Week 5 HW: Protein Design Part II
SOD1 Binder Peptide Design (From Pranam)
Generate Binders with PepMLM
SOD1 sequence: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Sequence with A4V mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binders, perplexity score:
- WRVPPAALRHKE, 22.653588
- HRSPPVAAEHWK, 19.512332
- WRYYPVAAAWKK, 11.081843
- WRYYVAALRHGK, 15.691672
- known SOD1 binder: FLYRWLPSRRGG, 20.635231
Evaluate Binders with AlphaFold3
| Binder | ipTM Score | Binding Location |
|---|---|---|
| WRVPPAALRHKE | 0.39 | near β-barrel and globular part |
| HRSPPVAAEHWK | 0.26 | near β-barrel, across the seam and onto globular part |
| WRYYPVAAAWKK | 0.28 | near β-barrel, across the seam |
| WRYYVAALRHGK | 0.30 | near β-barrel, across the seam |
| known: FLYRWLPSRRGG | 20.635231 | 0.31 |
The ipTM score is highest for the first binder, but none of the binders bind in a similar location compared to the known binder. All of the predicted binders bound a similar part of the protein that was very different from where the known binder is predicted to bind.
Evaluate Properties of Generated Peptides in the PeptiVerse
| Binder | Binding Affinity (pKd/pKi) | Solubility Probability | Hemolysis Probability | Net Charge | Molecular Weight (Da) |
|---|---|---|---|---|---|
| WRVPPAALRHKE | Weak, 5.114 | Soluble, 1 | Non-hemolytic, 0.020 | 1.85 | 1450.7 |
| HRSPPVAAEHWK | Weak, 4.576 | Soluble, 1 | Non-hemolytic, 0.014 | 0.94 | 1414.6 |
| WRYYPVAAAWKK | Weak, 6.124 | Soluble, 1 | Non-hemolytic, 0.021 | 2.76 | 1538.8 |
| WRYYVAALRHGK | Weak, 6.474 | Soluble, 1 | Non-hemolytic, 0.023 | 2.84 | 1519.8 |
| known: FLYRWLPSRRGG | Weak, 5.968 | Soluble, 1 | Non-hemolytic, 0.047 | 2.76 | 1507.7 |
Binding affinity is weak for all peptides. There does not seem to be a strong correlation between ipTM score and binding affinity. From PeptiVerse results, it appears that all peptides are soluble and non-hemolytic. The ones with stronger charges have a higher binding affinity.
I’m choosing to continue with the 4th generated peptide, WRYYVAALRHGK, because it has the highest binding affinity from PeptiVerse and it has a ipTM score similar to the known peptide from AlphaFold.
Generate Optimized Peptides with moPPIt
Motif positions used: 95-100, 85-90
Peptide generated: PKHCLQRLLSKH
- ipTM score: 0.45
- Binding location: end of the β-barrel that is closer to the termini
- Binding affinity (pKd/pKi): weak, 6.408
- Solubility probability: soluble, 1
- Hemolysis probability: non-hemolytic, 0.057
- Net charge: 3.12
- Molecular weight (Da): 1459.8
This peptide has one of the strongest binding affinities according to PeptiVerse along with the highest ipTM score from AlphaFold. Like the other peptides, it is soluble and non-hemolytic. Consistent with previous observations, it has the strongest charge, which could be why it binds mutated SOD1 relatively strongly.
Final Project: L-Protein Mutants
L-protein sequence: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Last 35 residues are the transmembrane domain
DnaJ sequence: MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Mutagenesis
L-protein mutation likeihood heatmap:
Experimental data of L-protein mutations
For the most part, experiments where lysis still occured correlates to a yellowish green point on the heatmap, suggesting that the heatmap is somewhat reliable.
Proposed mutations:
| Mutation | Domain | Reasoning | L-protein Multimer |
|---|---|---|---|
| R19S | Soluble | This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative. |  Multimer forms that appears circular and pore-like. However, the ipTM score is 0.14, which is low, suggesting low confidence in this assembly of a multimer.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.14, which is low, suggesting low confidence in this assembly of a multimer. |
| R31I | Soluble | This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative. |  Multimer forms that appears circular and pore-like. However, the ipTM score is 0.16, which is low, suggesting low confidence in this assembly of a multimer.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.16, which is low, suggesting low confidence in this assembly of a multimer. |
| H24L | Soluble | This amino acid is shown to change in BLASTp results, and the heatmap shows a very positive score for this mutation. There is no experimental data, so it is possible this mutation would result in a functional protein, even though other amino acid substitutions at this position have not, especially since histidine and lysine are both positively charged amino acids. | 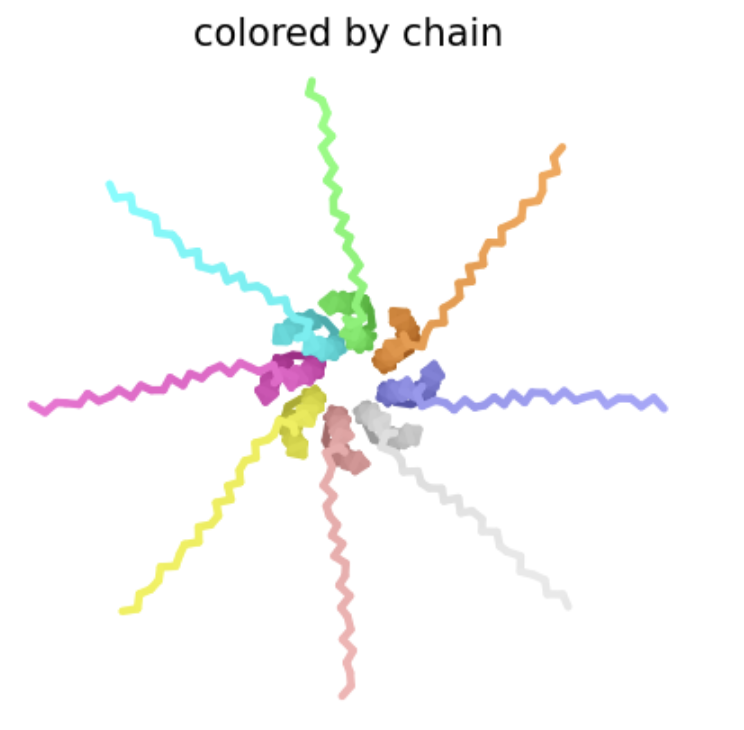 Multimer forms that appears circular and pore-like. However, the ipTM score is 0.15, which is low, suggesting low confidence in this assembly of a multimer.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.15, which is low, suggesting low confidence in this assembly of a multimer. |
| T69S | Transmembrane | This amino acid is shown to change in BLASTp results, and the heatmap shows a score near 0, so it’s not strongly negative. However, experimental data suggests this protein does not result in lysis. This makes sense because if the transmembrane domain is mutated, the L-protein will not be able to oligomerize in the membrane and create pores. | 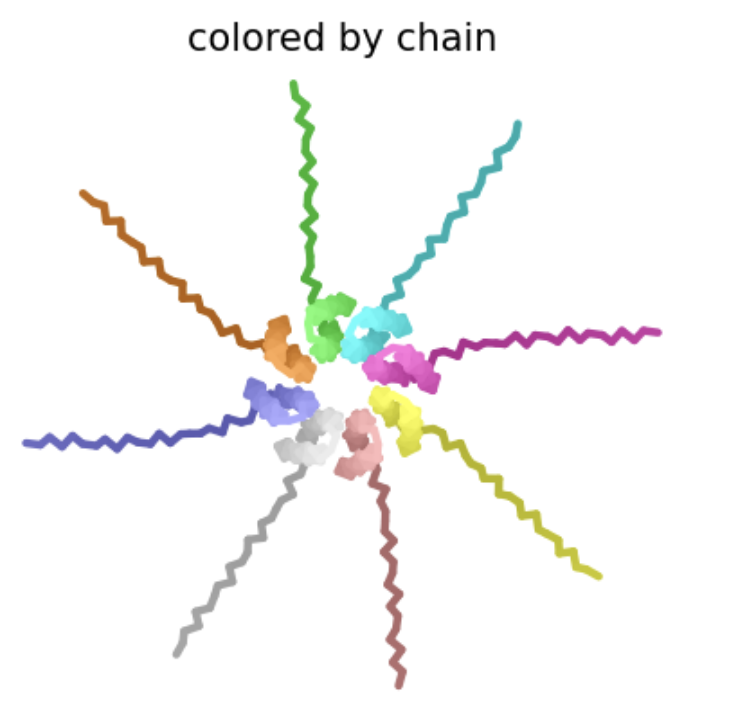 Multimer forms that appears circular and pore-like. However, the ipTM score is 0.15, which is low, suggesting low confidence in this assembly of a multimer.
Multimer forms that appears circular and pore-like. However, the ipTM score is 0.15, which is low, suggesting low confidence in this assembly of a multimer. |
| N53T | Transmembrane | This amino acid is shown to change in BLASTp results, and the heatmap shows a very positive score for this mutation. There is no experimental data, so it is possible this mutation would result in a functional protein, even though other amino acid substitutions at this position have not. Also, this mutation is the only polar-to-polar mutation that was not experimentally tested. | 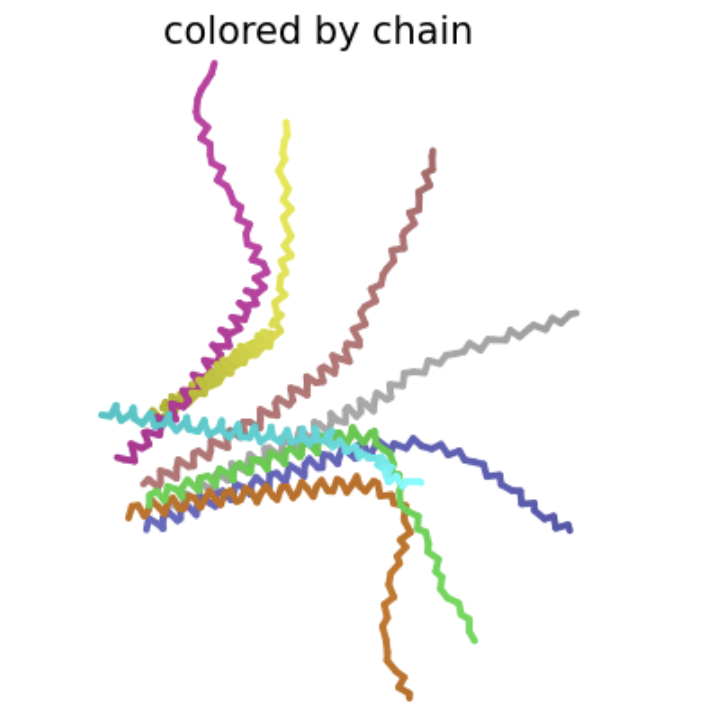 Multimer forms but appears asymmetrial. ipTM score is 0.16, which is low, suggesting low confidence in this assembly of a multimer.
Multimer forms but appears asymmetrial. ipTM score is 0.16, which is low, suggesting low confidence in this assembly of a multimer. |
Mutagenesis using Af2-Multimer
Proposed mutations:
| Mutation | Domain | Reasoning | L-protein-DnaJ Multimer |
|---|---|---|---|
| R19S | Soluble | This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative. | 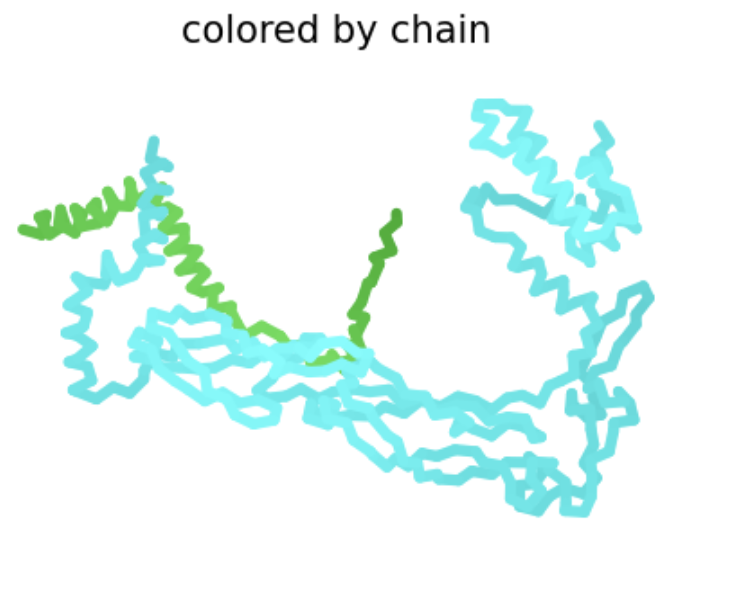 The L-protein and DnaJ appear to make contact. ipTM is 0.17, which is low, suggesting low confidence in this interaction.
The L-protein and DnaJ appear to make contact. ipTM is 0.17, which is low, suggesting low confidence in this interaction. |
| R31I | Soluble | This amino acid is shown to change in BLASTp results, experimental evidence suggests this results in a lytic protein. The heatmap score does not appear to be strongly negative. | 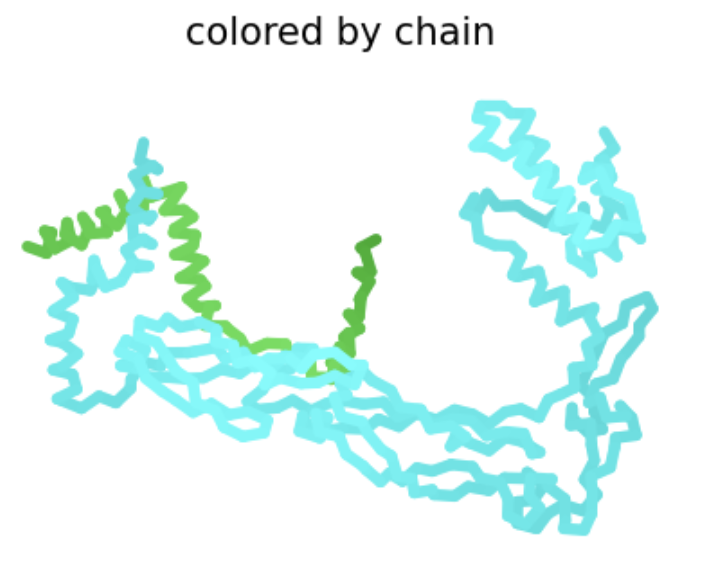 The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction.
The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction. |
| H24L | Soluble | This amino acid is shown to change in BLASTp results, and the heatmap shows a very positive score for this mutation. There is no experimental data, so it is possible this mutation would result in a functional protein, even though other amino acid substitutions at this position have not, especially since histidine and lysine are both positively charged amino acids. | 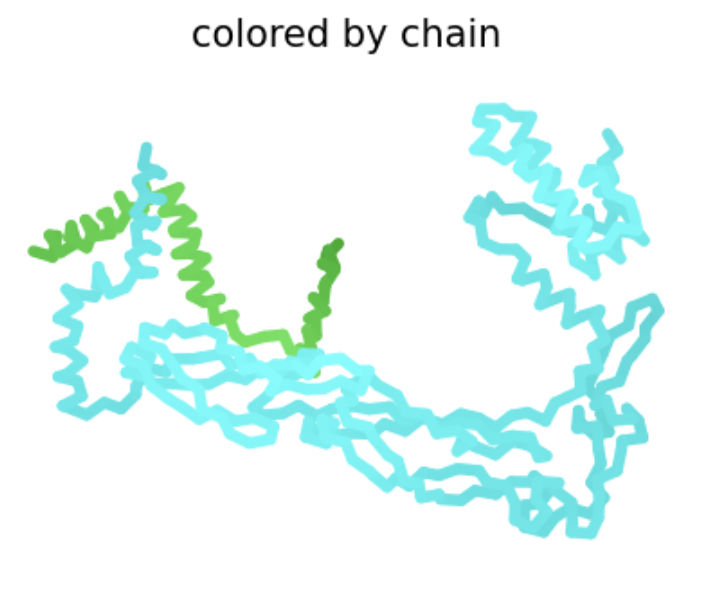 The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction.
The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction. |
| F5S | Soluble | This particular substitution exists in BLASTp results, and the heatmap shows a very positive score. There is no experimental data, so it is possible this mutation would result in a functional protein. | 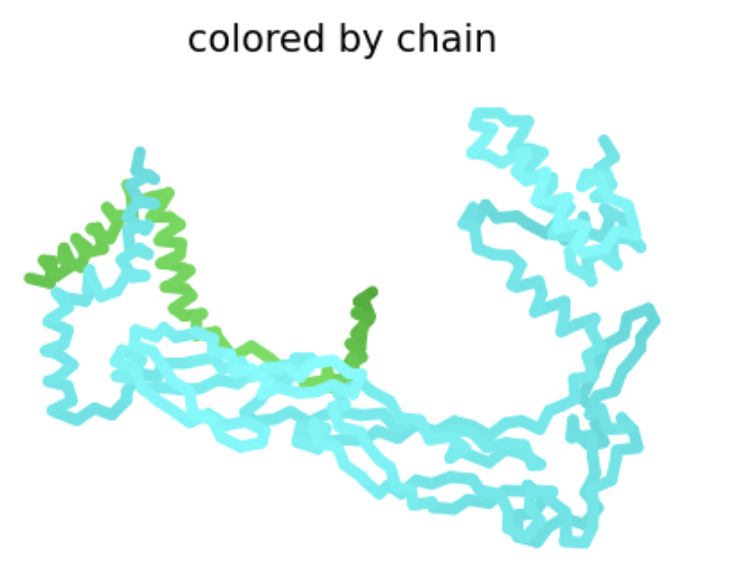 The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction.
The L-protein and DnaJ appear to make contact. ipTM is 0.16, which is low, suggesting low confidence in this interaction. |
| A14S | Soluble | This amino acid is shown to change in BLASTp results, and the heat map shows a positive score. There is no experimental data, so it is possible this mutation would result in a functional protein. | 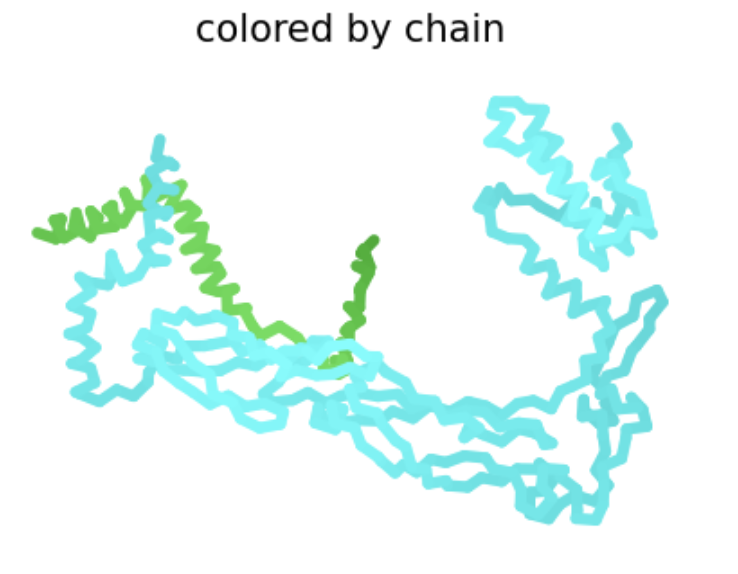 The L-protein and DnaJ appear to make contact ipTM is 0.16, which is low, suggesting low confidence in this interaction.
The L-protein and DnaJ appear to make contact ipTM is 0.16, which is low, suggesting low confidence in this interaction. |