Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Background
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine to Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Generate Binders with PepMLM
UNIPROT
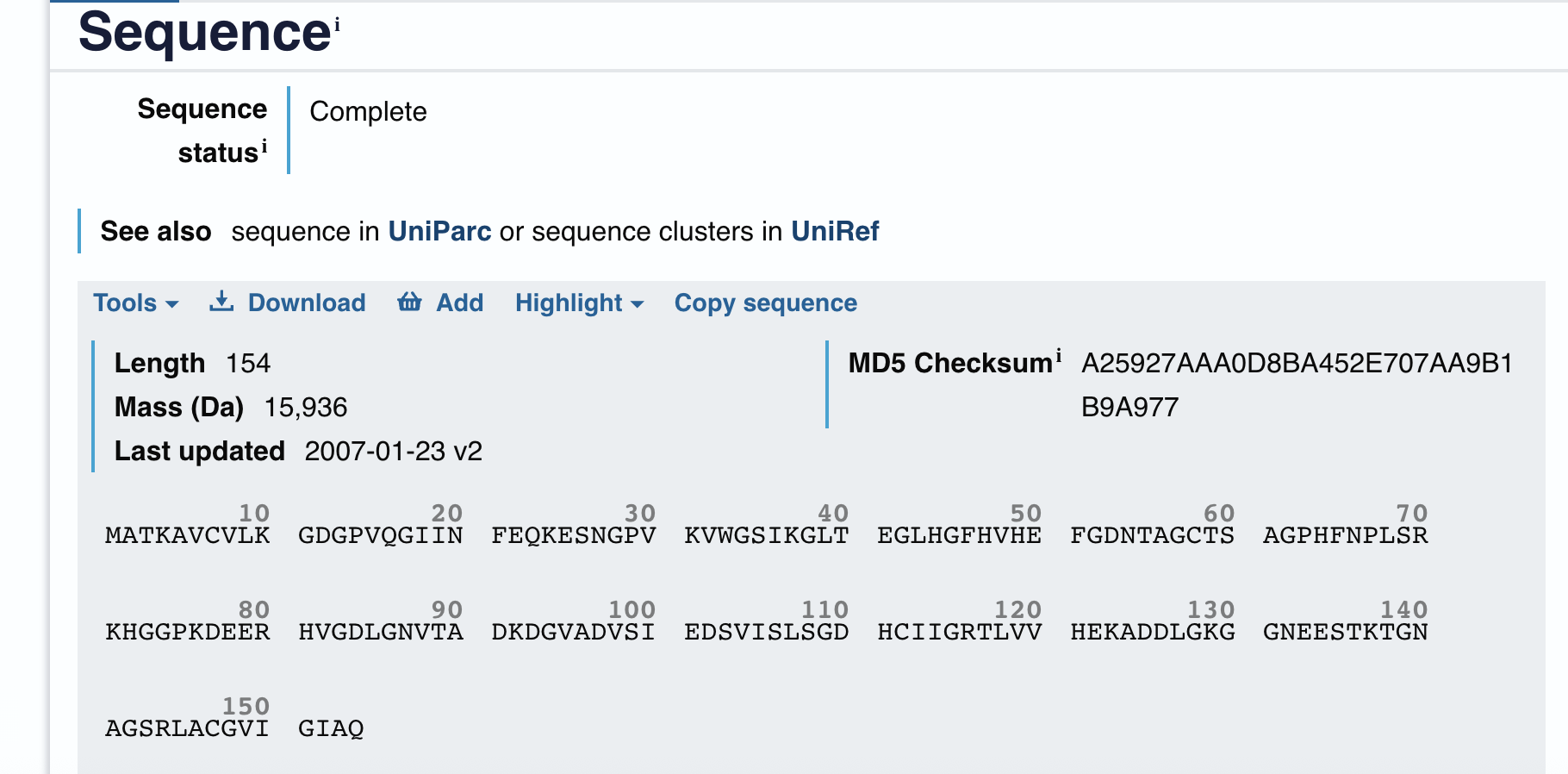
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
CHANGED TO MUTATED VERSION (4.K to V)
MATVAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
SETTING UP AND RUNNING PepMLM IN COLAB
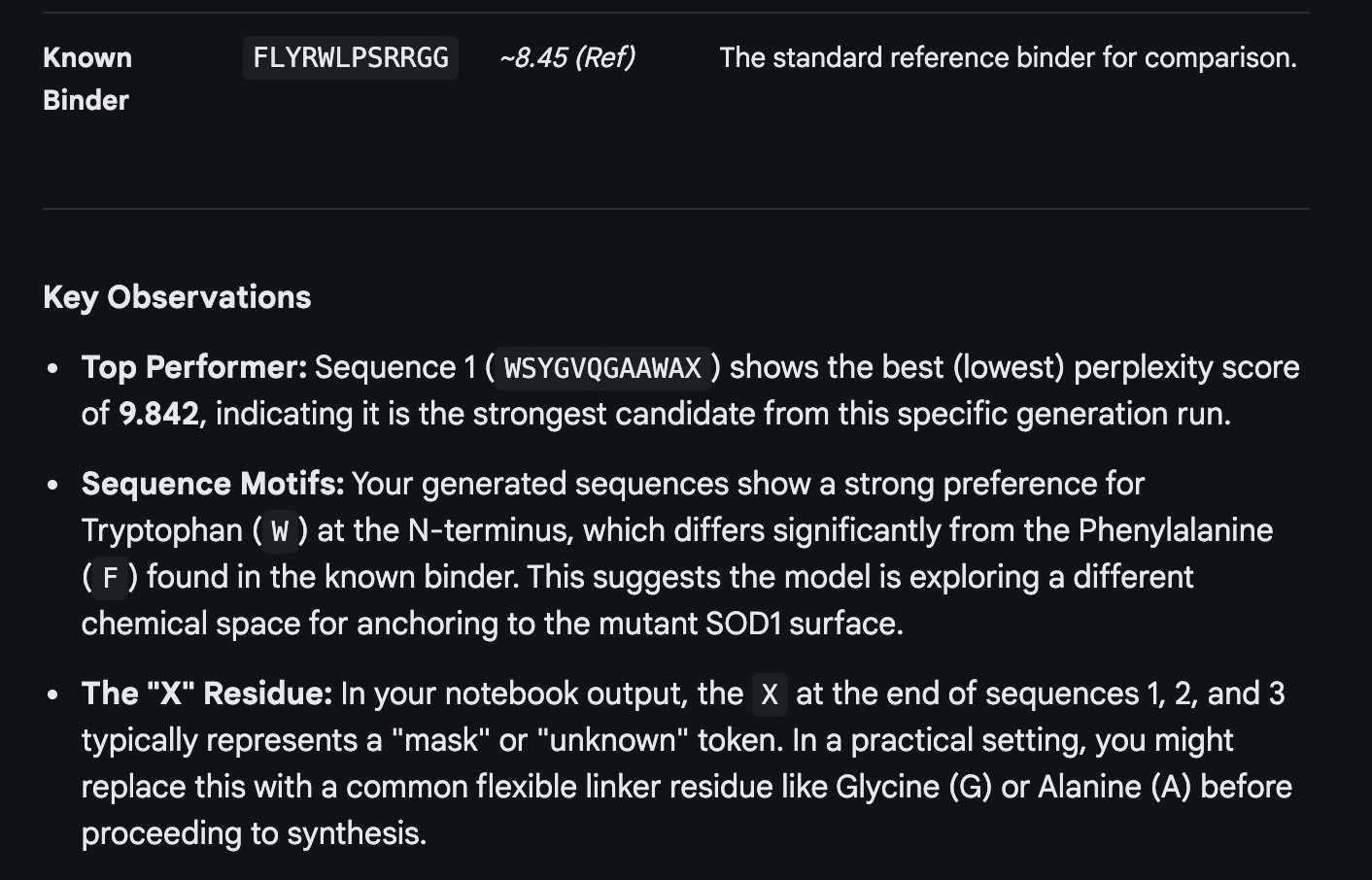
GENERATED 4 PEPTIDES AT 12 SEQUENCE LENGTH
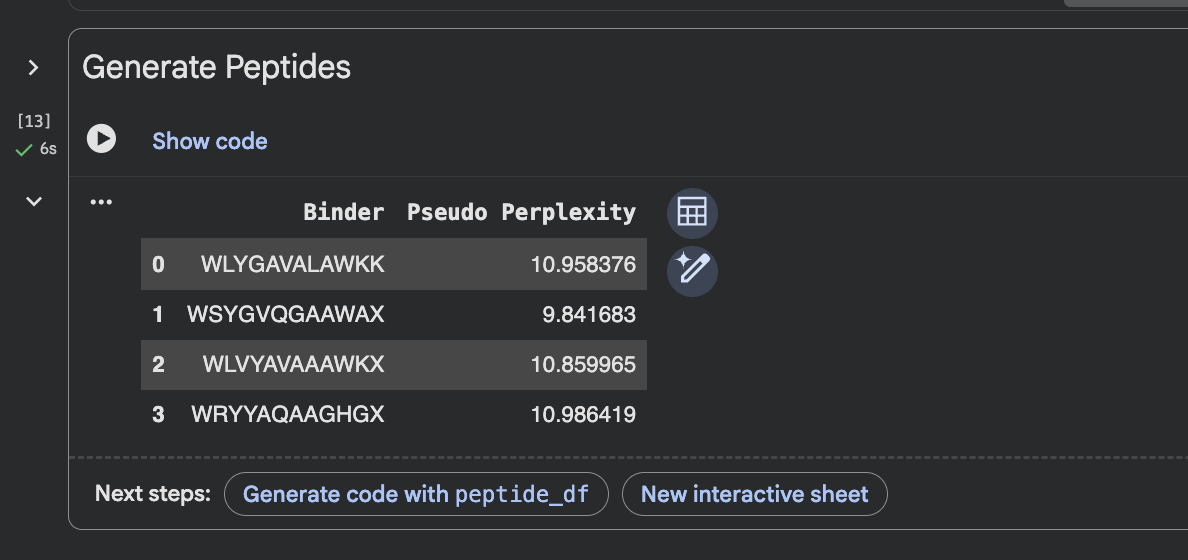
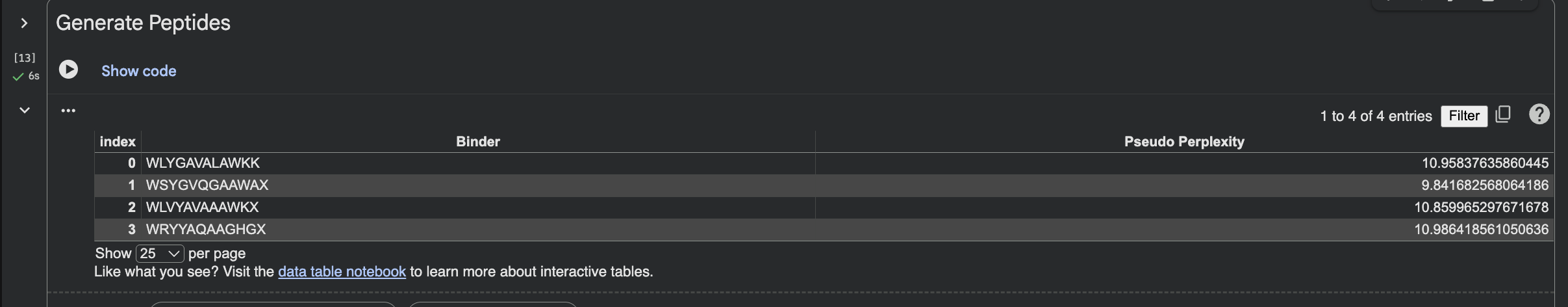
MORE DETAIL IN INTERACTIVE VIEW BELOW
Used GOOGLE GEMINI WITH INTERPRETING AND UNDERSTANDING THE OUTPUT

Evaluate Binders with AlphaFold3
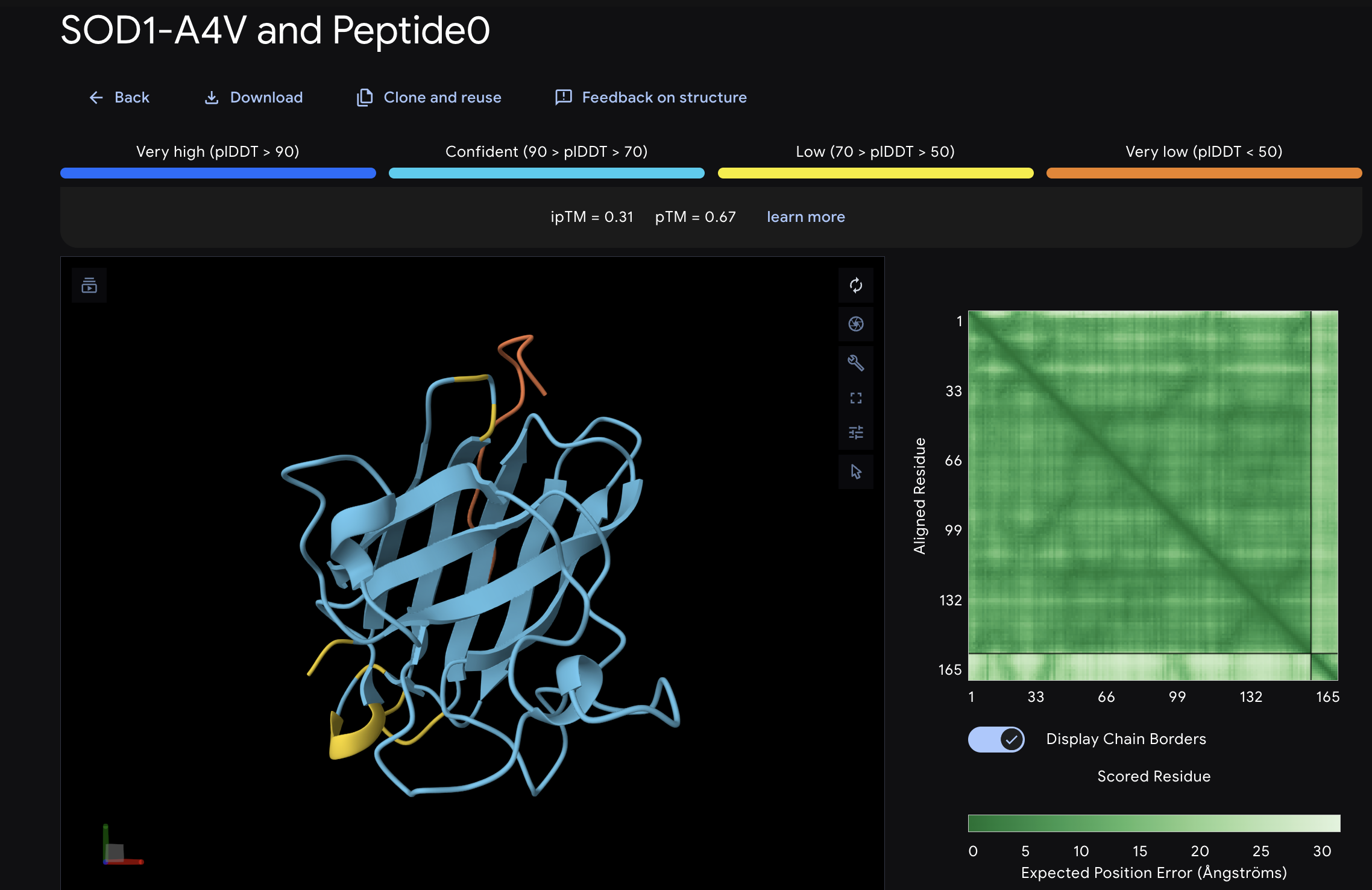

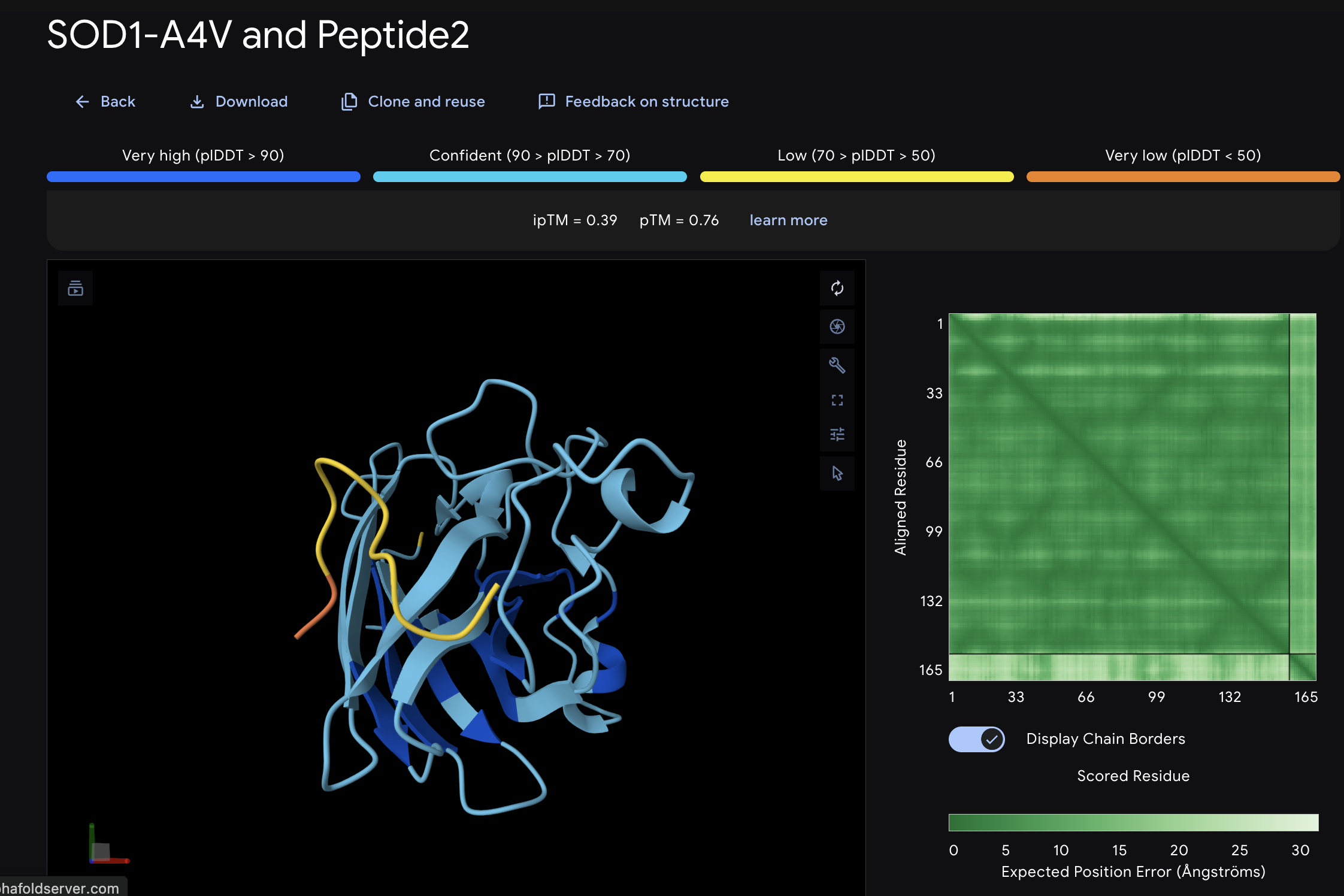


When I looked at the five structures, the known binder really stood out as it sat closely tucked against SOD1 and scored 0.73, which was by far the highest. You could see it engaging deeply with the protein. The PepMLM peptides told a different story. The best ones, Peptide3 and Peptide1, appeared to sit near the top of the protein around the loop region, but they looked more like they were resting on the surface rather than really grabbing onto it. Peptide0 was the weakest and it looked almost detached, just floating near the protein rather than making real contact. None of the generated peptides came close to the known binder, which shows that while PepMLM gave us a starting point, the peptides still need improvement to properly engage SOD1-A4V. (This was my favorite part due to the visuals).
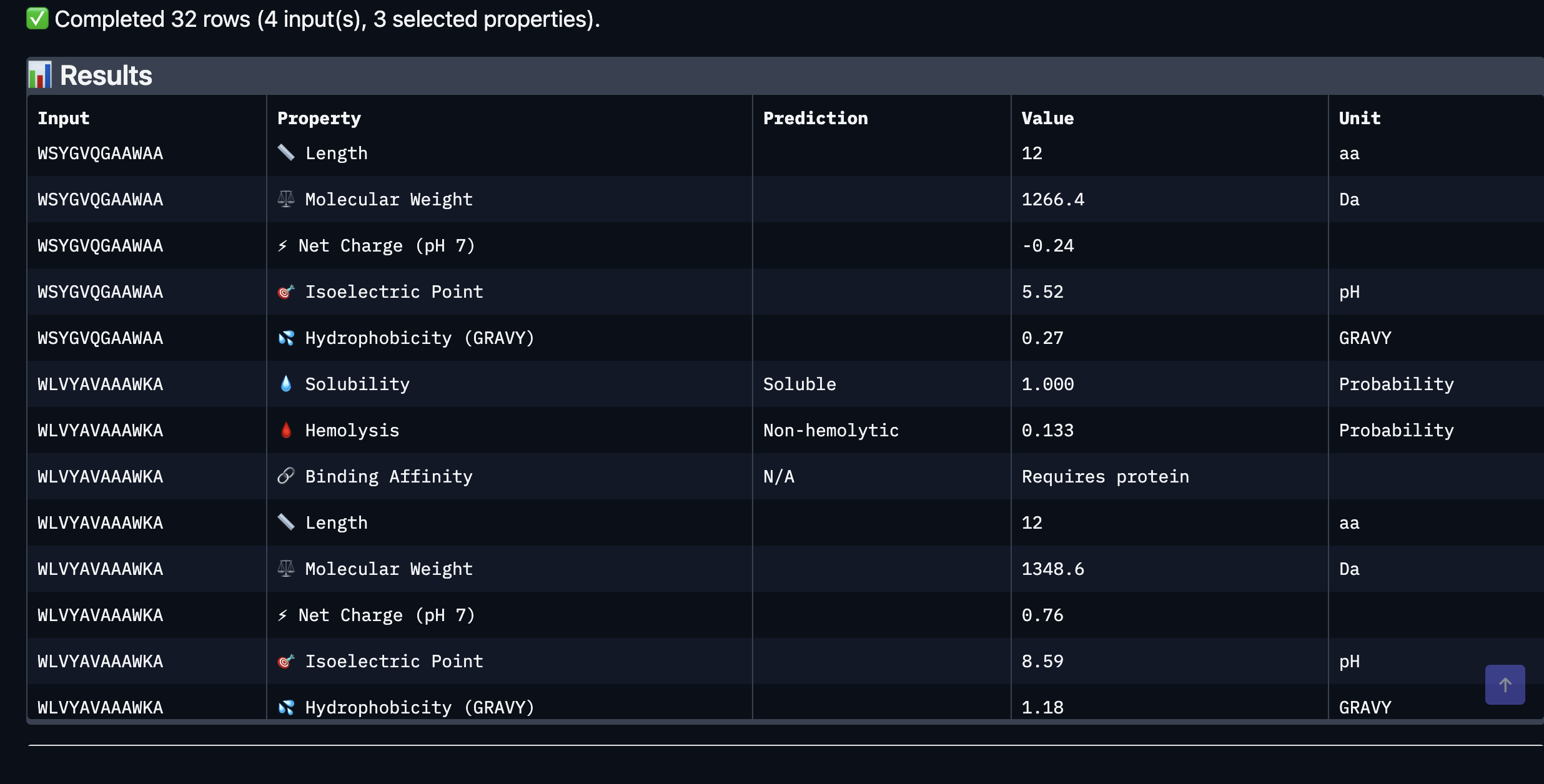
Part 3: Evaluate Properties with PeptiVerse
After evaluating the results below I would advance WLVYAVAAAWKA because it is the only peptide with medium binding affinity (7.247 pKd/pKi), compared to weak binding for the others. It is also well-balanced in terms of hemolysis risk with a low probability of 0.133, and its net charge of 0.76 at pH 7 is nearly neutral, which should help with both solubility and cellular uptake without causing charge-related toxicity. Although its ipTM score of 0.39 in AlphaFold3 was not the highest, the combination of improved predicted binding affinity and favorable therapeutic properties makes it the strongest candidate for further development. While Peptide3 had a slightly higher ipTM of 0.44, it showed the weakest predicted binding affinity of 5.498 and therefore does not balance structural and therapeutic properties as well


Part 4: Generate Optimized Peptides with moPPIt

It took 39min to run.
The moPPIt peptides differ from PepMLM in a key way: moPPIt allowed me to specify exactly which region of SOD1 I wanted to target, whereas PepMLM just generated peptides that looked plausible without that control. With Peptide 2 from moPPIt, I can see it’s actually engaging the N-terminal region where A4V sits, which is exactly what I designed it to do. PepMLM couldn’t guarantee that level of specificity.
Before advancing any peptide to clinical studies, I would need to do much more work. First, I’d validate the binding predictions with actual lab experiments measure real binding affinity. Most importantly, I’d likely run moPPIt again with different target regions on SOD1 in order to generate a larger panel of candidates and pick the best performers across all validation steps. No single computational prediction is enough to move forward to the clinical setting.
Part B: Optional
Part C: Final Project: L-Protein Mutants
The objective of this assignment is to improve the stability and auto-folding of the lysis protein of an MS2-phage. This mechanism is key to understanding how phages may help address antibiotic resistance.
After going through the readings, including the group final project document a Plan A would be: (This stays within scope, MurJ and multi-target approaches seem intersting though…)
1 Use computational tools like AlphaFold2 or ProteinMPNN to identify mutations that improve intrinsic stability and auto-folding of the lysis protein
2 Target mutations that strengthen the hydrophobic core, eliminate aggregation-prone regions, or introduce stabilising interactions like salt bridges
3 Engineer the lysis protein to fold correctly without requiring DnaJ or any other bacterial chaperone
4 Design mutations that also accelerate oligomerisation or enhance membrane pore-forming activity for faster lysis
5 Synthesise the mutant gene via Twist, clone into plasmid using Gibson Assembly, validate structural integrity with Nuclera, then test in E. coli.
References & Resources
Lecture Materials
- Week 5 Lecture - Protein Design Part II, Pranam Chatterjee, Gabriele Corso
- Week 5 Lab - Protein Design Part II Lab, March 5-6, 2026
Software & Tools Used
- UNIPROT
- PepMLM
- Alphfold
- Peptiverse
- moPPIt
AI Assistance
- Claude (Anthropic) - Protein design concepts
- Model: Claude Sonnet 4.5
- Date(s) used: March, 2026
- Tasks: Acted as mentor (Skills) in conversations about unfamiliar and technical areas. Checked homework was correct.
Additional Resources
- Advanced protein design literature
- Computational protein engineering tools
Acknowledgments
- Course instructors and TAs