Week 4 HW: Protein Design Part 1
Part A: Conceptual Questions
How many molecules of amino acids do you take with a piece of 500g of meat? (avg amino acid ~100 Daltons)
Since I am a visual learner, I needed an analogy to try to grasp Daltons, grams, and moles. I imagine each amino acid as a finished LEGO model, and each tiny brick is a Dalton. When I weigh all the models together in a cupboard, I have 500 grams. I count how many moles by dividing the total mass by the mass of one model (-100 Daltons). Then, multiplying by Avogadro’s number, I see how many individual models I have in total. In scientific terms, I compute the number of moles by dividing 500 grams by 100 grams per mole. Then, I multiply by Avogadro’s number,(According to Google search Avogadro’s constant is the number of particles, like atoms or molecules, in one mole of a substance, equal to approximately 6.022 times 10 to the 23) 6.022 times 10 to the 23, and that yields approximately 3.0 times 10 to the 24 molecules of amino acid.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We don’t become a cow or a fish, because we’re only using parts of the cow and fish to continue building on what already exists, which is our human body. In other words, digestion breaks down the proteins into amino acids, and then our body uses its own genetic instructions to reassemble those pieces into human proteins, ensuring we stay uniquely ourselves.
Why are there only 20 natural amino acids?
There are only 20 natural amino acids because, although their combinations can form infinite possibilities, evolution only needed these 20 to create all the proteins we rely on. Their chemical properties allow for immense diversity in protein structure, and this set is perfectly suited to the way DNA encodes and guides their assembly, giving us the versatility we need without adding more complexity.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed from simple chemicals dissolved in warm ocean water on early Earth, where energy sources like heat or lightning sparked chemical reactions. In a way, you can think of a modern dam as a kind of micro-ecosystem—just as water and energy flow through a dam, creating pockets of life, early oceans created the right conditions for these amino acids to form, eventually leading to the first building blocks of life.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Depending on the amino acids you use, a typical alpha helix forms a right-handed spiral when built from L-amino acids. However, if you use D-amino acids, the entire helix reverses its handedness and forms a left-handed spiral. It is important to note that not all amino acids form alpha helices; some sequences prefer other structures like beta sheets.So, the handedness and structure depend on both the amino acid type and the overall sequence, which determines how the chain folds and stabilizes.
Can you discover additional helices in proteins?
Because proteins have long chains of different amino acids, they can fold in all sorts of ways, and that means new helices or other patterns can be identified within them, especially with advanced tools like structural prediction or experimental techniques.
Why are most molecular helices right-handed?
The right-handedness is actually favored in DNA due to the natural chirality of its sugar backbone and the way it interacts with water and other molecules. So, that structural preference is built right into how the backbone forms. Keratin and hair curl were helpful for me to think about.
Why do β-sheets tend to aggregate?
So, beta sheets tend to aggregate because, like silk fibroin, they form straight, aligned strands that stack side by side. In silk, these parallel sheets create strong, stable fibers, but in proteins, this same alignment lets the sheets stack excessively, exposing those hydrogen bonds and promoting aggregation. So, just like silk’s strength comes from its sheet alignment, aggregation in proteins happens when these sheets stack and bind too readily.
What is the driving force for β-sheet aggregation? Why do many amyloid diseases form β-sheets?
Because these beta sheets stack so easily, they misfold and form these stable aggregates. In diseases like Alzheimer’s or Parkinson’s, these aggregated beta sheets build up, disrupting normal cell function and triggering the disease process. Tau tangles are a classic example of beta sheet misfolding driving disease.
Can you use amyloid β-sheets as materials?
Since silk fibroin is based on beta sheets and is already a natural, strong material, researchers have been exploring ways to harness amyloid beta sheets similarly. Amyloid structures are extremely stable, so with careful design, scientists are looking at them as potential biomaterials
Part B: Protein Analysis and Visualization**
Briefly describe the protein you selected and why you selected it.
sp|P60520|GBRL2_HUMAN MKWMFKEDHSLEHRCVESAKIRAKYPDRVPVIVEKVSGSQIVDIDKRKYLVPSDITVAQFMWIIRKRIQLPSEKAIFLFVDKTVPQSSLTMGQLYEKEKDEDGFLYVAYSGENTFGF
Identify the amino acid sequence of your protein.
Length 117 amino acids. Most frequent amino acid (image still needed from Colab output)
How long is it? What is the most frequent amino acid?
Homologs 250 homologs found via UniProt BLAST. Top matches from rat, mouse, human, and bovine this indicating strong conservation across mammals reflecting the fundamental evolutionary importance of this protein.
How many protein sequence homologs are there? (Use UniProt BLAST)
Protein family GABARAPL2 belongs to the ATG8 family, part of the broader GABARAP subfamily of autophagy-related proteins.
Does your protein belong to any protein family?
RCSB structure page PDB entry 7LK3. Crystal structure of untwinned human GABARAPL2.
Identify the structure page of your protein in RCSB.
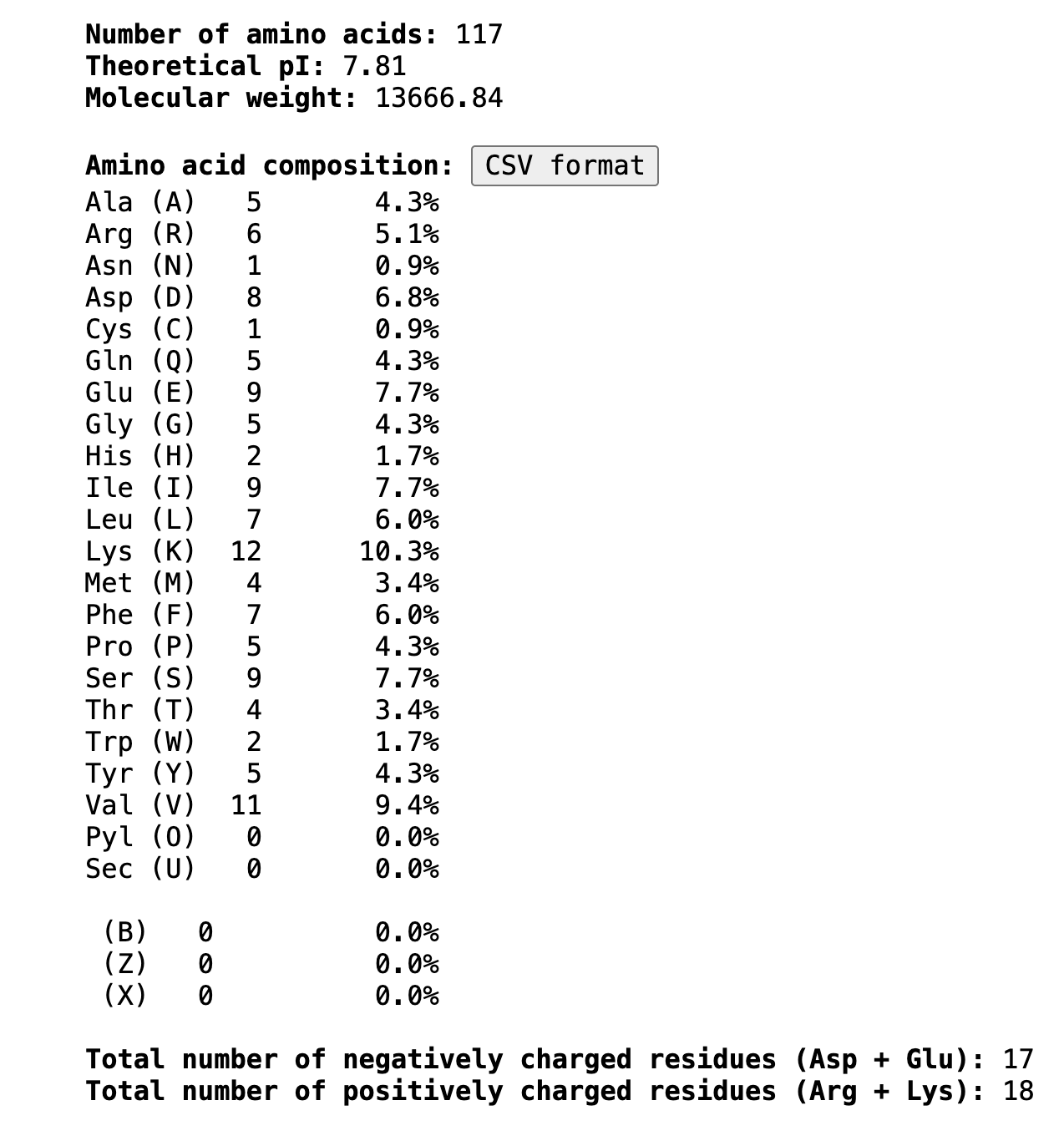
The most frequent amino acid is Lysine (K), appearing 12 times (10.3% of the sequence).
- When was the structure solved? Is it good quality? (Resolution: smaller = better, aim < 2.70 Å)
Structure quality Deposited February 1, 2021, released May 12, 2021. Resolution of 1.90 Å — excellent quality, well below the 2.70 Å threshold.
Are there any other molecules in the solved structure apart from protein?
Other molecules Yes. 1,2-Ethanediol (EDO) is present as a ligand.
Does your protein belong to any structure classification family?
GABARAPL2 belongs to the ubiquitin-like superfamily under the beta-grasp fold in SCOP classification. Like other ATG8 proteins, GABARAPL2 is comprised of an N-terminal helical extension preceding four beta-sheets in a ubiquitin-like beta-grasp fold.
Open the structure in 3D visualization software (PyMol):
- Visualize as “cartoon”, “ribbon”, and “ball and stick”
- Color by secondary structure — more helices or sheets?
- Color by residue type — hydrophobic vs hydrophilic distribution?
- Visualize the surface — any binding pockets?
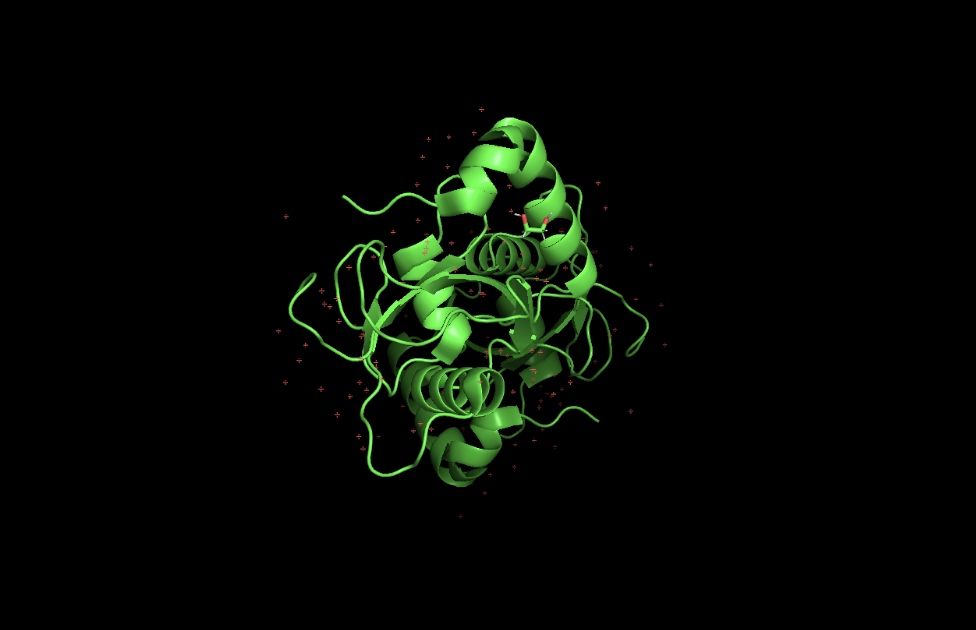
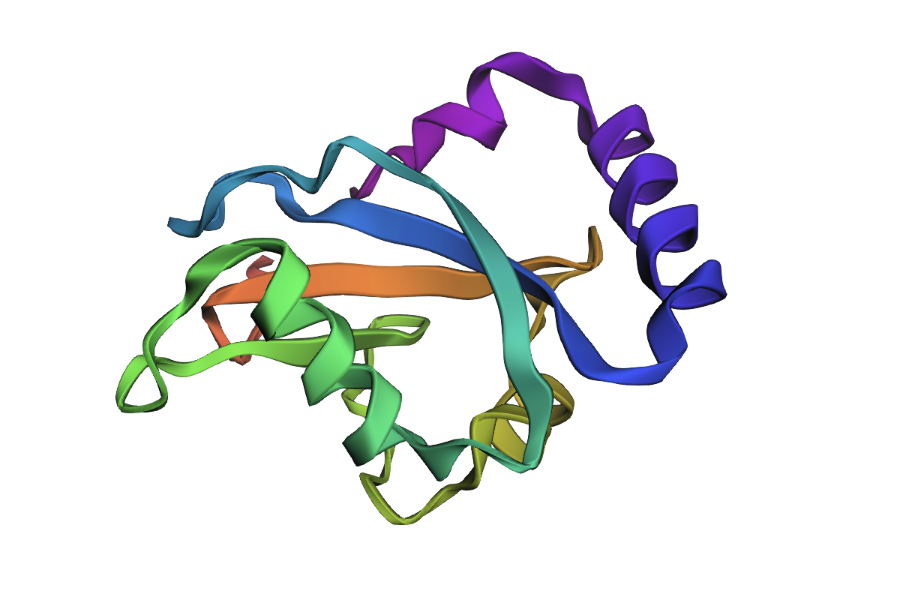
 When colored by secondary structure, GABARAPL2 shows a clear dominance of red (helices) over yellow (beta sheets). Green loops connect these elements throughout the structure.
When colored by secondary structure, GABARAPL2 shows a clear dominance of red (helices) over yellow (beta sheets). Green loops connect these elements throughout the structure.

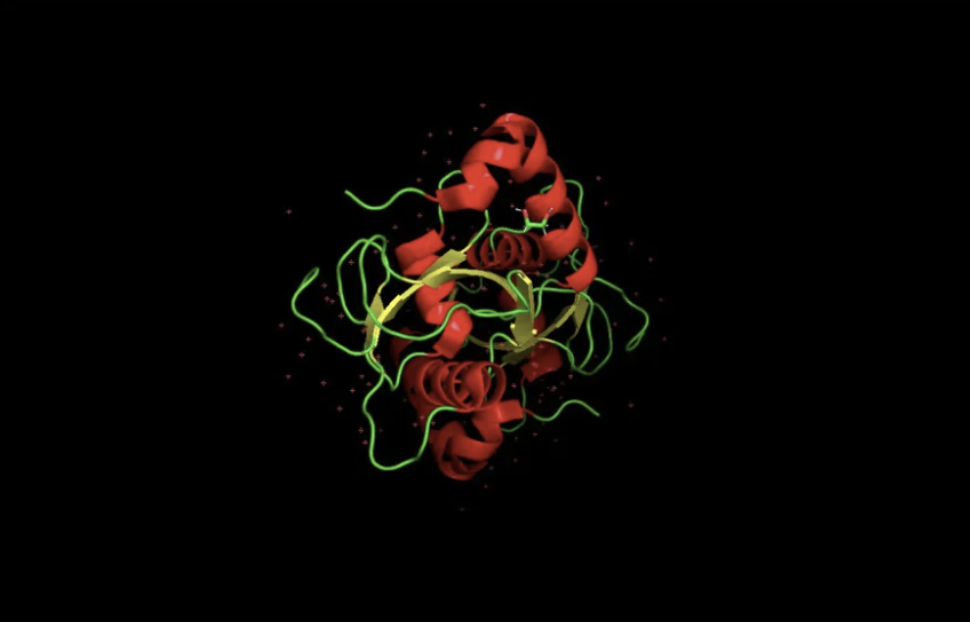
Hydrophobic residues (orange) concentrate in the protein core, while blue dominates the outer surface. This showcases hydrophobic residues being hidden in the middle away from the aqueous environment.
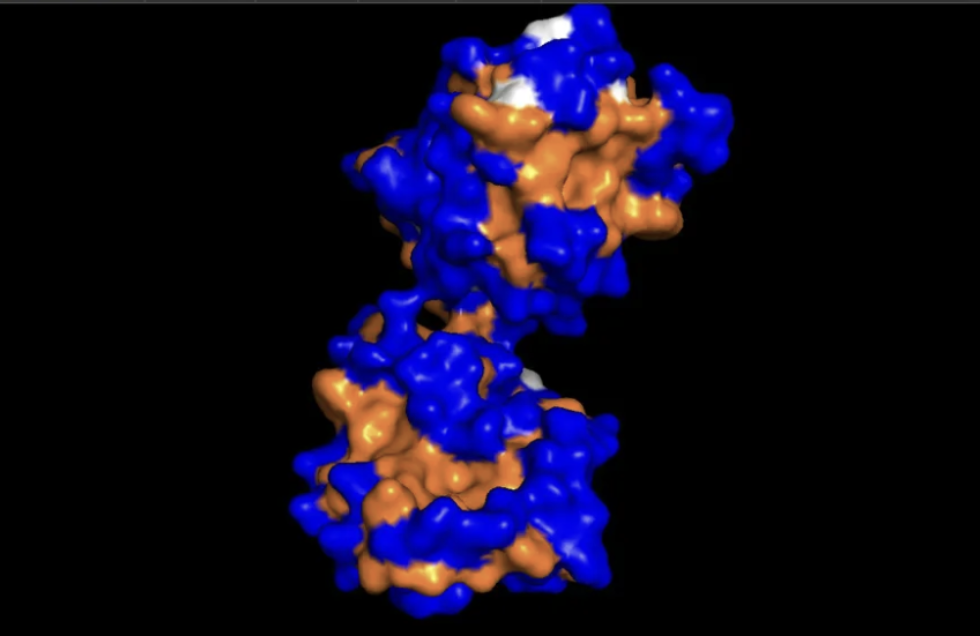
This surface visualization reveals a clear hydrophobic indentation in the middle of the structure, corresponding to the LIR docking site where GABARAPL2 interacts with autophagy receptors.


Part C: Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan based on language model likelihoods
- Can you explain any particular pattern? (choose a residue and mutation that stands out)
- (Bonus) Compare language model predictions to experimental scans

The brightest yellow spot in the heatmap appears at position 60, mutation to Glutamate (E), meaning the model predicts this change would be highly favorable. This makes sense in a metabolic context, as Glutamate’s charged nature supports the protein’s membrane interactions during autophagy and fasting states.
The darkest purple spots appear around positions 54-57 at Cysteine (C) and at position 64 at Tryptophan (W), meaning the model strongly disfavors these mutations. Cysteine in particular stands out as consistently disfavored
Latent Space Analysis
- Embed proteins in reduced dimensionality using the provided sequence dataset
- Analyze neighborhoods — do they approximate similar proteins?
- Place your protein in the map and explain its position and similarity to neighbors

The 3D t-SNE plot shows a large dense central cluster of proteins with outliers scattered at the edges. Proteins in the same neighborhood share similar sequence embeddings, suggesting structural and functional similarity. GABARAPL2, as a member of the highly conserved ubiquitin-like superfamily, would likely position itself near the central core of the cloud, close to other small globular autophagy and ubiquitin-related proteins. Its neighbors would likely include other ATG8 family members
C2. Protein Folding
- Fold your protein with ESMFold — do predicted coordinates match the original structure?
- Try mutations, then larger sequence changes — is the structure resilient?

The ESMFold predicted structure closely matches the original crystal structure. Both show the characteristic beta-grasp fold with a central beta sheet core surrounded by helices, and the overall globular compact shape is preserved.
C3. Protein Generation
- Use ProteinMPNN to inverse-fold your protein backbone and propose sequence candidates
- Analyze predicted sequence probabilities vs the original sequence
- Input the new sequence into ESMFold and compare the predicted structure to original
 ___
___
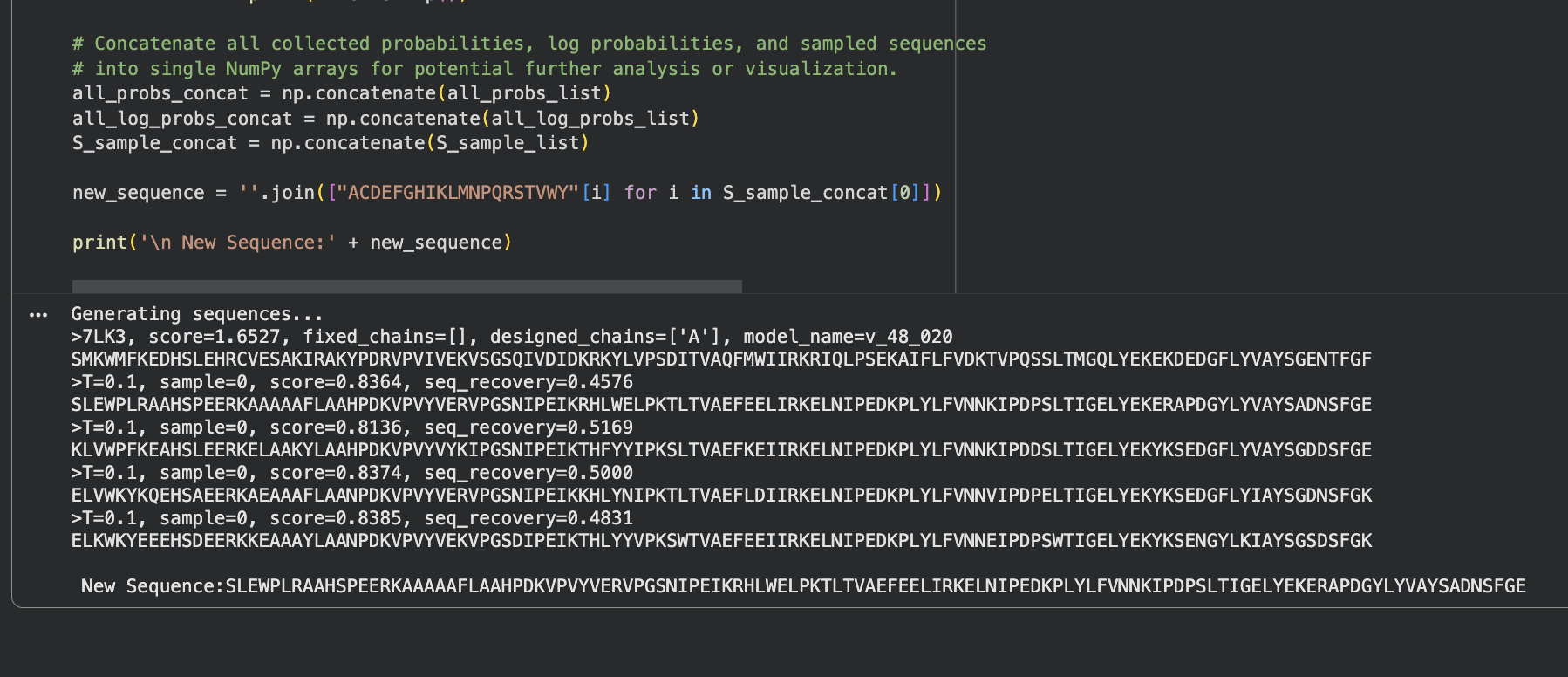
ProteinMPNN generated 4 sequence candidates from the 7LK3 backbone with sequence recovery rates between 46–52% and consistent scores around 0.81–0.84. The probability heatmap shows scattered high-confidence positions (yellow) where the backbone strongly constrains the amino acid choice, surrounded by flexible positions with lower confidence. Despite roughly half the sequence changing, the backbone fold is preserved suggesting that many different sequences can encode the same GABARAPL2 structure.
When the new ProteinMPNN sequence was folded with ESMFold, the overall shape stayed the same. But there were some small differences: the helices shifted slightly, the beta sheets moved a little, and the central loop region pulled closer together. This suggests that even though roughly half the amino acids changed, the protein still folds into essentially the same shape. The structure is resilient.
Part D: Group Brainstorm on Bacteriophage Engineering
Decided to try option 3, as if it fails, it still could help eliminate a possible pathway to end goal and just seemed more interesting. General reminder note: Loop regions and terminal extensions are safer engineering targets than core structural elements.
Higher Toxicity of the MS2 Lysis Protein:
Goal: Increase the toxicity of the MS2 L protein so it lyses bacterial cells faster and more completely.
Approach:
Use a protein language model (ESM or similar) to identify which amino acid positions in the L protein are most likely involved in membrane disruption Propose mutations at those positions using ProteinMPNN to suggest alternative amino acids that might make membrane interaction more aggressive Use AlphaFold-Multimer to model how the mutant L protein interacts with its bacterial target (DnaJ and the membrane) Compare predicted binding strength and structural changes between original and mutant versions
Why these tools help:
Language models capture evolutionary patterns across many proteins, helping identify positions where changes are most likely to matter AlphaFold-Multimer lets you check if your proposed mutations actually change how the protein docks with its bacterial target
Potential pitfalls:
The exact mechanism of membrane disruption by the L protein is not fully understood, so mutations may target the wrong part of the protein or “drill” for my analogy reference. Limited training data exists specifically for phage-bacteria lysis interactions, so predictions may be less reliable than for well-studied proteins
Pipeline schematic first draft:
L protein sequence → ESM (identify key positions) → ProteinMPNN (propose mutations) → AlphaFold-Multimer (validate structure and interaction) → compare mutant vs original
References & Resources
Lecture Materials
- Week 4 Lecture - Protein Design Part I, Thras Karydis, Jon Kaufman
- Week 4 Lab - Protein Design I, February 26-27, 2026
Protein Analyzed
- GABARAPL2 (GABA Type A Receptor Associated Protein Like 2)
- UniProt ID: sp|P60520|GBRL2_HUMAN
- PDB Structure: 7LK3 (1.90 Å resolution, deposited Feb 2021, released May 2021)
- 117 amino acids, ATG8 family, ubiquitin-like superfamily
Software & Tools Used
- UniProt - Protein sequence database and BLAST homolog searches
- RCSB Protein Data Bank - Protein structure database (PDB: 7LK3)
- PyMOL - 3D protein structure visualization and analysis
- Google Colab - Running ESM2, ProteinMPNN, ESMFold analyses
- ESM2 - Protein language model for deep mutational scanning and sequence embeddings
- ESMFold - Protein structure prediction
- ProteinMPNN - Inverse folding and sequence design
- t-SNE - Dimensionality reduction for latent space analysis
- Imgur - Image hosting for visualization documentation
Required Readings
- GABARAPL2 autophagy function literature
- ATG8 family protein structure and function papers
- Protein folding and stability principles
- Amyloid formation and beta-sheet aggregation mechanisms
AI Assistance
- Claude (Anthropic) - Protein analysis and ML tool interpretation
- Model: Claude Sonnet 4.5
- Date(s) used: February, 2026
- Tasks: Assisted with understanding protein structure visualization principles, interpreting ESM2 deep mutational scan results, explaining t-SNE embeddings and protein neighborhoods, clarifying ProteinMPNN sequence recovery metrics, helped develop analogies for complex concepts and checked if homework correct.
Bacteriophage Engineering Project
- Option 3: Increase MS2 lysis protein (L protein) toxicity
- Tools: ESM language model, ProteinMPNN, AlphaFold-Multimer
- Target: Enhanced membrane disruption and faster bacterial lysis
Additional Resources
- SCOP protein structure classification database
- MS2 bacteriophage literature
- Membrane disruption mechanism papers
- DnaJ protein interaction studies
Acknowledgments
- Course instructors for protein design tutorials
- TAs for PyMOL visualization assistance
- Colab community for ML tool notebooks