Week 6 HW: Genetic Circuits Part I
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion HF PCR Master Mix contains several key components that are already pre-combined for convenience. Phusion DNA Polymerase is the core enzyme responsible for copying the DNA template, and it has a built-in proofreader to ensure it is high-fidelity, meaning it reduces errors during amplification. The dNTPs provide the nucleotide building blocks that get incorporated into the new DNA strand. MgCl₂ (magnesium) acts as an essential cofactor that activates the polymerase. The reaction buffer (oven conditions in my analogy) maintains the correct pH and ionic environment for the reaction to work. For this particular lab, precise mutagenesis of the amilCP chromophore region was required, so the high-fidelity polymerase is especially important; it ensures there are no unintended amino acid changes beyond the designed mutation.
What are some factors that determine primer annealing temperature during PCR?
Several factors affect the temperature at which a primer successfully binds to its target on the DNA template. First, secondary structure is something to avoid. If a primer folds back on itself it is like a blurry photograph that cannot be read properly, meaning it cannot find its matching location on the template regardless of temperature.
Second, GC content affects annealing temperature. Primers with more G and C bases require higher temperatures because GC pairs bond more strongly than AT pairs. In this lab the backbone primers anneal at 57°C while the color insert primers anneal at 53°C, reflecting differences in their GC content.
Third, primer length matters. A longer primer is like a photograph that also shows the surrounding context, making it a more specific match. Longer primers bind more strongly and therefore require higher annealing temperatures. These factors were carefully balanced during primer design, aiming for a Tm range of 52–58°C with primer pairs kept within 5°C of each other.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digests differ in terms of precision and flexibility. PCR is more flexible and suitable for bespoke mutation designs, giving you control over exactly where a fragment begins and ends by designing the primers yourself. Restriction enzyme digests are more limited in that they can only cut where their recognition sequence naturally exists in the DNA, but this makes them faster and more straightforward when you already know exactly which sequence you need.
I would use PCR when attempting to design a mutation, as in this lab where the chromophore color changes were introduced through deliberate primer mismatches. I would use restriction enzyme digests when the recognition sites are already conveniently placed and the desired sequences are already known, as this would save time.
In terms of protocol, PCR requires designing primers, running denature, anneal and extend cycles in a thermocycler, cleaning up the original template with a DpnI digest, and then purifying the DNA. Restriction enzyme digests are more straightforward, requiring only choosing the right enzyme for the recognition site, incubating the DNA with the enzyme at 37°C, and running a gel to confirm the correct cut. No heating cycles or template cleanup are needed.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
There are several ways to ensure fragments are appropriate for Gibson cloning.
First, correct overlaps must be present. In this lab the primers were designed from the start with 20-40bp overhangs complementary to the adjacent fragment, ensuring the fragments can recognise and join each other during assembly.
Second, fragment size should be confirmed by running a diagnostic gel. If the band appears at the wrong size then the PCR was unsuccessful and the fragment would not be appropriate for Gibson cloning.
Third, the DNA must be clean and concentrated enough. The Nanodrop measurement confirms concentration is above 30ng/µL. Contaminants from PCR can inhibit the Gibson Assembly reaction.
Fourth, the original template must be removed. The DpnI digest ensures the original methylated mUAV plasmid is not carried over, which would otherwise produce background colonies of the unmutated purple protein. Finally, the correct molar ratio must be used. Gibson Assembly works best at a 2:1 insert to vector ratio to ensure efficient and complete assembly.
How does the plasmid DNA enter the E. coli cells during transformation?
The plasmid enters the E. coli cells through a process called heat shock transformation.
First, the cells are made chemically competent using CaCl₂. This partially neutralizes the repulsion between the negatively charged cell membrane and the negatively charged DNA, allowing the DNA to associate with the cell surface.
Next the cells are kept on ice, which makes the membrane more rigid and stable. Then the cells are heat shocked at exactly 42°C for 45 seconds, which temporarily disrupts the membrane and allows the plasmid to enter the cell by diffusion. The cells are then immediately returned to ice so the membrane stabilizes and closes again.
So essentially the process is: make the membrane rigid with ice, give it a heat shock to open it briefly, then put it back on ice to close it again with the plasmid now inside.
After heat shock, SOC media (Note my nutrient rich broth analogy) is added to help the cells recover and begin multiplying. Finally the cells are plated on chloramphenicol agar, where only cells that successfully received the plasmid will survive and grow.
Describe another assembly method in detail, such as Golden Gate Assembly. Explain the other method in 5 to 7 sentences plus diagrams, either handmade or online.
Golden Gate Assembly is a method of connecting DNA fragments together using custom 4 base sticky ends. It works by sending in a Type IIS restriction enzyme that acts like a self destructing instruction manual, cutting at a defined location outside its recognition site and then removing itself in the process, leaving behind unique sticky ends that have been designed to only connect to one specific matching partner. These sticky ends are self sorting, acting like magnets that can only attract their intended match and nothing else. Once the fragments are correctly joined the assembly is scarless, meaning no trace of the recognition site remains in the final product.

This differs from Gibson Assembly which uses an exonuclease, polymerase and ligase, and requires longer overlaps of 20-40bp between fragments rather than the 4 base sticky ends of Golden Gate. Golden Gate cycles between cutting and ligation temperatures repeatedly, whereas Gibson Assembly runs isothermally at 50°C. Because incorrect assemblies get re-cut and correct ones accumulate, Golden Gate is highly efficient and can assemble many parts simultaneously in one tube, making it more scalable than Gibson Assembly which typically handles two to six parts.
Model this assembly method with Benchling.
Golden Gate Assembly modeled in Benchling using mUAV plasmid (MG252981.1) as starting reference:

Figure 1: Backbone fragment (38bp) with BsaI recognition site (GGTCTCN) annotated at its end. BsaI cuts here, removing itself and exposing the sticky end.
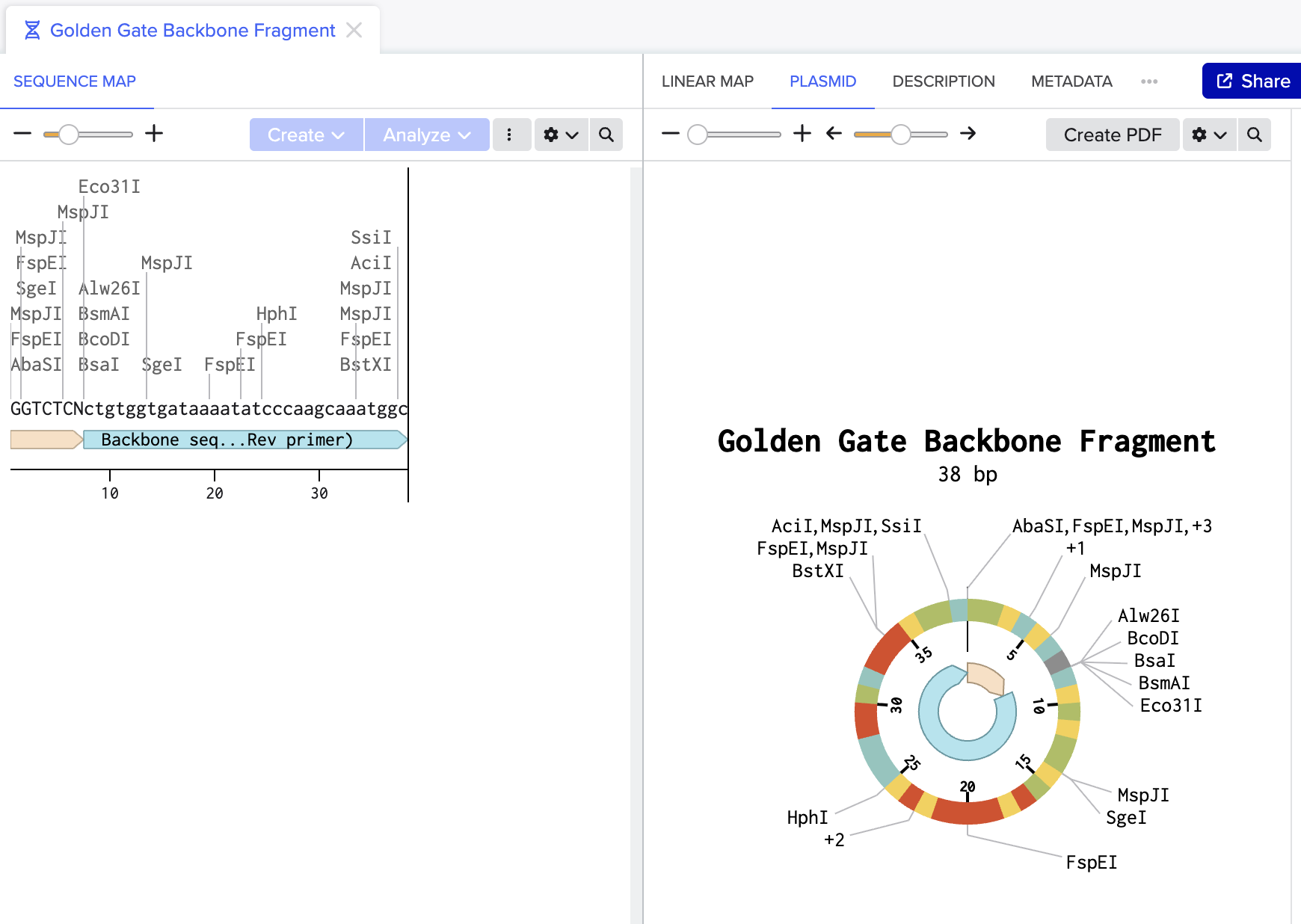
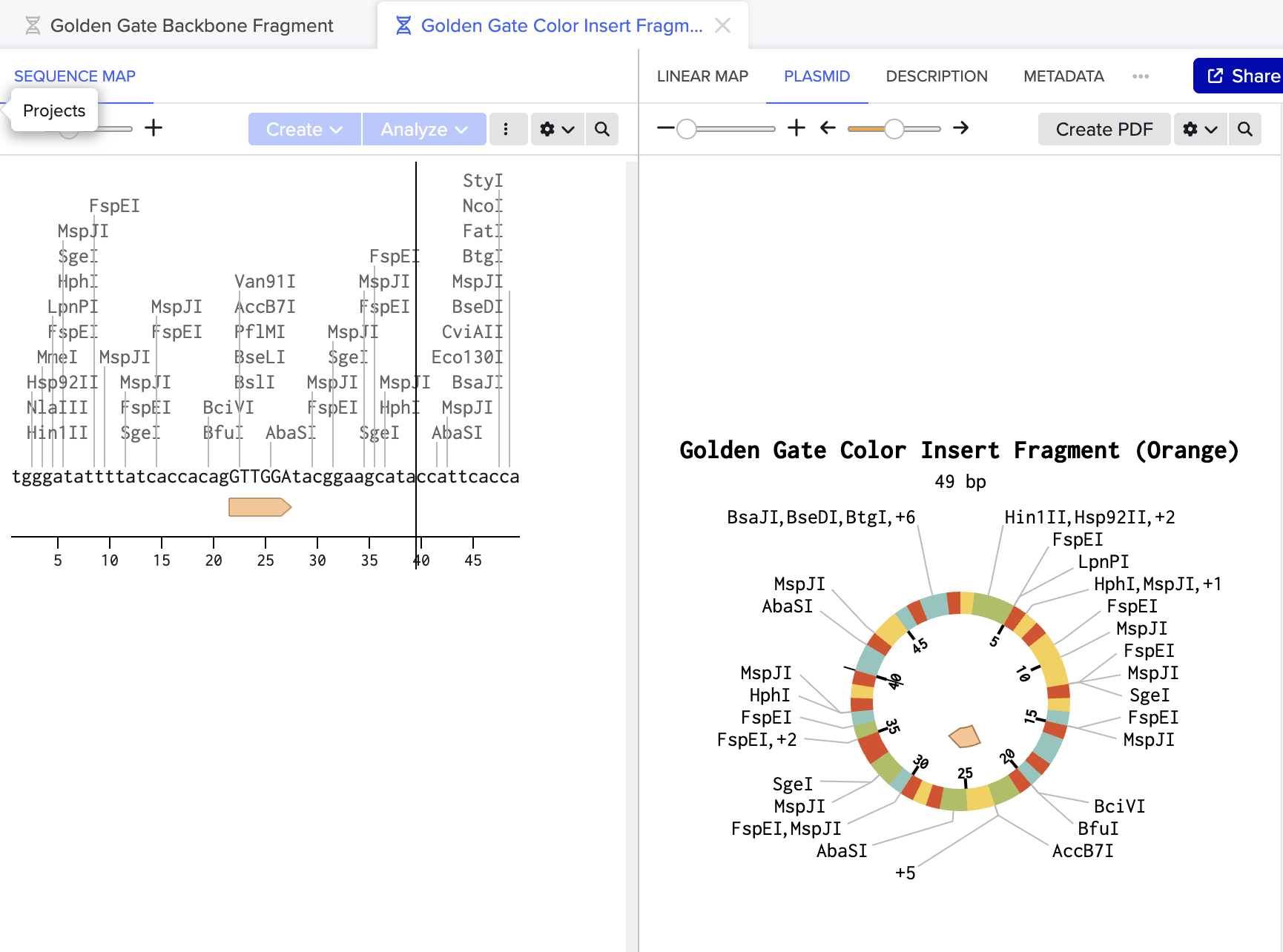
Figure 2: Color insert fragment (49bp) containing the orange chromophore mutation GTTGGA replacing original TGTCAG. This sequence changes the chromophore amino acids to produce orange instead of purple. Together these two fragments would be combined with BsaI and ligase in one tube to produce a scarless circular plasmid carrying the orange mutation.
Assignment: Asimov Kernel
Did not have access to Asimov Kernel. (Did attend the MIT Review and not sure if Nodes have access. Also, signed up to be beta tester when availible)
References & Resources
Lecture Materials
- Week 6 Lecture - Genetic Circuits Part I: Assembly Technologies, Doug Densmore, Traci Haddock
- Week 6 Lab - Gibson Assembly, March 12-13, 2026
Software & Tools Used
- Benchling
AI Assistance
- Claude (Anthropic) - Genetic circuit design
- Model: Claude Sonnet 4.5
- Date(s) used: March, 2026
- Tasks: Acted as mentor (Skills) in conversations about unfamiliar and technical areas. Checked homework was correct.
Additional Resources
- Gibson assembly protocol documentation
- Genetic circuit assembly technologies literature
Acknowledgments
- Course instructors and TAs