, but in our “system” of life, these 20 are enough.
Can you make other non-natural amino acids? Design some new amino acids.
Theoretically, yes. The R group can be anything, but only some will be functional with the 20 natural amino acids. Below are designs of new amino acids; I based them off of preexisting ones and added/removed atoms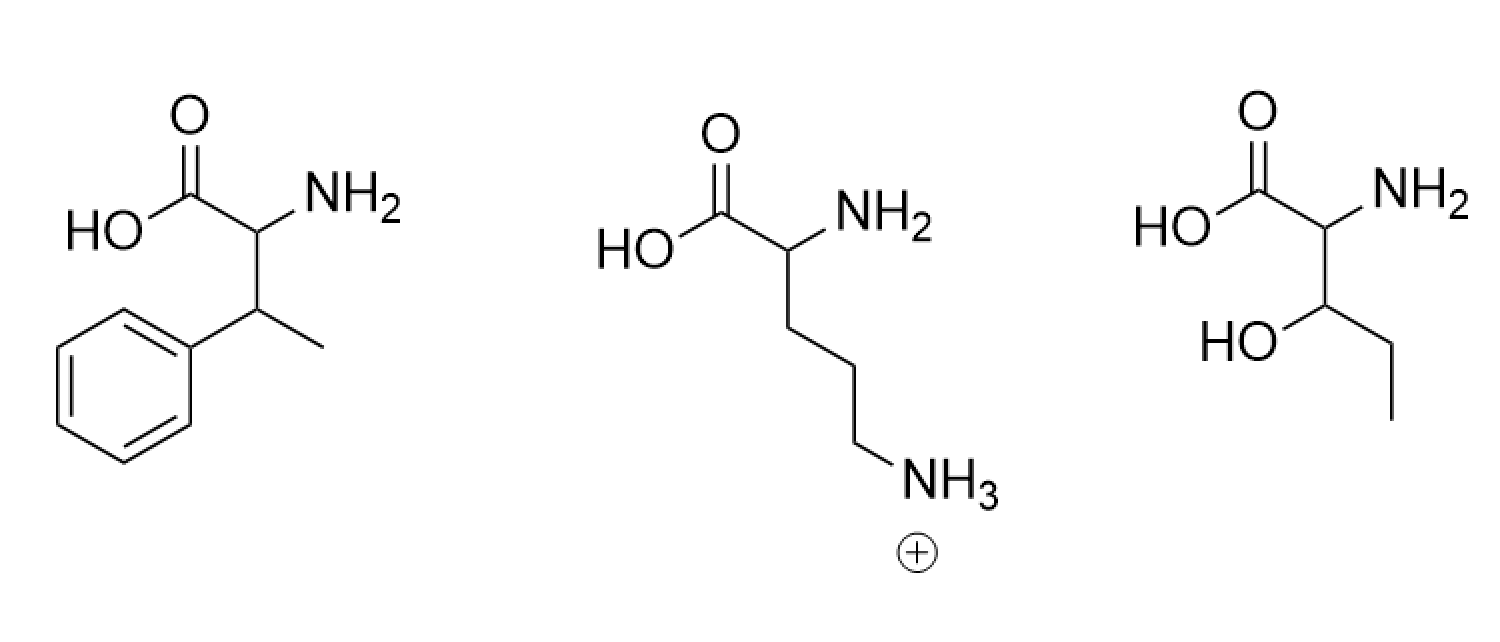
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids formed under natural conditions on Earth. One example is demonstrated by the Miller-Urey experiments where they demonstrated that atmospheric gases could form amino acids spontaneously when energy is put in in the form of lightning.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed because natural amino acids are L and create right-handed α-helixes. This means D-amino acids mirror L-amino acids, so the α-helix would also be mirrored.
Can you discover additional helices in proteins?
Yes, you can discover new α-helices that already exist in proteins using tools/techniques like AlphaFold and X-ray crystallography. Discovering novel types of helices would require changing the chemistry of the amino acids, so discovering pre-existing ones would be unlikely. Creating new types of helices would be interesting, though.
Why are most molecular helices right-handed?
This is because most biological molecules are D-enantiomers (right-handed), so they also create right-handed helices.
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets are formed using hydrogen bonds across backbones. β-sheet backbones “want” to form more hydrogen bonds with another peptide backbone, and can do that by stacking with other β-sheets.
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases form β-sheets because hydrogen bonding in β-sheets is extremely favorable, so if the protein is able to “misfold” into β-sheets, it will do that. β-sheets can be used as materials. In fact, silk is rich in β-sheets, which is why it has so much tensile strength.
Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I selected the antirepressor protein in phage Phi106 bound to a transcriptional activator in Bacillus. In the Saha Lab, we are interested in how Extracellular Prophage-Inducing Particles (EPIPs) are able to induce a prophage, HerberWM, that has a repressor. We hypothesize that the EPIPs may have an antirepressor, but the antirepressors are not in RCSB.
Identify the amino acid sequence of your protein.
Amino acid sequence of transcriptional activator: MSNNTGVKLKKLRKSKKLTLRDLADKLGVTHSYLSKIERGVTNPSLKMINSLAEFFDVDQSYFFTDEKNLDNFTDEELELTFERDLSIENLREKYNLTLGGKEVSDDEIKVMLEVLKAYRESKGGSRSD
Amino acid sequence of antirepressor:
MKKEISLDEYLEKLKQLLENESVGTRAAL
How long is it? What is the most frequent amino acid?
Transcriptional activator: 129 amino acids, most frequent is leucine.
Antirepressor: 29 amino acids, most frequent is leucine.
How many protein sequence homologs are there for your protein?
Transcriptional activator: 250 found in UniProt
Antirepressor: 247 found in UniProt
Does your protein belong to any protein family?
Yes, transcriptional activator and antirepressor.
Identify the structure page of your protein in RCSB
PDB ID: pdb_00009sf2
When was the structure solved? Is it a good quality structure?
Deposited 8/19/2025, released 1/14/2025. The resolution is 1.70 Å, which is small, so it’s a good quality structure.
Are there any other molecules in the solved structure apart from protein?
No.
Does your protein belong to any structure classification family?
No results showed up when I searched the PDB ID in SCOP, so no.
Open the structure of your protein in any 3D molecule visualization software
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon:
Ribbon:

Ball and stick:

Color the protein by secondary structure. Does it have more helices or sheets?
Red: helices
Yellow: sheets
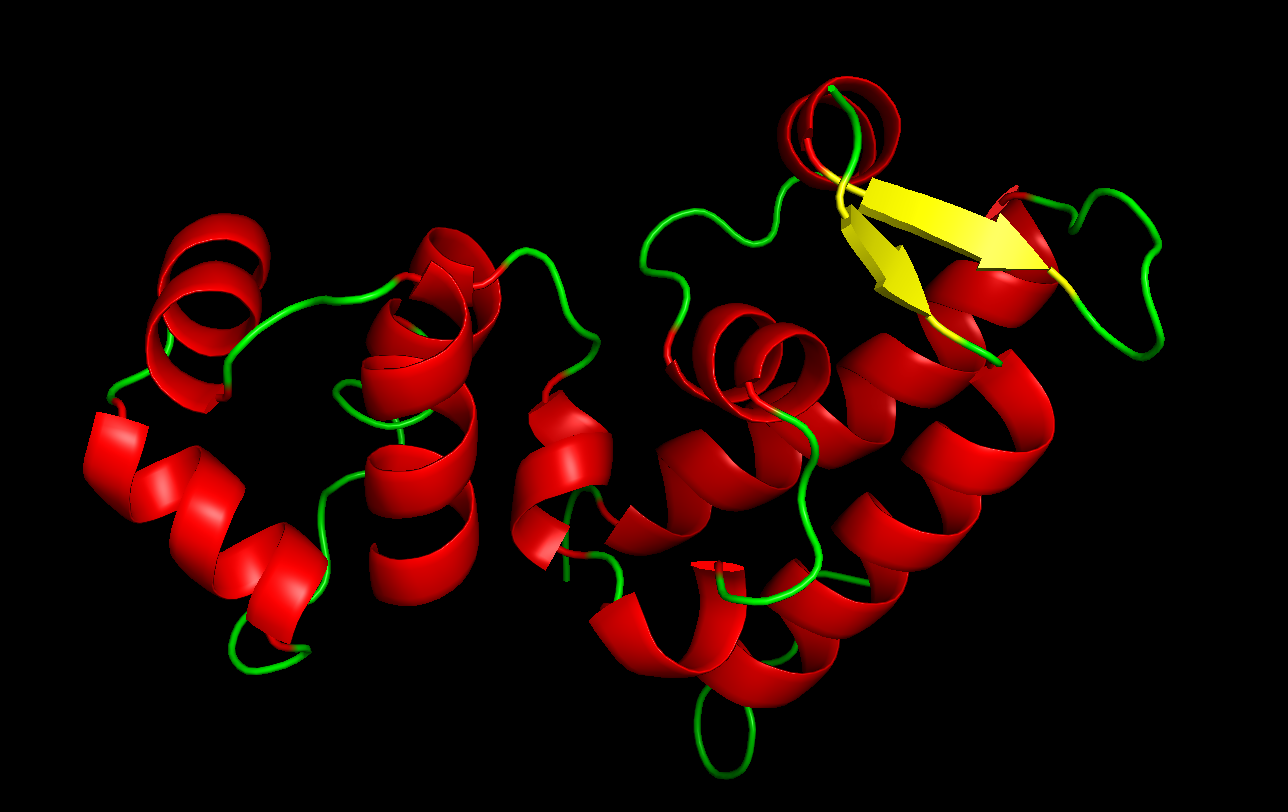
The protein has more helices.
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Used ChimeraX because pyMOL was difficult to work with.
Blue: hydrophobic
White: hydrophilic
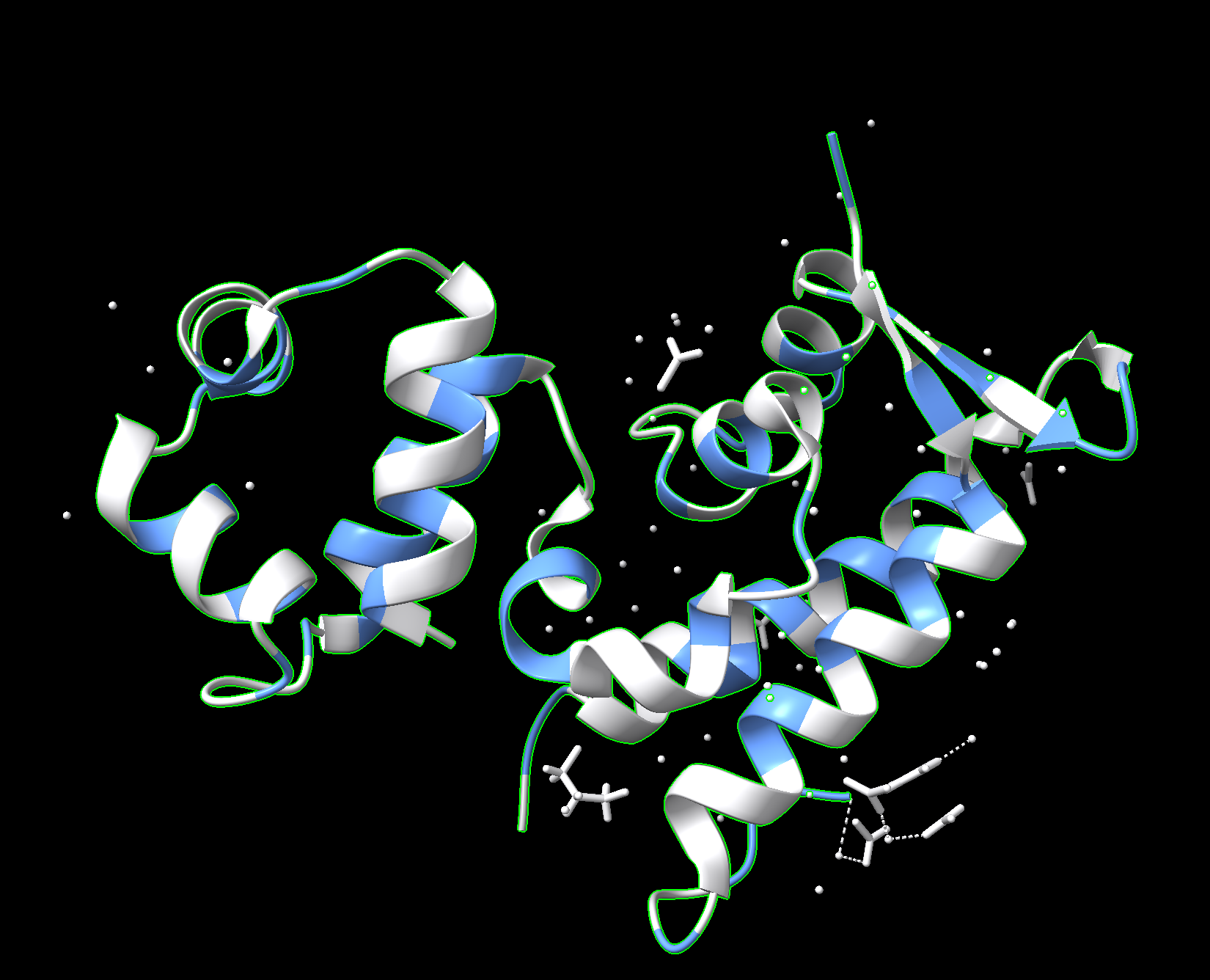
The proteins mostly have hydrophobic residues on the inside and hydrophobic residues on the outside.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
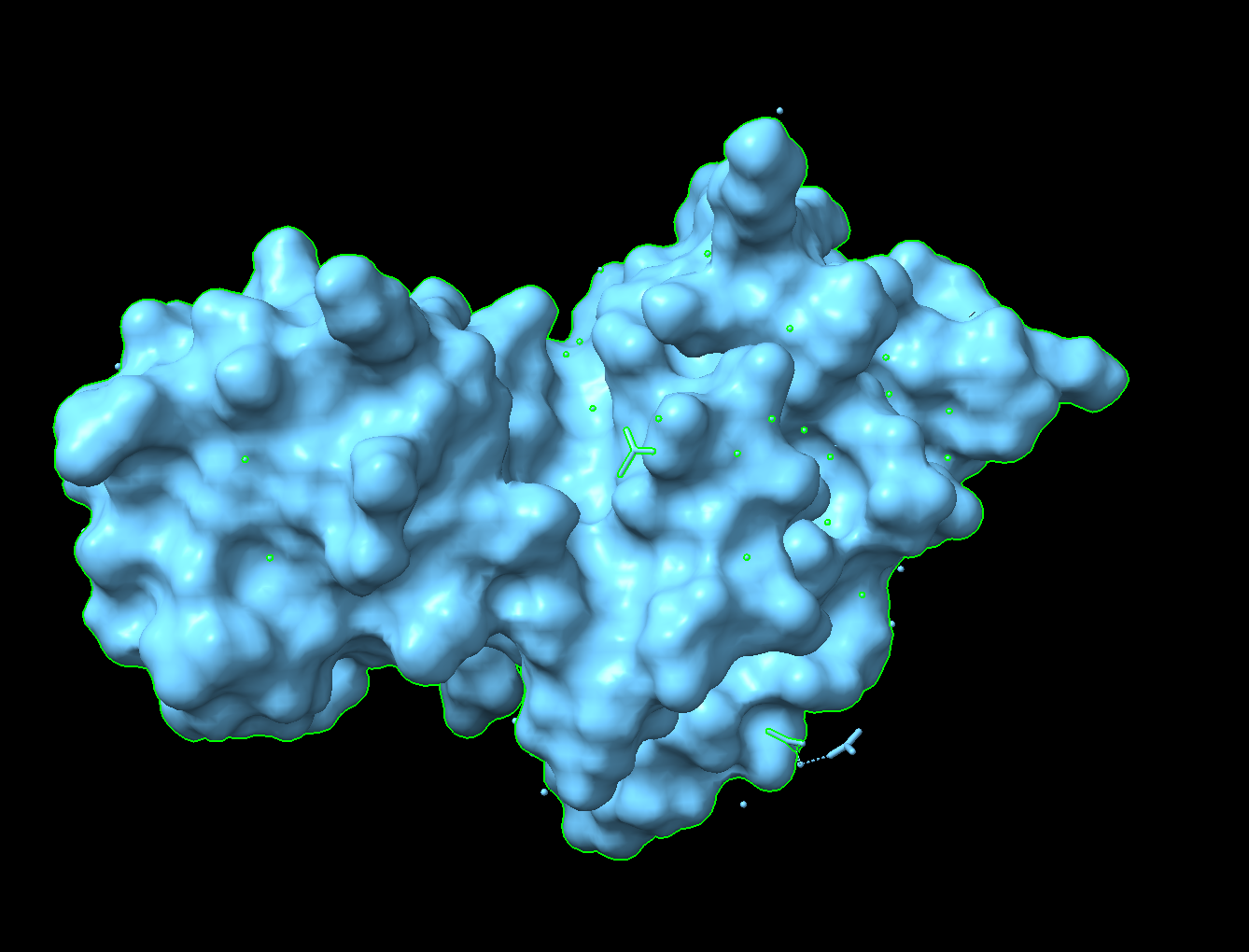
There is a “hole” that could be a binding pocket in the middle of this image.
Using ML-Based Protein Design Tools
Chose to use the same proteins as above (antirepressor in phage Phi106) for the same reasons.
Protein Language Modeling
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
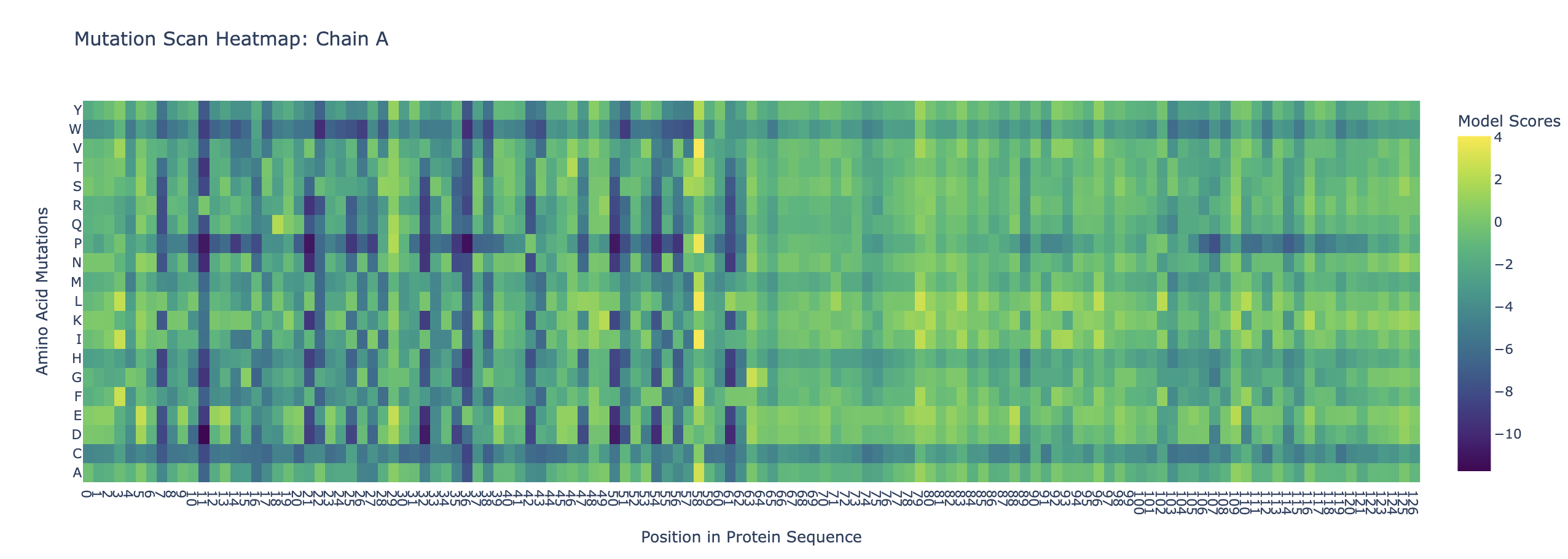
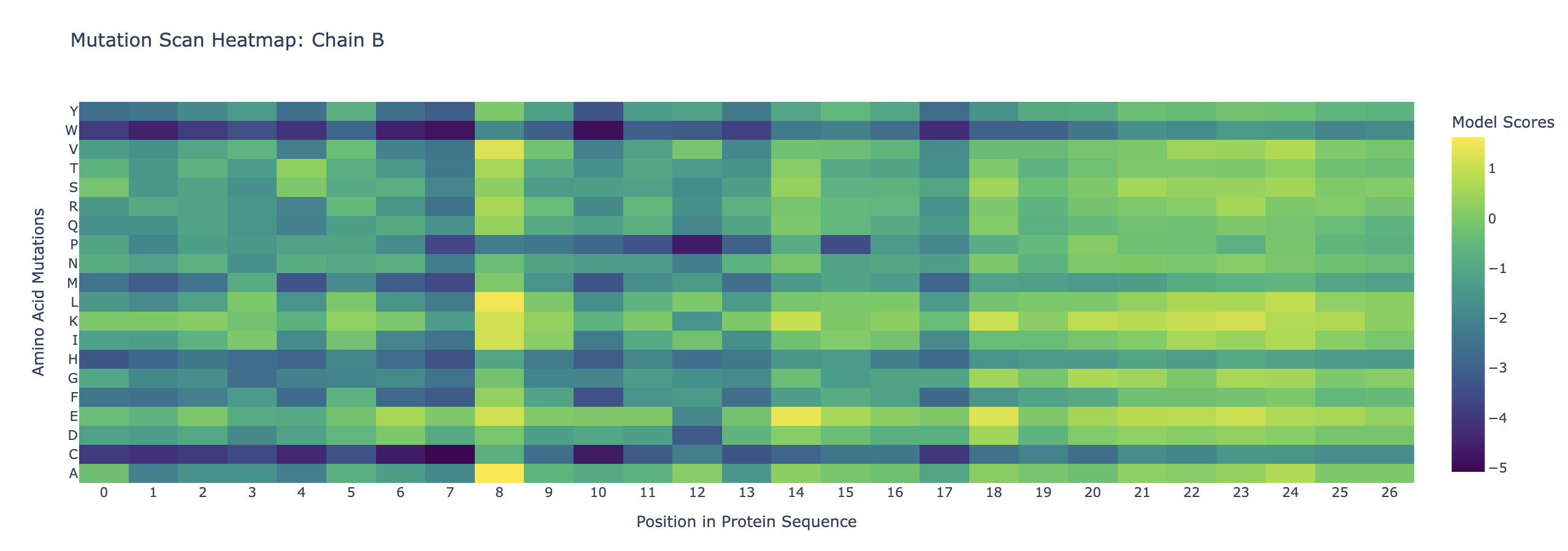
Can you explain any particular pattern?
Amino acids at the beginning of the two sequences appear to be more conserved. This could be because it is a binding site either for a ligand or for the other protein. Also, cystine and tryptophan are less likely to be substituted into the protein throughout the chains. This could be that cystine is unique as it is able to form disulfide bonds, which could disrupt the structure of the proteins. Tryptophan is large and bulky, so substituting it in could also disrupt the proteins’ structure.
Latent Space Analysis
Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, I have found clusters of proteins of similar function (eg. dehydrogenases, phosphatases).
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Modified Colab file with my protein inserted in the figure
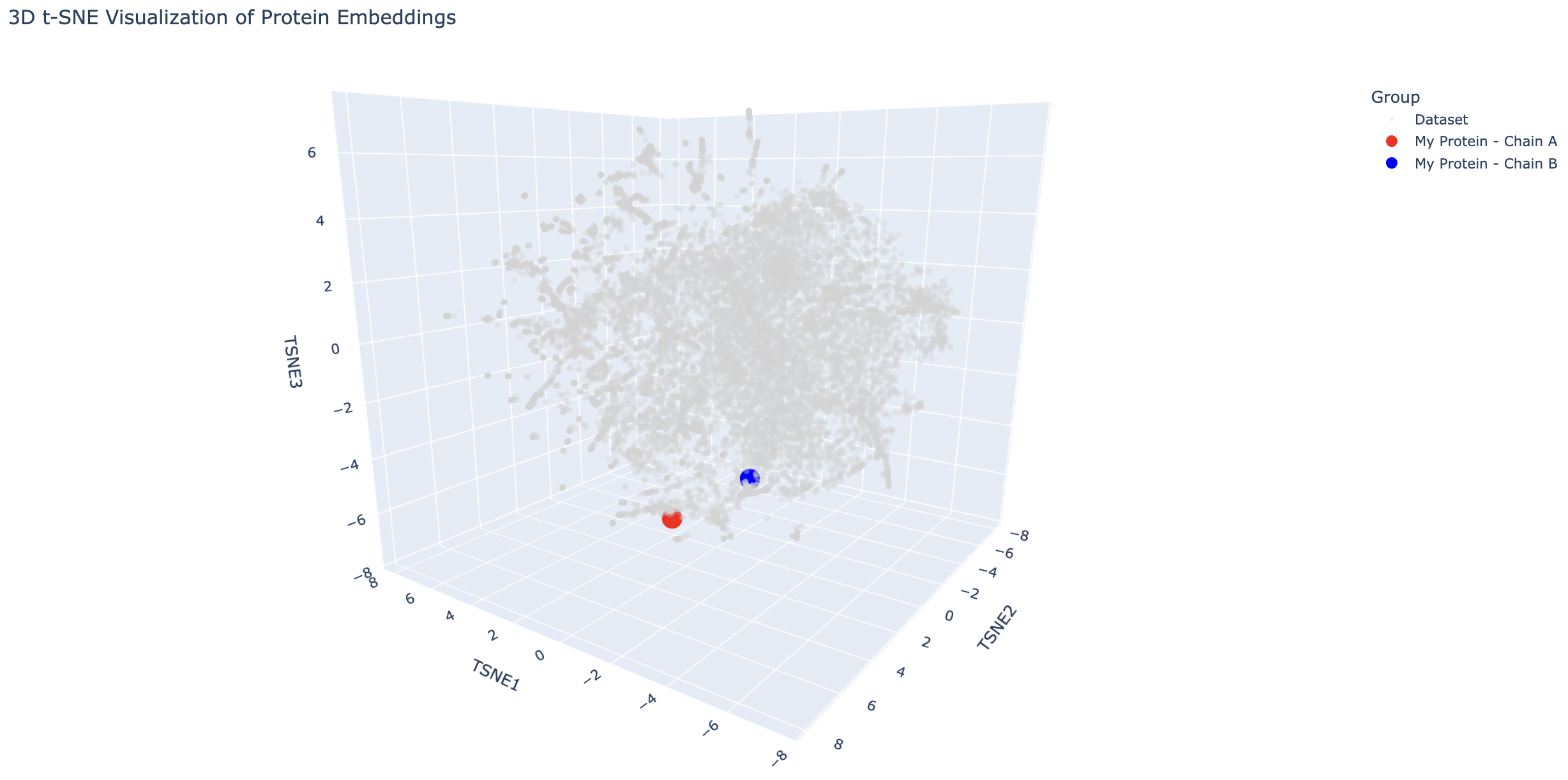
The transcriptional activator is close to proteins such as a transcriptional activator and a hypothetical protein with a DNA-binding domain, which makes sense because these are all DNA-binding proteins. The antirepressor is also near DNA-binding proteins such as transcription factors and a helicase. This is interesting because it suggests the antirepressor may also bind to DNA.
Protein Folding
Only did the transcriptional activator for simplicity and because it has a more interesting fold.
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
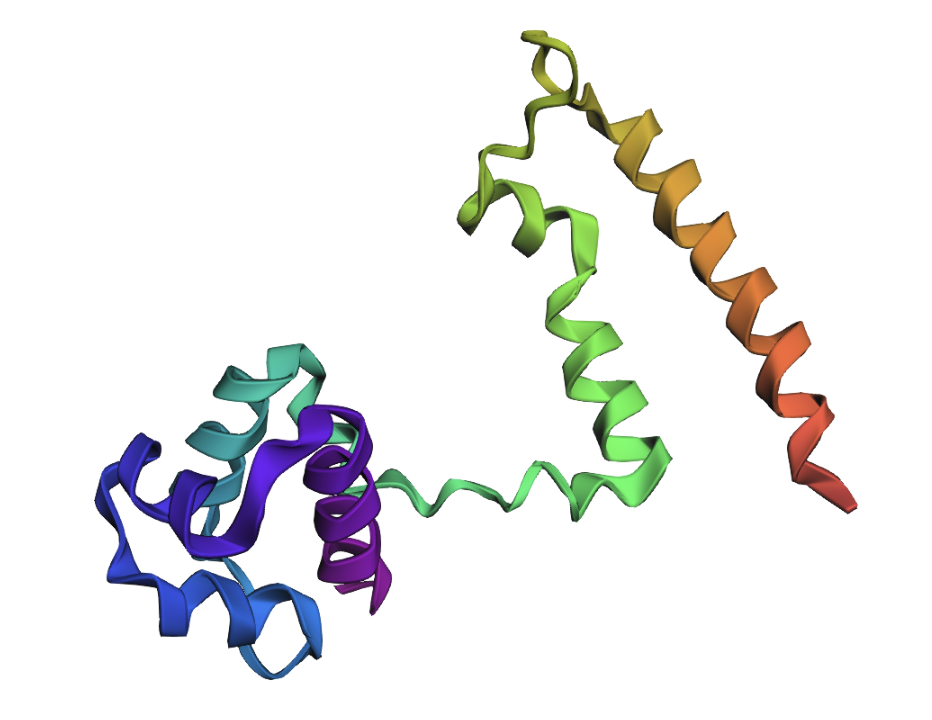 This predicted structure matches the original structure well.
This predicted structure matches the original structure well.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Transcriptional activator G6P substitution:
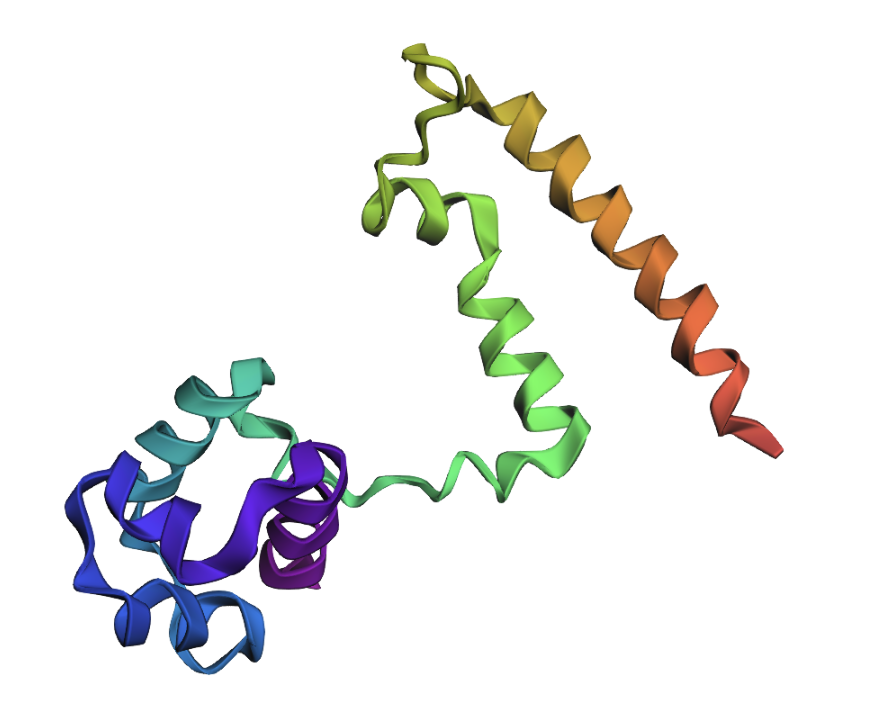
Protein appears more or less the same as the native protein, so this substitution did not have an impact.
Transcriptional activator - moved middle chunk of sequence to end:
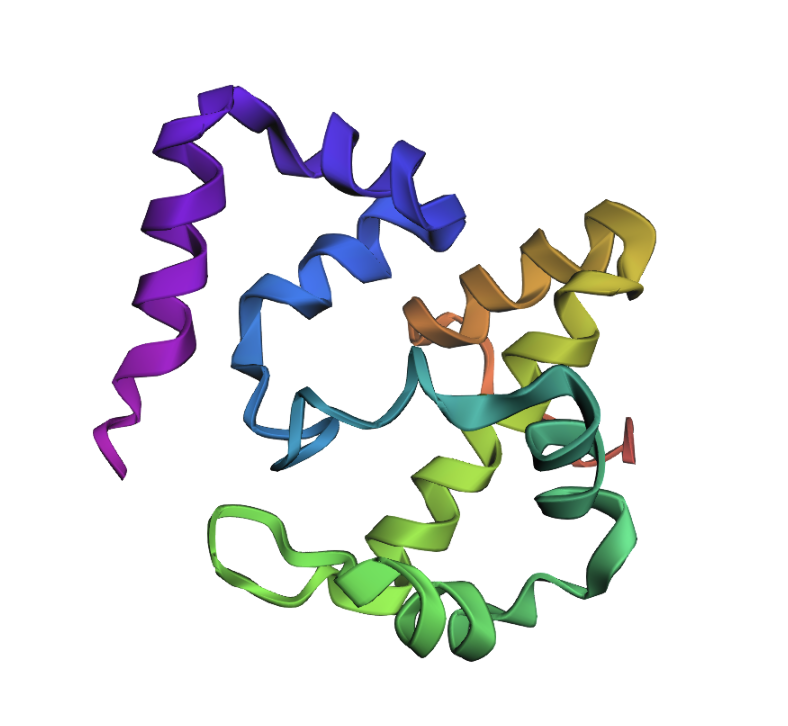
Protein appears different compared to native protein. It appears more scrunched, so translocating a segment of sequence had a large impact.
Protein Generation
I used the example sequence for this because I couldn’t figure out how to upload the PDF file and get the output for both chains for the protein I had been looking at.
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Output:
5MBA, score=1.3372, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020 SLSAAEADLAGKSWAPVFANKNANGLDFLVALFEKFPDSANFFADFKGKSVADIKASPKLRDVSSRIFTRLNEFVNNAANAGKMSAMLSQFAKEHVGFGVGSAQFENVRSMFPGFVASVAAPPAGADAAWTKLFGLIIDALKAAGA
T=0.1, sample=0, score=0.7425, seq_recovery=0.5137 ALSPEEAALLAAAWAPVAADREANGKAFLLTLFAEYPELAEYFPEFKGKSLAEIAASPALPAIAGAVFDALDALVAAAADAAAMAAALAALAAAHVARGIGAAHIERVAAIFPGFVASVAAPPPGADAAWAALLGGVIAALRAAGA
The score for the predicted sequence is much lower than the score for the original one - the predicted sequence matches the fold better. This could be due to evolutionary constraints on the original one. Also, the sequence recovery was 0.5137, meaning ~51% of the amino acids from the original sequence were correctly recovered.
Input this sequence into ESMFold and compare the predicted structure to your original.
Original (backbone in PDB file):
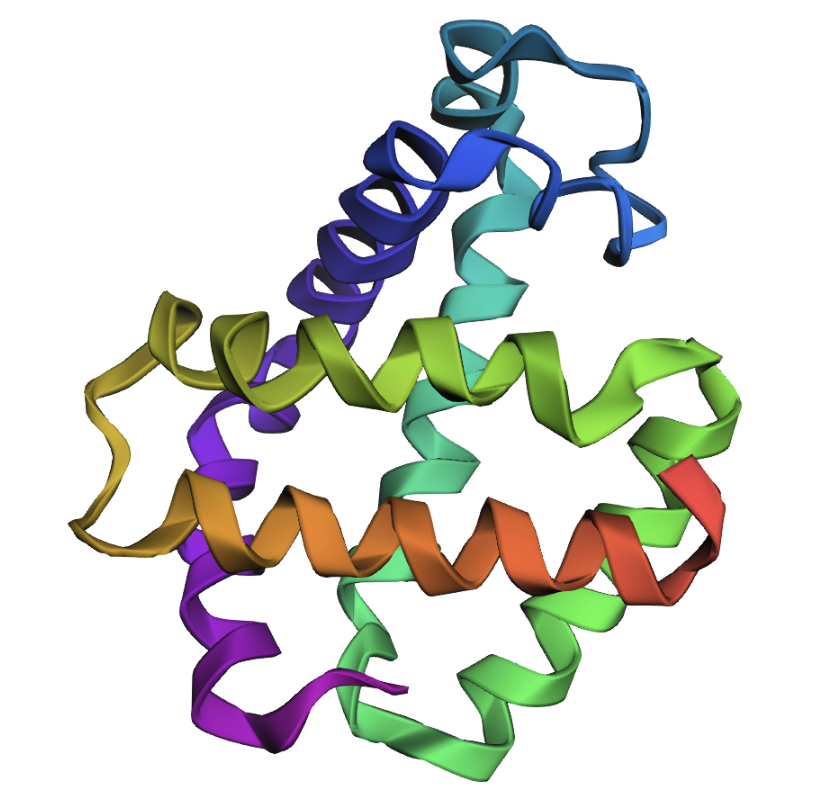
Predicted (based on input sequence and PDB file backbone):
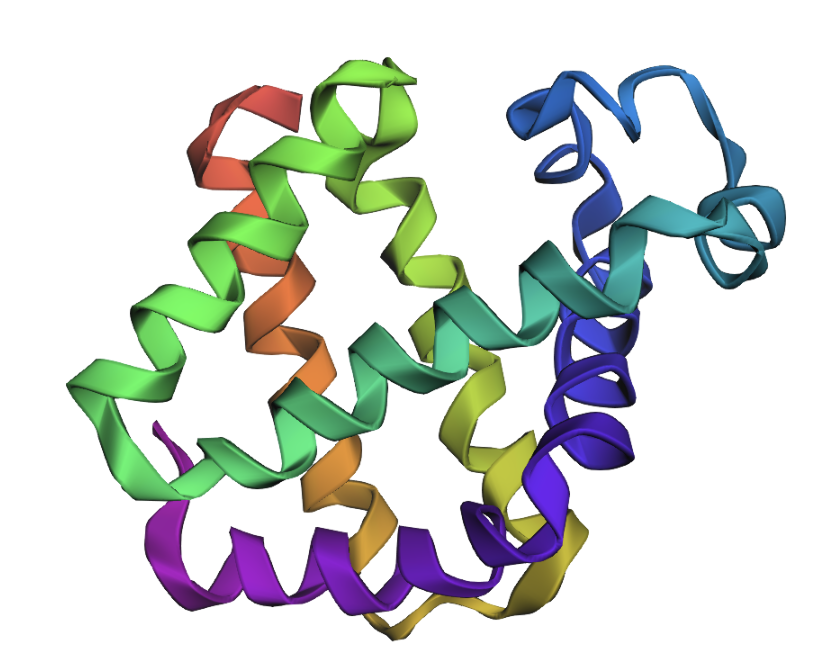
The folds appear very similar. Inverse folding appears to have worked well.
Group Brainstorm on Bacteriophage Engineering
I worked with Grace Hussey for this.
Computational Goal: We will attempt to increase the titer of the L protein expressed by MS2 in the E. coli host.
Overall solution: create a new transcription factor that binds very tightly to the promoter, increasing expression of the L protein
Inverse protein folding using ProteinMPNN
- Use structure of a transcription factor (a PDB file) that binds with very high affinity to a promoter that is highly homologous to the L protein promoter
- Input the structure of the aforementioned transcription factor into ProteinMPNN along with the amino acid sequence of a native transcription factor that binds to the L protein promoter
- This will theoretically generate the sequence of a protein that is structurally similar to a transcription factor with high DNA-binding affinity but is specific to the L protein promoter
Confirm the binding affinity between our designed transcription factor and the MS2 L protein promoter with a ligand-binding AI model
Why will these tools accomplish our computational goal?
- Protein MPNN is an inverse-folding algorithm. The sequence of the L protein promoter and other transcription factors that bind to this promoter are known. However, to generate a transcription factor with higher affinity for this promoter sequence than the native L protein transcription factors, we will model our inverse-folding after an existing transcription factor that behaves in the manner we envision (binds with high affinity) for our engineered transcription factor. As discussed in this week’s lecture, existing AI algorithms are good at designing structures similar to previously-characterized structures but less good at designing different (novel) structures. As such, our inverse-folding approach hinges on the existence of another high affinity transcription factor for a homologous promoter
- Using a ligand-binding AI model will provide a computational indication of the success of our engineering without the need to use an in vitro or in vivo model
Possible pitfalls:
- We are unable to find a transcription factor that binds to a highly homologous promoter sequence with high affinity :(
- Inaccuracies in AI predictions :(
Schematic:
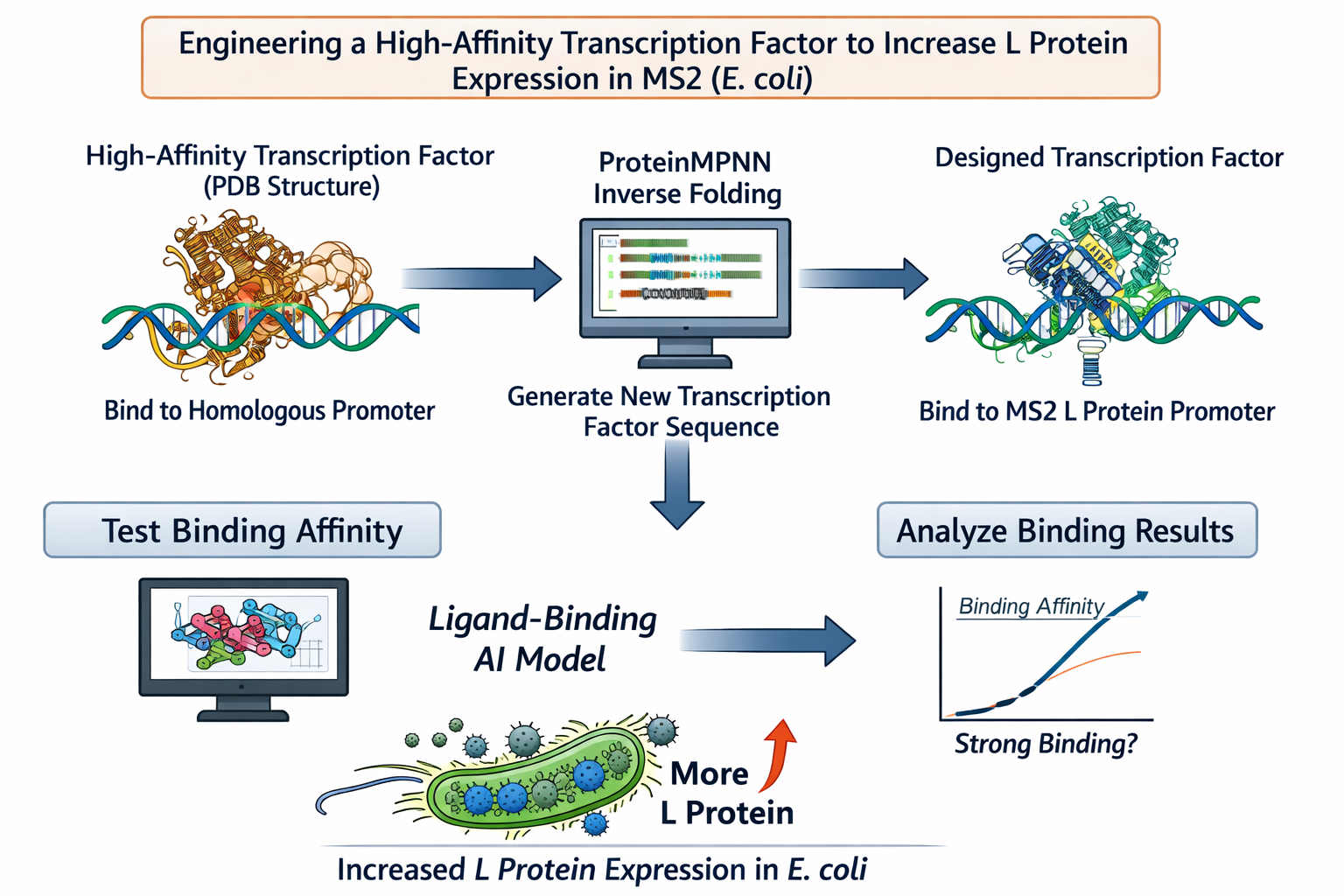 Generated by ChatGPT
Generated by ChatGPT