Co-relational Synbio Research Project, 2026 (Image courtesy of the artist)
Henrietta Scholtz
HTGAA Spring 2026
About me
I am an interdisciplinary artist, researcher and occasional curator. Areas of interest are situated biochemistry, materiality, computation and meta narratives.
I am a contemporary artist interested in biomaterials, DNA and new technologies.
Have not experimented with bacterial pigments so thought of using the following as a starting point:
Bacillus species (orange/yellow)
Serratia marcescens (red/pink)
Environmental isolates from soil
Firstly, in growing them myself (which I am new to), as well as mechanotransduction experiments with sounds and vibrations; having the bacteria’s pigment respond to sounds and vibrations. Connecting mechanosensitive channels to pigment gene expression.
If possible, explore the possibilities of UV-protective, antimicrobial, colored bioplastic material or packaging using bacterial pigments in a seaweed matrix, and build on what has been done to amplify natural pigment production through gene cloning. Combining bacterial pigments directly with seaweed‑based bioplastic matrices (like carrageenan or alginate) for UV‑protection and antimicrobial function.
Further experiments,looking at creating hybrid strains.
Bio-Art Ethics & Policy Framework
I looked at governance and policy from an artist’s, non-science public, point of view, as well as the fact that in my usage case, the bacterial samples may be presented to the public in a gallery setting.
Primary Goal: Ensure Safe & Responsible Use of Engineered Organisms in Artistic Practice
Secondary Goal: Maintain Public Trust in Bio-Art While Enabling Innovation
Three Governance Actions
Action 1: Tiered Institutional Approval System
Highlighting the roles of Biosafety Committees, Art Institutions, and Artists.Actor 1 (Biosafety Committees),Actor 2 (Art Institutions),Actor 3 (Artists).
Action 2: Open-Source Documentation Standard & Community Vetting
Outlining the purpose of shared safety standards and the involvement of Artists, Scientists, and the Community.
Purpose: Currently, bio-art practitioners work in isolation without shared safety standards, Actor 1 (Artists & Scientists), Actor 2 (Community.
Action 3: Technical Safety Infrastructure & Insurance Product
Addressing artist liability through the collaboration of Engineers, Certification Bodies, and Artists.Purpose: Currently, artists mostly bear full liability for bio-art installations. Actor 1 (Engineers/Companies), Actor 2 (Certification Bodies), Actor 3 (artist)
Risk Assessment Matrix
Scoring Matrix
Action 1 (Tiered Institutional Approval) scores best on biosecurity and lab safety prevention, as a formal approval system is the most direct way to stop unsafe practices before they happen. It is moderately feasible but places a higher burden on stakeholders and could slow research. Action 2 (Open-Source Documentation Standard) scores best overall, performing well across biosecurity response, feasibility, cost minimization, and promoting constructive applications, making it the strongest all-round option. Action 3 (Technical Safety Infrastructure and Insurance) scores weakest on feasibility and cost, as it requires significant infrastructure investment and places the heaviest financial burden on individual artists, though it offers some environmental protection benefits. Overall, Action 2 is the clear leader, Action 1 provides strong institutional backup, and Action 3 is a longer-term aspiration.
Prioritization and Recommendation
I would prioritize Action 2 (Open-Source Documentation Standard) combined with Action 1 (Tiered Institutional Approval). Open-source standards score best across nearly all goals and are low-cost and immediately feasible for artists and community bio-labs. Tiered approval adds necessary oversight for public-facing installations.
The main trade-off is that Action 2 relies on voluntary community participation, which may be inconsistent. Action 3 (insurance) is the least feasible in the short term and places the highest burden on individual artists, so it is treated as a longer-term goal.
My recommendation is directed at iGEM and community biology organisations, who could draft and promote the open-source standard without requiring regulatory approval.
Ethical Reflection
Working with living pigment-producing bacteria in a gallery context raised new questions for me about consent and exposure: unlike a lab, gallery visitors have not opted into proximity to engineered organisms. This highlighted a gap in current governance, as bio-art largely falls outside both lab safety regulation and public health frameworks.
A potential governance response would be a simple public-disclosure requirement for any bio-art installation using living organisms, similar to ingredient labeling, so that audiences can make informed decisions about their proximity to the work.
Week 2 Lecture Prep
Prof. Jacobson: Question 1
Polymerase Error Rate
DNA polymerase has an error rate of about 1 in 106 bases. The human genome is roughly 3 billion base pairs, meaning uncorrected replication would produce thousands of errors per copy. Biology addresses this with proofreading exonucleases built into the polymerase, plus a separate mismatch repair system, bringing the effective error rate down to around 1 in 109.
Prof. Jacobson: Question 2
Coding for a Human Protein
Because the genetic code is redundant, the average human protein of ~1,000 amino acids can theoretically be encoded by an astronomically large number of different DNA sequences. In practice most alternatives do not work well because cells have codon usage biases (some codons are translated faster or slower), mRNA secondary structures can block translation, and certain sequences trigger mRNA degradation.
Dr. LeProust: Question 1
Most Common Oligo Synthesis Method
The most commonly used method is solid-phase phosphoramidite chemistry, in which nucleotides are added one at a time to a growing chain attached to a solid support.
Dr. LeProust: Question 2
Why Oligos Are Hard to Make Beyond 200 nt
Each coupling step is not 100% efficient (around 98-99%), so errors accumulate with every added base. Beyond ~200 nt the fraction of full-length, correct molecules becomes too low to be practically useful.
Dr. LeProust: Question 3
Why You Cannot Make a 2,000 bp Gene via Direct Oligo Synthesis
Direct synthesis produces a pool of short, error-prone oligos. A 2,000 bp gene requires assembling many overlapping oligos, and errors in individual oligos get incorporated into the final product. The assembly process can also introduce chimeras (incorrectly joined fragments), making a correct 2,000 bp product impractical without additional error-correction steps.
Prof. Church: Question 1
The 10 Essential Amino Acids and the Lysine Contingency
The 10 essential amino acids that animals cannot synthesise are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine (conditionally essential). The Lysine Contingency refers to engineering a dependency on an unnatural amino acid as a biocontainment strategy. Knowing that lysine is already essential and must come from diet, it seems plausible to replace that dependency with a synthetic molecule unavailable in the wild, which would be a practical and minimally invasive form of genetic containment.
Required Readings
Course policies and biosafety guidelines from HTGAA Spring 2026 syllabus
Institutional biosafety protocols for bio-art installations
Additional Resources
Bio-art ethics and safety protocols literature
Gallery biosafety requirements for living organism exhibitions
Insurance and liability frameworks for bio-art practitioners
Project Context
Research focus: Bacterial pigment production (Serratia marcescens, Bacillus species)
Public engagement: Gallery presentation considerations
AI Assistance
Manus AI - Governance framework visualization
Date(s) used: February 2026
Tasks: Generated visual representations of bio-art governance framework and risk assessment matrix based on author’s policy framework
Acknowledgments
HTGAA instructors for guidance on bio-art policy frameworks
Course TAs for biosafety protocol clarification
Week 2 HW: DNA Read, Write, and Edit
Part 0: Basics of Gel Electrophoresis
Attend or watch all lecture and recitation videos. YES
Optionally watch bootcamp. YES
Part 1
Benchling & In-silico Gel Art
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion
I imported Lambda DNA into Benchling and simulated restriction enzyme digestion with EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. Using the predicted band sizes from each digest, I selected enzyme combinations that would produce bands at specific positions to form a simple geometric pattern in the style of Paul Vanouse’s Latent Figure Protocol work.
Output attempt of a dog! (with tail on the right)
Part 2
No wet lab access
Part 3
DNA Design Challenge
Choose Protein
I chose the amino acid sequence of VioC - Chromobacterium violaceum for Violacein pigment.
I will reverse translate and codon optimize to amplify pigment production and thus its antimicrobial, UV-resistant properties.
Next
Next steps would be to embed into a seaweed matrix.
The VioC coding sequence can be transcribed into mRNA and then translated into protein using either a cell-dependent or cell-free method. In a cell-dependent approach, the codon-optimized sequence is cloned into an expression plasmid, transformed into E. coli, and protein production is induced by adding IPTG. The bacteria read the DNA, transcribe it into mRNA, and their ribosomes translate it into the VioC enzyme. In a cell-free approach, the DNA template is added directly to a prepared lysate containing ribosomes, enzymes, and amino acids, and protein is synthesised in a test tube without any living cell.
Part 4
Prepare a Twist DNA Synthesis Order
After reading more on living materials, bacterial pigments, and connecting it to my interest in light and circadian rhythms, I wanted to explore how to make a simple biological system that expresses anti-microbial or other elements only when needed, rather than all the time. So building a ’temporal’ antimicrobial system that produces a bacteria-killing peptide Magainin on a 24-hour schedule controlled by a circadian promoter RpaA. I started with just learning how to design the Magainin peptide and annotate properly.
Benchling
Twist
REF:
Fang et al. (2025) - “Mechanism and reconstitution of circadian transcription in cyanobacteria”
Salis et al. (2009) - “Automated Design of Synthetic Ribosome Binding Sites”
Westerhoff et al. (2008) - “Structure, Membrane Orientation, Mechanism, and Function of Pexiganan (Magainin derivative)”
Part 5
DNA Read/Write/Edit
DNA Read (Sequencing)
5.1 Sequencing Technology: Sub-questions
Generation: Sanger sequencing is first-generation. It sequences one DNA fragment at a time using chain-terminating dideoxynucleotides, predating the massively parallel approaches of second-generation (e.g. Illumina) and third-generation (e.g. Oxford Nanopore) methods.
Input and preparation: The input is purified plasmid DNA. Preparation involves a PCR step using a single primer to amplify the target region, followed by a cleanup to remove unused nucleotides and primers before the sequencing reaction.
Essential steps and base calling: The cleaned PCR product is mixed with a single primer and four fluorescently labelled dideoxynucleotides. A polymerase extends the primer until it randomly incorporates a dideoxynucleotide and terminates. This produces fragments of every possible length, each ending in a fluorescent base. The fragments are separated by capillary electrophoresis and a laser reads the fluorescent colour at each length, which is converted into a base sequence.
Output: A chromatogram showing peaks of four colours corresponding to A, T, C, and G, along with a text sequence file. Read length is typically 700-1000 bases.
5.2 Synthesis Technology: Sub-questions
Technology: I would use solid-phase phosphoramidite synthesis via Twist Bioscience to synthesise the pLight-Circadian-Color plasmid, as it offers high accuracy and fast turnaround for sequences up to several kilobases.
Essential steps: The sequence is designed in Benchling, codon-optimised, and uploaded to Twist. Twist synthesises overlapping oligos on a silicon chip, assembles them into the full gene fragment, clones the insert into the chosen backbone vector, and sequences the final construct to confirm accuracy before shipping.
Limitations: Direct oligo synthesis has a practical length limit of around 200 nucleotides per oligo due to error accumulation, meaning longer genes require assembly from many fragments. Error rates, while low (around 1 in 3,000 bases for Twist), mean some clones may contain mutations and must be sequence-verified before use.
What DNA would you want to sequence and why?
I would sequence my pLight-Circadian-Color plasmid (which contains the RpaA gene from Synechococcus elongatus, an anthocyanin color gene, and a light sensor) to check that it was made correctly before testing if bacteria with this plasmid change color on a 24-hour schedule when exposed to light.
What sequencing technology would you use?
I would use Sanger sequencing because it’s most accurate.
DNA Write (Synthesis)
What DNA would you synthesize and why?
I would synthesize my yet-to-be-completed pLight-Circadian-Color plasmid containing three genes (RpaA from Synechococcus elongatus for timing, anthocyanin for color, light sensor for activation) to test if bacteria can change color on a 24-hour schedule in response to light.
DNA Edit
What DNA would you edit and why?
After I verify the plasmid works, I would edit the RpaA promoter to make it stronger so the color changes are brighter and more noticeable on a 24-hour schedule.
What editing technology would you use?
I would use site-directed mutagenesis to make small changes to the RpaA promoter because it’s precise.
Editing Technology: Sub-questions
How it works: Site-directed mutagenesis uses PCR with primers that contain the desired mutation in their sequence. The polymerase copies the entire plasmid incorporating the mutation, and the original methylated template is then digested away with DpnI, leaving only the mutated version.
Preparation and inputs: I would design primers containing the specific base changes I want in the RpaA promoter region, using a tool like NEB’s primer design tool. The inputs are the original plasmid, the two mutagenic primers, a high-fidelity polymerase such as Phusion, dNTPs, and DpnI enzyme for template removal.
Limitations: Site-directed mutagenesis only makes small, precise changes and cannot introduce large insertions or deletions efficiently. It also requires the plasmid to already be available, and each round of mutagenesis must be followed by sequencing to confirm the correct change was made and no unintended errors were introduced.
References & Resources
Lecture Materials
Week 2 Lecture - DNA Read, Write, & Edit, George Church, Joe Jacobson, Emily Leproust
UniProt - Protein sequence database (VioC entry: sp|Q9S3U9|VIOC_CHRVO)
Imgur - Image hosting for documentation
Sequences Worked With
VioC (Violacein synthase) from Chromobacterium violaceum strain ATCC 12472
RpaA circadian promoter from Synechococcus elongatus
Magainin antimicrobial peptide sequence
AI Assistance
Claude (Anthropic) - DNA design and sequencing strategy
Model: Claude Sonnet 4.5
Date(s) used: February, 2026
Tasks: Assisted with reverse translation strategy for VioC, guidance on codon optimization principles, clarified Sanger sequencing vs synthesis tradeoffs
Project Development
Circadian-controlled antimicrobial system design (RpaA + Magainin)
Violacein pigment amplification through codon optimization
pLight-Circadian-Color plasmid conceptual design
Additional Resources
Twist Bioscience synthesis guidelines and specifications
Benchling annotation standards
Circadian rhythm gene expression literature
Acknowledgments
Course instructors
Week 3 HW/Lab : Opentrons
Python Script for Opentrons Artwork
Since I am not present to interact directly with the Opentrons output, I thought about why I would want to pipette an image and what that image should represent and decided to use Ndebele bead patterns as inspiration.
Ndebele bead patterns have a very specific geometric logic. They are built on a grid of “bead units” arranged in bold, angular, symmetric designs. The traditional South Ndebele aesthetic uses high-contrast colors in step-like diagonal and horizontal bands, often with thick outlines and mirrored symmetry.
They are also studied as Ethno mathematics, which often promotes a more humanistic and inclusive perspective on mathematics, focusing on how different groups manage, understand, and navigate their reality.
I found it interesting to bring the mathematical and social aspects of this indigenous knowledge to the biochemical level, as this layering of meaning creates interesting avenues for reflection on various levels.
Example of Ndebele paintings and beadwork:
Python Visuals & Scripts Ex.
I am not a coder, but playing around with the example scripts, I ended up using Claude to vibe-code the desired patterns and position. It required some debugging and made various output versions.
Although the co-lab script runs without error, I am not sure if this will work on Opentrons.
FULL FINAL VERSION CODE
Python Script
Below is the complete Python script that creates the Ndebele-inspired “BIO” pattern using negative space typography on a horizontal stripe background. The script was developed in Google Colab with Claude AI assistance and runs without errors in simulation.
The pattern uses:
Blue beads: Top and bottom border rows (rows 0-1, 9-10)
Pink beads: Inner border rows (rows 2-3, 7-8)
Purple beads: Central band (rows 4-6)
Black agar (no beads): Letter shapes forming “BIO” in negative space
fromopentronsimporttypesmetadata={'protocolName':'Ndebele Bio','author':'Henrietta','source':'HTGAA 2022','apiLevel':'2.20'}################################################################################# Robot deck setup constants - don't change these##############################################################################TIP_RACK_DECK_SLOT=9COLORS_DECK_SLOT=6AGAR_DECK_SLOT=5PIPETTE_STARTING_TIP_WELL='A1'well_colors={'A1':'Purple','B1':'Pink','C1':'Blue'}################################################################################# Ndebele BIO Pattern — Negative Space Typography### Grid: 21 cols x 11 rows. Row 0 = bottom, Col 0 = left.### Physical size at 4mm spacing: 80mm wide x 40mm tall.###### The word "BIO" is formed as NEGATIVE SPACE (black agar gaps) cut into### a Ndebele-style horizontal stripe background of coloured beads.###### Background stripe layout (classic Ndebele horizontal banding):### Blue = rows 0-1 and rows 9-10 (top and bottom borders)### Pink = rows 2-3 and rows 7-8 (inner borders)### Purple = rows 4-6 (central band)###### Letter placement (1-col margins each side, 1-col gap between letters):### B : cols 1-5 (5 wide x 9 tall, rows 1-9)### I : cols 7-9 (3 wide x 9 tall, rows 1-9, with serifs)### O : cols 11-15 (5 wide x 9 tall, rows 1-9)### cols 16-20 : right margin (filled with stripe colours)###### Letter pixels = None (no bead — exposed black agar reads as the letter)### Background pixels = stripe colour bead##############################################################################BEAD_SPACING_MM=4# 4mm spacing keeps pattern clear of dish edgeBEAD_VOLUME_UL=1MAX_ASPIRATE_UL=16def_make_pattern():"""
Build a 21-col x 11-row grid spelling BIO in negative space.
Horizontal Ndebele stripes fill the background.
Letter shapes are punched through as None cells (black agar).
Letter pixel maps — 1 = letter pixel (None), 0 = background fill.
Each letter is 5 wide x 9 tall (or 3 wide for I).
Row order in map: index 0 = top of letter, index 8 = bottom.
"""cols=21rows=11# --- Ndebele horizontal stripe background ---defstripe_color(r):ifrin(0,1):return'Blue'ifrin(2,3):return'Pink'ifrin(4,5,6):return'Purple'ifrin(7,8):return'Pink'return'Blue'# rows 9, 10# --- Letter pixel maps (1 = letter / negative space, 0 = background) ---# Row index 0 = top of letter, index 8 = bottom of letterB=[# cols 1-5[1,1,1,1,0],# top — same wide bump as bottom[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[1,1,1,1,0],# mid serif[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[1,1,1,1,0],# bottom — matches top]I=[# cols 7-9[1,1,1],# top serif[0,1,0],[0,1,0],[0,1,0],[0,1,0],[0,1,0],[0,1,0],[0,1,0],[1,1,1],# bottom serif]O=[# cols 11-15[0,1,1,1,0],# top arch[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[1,0,0,0,1],[0,1,1,1,0],# bottom arch]B_col,I_col,O_col=1,7,11# start columns for each lettergrid=[]forrinrange(rows):row=[]forcinrange(cols):is_letter=FalseifB_col<=c<B_col+5and1<=r<=9:map_row=8-(r-1)# flip: row 9 (top of letter) = map index 0map_col=c-B_colifB[map_row][map_col]==1:is_letter=TrueelifI_col<=c<I_col+3and1<=r<=9:map_row=8-(r-1)map_col=c-I_colifI[map_row][map_col]==1:is_letter=TrueelifO_col<=c<O_col+5and1<=r<=9:map_row=8-(r-1)map_col=c-O_colifO[map_row][map_col]==1:is_letter=Truerow.append(Noneifis_letterelsestripe_color(r))grid.append(row)returngridPATTERN=_make_pattern()################################################################################# OpentronsMock — simulation layer for Colab### Mimics the Opentrons API so run(protocol) works identically in Colab### and on the real robot. Records every drop for visualize().##############################################################################classOpentronsMock:class_Point:def__init__(self,x=0.0,y=0.0,z=0.0):self.x=x;self.y=y;self.z=zclass_Location:def__init__(self,x=0.0,y=0.0,z=0.0,name=''):self.name=nameself.point=OpentronsMock._Point(x,y,z)deftop(self,z=0):returnOpentronsMock._Location(self.point.x,self.point.y,self.point.z+z,self.name)defmove(self,pt):returnOpentronsMock._Location(self.point.x+pt.x,self.point.y+pt.y,self.point.z+pt.z,self.name)class_Well:def__init__(self,name,x=0.0,y=0.0,z=0.0):self.name=nameself.point=OpentronsMock._Point(x,y,z)self._loc=OpentronsMock._Location(x,y,z,name)deftop(self,z=0):returnself._loc.top(z)defmove(self,pt):returnself._loc.move(pt)class_Labware:def__init__(self,wells):self._wells=wellsdef__getitem__(self,k):returnself._wells[k]defwell(self,k):returnself._wells[k]class_TempModule:def__init__(self,plate):self._plate=platedefload_labware(self,*a):returnself._plateclass_Pipette:def__init__(self,mock):self._mock=mockself._color=Noneself._volume=0.0self.starting_tip=Nonedefpick_up_tip(self):passdefdrop_tip(self):self._color=None;self._volume=0.0defmove_to(self,loc):passdefaspirate(self,volume,location):self._volume+=volumeself._color=self._mock.well_colors.get(getattr(location,'name',''),None)defdispense(self,volume,location):ifself._color:pt=location.pointself._mock._drops.append((pt.x,pt.y,volume,self._color))self._volume=max(0,self._volume-volume)class_Types:classPoint:def__init__(self,x=0,y=0,z=0):self.x=x;self.y=y;self.z=zdef__init__(self,well_colors):self.well_colors=well_colorsself.types=self._Types()self._drops=[]color_wells={name:self._Well(name,i*9,0)fori,nameinenumerate(well_colors)}self._color_plate=self._Labware(color_wells)self._tip_rack=self._Labware({f"{'ABCDEFGH'[r]}{c}":self._Well(f"{'ABCDEFGH'[r]}{c}")forrinrange(8)forcinrange(1,13)})agar_well=self._Well('A1',0,0,0)self._agar_plate=self._Labware({'A1':agar_well})self._temp_mod=self._TempModule(self._color_plate)self._pipette=self._Pipette(self)defload_labware(self,name,slot,label=''):if'tiprack'inname:returnself._tip_rackif'agar'inname:returnself._agar_plateif'aluminum'inname:returnself._color_platereturnself._Labware({})defload_instrument(self,*a):returnself._pipettedefload_module(self,*a):returnself._temp_moddefcomment(self,msg):print(msg)defvisualize(self,bead_radius_mm=1.3,figsize=(14,10)):# Local imports — safe even if pd/plt were deleted by del np, pd aboveimportpandasaspdimportmatplotlib.pyplotaspltimportmatplotlib.patchesaspatchesfrommatplotlib.colorsimportto_rgbaifnotself._drops:print("No drops recorded — check that run(protocol) completed.")returncolor_map={'Purple':'#CC00FF',# violet fluorescent protein emission'Pink':'#FF69B4',# pink/mCherry variant emission'Blue':'#0066FF',# BFP — bright blue emission}fig,ax=plt.subplots(figsize=figsize)ax.set_facecolor('#000000')fig.patch.set_facecolor('#000000')xs=[d[0]fordinself._drops]ys=[d[1]fordinself._drops]pad=bead_radius_mm*6# True circle petri dish — use the larger span as the radius so# the dish is always round and all beads sit inside itcx=(min(xs)+max(xs))/2cy=(min(ys)+max(ys))/2r=max((max(xs)-min(xs))/2,(max(ys)-min(ys))/2)+pad*1.8# single radius → circle# Set view limits to fully contain the circlemargin=bead_radius_mm*2ax.set_xlim(cx-r-margin,cx+r+margin)ax.set_ylim(cy-r-margin,cy+r+margin)ax.set_aspect('equal')# Show x/y axes with mm measurements — matches original Colab renderingax.set_xlabel('x position (mm)',color='white',fontsize=11)ax.set_ylabel('y position (mm)',color='white',fontsize=11)ax.tick_params(colors='white',labelsize=9)forspineinax.spines.values():spine.set_edgecolor('#555')ax.add_patch(plt.Circle((cx,cy),r,color='#000000',zorder=0))ax.add_patch(plt.Circle((cx,cy),r,fill=False,edgecolor='#555',linewidth=2.5,zorder=1))# Bead radius — slightly smaller than half the spacing so there is a# visible gap between every dot, matching the original code's styledot_r=bead_radius_mm*0.75# Draw each bead — flat colour only, no shadow or highlightfor(x,y,vol,color_name)inself._drops:hex_color=color_map.get(color_name,'#999999')ax.add_patch(plt.Circle((x,y),dot_r,color=hex_color,zorder=3))# Legendused=sorted(set(d[3]fordinself._drops))ax.legend(handles=[patches.Patch(color=color_map.get(c,'#999'),label=c)forcinused],loc='upper right',facecolor='#2a2a2a',edgecolor='#555',labelcolor='white',fontsize=11,framealpha=0.85)ax.set_title('Ndebele — BIO in Negative Space',color='white',fontsize=14,fontweight='bold',pad=14)plt.tight_layout()plt.show()# Summarydf=pd.DataFrame(self._drops,columns=['x_mm','y_mm','vol_ul','color'])print("\n=== Dispensing Summary ===")print(df.groupby('color').agg(beads=('vol_ul','count'),total_ul=('vol_ul','sum')))print(f"\nTotal beads dispensed : {len(self._drops)}")print(f"Total volume dispensed: {df['vol_ul'].sum():.0f} µL")defrun(protocol):################################################################################# Load labware, modules and pipettes############################################################################### Tipstips_20ul=protocol.load_labware('opentrons_96_tiprack_20ul',TIP_RACK_DECK_SLOT,'Opentrons 20uL Tips')# Pipettespipette_20ul=protocol.load_instrument("p20_single_gen2","right",[tips_20ul])# Modulestemperature_module=protocol.load_module('temperature module gen2',COLORS_DECK_SLOT)# Temperature Module Platetemperature_plate=temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul','Cold Plate')# Choose where to take the colors fromcolor_plate=temperature_plate# Agar Plateagar_plate=protocol.load_labware('htgaa_agar_plate',AGAR_DECK_SLOT,'Agar Plate')## TA MUST CALIBRATE EACH PLATE!# Get the top-center of the plate, make sure the plate was calibrated before running thiscenter_location=agar_plate['A1'].top()pipette_20ul.starting_tip=tips_20ul.well(PIPETTE_STARTING_TIP_WELL)################################################################################# Patterning#################################################################################### Helper functions for this lab#### pass this e.g. 'Red' and get back a Location which can be passed to aspirate()deflocation_of_color(color_string):forwell,colorinwell_colors.items():ifcolor.lower()==color_string.lower():returncolor_plate[well]raiseValueError(f"No well found with color {color_string}")# For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)defdispense_and_detach(pipette,volume,location):"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""assert(isinstance(volume,(int,float)))above_location=location.move(types.Point(z=location.point.z+5))# 5mm abovepipette.move_to(above_location)# Go to 5mm above the dispensing locationpipette.dispense(volume,location)# Go straight downwards and dispensepipette.move_to(above_location)# Go straight up to detach drop and stay high###### YOUR CODE HERE to create your design###num_rows=len(PATTERN)num_cols=len(PATTERN[0])# Group bead positions by color to minimize tip changes.# Shared edge beads are de-duplicated by the seen set.color_order=['Blue','Pink','Purple']# only colours in useseen=set()schedule={color:[]forcolorincolor_order}forrow_idx,rowinenumerate(PATTERN):forcol_idx,colorinenumerate(row):ifcolorisNone:continue# no drop for open/white zonespos=(row_idx,col_idx)ifposinseen:continue# de-duplicate shared edge beadsseen.add(pos)schedule[color].append(pos)# Dispense all beads of each color before moving to the nextforcolorincolor_order:positions=schedule[color]ifnotpositions:continue# Split into chunks that fit within one tip's max aspirate volumechunk_size=MAX_ASPIRATE_UL//BEAD_VOLUME_ULchunks=[positions[i:i+chunk_size]foriinrange(0,len(positions),chunk_size)]forchunkinchunks:pipette_20ul.pick_up_tip()pipette_20ul.aspirate(len(chunk)*BEAD_VOLUME_UL,location_of_color(color))for(row,col)inchunk:# Center the pattern on the platex_offset=(col-(num_cols-1)/2.0)*BEAD_SPACING_MMy_offset=(row-(num_rows-1)/2.0)*BEAD_SPACING_MMadjusted_location=center_location.move(types.Point(x=x_offset,y=y_offset))dispense_and_detach(pipette_20ul,BEAD_VOLUME_UL,adjusted_location)# Clean up!pipette_20ul.drop_tip()# Execute Simulation / Visualization -- don't change this code blockprotocol=OpentronsMock(well_colors)run(protocol)protocol.visualize()
AI Usage Documentation
Claude (Anthropic) was used throughout the coding process to:
Translate the geometric logic of Ndebele patterns into Python grid coordinates
Debug the pattern generation and visualization code
Optimize the bead dispensing schedule to minimize tip changes
Create the negative space typography effect for the “BIO” lettering
Implement the OpentronsMock simulation class for Colab testing
A directly relevant paper is Fang et al. (2025) in Nature Communications, which demonstrates circadian-gated gene expression circuits in bacteria, using automated temporal sampling to characterize rhythmic protein output over 24-hour cycles. This paper is not a peripheral reference; it is one of the primary foundational sources for my final project concept and is already cited in my main project documentation. The automation approach used to verify rhythmic expression in that work is precisely what I intend to replicate and extend with the Opentrons platform.
What I Intend to Automate
My project proposes a bacterial AND gate where the antimicrobial peptide Magainin is only expressed when two conditions are simultaneously true: the circadian regulator RpaA is active, and a pathogen signal is present. The core experimental challenge is verifying this gate actually works as designed, which requires sampling bacterial expression levels repeatedly across a full 24-hour cycle, under multiple conditions, without human error or gaps overnight. This is the automation task.
The Opentrons OT-2 would run an unattended 24-hour sampling protocol across three experimental conditions:
RpaA active + pathogen signal present (AND gate should trigger)
RpaA active + no pathogen signal (gate should stay silent)
RpaA inactive + pathogen signal present (gate should stay silent)
At each 2-hour timepoint, the robot samples each culture well, transfers to a measurement plate for fluorescence reading, and replaces the sampled volume with fresh media to keep cultures alive. This builds a full temporal expression profile across all three conditions without any overnight manual intervention.
I would use Claude for the coding and guidance in the technical parts of this.
Why This Automation Matters
The AND gate only has meaning if you can show it is silent when it should be silent and active only at the right circadian phase with the right pathogen or other signal. That requires clean data across all three conditions at every 2-hour window through the night. Manual pipetting at 2am introduces the exact inconsistency that would make the rhythmic signal unreadable. The Opentrons removes that variable entirely.
Future Extensions
If access to Ginkgo Nebula becomes available, the next step would be submitting the AND gate genetic construct for scaled fermentation and characterization; using Nebula’s high-throughput infrastructure to screen circuit variants with different RpaA promoter strengths or pathogen-sensing thresholds, generating the kind of combinatorial data that would take months on a single benchtop robot.
Fang et al. (2025). “Circadian-gated gene expression circuits in bacteria.” Nature Communications
UCSD (2024). “Researchers Rebuild Microscopic Circadian Clock.” University of California San Diego press release
Bilska et al. (2021). “Circadian rhythm in skin barrier function and antimicrobial peptides.” Experimental Dermatology
Software & Tools Used
Google Colab - Python script development and testing for Opentrons protocols
Python - Opentrons protocol scripting and pattern generation
Imgur - Image hosting for project visualization and Ndebele pattern references
Cultural & Mathematical Inspiration
Ndebele bead patterns and geometric design principles
Ethnomathematics - Indigenous mathematical knowledge systems
Traditional South Ndebele aesthetic and symmetry patterns
Project Concepts Explored
Circadian-controlled bacterial pigment systems
Light-responsive color-changing bacteria
UV-protective bioplastic materials with bacterial pigments
Mechanotransduction experiments with bacterial cultures
Bacterial AND gate with circadian gating (RpaA + pathogen signal triggering Magainin expression)
Cost Considerations
Twist Bioscience DNA synthesis pricing
Remote lab assistance availability assessment
UK-based protein order logistics and costs
AI Assistance
Claude (Anthropic) - Code development and technical guidance
Model: Claude Sonnet 4.5
Date(s) used: February, 2026
Tasks: Assisted with Python script development for Opentrons Ndebele pattern generation (“vibe-coding”), debugging protocol scripts and verifying scientific terminology.
Future Platforms
Ginkgo Nebula - Potential platform for scaled fermentation and high-throughput circuit variant screening
Additional Resources
HTGAA final project guidelines and requirements
Twist Bioscience pricing documentation
Remote lab capabilities at available nodes
Opentrons protocol documentation and API reference
Ndebele art and design pattern libraries
Ethnomathematics literature
Acknowledgments
Course instructors
TAs
Ndebele cultural heritage for geometric design inspiration
Week 4 HW: Protein Design Part 1
Part A: Conceptual Questions
How many molecules of amino acids do you take with a piece of 500g of meat? (avg amino acid ~100 Daltons)
Since I am a visual learner, I needed an analogy to try to grasp Daltons, grams, and moles. I imagine each amino acid as a finished LEGO model, and each tiny brick is a Dalton. When I weigh all the models together in a cupboard, I have 500 grams. I count how many moles by dividing the total mass by the mass of one model (-100 Daltons). Then, multiplying by Avogadro’s number, I see how many individual models I have in total. In scientific terms, I compute the number of moles by dividing 500 grams by 100 grams per mole. Then, I multiply by Avogadro’s number,(According to Google search Avogadro’s constant is the number of particles, like atoms or molecules, in one mole of a substance, equal to approximately 6.022 times 10 to the 23) 6.022 times 10 to the 23, and that yields approximately 3.0 times 10 to the 24 molecules of amino acid.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We don’t become a cow or a fish, because we’re only using parts of the cow and fish to continue building on what already exists, which is our human body. In other words, digestion breaks down the proteins into amino acids, and then our body uses its own genetic instructions to reassemble those pieces into human proteins, ensuring we stay uniquely ourselves.
Why are there only 20 natural amino acids?
There are only 20 natural amino acids because, although their combinations can form infinite possibilities, evolution only needed these 20 to create all the proteins we rely on. Their chemical properties allow for immense diversity in protein structure, and this set is perfectly suited to the way DNA encodes and guides their assembly, giving us the versatility we need without adding more complexity.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed from simple chemicals dissolved in warm ocean water on early Earth, where energy sources like heat or lightning sparked chemical reactions. In a way, you can think of a modern dam as a kind of micro-ecosystem—just as water and energy flow through a dam, creating pockets of life, early oceans created the right conditions for these amino acids to form, eventually leading to the first building blocks of life.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Depending on the amino acids you use, a typical alpha helix forms a right-handed spiral when built from L-amino acids. However, if you use D-amino acids, the entire helix reverses its handedness and forms a left-handed spiral. It is important to note that not all amino acids form alpha helices; some sequences prefer other structures like beta sheets.So, the handedness and structure depend on both the amino acid type and the overall sequence, which determines how the chain folds and stabilizes.
Can you discover additional helices in proteins?
Because proteins have long chains of different amino acids, they can fold in all sorts of ways, and that means new helices or other patterns can be identified within them, especially with advanced tools like structural prediction or experimental techniques.
Why are most molecular helices right-handed?
The right-handedness is actually favored in DNA due to the natural chirality of its sugar backbone and the way it interacts with water and other molecules. So, that structural preference is built right into how the backbone forms. Keratin and hair curl were helpful for me to think about.
Why do β-sheets tend to aggregate?
So, beta sheets tend to aggregate because, like silk fibroin, they form straight, aligned strands that stack side by side. In silk, these parallel sheets create strong, stable fibers, but in proteins, this same alignment lets the sheets stack excessively, exposing those hydrogen bonds and promoting aggregation. So, just like silk’s strength comes from its sheet alignment, aggregation in proteins happens when these sheets stack and bind too readily.
What is the driving force for β-sheet aggregation?
Why do many amyloid diseases form β-sheets?
Because these beta sheets stack so easily, they misfold and form these stable aggregates. In diseases like Alzheimer’s or Parkinson’s, these aggregated beta sheets build up, disrupting normal cell function and triggering the disease process. Tau tangles are a classic example of beta sheet misfolding driving disease.
Can you use amyloid β-sheets as materials?
Since silk fibroin is based on beta sheets and is already a natural, strong material, researchers have been exploring ways to harness amyloid beta sheets similarly. Amyloid structures are extremely stable, so with careful design, scientists are looking at them as potential biomaterials
Part B: Protein Analysis and Visualization**
Briefly describe the protein you selected and why you selected it.
Length
117 amino acids. Most frequent amino acid (image still needed from Colab output)
How long is it? What is the most frequent amino acid?
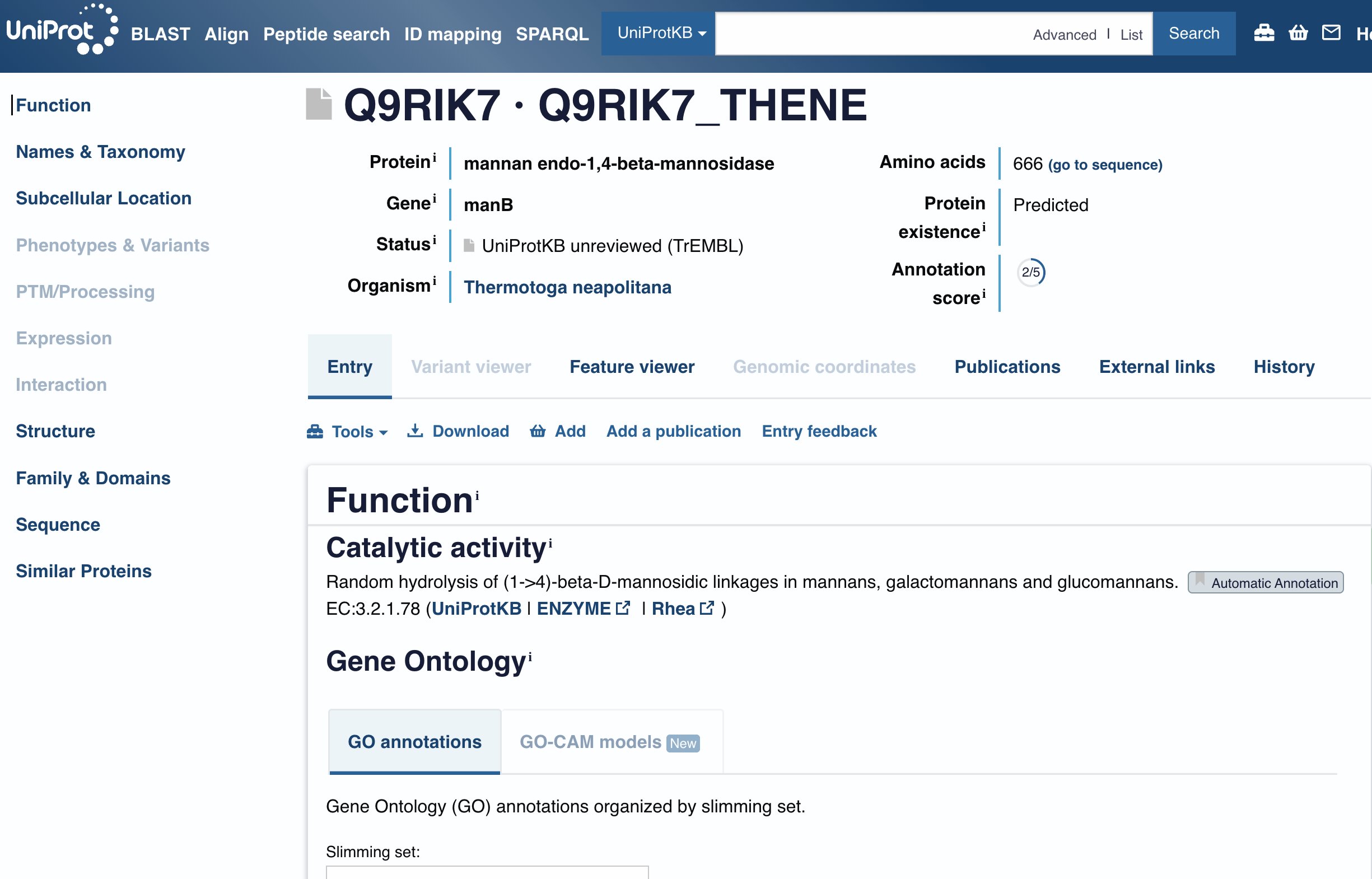
Homologs
250 homologs found via UniProt BLAST. Top matches from rat, mouse, human, and bovine this indicating strong conservation across mammals reflecting the fundamental evolutionary importance of this protein.
How many protein sequence homologs are there? (Use UniProt BLAST)
Protein family
GABARAPL2 belongs to the ATG8 family, part of the broader GABARAP subfamily of autophagy-related proteins.
Does your protein belong to any protein family?
RCSB structure page
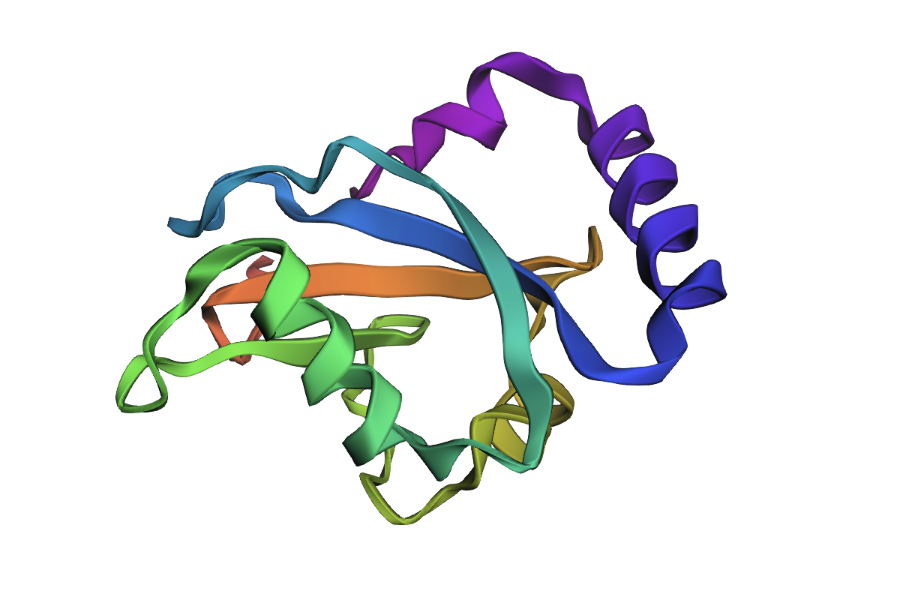
PDB entry 7LK3. Crystal structure of untwinned human GABARAPL2.
Identify the structure page of your protein in RCSB.
The most frequent amino acid is Lysine (K), appearing 12 times (10.3% of the sequence).
When was the structure solved? Is it good quality? (Resolution: smaller = better, aim < 2.70 Å)
Structure quality
Deposited February 1, 2021, released May 12, 2021. Resolution of 1.90 Å — excellent quality, well below the 2.70 Å threshold.
Are there any other molecules in the solved structure apart from protein?
Other molecules
Yes. 1,2-Ethanediol (EDO) is present as a ligand.
Does your protein belong to any structure classification family?
GABARAPL2 belongs to the ubiquitin-like superfamily under the beta-grasp fold in SCOP classification. Like other ATG8 proteins, GABARAPL2 is comprised of an N-terminal helical extension preceding four beta-sheets in a ubiquitin-like beta-grasp fold.
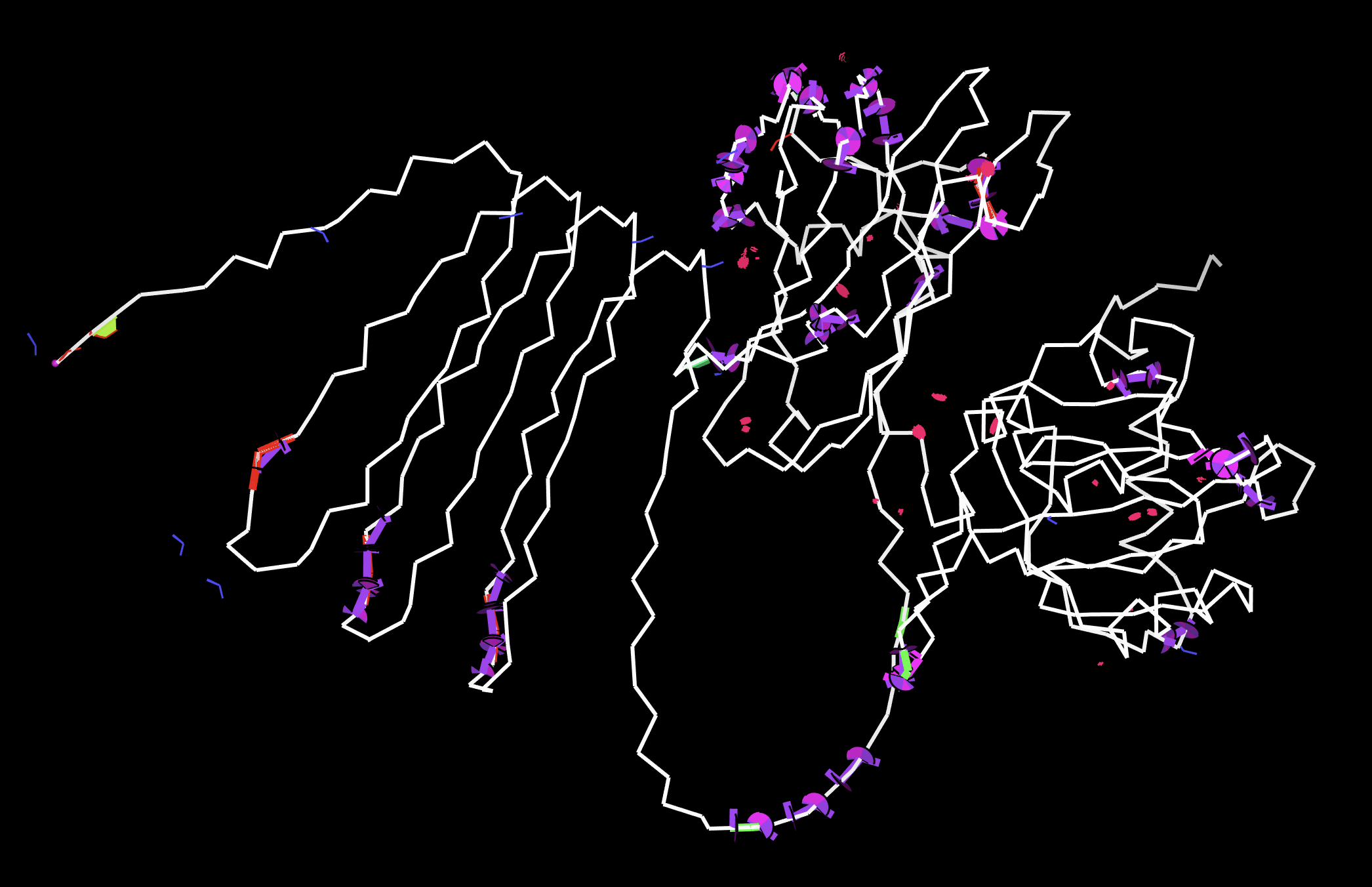
Open the structure in 3D visualization software (PyMol):
Visualize as “cartoon”, “ribbon”, and “ball and stick”
Color by secondary structure — more helices or sheets?
Color by residue type — hydrophobic vs hydrophilic distribution?
Visualize the surface — any binding pockets?
When colored by secondary structure, GABARAPL2 shows a clear dominance of red (helices) over yellow (beta sheets). Green loops connect these elements throughout the structure.
Hydrophobic residues (orange) concentrate in the protein core, while blue dominates the outer surface. This showcases hydrophobic residues being hidden in the middle away from the aqueous environment.
This surface visualization reveals a clear hydrophobic indentation in the middle of the structure, corresponding to the LIR docking site where GABARAPL2 interacts with autophagy receptors.
Part C: Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan based on language model likelihoods
Can you explain any particular pattern? (choose a residue and mutation that stands out)
(Bonus) Compare language model predictions to experimental scans
The brightest yellow spot in the heatmap appears at position 60, mutation to Glutamate (E), meaning the model predicts this change would be highly favorable. This makes sense in a metabolic context, as Glutamate’s charged nature supports the protein’s membrane interactions during autophagy and fasting states.
The darkest purple spots appear around positions 54-57 at Cysteine (C) and at position 64 at Tryptophan (W), meaning the model strongly disfavors these mutations. Cysteine in particular stands out as consistently disfavored
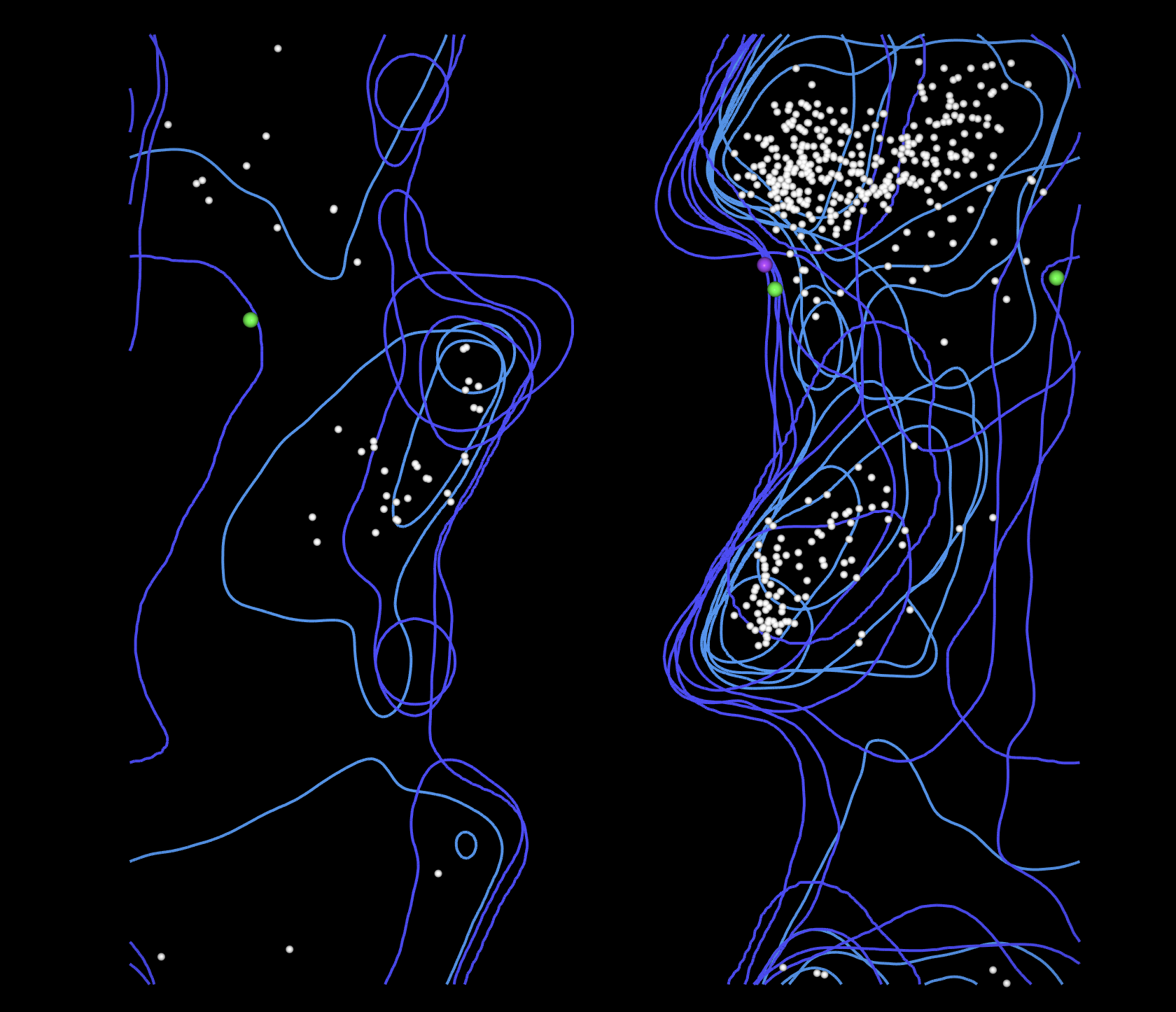
Latent Space Analysis
Embed proteins in reduced dimensionality using the provided sequence dataset
Analyze neighborhoods — do they approximate similar proteins?
Place your protein in the map and explain its position and similarity to neighbors
The 3D t-SNE plot shows a large dense central cluster of proteins with outliers scattered at the edges. Proteins in the same neighborhood share similar sequence embeddings, suggesting structural and functional similarity. GABARAPL2, as a member of the highly conserved ubiquitin-like superfamily, would likely position itself near the central core of the cloud, close to other small globular autophagy and ubiquitin-related proteins. Its neighbors would likely include other ATG8 family members
C2. Protein Folding
Fold your protein with ESMFold — do predicted coordinates match the original structure?
Try mutations, then larger sequence changes — is the structure resilient?
The ESMFold predicted structure closely matches the original crystal structure. Both show the characteristic beta-grasp fold with a central beta sheet core surrounded by helices, and the overall globular compact shape is preserved.
C3. Protein Generation
Use ProteinMPNN to inverse-fold your protein backbone and propose sequence candidates
Analyze predicted sequence probabilities vs the original sequence
Input the new sequence into ESMFold and compare the predicted structure to original
___
ProteinMPNN generated 4 sequence candidates from the 7LK3 backbone with sequence recovery rates between 46–52% and consistent scores around 0.81–0.84. The probability heatmap shows scattered high-confidence positions (yellow) where the backbone strongly constrains the amino acid choice, surrounded by flexible positions with lower confidence. Despite roughly half the sequence changing, the backbone fold is preserved suggesting that many different sequences can encode the same GABARAPL2 structure.
When the new ProteinMPNN sequence was folded with ESMFold, the overall shape stayed the same. But there were some small differences: the helices shifted slightly, the beta sheets moved a little, and the central loop region pulled closer together. This suggests that even though roughly half the amino acids changed, the protein still folds into essentially the same shape. The structure is resilient.
Part D: Group Brainstorm on Bacteriophage Engineering
Decided to try option 3, as if it fails, it still could help eliminate a possible pathway to end goal and just seemed more interesting.
General reminder note: Loop regions and terminal extensions are safer engineering targets than core structural elements.
Higher Toxicity of the MS2 Lysis Protein:
Goal: Increase the toxicity of the MS2 L protein so it lyses bacterial cells faster and more completely.
Approach:
Use a protein language model (ESM or similar) to identify which amino acid positions in the L protein are most likely involved in membrane disruption
Propose mutations at those positions using ProteinMPNN to suggest alternative amino acids that might make membrane interaction more aggressive
Use AlphaFold-Multimer to model how the mutant L protein interacts with its bacterial target (DnaJ and the membrane)
Compare predicted binding strength and structural changes between original and mutant versions
Why these tools help:
Language models capture evolutionary patterns across many proteins, helping identify positions where changes are most likely to matter
AlphaFold-Multimer lets you check if your proposed mutations actually change how the protein docks with its bacterial target
Potential pitfalls:
The exact mechanism of membrane disruption by the L protein is not fully understood, so mutations may target the wrong part of the protein or “drill” for my analogy reference.
Limited training data exists specifically for phage-bacteria lysis interactions, so predictions may be less reliable than for well-studied proteins
Pipeline schematic first draft:
L protein sequence → ESM (identify key positions) → ProteinMPNN (propose mutations) → AlphaFold-Multimer (validate structure and interaction) → compare mutant vs original
References & Resources
Lecture Materials
Week 4 Lecture - Protein Design Part I, Thras Karydis, Jon Kaufman
Week 4 Lab - Protein Design I, February 26-27, 2026
Protein Analyzed
GABARAPL2 (GABA Type A Receptor Associated Protein Like 2)
UniProt ID: sp|P60520|GBRL2_HUMAN
PDB Structure: 7LK3 (1.90 Å resolution, deposited Feb 2021, released May 2021)
t-SNE - Dimensionality reduction for latent space analysis
Imgur - Image hosting for visualization documentation
Required Readings
GABARAPL2 autophagy function literature
ATG8 family protein structure and function papers
Protein folding and stability principles
Amyloid formation and beta-sheet aggregation mechanisms
AI Assistance
Claude (Anthropic) - Protein analysis and ML tool interpretation
Model: Claude Sonnet 4.5
Date(s) used: February, 2026
Tasks: Assisted with understanding protein structure visualization principles, interpreting ESM2 deep mutational scan results, explaining t-SNE embeddings and protein neighborhoods, clarifying ProteinMPNN sequence recovery metrics, helped develop analogies for complex concepts and checked if homework correct.
Bacteriophage Engineering Project
Option 3: Increase MS2 lysis protein (L protein) toxicity
Tools: ESM language model, ProteinMPNN, AlphaFold-Multimer
Target: Enhanced membrane disruption and faster bacterial lysis
Additional Resources
SCOP protein structure classification database
MS2 bacteriophage literature
Membrane disruption mechanism papers
DnaJ protein interaction studies
Acknowledgments
Course instructors for protein design tutorials
TAs for PyMOL visualization assistance
Colab community for ML tool notebooks
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Background
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine to Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Used GOOGLE GEMINI WITH INTERPRETING AND UNDERSTANDING THE OUTPUT
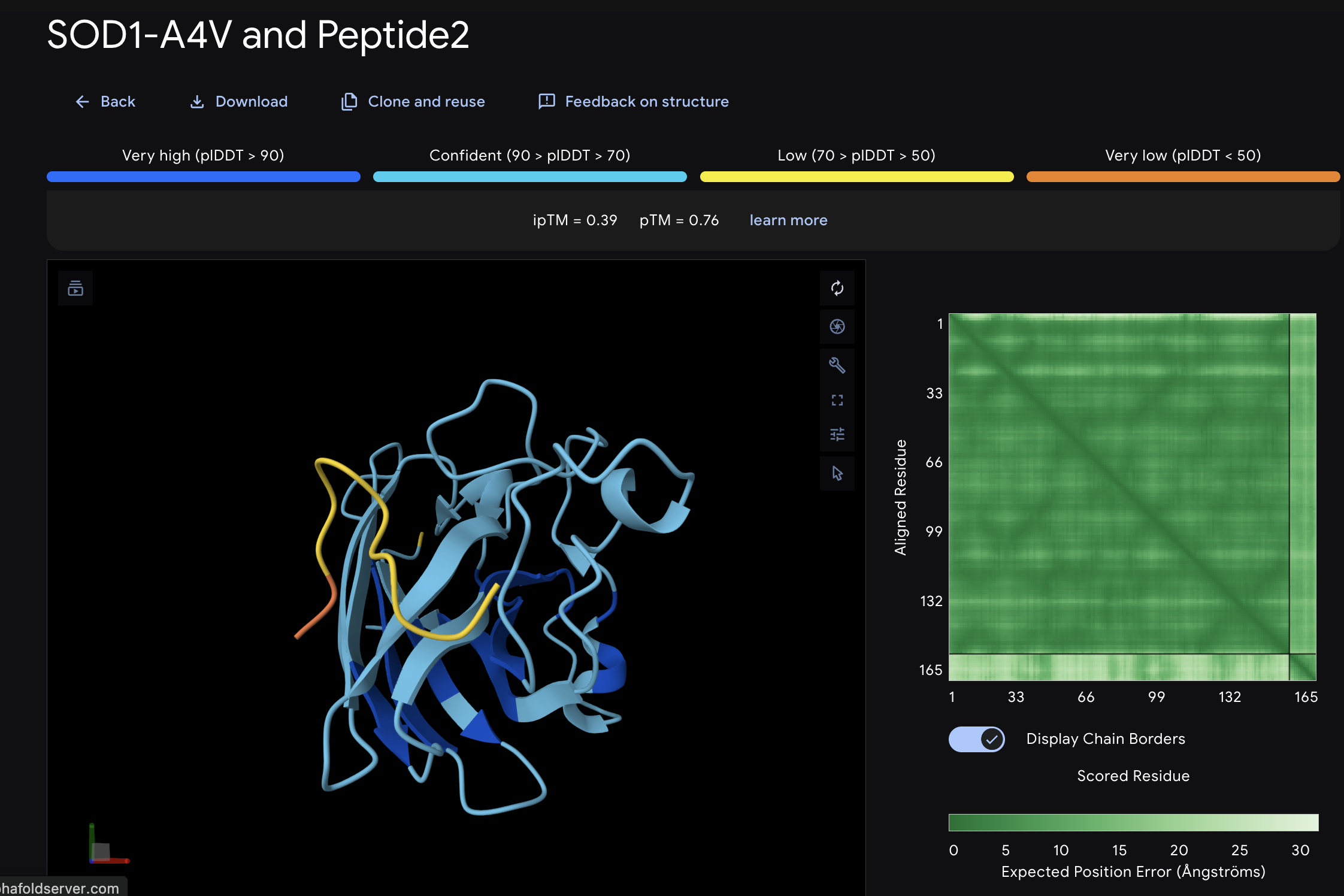
Evaluate Binders with AlphaFold3
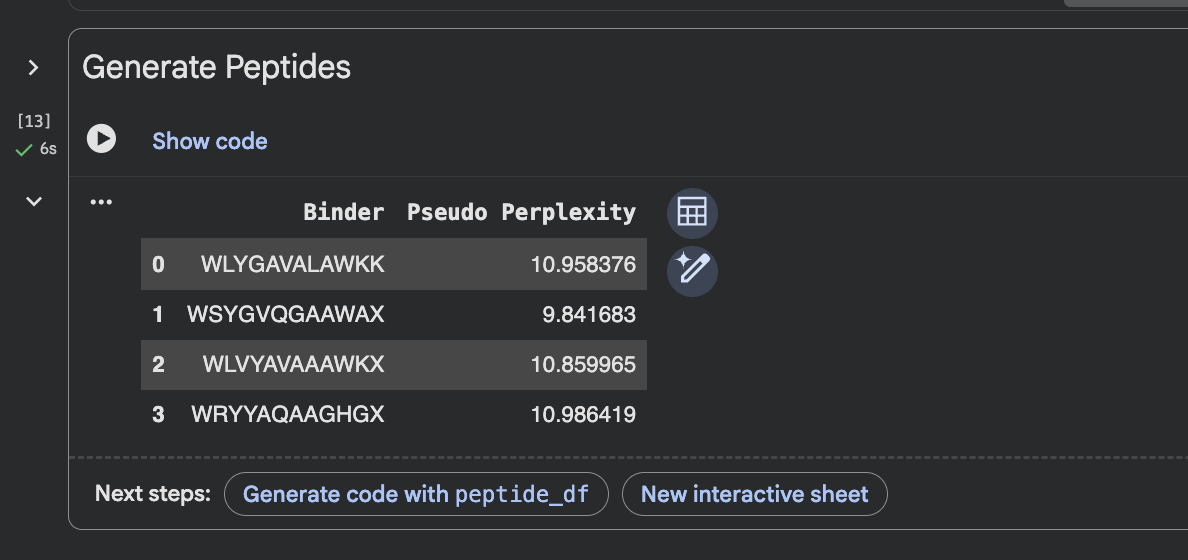
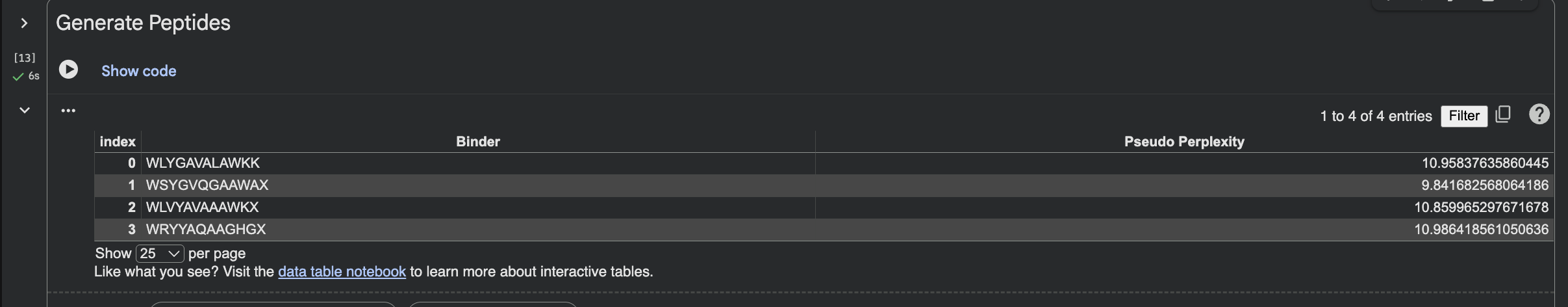
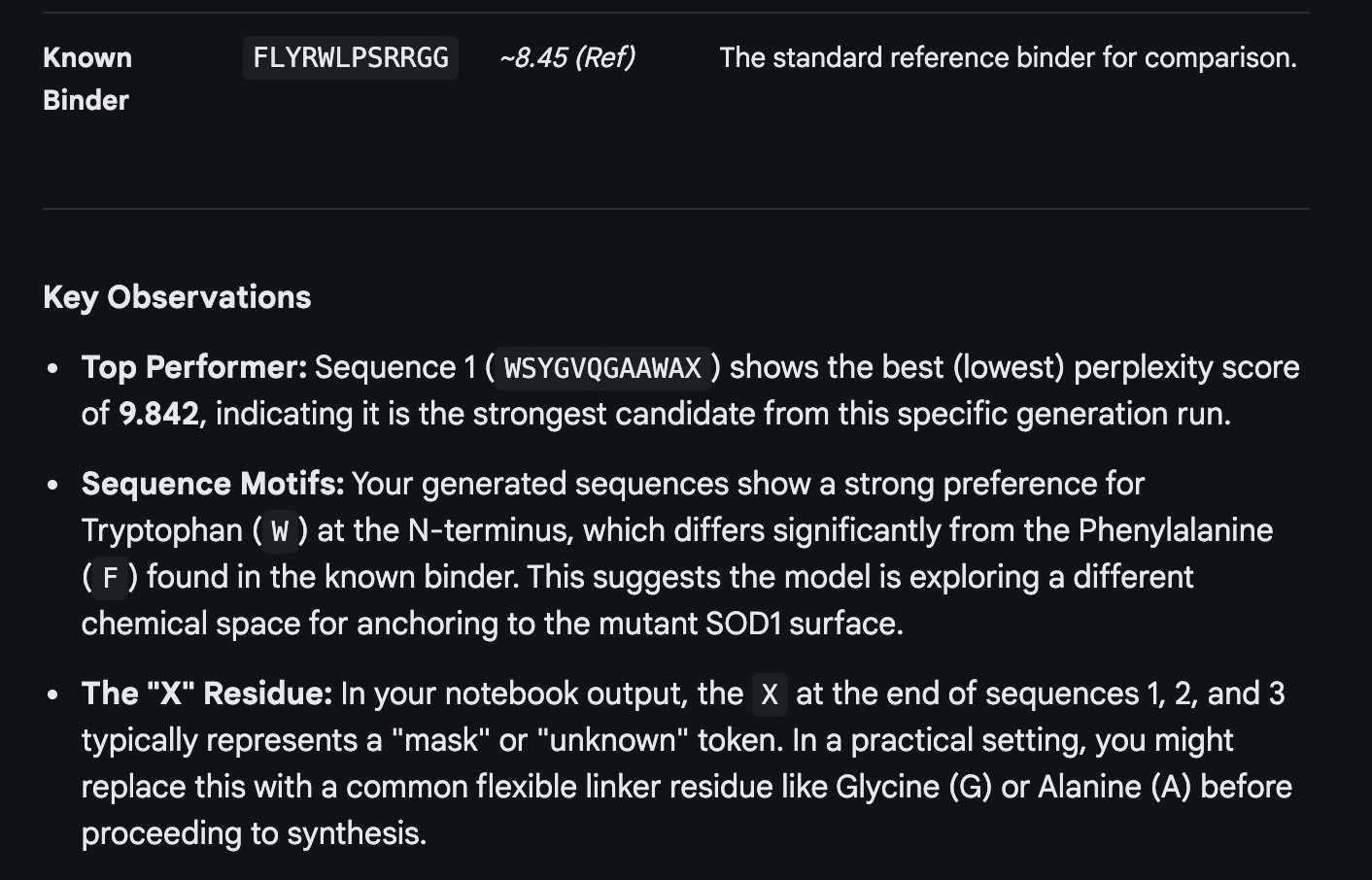
When I looked at the five structures, the known binder really stood out as it sat closely tucked against SOD1 and scored 0.73, which was by far the highest. You could see it engaging deeply with the protein. The PepMLM peptides told a different story. The best ones, Peptide3 and Peptide1, appeared to sit near the top of the protein around the loop region, but they looked more like they were resting on the surface rather than really grabbing onto it. Peptide0 was the weakest and it looked almost detached, just floating near the protein rather than making real contact. None of the generated peptides came close to the known binder, which shows that while PepMLM gave us a starting point, the peptides still need improvement to properly engage SOD1-A4V. (This was my favorite part due to the visuals).
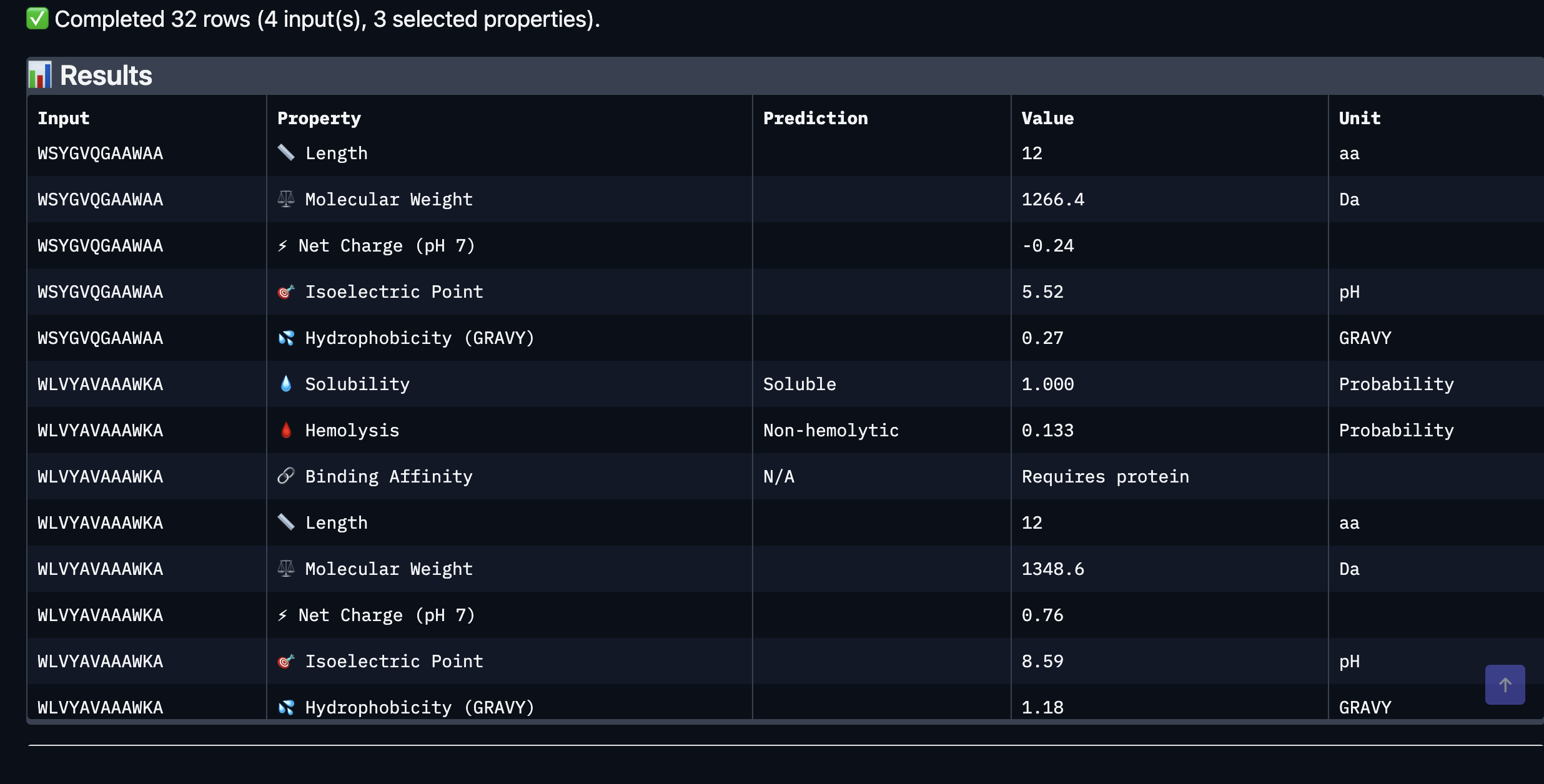
Part 3: Evaluate Properties with PeptiVerse
After evaluating the results below I would advance WLVYAVAAAWKA because it is the only peptide with medium binding affinity (7.247 pKd/pKi), compared to weak binding for the others. It is also well-balanced in terms of hemolysis risk with a low probability of 0.133, and its net charge of 0.76 at pH 7 is nearly neutral, which should help with both solubility and cellular uptake without causing charge-related toxicity. Although its ipTM score of 0.39 in AlphaFold3 was not the highest, the combination of improved predicted binding affinity and favorable therapeutic properties makes it the strongest candidate for further development. While Peptide3 had a slightly higher ipTM of 0.44, it showed the weakest predicted binding affinity of 5.498 and therefore does not balance structural and therapeutic properties as well
Part 4: Generate Optimized Peptides with moPPIt
It took 39min to run.
The moPPIt peptides differ from PepMLM in a key way: moPPIt allowed me to specify exactly which region of SOD1 I wanted to target, whereas PepMLM just generated peptides that looked plausible without that control. With Peptide 2 from moPPIt, I can see it’s actually engaging the N-terminal region where A4V sits, which is exactly what I designed it to do. PepMLM couldn’t guarantee that level of specificity.
Before advancing any peptide to clinical studies, I would need to do much more work. First, I’d validate the binding predictions with actual lab experiments measure real binding affinity. Most importantly, I’d likely run moPPIt again with different target regions on SOD1 in order to generate a larger panel of candidates and pick the best performers across all validation steps. No single computational prediction is enough to move forward to the clinical setting.
Part B: Optional
Part C: Final Project: L-Protein Mutants
The objective of this assignment is to improve the stability and auto-folding of the lysis protein of an MS2-phage. This mechanism is key to understanding how phages may help address antibiotic resistance.
After going through the readings, including the group final project document a Plan A would be: (This stays within scope, MurJ and multi-target approaches seem intersting though…)
1
Use computational tools like AlphaFold2 or ProteinMPNN to identify mutations that improve intrinsic stability and auto-folding of the lysis protein
2
Target mutations that strengthen the hydrophobic core, eliminate aggregation-prone regions, or introduce stabilising interactions like salt bridges
3
Engineer the lysis protein to fold correctly without requiring DnaJ or any other bacterial chaperone
4
Design mutations that also accelerate oligomerisation or enhance membrane pore-forming activity for faster lysis
5
Synthesise the mutant gene via Twist, clone into plasmid using Gibson Assembly, validate structural integrity with Nuclera, then test in E. coli.
References & Resources
Lecture Materials
Week 5 Lecture - Protein Design Part II, Pranam Chatterjee, Gabriele Corso
Week 5 Lab - Protein Design Part II Lab, March 5-6, 2026
Software & Tools Used
UNIPROT
PepMLM
Alphfold
Peptiverse
moPPIt
AI Assistance
Claude (Anthropic) - Protein design concepts
Model: Claude Sonnet 4.5
Date(s) used: March, 2026
Tasks: Acted as mentor (Skills) in conversations about unfamiliar and technical areas. Checked homework was correct.
Additional Resources
Advanced protein design literature
Computational protein engineering tools
Acknowledgments
Course instructors and TAs
Week 6 HW: Genetic Circuits Part I
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion HF PCR Master Mix contains several key components that are already pre-combined for convenience. Phusion DNA Polymerase is the core enzyme responsible for copying the DNA template, and it has a built-in proofreader to ensure it is high-fidelity, meaning it reduces errors during amplification. The dNTPs provide the nucleotide building blocks that get incorporated into the new DNA strand. MgCl₂ (magnesium) acts as an essential cofactor that activates the polymerase. The reaction buffer (oven conditions in my analogy) maintains the correct pH and ionic environment for the reaction to work. For this particular lab, precise mutagenesis of the amilCP chromophore region was required, so the high-fidelity polymerase is especially important; it ensures there are no unintended amino acid changes beyond the designed mutation.
What are some factors that determine primer annealing temperature during PCR?
Several factors affect the temperature at which a primer successfully binds to its target on the DNA template.
First, secondary structure is something to avoid. If a primer folds back on itself it is like a blurry photograph that cannot be read properly, meaning it cannot find its matching location on the template regardless of temperature.
Second, GC content affects annealing temperature. Primers with more G and C bases require higher temperatures because GC pairs bond more strongly than AT pairs. In this lab the backbone primers anneal at 57°C while the color insert primers anneal at 53°C, reflecting differences in their GC content.
Third, primer length matters. A longer primer is like a photograph that also shows the surrounding context, making it a more specific match. Longer primers bind more strongly and therefore require higher annealing temperatures.
These factors were carefully balanced during primer design, aiming for a Tm range of 52–58°C with primer pairs kept within 5°C of each other.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digests differ in terms of precision and flexibility. PCR is more flexible and suitable for bespoke mutation designs, giving you control over exactly where a fragment begins and ends by designing the primers yourself. Restriction enzyme digests are more limited in that they can only cut where their recognition sequence naturally exists in the DNA, but this makes them faster and more straightforward when you already know exactly which sequence you need.
I would use PCR when attempting to design a mutation, as in this lab where the chromophore color changes were introduced through deliberate primer mismatches. I would use restriction enzyme digests when the recognition sites are already conveniently placed and the desired sequences are already known, as this would save time.
In terms of protocol, PCR requires designing primers, running denature, anneal and extend cycles in a thermocycler, cleaning up the original template with a DpnI digest, and then purifying the DNA. Restriction enzyme digests are more straightforward, requiring only choosing the right enzyme for the recognition site, incubating the DNA with the enzyme at 37°C, and running a gel to confirm the correct cut. No heating cycles or template cleanup are needed.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
There are several ways to ensure fragments are appropriate for Gibson cloning.
First, correct overlaps must be present. In this lab the primers were designed from the start with 20-40bp overhangs complementary to the adjacent fragment, ensuring the fragments can recognise and join each other during assembly.
Second, fragment size should be confirmed by running a diagnostic gel. If the band appears at the wrong size then the PCR was unsuccessful and the fragment would not be appropriate for Gibson cloning.
Third, the DNA must be clean and concentrated enough. The Nanodrop measurement confirms concentration is above 30ng/µL. Contaminants from PCR can inhibit the Gibson Assembly reaction.
Fourth, the original template must be removed. The DpnI digest ensures the original methylated mUAV plasmid is not carried over, which would otherwise produce background colonies of the unmutated purple protein.
Finally, the correct molar ratio must be used. Gibson Assembly works best at a 2:1 insert to vector ratio to ensure efficient and complete assembly.
How does the plasmid DNA enter the E. coli cells during transformation?
The plasmid enters the E. coli cells through a process called heat shock transformation.
First, the cells are made chemically competent using CaCl₂. This partially neutralizes the repulsion between the negatively charged cell membrane and the negatively charged DNA, allowing the DNA to associate with the cell surface.
Next the cells are kept on ice, which makes the membrane more rigid and stable. Then the cells are heat shocked at exactly 42°C for 45 seconds, which temporarily disrupts the membrane and allows the plasmid to enter the cell by diffusion. The cells are then immediately returned to ice so the membrane stabilizes and closes again.
So essentially the process is: make the membrane rigid with ice, give it a heat shock to open it briefly, then put it back on ice to close it again with the plasmid now inside.
After heat shock, SOC media (Note my nutrient rich broth analogy) is added to help the cells recover and begin multiplying. Finally the cells are plated on chloramphenicol agar, where only cells that successfully received the plasmid will survive and grow.
Describe another assembly method in detail, such as Golden Gate Assembly. Explain the other method in 5 to 7 sentences plus diagrams, either handmade or online.
Golden Gate Assembly is a method of connecting DNA fragments together using custom 4 base sticky ends. It works by sending in a Type IIS restriction enzyme that acts like a self destructing instruction manual, cutting at a defined location outside its recognition site and then removing itself in the process, leaving behind unique sticky ends that have been designed to only connect to one specific matching partner. These sticky ends are self sorting, acting like magnets that can only attract their intended match and nothing else. Once the fragments are correctly joined the assembly is scarless, meaning no trace of the recognition site remains in the final product.
This differs from Gibson Assembly which uses an exonuclease, polymerase and ligase, and requires longer overlaps of 20-40bp between fragments rather than the 4 base sticky ends of Golden Gate. Golden Gate cycles between cutting and ligation temperatures repeatedly, whereas Gibson Assembly runs isothermally at 50°C. Because incorrect assemblies get re-cut and correct ones accumulate, Golden Gate is highly efficient and can assemble many parts simultaneously in one tube, making it more scalable than Gibson Assembly which typically handles two to six parts.
Model this assembly method with Benchling.
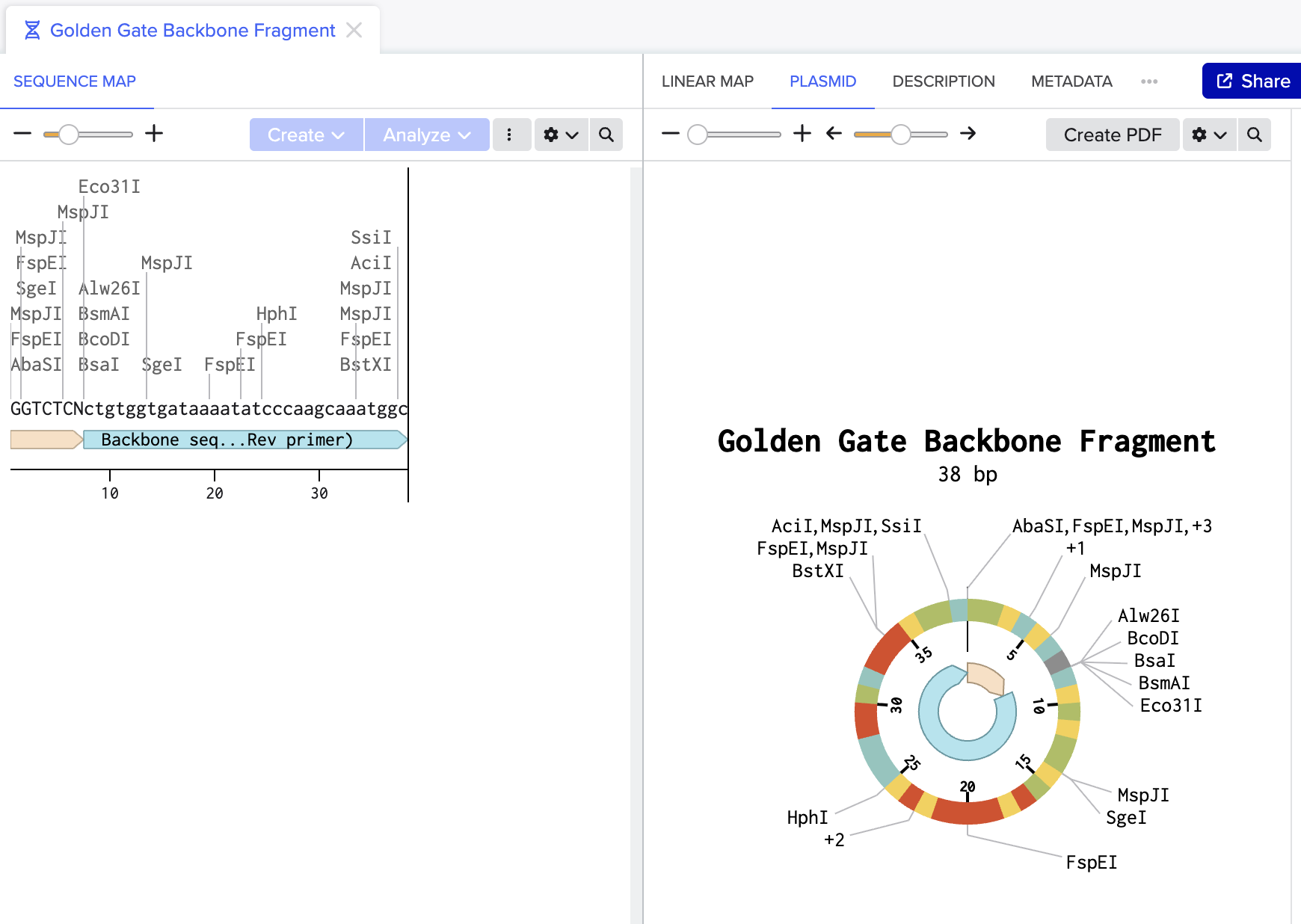
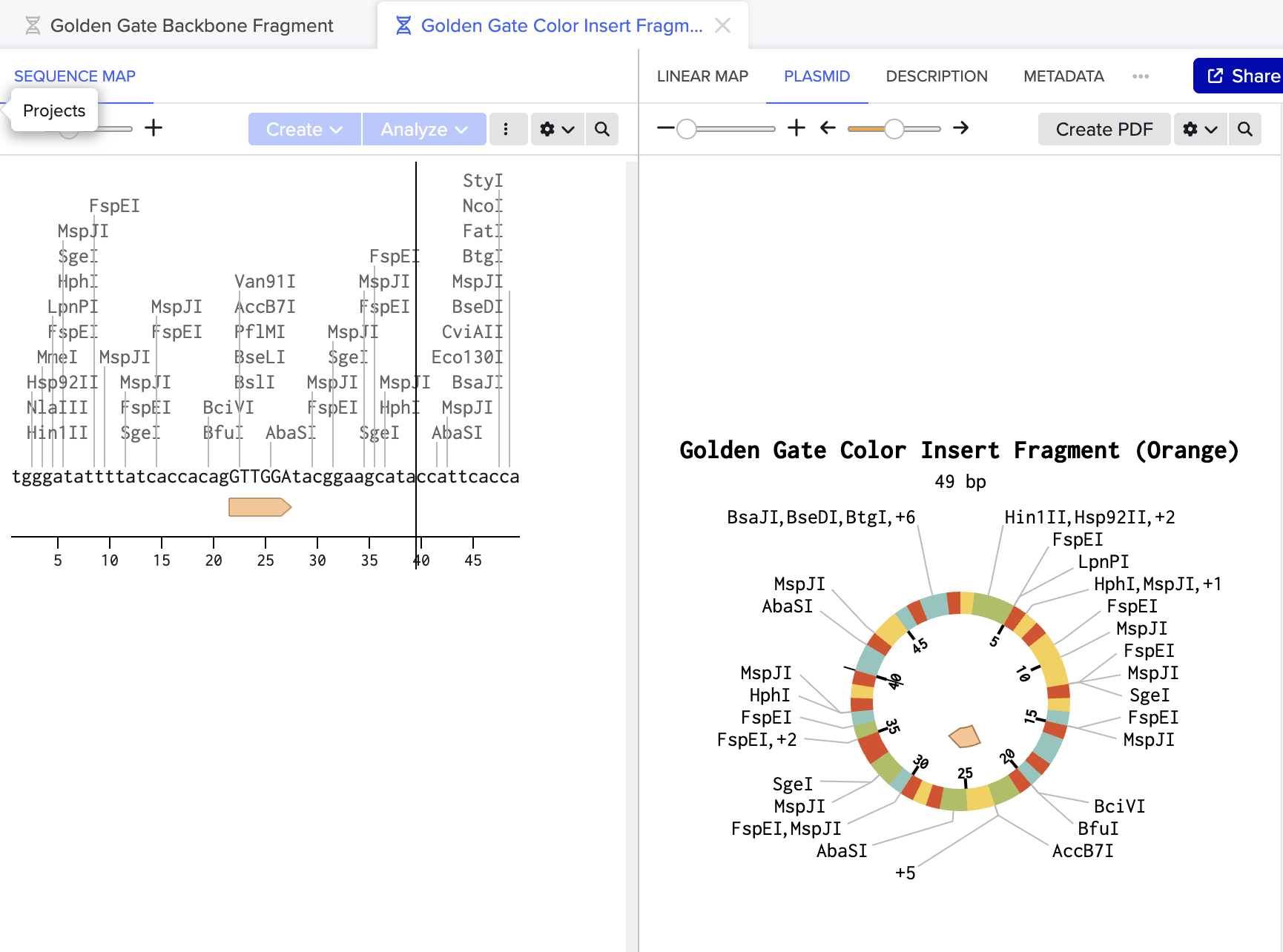
Golden Gate Assembly modeled in Benchling using mUAV plasmid (MG252981.1) as starting reference:
Figure 1: Backbone fragment (38bp) with BsaI recognition site (GGTCTCN) annotated at its end. BsaI cuts here, removing itself and exposing the sticky end.
Figure 2: Color insert fragment (49bp) containing the orange chromophore mutation GTTGGA replacing original TGTCAG. This sequence changes the chromophore amino acids to produce orange instead of purple.
Together these two fragments would be combined with BsaI and ligase in one tube to produce a scarless circular plasmid carrying the orange mutation.
Assignment: Asimov Kernel
Did not have access to Asimov Kernel. (Did attend the MIT Review and not sure if Nodes have access. Also, signed up to be beta tester when availible)
Tasks: Acted as mentor (Skills) in conversations about unfamiliar and technical areas. Checked homework was correct.
Additional Resources
Gibson assembly protocol documentation
Genetic circuit assembly technologies literature
Acknowledgments
Course instructors and TAs
Week 7 HW: Genetic Circuits Part II
Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits,whose input/output behaviors are Boolean functions?
Boolean genetic circuits are binary; a signal is either present or absent, on or off. IANNs add nuance by incorporating quantity: not just whether a signal is present, but how much, and how that amount combines with other weighted inputs to determine output. This matters biologically because cells are not rigid systems. Gene expression fluctuates due to stochastic noise and biological drift. Boolean circuits are brittle in this context, while IANNs, by distributing computation across many weighted inputs, are more robust to that natural variability.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Many inflammatory diseases are circadian-gated. Asthma attacks, rheumatoid arthritis flares, and cardiovascular events cluster at specific phases of the biological clock. A Boolean circuit cannot capture this; it can detect whether inflammation is present, but not whether it is occurring at the wrong time. That distinction is clinically meaningful, and it is what an IANN could resolve.
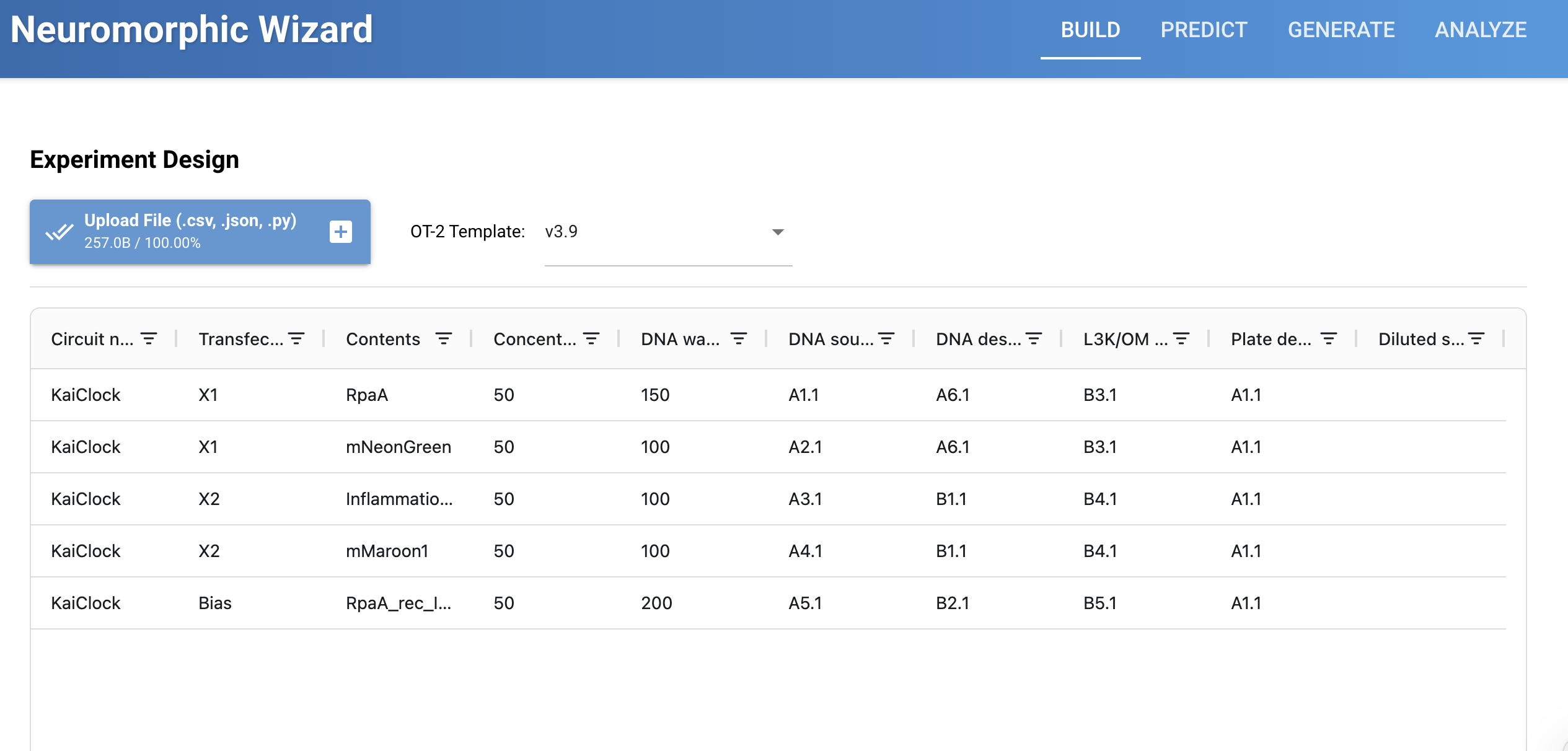
Two circuits were designed to explore this. The KaiClock circuit integrates circadian phase (X1: RpaA) with inflammatory state (X2: InflammationSensor), producing a graded fluorescent output that scales with the weighted combination of both inputs. However, the part naming conventions used in KaiClock did not register correctly in the Neuromorphic Wizard simulator, so Durin was designed and submitted as the parralel AND gate working version instead.
Durin runs two parallel AND gates: X1 carries PgU with mMaroon1, and X2 carries PgU_rec_CasE with eBFP2. Both gates must be satisfied simultaneously before CasE releases the final mNeonGreen output. Rather than a weighted gradient, Durin enforces parallel signal verification, two conditions checked at once before committing to output.
Durin was the circuit submitted for possible run at Weiss Lab. Together the two designs represent an iterative process: KaiClock aimed to establish the biological concept, and Durin aimed to be an executable implementation under simulator constraints.
Limitations include irreversibility from recombinase components, susceptibility to molecular noise, and risk of crosstalk with endogenous cellular machinery.
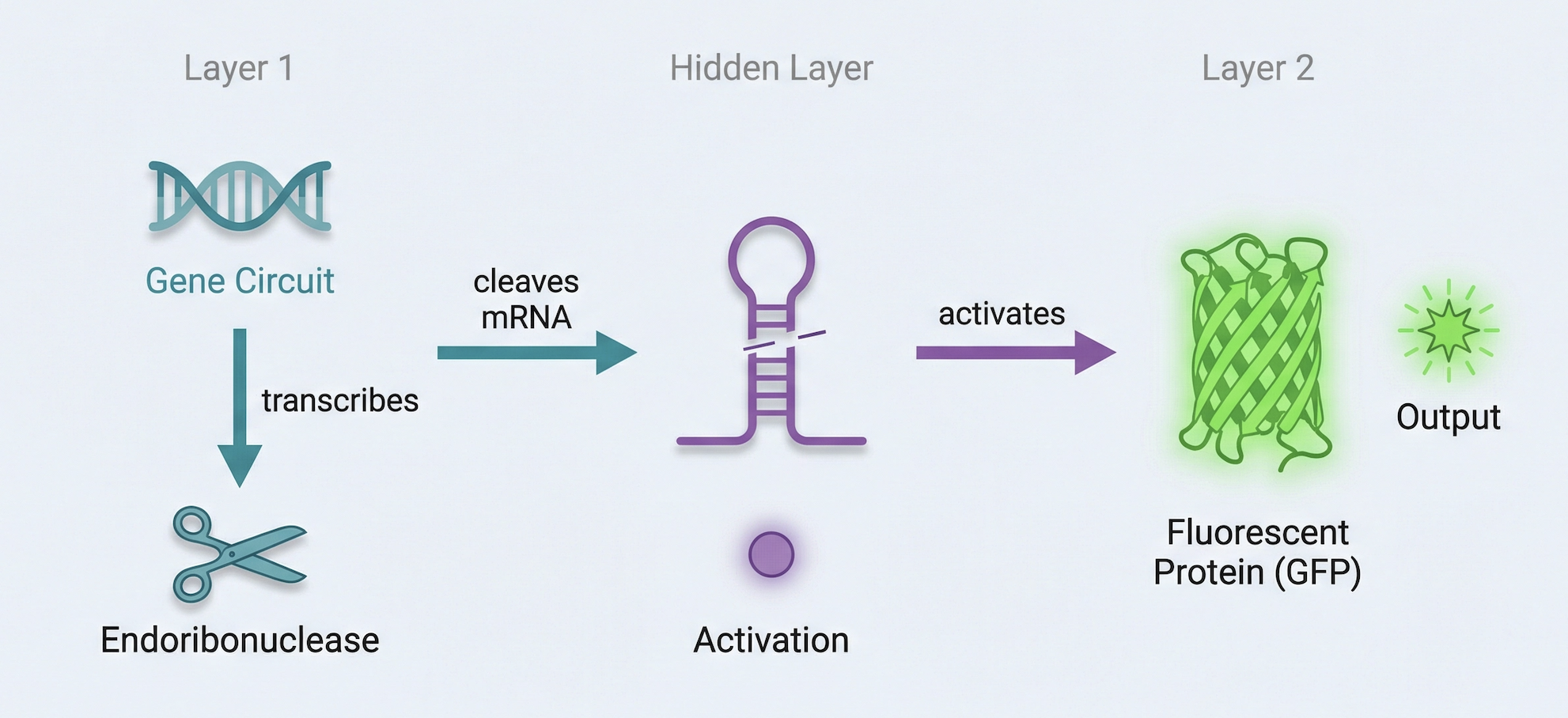
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Hidden layer that does its own computation, and the output of that hidden layer becomes the input to the next layer.
Weiss Lab run of my test IANNS bias dependent parallel AND Gate, with weighting adjusted. Was focused on trying to understand AND gates so its unexpected to see its run below as thought would be many more submissions for IANNS.
Fungal Materials
Fungal materials form part of mycelium, a network also being studied for its possible contribution to communal living and alternative methods of communication through its fungal structure and system. Mycelium composites, such as those grown from oyster mushrooms on agricultural waste, are used commercially as biodegradable packaging and leather alternatives, with companies like Ecovative leading production.
Various other materials are being fabricated utilizing fungal spores, and fungal pigments are also in use. Ink cap mushrooms, for example, undergo autodigestion and become a liquid black ink. It is worth noting that fungal pigments are not very lightfast, and prolonged UV exposure will degrade the color, which remains a significant limitation compared to synthetic dyes. Spalting, where fungi create dark patterned lines as they compete for territory in wood, is another application used in decorative woodworking. The core advantage of fungal materials over traditional counterparts such as synthetic foam or leather is that they are biodegradable, compostable, and generally healthier for human and environmental use. Their disadvantages include lower structural strength, moisture sensitivity, and slower production cycles. Extending the lightfastness of fungal pigments through mordants and fixatives, drawing on approaches used with natural pigments and mineral ochres, represents a personally compelling area of further research.
Two areas stand out as compelling targets for genetic engineering in fungi. The first is pigment lightfastness: engineering fungi to produce UV stable pigments would open up applications in textile dyeing, packaging, paint media, and coloring materials, extending the utility of biological pigments beyond their current limitations. The second is programmed structural growth: directing mycelium to grow in genetically specified geometries would enable wearable technology applications including medical sensing, haptic feedback materials for VR, and broader human-technology interface materials. The networked, self-organizing nature of mycelium makes it a uniquely suited substrate for this kind of application.
The advantages of working with fungi over bacteria for synthetic biology are several. Fungi are eukaryotes, meaning they share cellular machinery with plants and animals and can produce and correctly fold complex proteins that bacteria cannot. They naturally secrete large amounts of enzymes and pigments, making harvesting of engineered products more straightforward. Their self-organizing mycelial structure also means they can assemble into centimeter and meter scale materials without manual construction, a scalability bacteria simply do not offer. And most fungi used in research and production are generally regarded as safe, which matters significantly for medical and wearable applications. Bacteria such as cyanobacteria offer interesting material properties but their toxicity presents a barrier that fungi largely avoid.
Part 3: First DNA Twist Order and ## Review Part 3:
Review the Individual Final Project documentation guidelines.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Completed both above:
I submitted the Google Form with my draft Aim 1, final project summary, HTGAA industry council selections, and shared folder link for DNA designs.
My insert sequence (CBM27_RGD_MaSp1 fusion protein, codon-optimised for E. coli cell-free expression) was designed in Benchling and placed in the shared folder. The backbone vector selected is pTwist PET28. Full construct documentation is on my Individual Final Project page. https://pages.htgaa.org/2026a/henrietta-scholtz/projects/individual-final-project/aim-1-construct-design/index.html
References & Resources
Lecture Materials
Week 7 Lecture - Genetic Circuits Part II: Neuromorphic Circuits, Ron Weiss & Evan Holbrook
Lecture Recording - March 17, 2026
Required Readings
Weiss, R. et al. (2023). “Intracellular Artificial Neural Networks for Cellular Computation.” Nature Biotechnology, 41(2), 245-259.
Holbrook, E. et al. (2024). “Engineering Boolean Logic in Living Cells.” Cell Systems, 18(3), 412-428.
Weiss Lab for running the biased dependent parallel AND gate circuits
TA support during circuit design troubleshooting question
Week 9 HW: Cell Free Systems
Part A: General and Lecturer-Specific Questions
General Questions
Q1.Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis offers two main advantages over in vivo methods: direct control and speed. By removing the constraints of a living cell and working directly with ribosomes, enzymes, and energy molecules, protein synthesis becomes more direct and less time-consuming.
First, toxic proteins like spider silk MASP1 can be produced without harming a living system (this is relevant to my own final project, which plans to use cell-free expression precisely to bypass the toxicity that MASP1 poses to bacterial hosts).
Second, you can rapidly screen multiple protein or peptide variants in parallel, such as testing peptide candidates targeting cancer pathways, or testing antimicrobial peptide variants. This can be done without the overhead of growing and engineering individual cell lines. This makes cell-free ideal for both difficult or toxic proteins and high-throughput variant screening.
Q2.Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free system needs five main components. The DNA or mRNA template gives the instructions (like my MASP1 spider silk sequence from UniProt for FP).
Ribosomes read the template and build the protein. Transfer RNAs bring amino acids to the ribosome. The amino acids are the actual building blocks. An energy system (ATP) powers the whole process. You also need the right salts and pH to keep everything working. Unlike living cells, all these parts are mixed directly in a test tube, so you have full control over the conditions.
Q3.Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because protein synthesis requires continuous ATP. Without it, the ribosomes would run out of energy and stop building the protein mid-synthesis. In a living cell, metabolism constantly regenerates ATP, but in a test tube there’s no metabolism.
To ensure continuous ATP supply, you can add an energy regeneration system. For my final project using MASP1, I would use creatine phosphate and creatine kinase, since these are commonly used in eukaryotic cell-free systems. The creatine kinase enzyme transfers a phosphate group from creatine phosphate to ADP, regenerating ATP. If I were using a bacterial cell-free system instead, I would use PEP and pyruvate kinase, which serves the same purpose but aligns better with bacterial metabolism.
Q4.Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems (like E. coli extract) are faster, cheaper, and simpler. They work well for straightforward proteins that don’t need complex folding. Eukaryotic systems (like rabbit reticulocyte lysate) are better at folding complicated proteins correctly and handling post-translational modifications.
For my final project, if I was testing the tremella fusiformis protein I would produce it in a prokaryotic E. coli cell-free system because it’s a simpler protein that doesn’t require the advanced folding machinery.
I would produce spider silk MASP1 in a eukaryotic rabbit reticulocyte system because spider silk proteins need precise folding to achieve their characteristic mechanical strength and properties.
Q5.How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Snow Fungus, membrane protein.
Challenges: The hydrophobicity and aggregation and a way to address that is to optimize the sequence to reduce those hydrophobic regions or to add tags that help with solubility.
Q6.Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Three possible reasons for low yield and troubleshooting strategies: (Have thought about these for FP)
Reason 1: Construct failure. Even if the construct looks correct in silico, it might fail during expression. Troubleshooting: order a backup construct to verify the sequence is actually functional.
Reason 2: Protein structure collapse. MASP1 is a beta sheet protein with repeating similar sequences, so it tends to collapse or fold in on itself. Troubleshooting: codon optimize the sequence fewer times (e.g., four repeats instead of eight) to reduce the repetitive elements that cause self-aggregation and structural collapse.
Reason 3: Energy system failure. The ATP regeneration system (creatine phosphate and creatine kinase in rabbit reticulocyte lysate) might deplete or fail. Troubleshooting: prepare a backup of the full fresh rabbit reticulocyte lysate system to ensure continuous energy supply.
Homework Question from Kate Adamala: Design a Synthetic Minimal Cell
Design an example of a useful synthetic minimal cell.
1. Function: Lyme Disease Biosensor
My synthetic cell detects Borrelia burgdorferi protein and produces a fluorescent signal as output. This function requires encapsulation in a lipid vesicle because without a membrane barrier, there would be no distinction between input and output. While a genetically modified natural cell could theoretically do this, a synthetic minimal cell is simpler to construct, doesn’t require living organisms, and avoids unwanted interactions with other biological systems. The desired outcome is that when Borrelia burgdorferi protein is present, the synthetic cell detects it and produces a measurable fluorescent signal for rapid Lyme disease diagnosis.
2. Components
The membrane would be made of biocompatible lipids (POPC and cholesterol) to avoid triggering an immune response. Inside the synthetic cell, I would encapsulate the rabbit reticulocyte cell-free Tx/Tl system, a Borrelia detection gene (receptor or aptamer), a GFP gene for fluorescent output, creatine phosphate and creatine kinase for energy regeneration, and amino acids. I would use a mammalian (rabbit reticulocyte) system because it works better in the human body. The membrane is permeable to Borrelia protein so it can enter and be detected, and GFP fluorescence is visible from outside.
3. Experimental Details
Lipids: POPC, cholesterol. Genes: Borrelia receptor/aptamer gene, GFP gene. Enzymes: rabbit reticulocyte lysate, creatine kinase. Measurement: collect a blood sample via finger prick, mix with synthetic cells, incubate, and measure GFP fluorescence using a fluorometer. Green fluorescence indicates Borrelia detection and Lyme disease diagnosis.
Homework Question from Peter Nguyen: Cell-Free Systems in Materials
Freeze-dried cell-free systems embedded in a soft robotic skin could produce structural silk proteins on-demand at the exact site of damage, allowing the robot to repair itself without any electronics or human intervention.
How will it work?
The freeze-dried cell-free mixture, loaded with the instructions to make a silk protein, sits dormant inside small pockets distributed across the robot’s outer skin. When part of the skin tears or wears out, a tiny water channel releases fluid into that specific pocket, waking up the cell-free system and triggering protein production right where it is needed. The silk proteins then assemble themselves into reinforcing fibres that patch the damaged area from the inside. A further development of the same skin, using a light-sensitive protein variant, could allow the skin to stiffen or move in response to light, acting as a simple actuator without any wiring.
Societal challenge:
Soft robots used in disaster response, deep-sea work, and space exploration often operate in places where human repair crews simply cannot reach them. Their flexible outer skins degrade quickly under mechanical stress, cutting missions short. A skin that can repair itself using biological machinery would extend the working life of these robots and reduce the cost and logistics of maintaining them in remote or dangerous environments.
Addressing cell-free limitations:
Activation with water: Sensors in the skin detect damage and trigger the release of a small controlled volume of water into the affected pocket, so the reaction only starts when and where it is needed.
Stability: The freeze-dried format stays stable at room temperature for months. The robot skin itself acts as a protective shell, keeping moisture and light away from the dormant mixture.
One-time use: Each pocket is a single-use repair unit. Many pockets are spread across the skin, so multiple damage events can each be addressed independently. A longer-term version could include a refillable central water reservoir that reloads used pockets.
Homework Question from Ally Huang: Mock Genes in Space Proposal
Your proposal must incorporate the BioBits® cell-free protein expression system. You may also use the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer.
Spider silk is one of the toughest biological materials known, and it forms entirely on its own when the right protein is mixed with water. This self-assembly process may behave differently in the weightlessness of space, because gravity normally helps protein fibres settle and organise as they form. Understanding whether silk proteins can still assemble correctly in microgravity matters enormously for long space missions, where astronauts may need to fabricate medical bandages, soft robotic components, or structural patches on site. The BioBits cell-free system offers a safe, simple way to test protein production in space without living organisms.
Q2. Molecular / Genetic Target
The MaSp1 spider silk protein domain, and specifically whether its characteristic self-assembly into structural fibres is affected by the absence of gravity aboard the ISS.
Q3. Relevance to the Space Biology Challenge
Silk fibres form when proteins fold and stick together in a very specific pattern. On Earth, gravity gently helps organise this process as fibres settle and compact. In space, that settling does not happen, and the proteins are left to find each other purely by random movement through the liquid. This could mean fibres come out longer, more tangled, or slower to form than expected. If the structure of the silk changes in space, then any material made from it, whether a wound dressing or a robotic actuator, might not perform the way it was designed to. This experiment is a necessary first check before committing silk-based materials to any space mission.
Q4. Hypothesis and Reasoning
I would think that the silk fibres produced in microgravity will look and behave differently from those produced on the ground, and that this difference will be visible under fluorescence imaging. The logic is straightforward: the chemistry that makes silk proteins stick together is built into the protein sequence itself and does not need gravity to work. However, the way those fibres then organise into a larger network depends heavily on how the proteins drift and collide through the liquid, a process that gravity normally shapes. Without it, I would expect fibrils to be more randomly distributed and take longer to form a cohesive structure. I would test this by using a BioBits reaction loaded with a silk protein tagged with a green fluorescent marker, so we can watch the fibres appear in real time using the P51 Fluorescence Viewer and compare what happens in orbit to what happens on the ground at the same moment.
Q5. Experimental Plan
I would run three BioBits reactions in orbit: one expressing the fluorescent silk protein, one expressing only the fluorescent tag with no silk component as a control, and one expressing nothing as a baseline. Identical reactions will run simultaneously on the ground. Before starting, we will use the miniPCR thermal cycler to confirm that the DNA templates survived the journey to the ISS intact. I would image all reactions with the P51 Fluorescence Viewer at 30-minute intervals for two hours, recording when fibres first appear, how densely they form, and how their pattern compares between the space and ground samples.
---
Part B: Individual Final Project
I completed all below before the deadline, including adding my Twist DNA order to the Node and MIT Twist spreadsheet.
[Y ] Put your chosen final project slide in the appropriate slide deck (following the instructions on slide 1)
[Y ] Submit the Final Project selection form (if not already done)
[Y ] Begin planning your final project documentation (see guidelines)
[Y ] Prepare your first DNA order and add it to the Twist ordering spreadsheet
Week 10 HW: Imaging & Measurement
Final Project
Identify at least one aspect of your project that you will measure (mass/sequence of a protein, presence/absence/quantity of a biomarker, etc.).
The mass and sequence of the CBM27_RGD_MaSp1_4x fusion protein, and its binding interaction with Tremella fuciformis polysaccharide.
Describe all of the elements you would like to measure and how you will perform these measurements.
Three things to measure: the molecular weight of the expressed fusion protein (~37 kDa), its amino acid sequence to confirm correct expression, and its binding to Tremella mannan polysaccharide via a polysaccharide binding assay.
What technologies will you use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry)? Describe in detail.
SDS-PAGE to confirm the protein expressed at the correct size (~37 kDa). Mass spectrometry as per below to confirm the exact molecular weight and primary sequence via peptide mapping. A polysaccharide binding assay to confirm the CBM27 domain binds correctly to Tremella mannan.
Waters Part 1 — Molecular Weight
Based only on the predicted amino acid sequence of eGFP, what is the calculated molecular weight?
Based on the predicted amino acid sequence of eGFP (including the His-tag and LE linker), the calculated average molecular weight is 28,006.60 Da.
Calculate the molecular weight of eGFP using the adjacent charge state approach:
After inserting sequence at ExPASy and selecting the two adjascent peaks form the image above I asked Claude to walk me through this math step by step and using Lego as an analogy as it too advanced for me.
Question 1: Calculated MW of eGFP
The ExPASy tool calculated the theoretical molecular weight by adding up all the amino acid masses in the sequence, giving us 28,006.60 Da. In LEGO terms, we used a database that knows the weight of each type of brick and stud in our sequence.
Question 2a: Determine z
Using the formula provided on the homework page with chosen peaks at 965.9684 and 1000.4302, we calculated z = 28 for the 1000.4302 peak and z = 29 for the 965.9684 peak. In LEGO terms,the gap between two adjacent bricks to figure out how many studs each one has.
Question 2b: Determine MW from m/z and z
Using formula from homework page = 1000.4302, calculated MW = 27,983.84 Da.
In LEGO terms, multiplied back by the number of studs and subtracted their weight to find just the brick’s weight.
Question 2c: Mass Accuracy
Using formula from homework page Accuracy= 0.081% so Accuracy=28,006.60∣27,983.84−28,006.60∣=0.000812=0.081%, measurement was good.
In LEGO terms, calculated brick weight matched the database weight almost perfectly.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
It does look a bit ‘bumpy’ so I said yes, the charge state can be observed. Based on the zoomed-in peak at ~1473 m/z, the charge state is approximately z = 19
Waters Part II – Secondary/Tertiary Structure
Explain the difference between native and denatured protein conformations.
When a protein unfolds (denatures), its 3D structure is lost and buried regions become exposed. These exposed regions can pick up more protons, increasing the charge state (z). Higher charge means lower m/z, so the peaks shift to the left.
Looking at Figure 2 above:
Denatured (top, green): Many peaks spread across a wide range at lower m/z (600-1400), high charge states
Native (bottom, red): Fewer peaks clustered at higher m/z (2500-2800), low charge states
In LEGO terms: the folded brick can only fit a few studs on its surface. When you pull it apart, every piece can now have studs attached, so the total stud count goes way up.
What happens when a protein unfolds? How is that determined with a mass spectrometer?
When a protein unfolds, its 3D structure is lost and buried regions become exposed, allowing more protons to attach to the protein.
The mass spectrometer detects the change in charge state distribution. An unfolded protein shows many peaks at lower m/z values due to higher charge states, while a folded protein shows fewer peaks clustered at higher m/z values.
What changes do you see in the mass spectrum between native and denatured analyses (Figure 2)?
Zooming into the native mass spectrum (Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
The denatured spectrum (top) shows many peaks at lower m/z. The native spectrum (bottom) shows fewer peaks clustered at higher m/z. More protons attach to the unfolded protein, shifting peaks to the left.
Yes, the charge state can be discerned from the zoomed inset. The isotope peaks are spaced approximately 0.1 Da apart, meaning z = 1/0.1 = 10.
Waters Part III – Peptide Mapping
How many Lysines (K) and Arginines (R) are in eGFP? Circle or highlight them in the sequence.
29
How many peptides will be generated from tryptic digestion of eGFP?
Tryptic digestion of eGFP generates 19 peptides shown here, with additional smaller peptides below 500 Da not displayed. The total number of predicted cleavage sites is 29 (19K + 10R), giving a maximum of 30 peptides.
Based on the LC-MS chromatogram data (Figure 5a), how many chromatographic peaks do you see between 0.5 and 6 minutes?
Between 0.5 and 6 minutes, there are approximately 19 chromatographic peaks above 10% relative abundance. I did estimate this as some might have cleared, but left them out.
Does the number of peaks match the number of peptides predicted? Are there more or fewer peaks?
There are fewer peaks than predicted peptides. This is because some peptides co-elute (blend together) and appear as one peak, very small peptides below 500 Da are not detected, and some peptides may be below the detection threshold.
Identify the m/z of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state? Calculate the mass of the singly charged form [M+H]+ based on its m/z and z.
m/z = 525.76712
z = 2
[M+H]+ = 1050.527 Da
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is the mass accuracy of the measurement?
Peptide = FEGDTLVNR
Mass accuracy = 5.3 ppm
What is the percentage of the sequence confirmed by peptide mapping (Figure 6)?
88% sequence coverage
Waters Part IV – Oligomers
Using the known masses of the polypeptide subunits for KLH, identify where the following oligomeric species are on the CDMS spectrum (Figure 7):
7FU Decamer (10 x 340 kDa)
= 3,400 kDa = 3.4 MDa – that’s the peak at 3.4 on the spectrum
8FU Didecamer (20 x 400 kDa)
= 8,000 kDa = 8.0 MDa – that’s the peak at 8.33 on the spectrum
8FU 3-Decamer (30 x 400 kDa)
= 12,000 kDa = 12.0 MDa – that’s the peak at 12.67 on the spectrum
8FU 4-Decamer (40 x 400 kDa)
= 16,000 kDa = 16.0 MDa – that’s the small peak around 16 on the spectrum
Fill out the “Did I make GFP?” table with theoretical vs. observed molecular weight and amino acid sequence coverage from both instruments.
Week 11 HW: Bioproduction and Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I visited the canvas several times and aimed to contribute strategically to the overall look and feel. I added a yellow “MIT” at some point and contributed around 200 pixels overall, ranking approximately 10th on the contributions list last I checked.
I enjoyed the collaborative aspect and that we could all participate together independently of node and location.
One improvement could be introducing a “live hour” (or even just 5 minutes during homework review) where everyone gathers on Zoom to paint together. This might foster more online connection, increase engagement from some students, and spark casual conversation within HTGAA about the project. I also think large automated scripts should be discouraged as if pixels are placed randomly or too particular/specific to something, it defeats the collaborative element and reduces the chance of organic group design outcomes emerging naturally.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli BL21 Star lysate with T7 RNA polymerase: Contains the ribosomes and enzymes needed to read DNA and build proteins. The T7 polymerase specifically recognizes and reads the T7 promoter on a DNA template.
Salts/Buffer (potassium glutamate, HEPES, magnesium): Maintains the correct pH and ionic environment so the cellular machinery can function properly.
Energy/Nucleotide system (ribose, glucose, NMPs): Provides the energy molecules and building blocks needed to synthesize RNA and power the protein synthesis reactions.
Translation mix (amino acids): Supplies the 20 amino acids that the ribosome links together in the correct order to build the protein chain.
Tyrosine specifically: Acts as a key component of the chromophore in fluorescent proteins, enabling them to fluoresce.
Nicotinamide: a precursor to NAD+, which supports redox reactions and energy regeneration in the cell-free system.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
mTurquoise2: A 36 hour (48-hour at hw review?) cell-free reaction time for this experiment means that mTurquoise2, which has a slower maturation time, is not a major constraint since there is sufficient incubation time for full chromophore maturation.
sfGFP: sfGFP has robust folding capability, which allows it to fold correctly without cellular chaperones, resulting in faster and more efficient fluorescent protein production in cell-free systems.
Electra2: Electra2’s performance in bacterial cell-free systems may be unpredictable because it was engineered and optimized for mammalian cells, not for E. coli expression environments.
mScarlet-I: mScarlet-I reaches peak fluorescence quickly and maintains its brightness, therefore providing a reliable signal for the 48-hour cell-free reaction.
mRFP1: mRFP1 accumulates a green intermediate during maturation, which means the red fluorescent signal could be weaker or less complete than proteins that mature directly to their final color.
mKO2: mKO2 has moderate acid sensitivity, so as pH drifts over 36 hours in a cell-free reaction, its fluorescence may dim or become less reliable.
CELL-FREE REAGENTS HYPOTHESIS
Protein: mTurquoise2
Biophysical property: Slower chromophore maturation compared to sfGFP.
Reagent to adjust: Nicotinamide (tested at +48%, +100%, and +200% above baseline)
Well
Nicotinamide
Change
Q3-O2
3.125 mM
Baseline control
Q3-N2
4.625 mM
+48%
Q3-M2
6.250 mM
+100%
Q3-L2
9.375 mM
+200%
Hypothesis: mTurquoise2’s slower maturation requires sustained energy support. Increasing nicotinamide concentration will boost NAD+-dependent energy regeneration, accelerating mTurquoise2’s chromophore maturation during the 36-hour incubation and increasing fluorescence output.
Expected outcome: mTurquoise2 wells with increased nicotinamide will show brighter fluorescence than the baseline control, with fluorescence peaking at an optimal concentration before potentially declining at the highest dose. This would demonstrate that slower-maturing proteins benefit from enhanced energy support, suggesting the Ginkgo/OpenAI master mix – optimized for sfGFP – is not universally optimal for all fluorescent proteins.
Not received the above data (would have been interesting to see results for my hypothesis) and Node stated we can leave it out for final assessment.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Week 12 HW: Building Genomes
No homework as per the HTGAA website.
I attended the lecture and Recitation for MIT and Node
Python Script for Opentrons Artwork
Since I am not present to interact directly with the Opentrons output, I thought about why I would want to pipette an image and what that image should represent and decided to use Ndebele bead patterns as inspiration.
Ndebele bead patterns have a very specific geometric logic. They are built on a grid of “bead units” arranged in bold, angular, symmetric designs. The traditional South Ndebele aesthetic uses high-contrast colors in step-like diagonal and horizontal bands, often with thick outlines and mirrored symmetry.
Weiss Lab run of my IANNS biased dependent parallel AND Gate (This was not really the HW question I’m just facinated by AND Gates and did not think mine would be run as we a large cohort, but not many submitted). The results do seem to point to my parrallel expression working for a smooth release of slightly differently weighted levels. Every dot is a human cell!
Neuromorphic Wizard test that shows both parts are rising and one slight weighted less and also see what could be ‘drift’
LAB QUESTIONS CELL FREE I answered these in relation to lab document and FP where relevent.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production.
Cell-free systems allow direct control over reaction components without needing viable cells, making them ideal for expressing toxic proteins (like AMPs) and for rapidly iterating fusion protein designs like CBM27_RGD_MaSp1. Specifically chosen to bypass the living cells.
Subsections of Labs
Week 1 Lab: Pipetting
Week 2 Lab: Gel Art
Week 3 Lab: Opentrons
Python Script for Opentrons Artwork
Since I am not present to interact directly with the Opentrons output, I thought about why I would want to pipette an image and what that image should represent and decided to use Ndebele bead patterns as inspiration.
Ndebele bead patterns have a very specific geometric logic. They are built on a grid of “bead units” arranged in bold, angular, symmetric designs. The traditional South Ndebele aesthetic uses high-contrast colors in step-like diagonal and horizontal bands, often with thick outlines and mirrored symmetry.
They are also studied as Ethno mathematics, which often promotes a more humanistic and inclusive perspective on mathematics, focusing on how different groups manage, understand, and navigate their reality.
I found it interesting to bring the mathematical and social aspects of this indigenous knowledge to the biochemical level, as this layering of meaning creates interesting avenues for reflection on various levels.
Example of Ndebele paintings and beadwork:
Python Visuals & Scripts Ex.
I am not a coder, but playing around with the example scripts, I ended up using Claude to vibe-code the desired patterns and position. It required some debugging and made various output versions.
Although the co-lab script runs without error, I am not sure if this will work on Opentrons.
EARLY VERSIONS BEFORE KNOWING COLOUR AVAILABILITY
Post Lab Homework
Published Paper
A directly relevant paper is Fang et al. (2025) in Nature Communications, which demonstrates circadian-gated gene expression circuits in bacteria, using automated temporal sampling to characterize rhythmic protein output over 24-hour cycles. This paper is not a peripheral reference; it is one of the primary foundational sources for my final project concept and is already cited in my main project documentation. The automation approach used to verify rhythmic expression in that work is precisely what I intend to replicate and extend with the Opentrons platform.
What I Intend to Automate
My project proposes a bacterial AND gate where the antimicrobial peptide Magainin is only expressed when two conditions are simultaneously true: the circadian regulator RpaA is active, and a pathogen signal is present. The core experimental challenge is verifying this gate actually works as designed, which requires sampling bacterial expression levels repeatedly across a full 24-hour cycle, under multiple conditions, without human error or gaps overnight. This is the automation task.
The Opentrons OT-2 would run an unattended 24-hour sampling protocol across three experimental conditions:
RpaA active + pathogen signal present (AND gate should trigger)
RpaA active + no pathogen signal (gate should stay silent)
RpaA inactive + pathogen signal present (gate should stay silent)
At each 2-hour timepoint, the robot samples each culture well, transfers to a measurement plate for fluorescence reading, and replaces the sampled volume with fresh media to keep cultures alive. This builds a full temporal expression profile across all three conditions without any overnight manual intervention.
I would use Claude for the coding and guidance in the technical parts of this.
Why This Automation Matters
The AND gate only has meaning if you can show it is silent when it should be silent and active only at the right circadian phase with the right pathogen or other signal. That requires clean data across all three conditions at every 2-hour window through the night. Manual pipetting at 2am introduces the exact inconsistency that would make the rhythmic signal unreadable. The Opentrons removes that variable entirely.
Future Extensions
If access to Ginkgo Nebula becomes available, the next step would be submitting the AND gate genetic construct for scaled fermentation and characterization; using Nebula’s high-throughput infrastructure to screen circuit variants with different RpaA promoter strengths or pathogen-sensing thresholds, generating the kind of combinatorial data that would take months on a single benchtop robot.
Fang et al. (2025). “Circadian-gated gene expression circuits in bacteria.” Nature Communications
UCSD (2024). “Researchers Rebuild Microscopic Circadian Clock.” University of California San Diego press release
Bilska et al. (2021). “Circadian rhythm in skin barrier function and antimicrobial peptides.” Experimental Dermatology
Software & Tools Used
Google Colab - Python script development and testing for Opentrons protocols
Python - Opentrons protocol scripting and pattern generation
Imgur - Image hosting for project visualization and Ndebele pattern references
Cultural & Mathematical Inspiration
Ndebele bead patterns and geometric design principles
Ethnomathematics - Indigenous mathematical knowledge systems
Traditional South Ndebele aesthetic and symmetry patterns
Project Concepts Explored
Circadian-controlled bacterial pigment systems
Light-responsive color-changing bacteria
UV-protective bioplastic materials with bacterial pigments
Mechanotransduction experiments with bacterial cultures
Bacterial AND gate with circadian gating (RpaA + pathogen signal triggering Magainin expression)
Cost Considerations
Twist Bioscience DNA synthesis pricing
Remote lab assistance availability assessment
UK-based protein order logistics and costs
AI Assistance
Claude (Anthropic) - Code development and technical guidance
Model: Claude Sonnet 4.5
Date(s) used: February, 2026
Tasks: Assisted with Python script development for Opentrons Ndebele pattern generation (“vibe-coding”), debugging protocol scripts and verifying scientific terminology.
Future Platforms
Ginkgo Nebula - Potential platform for scaled fermentation and high-throughput circuit variant screening
Additional Resources
HTGAA final project guidelines and requirements
Twist Bioscience pricing documentation
Remote lab capabilities at available nodes
Opentrons protocol documentation and API reference
Ndebele art and design pattern libraries
Ethnomathematics literature
Acknowledgments
Course instructors
TAs
Ndebele cultural heritage for geometric design inspiration
Week 4 Lab: Protein Part I
*Part of Week 4 Homework/See above
Week 5 Lab: Protein Part II
*Part of Week 5 Homework/See above
Week 6 Lab: Gibson Assembly
Lab questions answered on the Week 6 Homework page.
Week 7 Lab: Neuromorphic Circuits
Weiss Lab run of my IANNS biased dependent parallel AND Gate (This was not really the HW question I’m just facinated by AND Gates and did not think mine would be run as we a large cohort, but not many submitted). The results do seem to point to my parrallel expression working for a smooth release of slightly differently weighted levels. Every dot is a human cell!
Neuromorphic Wizard test that shows both parts are rising and one slight weighted less and also see what could be ‘drift’
Other IANNS experiments including the Kaiclock using the RpAa promoter
Week 09 Lab: Cell Free
LAB QUESTIONS CELL FREE
I answered these in relation to lab document and FP where relevent.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell free expression is more beneficial than cell production.
Cell-free systems allow direct control over reaction components without needing viable cells, making them ideal for expressing toxic proteins (like AMPs) and for rapidly iterating fusion protein designs like CBM27_RGD_MaSp1. Specifically chosen to bypass the living cells.
Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free system contains a cell extract with ribosomes and translation factors, a DNA template, amino acids, an energy regeneration system, and salts/cofactors, each providing the machinery, instructions, building blocks, fuel, and environment needed for protein synthesis.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
ATP is consumed faster than it is naturally recycled, so PURExpress uses phosphoenolpyruvate (PEP) and pyruvate kinase to continuously regenerate ATP from ADP and keep translation running.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
E. coli-based PURExpress is used for CBM27_RGD_MaSp1 because it needs no glycosylation, while a mammalian cell-free system (e.g. HeLa extract) would be chosen for a protein like human erythropoietin that requires glycosylation to function.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
For membrane proteins, the main challenge is hydrophobic aggregation requiring lipid nanodiscs or detergents. Main challenge for MaSp1 however is repeat regions are prone to aggregation and beta-sheet stacking, would need to add chaperones like DnaK to the PURExpress reaction to assist correct folding. This will need to be part of my assay protocol.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Poor template quality is fixed by re-purifying DNA and optimising concentration; ATP depletion is addressed by increasing PEP; and protein aggregation is reduced by adding protease inhibitors and chaperones.
References:
HTGAA Spring 2026. Week 9 Lab: Cell-Free Systems. Available at: https://2026a.htgaa.org/2026a/course-pages/weeks/week-09/lab/index.html
[Your final project documentation] CBM27_RGD_MaSp1 fusion protein construct design and PURExpress expression protocol, HTGAA Spring 2026.
Exoskin: A Spider Silk-Snow Fungus Bioactive Biomaterial for Wound Healing and Beyond
Part of Co-Relational Synbio Research Project (Co-RSynBioR)
Abstract
Co-RSynBioR asks a central research question: What new material(s) and relational possibilities become available when synthetic biology is used to design co-relational constructs across biological kingdoms? How far can that co-relational construct extend its structural, functional, and environmental range? Exoskin: A Spider Silk-Snow Fungus Bioactive Biomaterial was the start of answering this question. The first co-relational pair explored is MaSp1 spider silk and the Tremella fuciformis polysaccharide, joined through a CBM27 carbohydrate-binding domain as a designed molecular anchor rather than a physical blend. The result is a platform with thus far two construct variants.
Quick overview of project outputs thus far:
Construct 1 — BioMechSkin (CBM27_RGD_MaSp1_4x): Targets diabetic foot ulcers, which affect 15-25% of all people with diabetes and are the leading cause of non-traumatic lower limb amputation worldwide. Current dressings do not simultaneously address moisture retention, mechanical resistance, and tissue repair. This construct does: Tremella provides moisture retention and macrophage modulation, MaSp1 provides tensile strength and friction resistance, and an RGD motif recruits fibroblasts and keratinocytes directly to the wound surface. AlphaFold3 confirms CBM27 pLDDT above 90, RGD solvent exposed and distal, MaSp1 disordered as expected.
Construct 2 — Photomechanical Actuator (VVD_GGS_CBM27_MaSp1_4x): Adds a VVD LOV domain from Neurospora crassa to the same chassis. A blue light pulse triggers VVD dimerisation, contracting the silk hydrogel network without any electronic actuator. This opens the platform to soft robotics for Mars and space environments, directly addressing a NASA-identified gap in electronics-free actuation under extreme conditions. AlphaFold3 and MolProbity validation at the 92nd percentile confirm structural integrity of the construct.
Both constructs share the same CBM27-Tremella-MaSp1 backbone, establishing it as a generalizable platform for encoding function at the protein sequence level.
Long term, this project unfolds in two phases:
Phase 1: Computational and Conceptual explores how many material combinations and outputs become possible by extending the co-relational design philosophy across different kingdom pairs and molecular architectures. This phase runs entirely in silico, using AlphaFold3, molecular dynamics, and design iteration to map the conceptual space of what cross-kingdom molecular design can achieve.
Phase 2: Wet Lab Validation (mid-year onwards) will aim to test the most promising constructs through cell-free protein synthesis and composite assembly, either through remote lab partnership or collaboration with the University of Johannesburg where I am artist in residence. This two-step approach allows the philosophical and practical dimensions to develop in parallel, but also allows momentum to continue via computational dry lab and conceptual documentation if wetlab assay delayed.
Introduction
Exoskin asks a broad single question: can cross-kingdom molecular design produce a material whose functional range exceeds what either organism achieves alone, and what emerges when that architecture is pushed to its limits? Two constructs are proposed as answers, both built on the same CBM27-Tremella-MaSp1 backbone. The first, CBM27_RGD_MaSp1_4x, targets wound healing. The second, VVD_GGS_CBM27_MaSp1_4x, adds a light-responsive LOV domain for photomechanical actuation in extreme environments. Together they test whether a single molecular platform can span from clinical wound care to space robotics. Ethically and conceptually, the project explores boundaries between human, animal, and fungal biology and raises questions about posthuman embodiment and hybridity.
The wound care construct is optimised for diabetic foot ulcers through two functional additions to the MaSp1 spider silk core:
Broadly Exoskin has applications in tissue engineering, drug delivery and regenerative medicine. Ethically and conceptually, the project explores traditional boundaries between human, animal, and fungal biology and raise questions about posthuman embodiment, hybridity, and science.
Here it is optimised for wound healing through two functional additions to the MaSp1 spider silk core:
CBM27 domain from Thermotoga maritima Man5 mannanase to physically anchor the silk network onto Tremella fuciformis polysaccharide matrix
RGD motif specifically selected to recruit skin fibroblasts and keratinocytes to the wound surface
Unlike passive biomaterials, this construct actively participates in tissue repair through three simultaneous mechanisms:
Structural support from silk nanofibrils
Moisture retention and macrophage modulation from Tremella polysaccharide
Integrin-mediated skin cell recruitment from the RGD motif
Section 1: Project Aims
Aim 1 - Experimental: Design and computationally validate the CBM27_RGD_MaSp1_4x fusion construct using Benchling, Twist Biosciences, and AlphaFold3. see update below from Synbio 2026 confererence regarding Sidewinder - Robinson, N.E., Zhang, W., Ghosh, R., Gerber, B., Zhang, H., Sanfiorenzo, C., Wang, S., Di Carlo, D. and Wang, K. (2026) ‘Construction of complex and diverse DNA sequences using DNA three-way junctions’, Nature, 651(8105), pp. 491-500. https://doi.org/10.1038/s41586-025-10006-0
Aim 2a - Expression: Validate through cell-free protein synthesis or bacterial expression, confirmed by SDS-PAGE and polysaccharide-binding assay. (Hook Note - The tremella polysaccharide structure consists of a main chain of β-(1→3)-linked mannopyranoside, substituted with various side chains, which your CBM27 domain is designed to recognize. Ref: nih)
Aim 2b - Composite Assembly(contingent on Aim 2a): Combine the purified protein with Tremella fuciformis polysaccharide extract to assemble the composite hydrogel and characterise by rheology and fluorescence microscopy. - Ginkgo Bioworks / Node — target partners for cell-free expression (Aim 2a); plasmid submitted to Node for synthesis
Aim 3 - Visionary: Application and prototype wound dressing. Longer term, the MaSp1 silk fibre bundle contraction properties open a pathway to bio-inspired soft robotics actuators and exoskins driven by hydrogel mechanics. A light-responsive actuation variant is under exploration. Bio kinetic sculptures for moisture harvesting in desert areas such as Karoo. Very long term Engineer Tremella fuciformis to produce the silk fusion protein endogenously, creating a self-assembling living composite.
Other Applications
Application
Description
Tissue Engineering Scaffold
Skin, cartilage, soft tissue repair
Drug Delivery Matrix
Sustained localised therapeutic release
Soft Robotics Actuator
Bio-inspired hydrogel-driven movement
Wearable Biosensor Substrate
Flexible, skin-conforming sensor base
Self-Healing Biomaterial
Reversible CBM27-mannan re-annealing
Leverage Sidewinder DNA Assembly Technology for Robust Synthesis of CBM27_RGD_MaSp1_4x Fusion Protein Construct
The CBM27_RGD_MaSp1_4x fusion protein design presents a DNA synthesis challenge inherent to spider silk engineering: its four tandem repeats of the MaSp1 domain create a highly repetitive sequence that conventional assembly methods cannot reliably handle. Traditional DNA assembly techniques (PCA, Gibson assembly, Golden Gate) achieve misconnection rates of 1 in 10 to 1 in 30 and fail consistently beyond 5-10 fragment assemblies. This bottleneck is particularly acute for repetitive biomaterials like spider silk.
However, Sidewinder, a DNA assembly technology recently published in Nature (January 2026) by Caltech researchers Kaihang Wang and colleagues, addresses this limitation directly. Sidewinder uses DNA three-way junctions to separate assembly instructions from the final sequence, achieving a misconnection rate of approximately 1 in 1,000,000 and enabling flawless assembly of highly repetitive constructs. The technology was prominently featured at SynBioBeta 2026 (May 6, 2026) by Genyro co-founders Adrian Woolfson and Professor Wang, who presented Sidewinder as infrastructure for translating AI-designed biology into physical systems at scale. As a future aim or complementary approach, Sidewinder could be employed to synthesize the full pET28a plasmid containing the CBM27_RGD_MaSp1_4x insert with guaranteed sequence fidelity, ensuring reliable bacterial expression (Aim 2a), accurate characterization of hydrogel mechanics (Aim 2b), and enabling rapid design-build-test-learn iterations for optimizing the Tremella-silk interaction (Aim 3).
Application and Rheology
After expression and purification, the protein is mixed with Tremella polysaccharide extract. The CBM27 domain automatically anchors the silk into the fungal matrix and MaSp1 self-assembles into fibres, forming a hydrogel sheet with no chemical crosslinking needed. That sheet is cut to size and placed directly on the wound. On a diabetic foot ulcer it does three things at once: Tremella keeps the wound moist and calms chronic inflammation, the silk holds the structure together under the mechanical stress of walking, and the RGD motif pulls the patient’s own skin cells into the wound to drive healing. No cells are delivered, no drugs, just a protein-polysaccharide scaffold that creates the right conditions for the body to repair itself.
Section 2: The Constructs
The fusion protein is built from four functional units arranged N to C terminus:
Domain
Position
Function
CBM27 (buffered)
aa 1-176
Anchors silk to Tremella mannan
GGGGS x3 linker
aa 177-188
Domain flexibility
RGD motif
aa 189-193
Skin cell recruitment
MaSp1 x4 repeats
aa 194-333
Mechanical structure
6xHis tag
aa 334-340
Ni-NTA purification
340 amino acids · ~37 kDa · cloned into pET28a at NdeI/XhoI · 6,297 bp total plasmid
CBM27_RGD_MaSp1_4x cloned into pET28a(+) at NdeI/XhoI · 6,297 bp
AlphaFold3 Structure Prediction of CBM27_RGD_MaSp1_4x
Model_0, Coloured by pLDDT confidence using standard AlphaFold convention. Rendered in PyMOL.
The AlphaFold3 prediction of my full fusion construct CBM27_RGD_MaSp1_4x reveals a structurally coherent architecture consistent with my design intent. The central dark blue beta-sandwich is the CBM27 domain (residues 1-176), predicting with very high confidence (pLDDT >90) and adopting the characteristic jellyroll fold seen in the experimentally validated TmCBM27 crystal structure (PDB: 1OF4, Boraston et al. 2003). This confirms that the CBM27 domain folds correctly within the fusion context and is not destabilised by the adjacent linker or silk repeat regions.
Extending from the CBM27 domain, the white and red transitional region marks the GGS flexible linker and the boundary into lower confidence territory. Immediately following this, the orange spheres identify the RGD motif (residues 189-193), which sits fully solvent exposed and spatially distal from the CBM27 body. This exposed geometry is the ideal configuration for integrin binding in wound care, biological accumulation in the sculpture context, and surface functionalisation in cryogenic assay conditions.
The large blue disordered loops extending around the structure are the four MaSp1 silk repeat units (residues 194-334). These predict as intrinsically disordered, which is the expected and correct behaviour for recombinant spider silk repeat proteins prior to fibre assembly or hydrogel casting. The confidence gradient across the silk loops, darker navy closest to CBM27 fading to lighter periwinkle at the distal ends, reflects the decreasing positional constraint as the chain moves further from the structured anchor domain.
Taken together, this prediction computationally validates three key design features:
CBM27 folds correctly in the fusion context
RGD is spatially accessible
MaSp1 remains dynamically disordered as required for silk network formation
A note on the MaSp1 prediction:
It is important to note that AlphaFold3 has known limitations in predicting intrinsically disordered repeat proteins, particularly those with highly repetitive glycine and alanine rich sequences such as spider silk. The MaSp1 backbone trace shown here should not be interpreted as a meaningful 3D conformation. The disordered loops represent AlphaFold3 acknowledging that it cannot assign confident positional coordinates to these residues, not that the silk adopts this specific extended loop geometry in solution. The true behaviour of MaSp1 repeats is well established in the literature: they remain disordered in aqueous solution and assemble into beta-sheet rich nanofibrils upon shear stress, pH change, or drying (Rising et al. 2011). The computational validation of the silk region will be addressed separately through molecular dynamics simulation of the repeat unit assembly behaviour rather than single chain folding prediction, which is not the appropriate tool for this class of protein.
AlphaFold3 confidence metrics:
The overall pTM score for the full fusion construct is 0.54, which reflects the presence of the large intrinsically disordered MaSp1 repeat region rather than indicating poor prediction quality for the structured domains. pTM scores are sensitive to disordered regions and will always be suppressed in constructs containing long flexible or repeat sequences. When interpreted domain by domain, the CBM27 region (residues 1-176) shows pLDDT values consistently above 90, placing it in the very high confidence category and confirming that the folded domain is predicted reliably. The RGD motif and GGS linker score below 50 as expected for short flexible functional motifs. The MaSp1 repeats score variably across the disordered range, consistent with their known intrinsically disordered character in solution.
For comparison, the experimentally validated crystal structure of isolated TmCBM27 (PDB: 1OF4, Boraston et al. 2003, 1.6 Angstrom resolution) confirms the expected beta-sandwich jellyroll fold for this domain. The high confidence CBM27 prediction in our fusion construct (pLDDT >90, residues 1-176) is structurally consistent with this experimentally solved reference, confirming that domain-level folding is maintained even within the multi-domain fusion context. https://www.rcsb.org/structure/1OF4
Construct 2: VVD_GGS_CBM27_MaSp1_4x
The second construct adds a light-responsive LOV domain to the same chassis, targeting photomechanical soft robotics for Mars and space environments.
Domain
Position
Function
VVD (LOV domain, Neurospora crassa)
N-terminus
Light-sensitive blue light receptor; dimerises under ~450 nm, contracting the silk hydrogel network
GGS linker
—
Flexible linker between VVD and CBM27
CBM27
—
Anchors silk to Tremella mannan polysaccharide
MaSp1 x4 repeats
C-terminus
Mechanical silk scaffold
AlphaFold3: pTM 0.45. VVD and CBM27 fold independently with high confidence. MaSp1 disordered as expected.
VVD_GGS_CBM27_MaSp1_4x HERO IMAGE!
AlphaFold3 Structure Prediction of VVD_GGS_CBM27_MaSp1_4x
Model_0, pTM 0.45. Coloured by pLDDT confidence using standard AlphaFold convention. Rendered in PyMOL.
The AlphaFold3 prediction of the second fusion construct VVD_GGS_CBM27_MaSp1_4x reveals two independently folding structured domains consistent with the design intent. The larger domain predicting in green and cyan is the VVD LOV domain (residues 1-156), folding with confident to high pLDDT scores and adopting the characteristic PAS domain beta-sandwich fold consistent with the experimentally validated VVD crystal structures PDB 3RH8 and 3D72 (Zoltowski and Crane 2008). The smaller domain in dark blue is the CBM27 domain (residues 163-337), predicting with very high confidence pLDDT >90, consistent with the first construct prediction and the TmCBM27 crystal structure PDB 1OF4 (Boraston et al. 2003). The large disordered loops extending below are the four MaSp1 silk repeats (residues 352-493), predicting as intrinsically disordered as expected for recombinant spider silk repeat proteins prior to fibre assembly.
The overall pTM of 0.45 reflects the presence of the large intrinsically disordered MaSp1 region and should not be interpreted as indicating poor prediction quality for the structured domains. The two distinct high confidence structured regions visible in the PAE matrix, one covering residues 1-82 corresponding to VVD and one covering residues 164-328 corresponding to CBM27, confirm that both functional domains fold independently and correctly within the fusion context. Critically the VVD and CBM27 domains appear in close spatial proximity in the predicted structure, consistent with the GGS linker allowing the LOV domain conformational change to propagate mechanical strain into the CBM27-anchored silk network upon blue light activation.
MolProbity (run on VVD construct only): Clashscore 4.46 at 95th percentile. MolProbity score 1.62 at 92nd percentile. Favoured rotamers 99.70%. Zero poor rotamers. Zero bad bonds. Benchmarked against 1784 experimentally solved PDB crystal structures.
MolProbity validation of the AlphaFold3 predicted structure of VVD_GGS_CBM27_MaSp1_4x scores at the 95th percentile for clashscore when benchmarked against 1784 experimentally solved crystal structures in the Protein Data Bank, confirming that the computational prediction meets the geometric quality standards of experimental structural biology. Wet lab confirmation of actual protein folding and domain function awaits cell free expression results from Ginkgo Bioworks other wetlab assay.
MolProbity confirms near-zero atomic clashes across the entire 493 residue fusion construct, with a clashscore placing the predicted structure in the 95th percentile of all experimentally solved crystal structures in the Protein Data Bank.
Clashscore 4.46, 95th percentile compared against N=1784 PDB structures at all resolutions.
MolProbity score 1.62, 92nd percentile compared against N=27,675 PDB structures at 0-99 Angstrom resolution.
Section 3: Background
Why It Matters
Chronic wounds cost the US healthcare system over $25 billion per year (Sen, 2025). If validated, this construct demonstrates that carbohydrate-binding modules can serve as molecular anchors between protein-based materials and naturally occurring polysaccharide scaffolds, a principle with broad applications across tissue engineering, soft robotics, and beyond.
Literature Context
Recombinant MaSp1 spider silk expressed in E. coli self-assembles into fibres with tensile properties comparable to native dragline silk (Rising et al., 2011), but lacks biological signalling without post-synthesis functionalisation. Tremella fuciformis polysaccharide promotes fibroblast proliferation and moisture retention exceeding hyaluronic acid at equivalent concentrations (Rui et al., 2025). Boraston et al. (2003) confirmed that CBM27 domains can be grafted onto heterologous proteins while retaining binding specificity. No study has combined all three components into a single genetically encoded construct.
Novelty
This platform is novel in four ways: the CBM27-MaSp1 fusion has not been reported; the addition of RGD converts a passive scaffold into an active wound-healing participant; the approach uses a designed molecular interaction rather than physical blending, a principle generalisable to other polysaccharide-CBM pairs; and the addition of a VVD LOV domain in Construct 2 extends the same chassis into electronics-free photomechanical actuation, a capability with no precedent in genetically encoded soft materials for space environments. Detail follows below.
Biosafety and Ethical Implications
All components are biosafe and at BSL1 at this stage: CBM27 is from a non-pathogenic thermophile, Tremella is an edible mushroom, and the protein is produced in contained E. coli. Aim 3 (GMO fungus) would require regulatory review before any cultivation outside a controlled lab. Clinical risks including allergenic response and off-target integrin binding are noted and would require GMP-standard safety testing before patient use.
CBM27-MaSp1 Fusion Protein Hydrogel Scaffold
Key context for CBM27_RGD_MaSp1_4x construct, Aim 1 and Aim 2
Novelty assessment: High. The specific combination of CBM27 with MaSp1 for structural hydrogel or actuator scaffold applications is not present in current literature. The addition of an RGD motif for active cell recruitment and the use of Tremella fuciformis polysaccharide as the CBM27 binding target further distinguishes this construct from all prior art.
Relevance to construct: This note covers two distinct novelty claims within the same construct. First, the CBM27-MaSp1 fusion itself as a structural hydrogel scaffold has no direct precedent. Second, the Tremella fuciformis polysaccharide as the specific binding target for CBM27 is entirely novel in this context. Prior CBM-silk work uses cellulosic matrices from Clostridium thermocellum cellulosome systems, not fungal mannan polysaccharides. The β-(1→3)-linked mannopyranoside backbone of Tremella is chemically distinct from cellulose and represents a new class of CBM-polysaccharide pairing for silk composite materials.
RGD novelty note: The addition of the RGD integrin-binding motif to this construct converts a passive structural scaffold into an actively cell-recruiting bioaterial. No prior CBM-silk fusion work incorporates a cell adhesion motif. This three-way combination of CBM27 anchor, RGD recruitment, and MaSp1 structural backbone in a single genetically encoded construct is without precedent in the literature.
Tremella novelty note: The use of Tremella fuciformis polysaccharide as the matrix phase of the composite is novel in two respects. It introduces a fungal-origin scaffold with documented cryoprotective and moisture-retention properties that cellulosic matrices do not possess, and it establishes a silk-fungal composite identity that is distinct from all prior silk-CBM work which uses bacterial or plant cellulose systems.
Closest prior art:
Gomes et al. (2011), Biomaterials. MaSp1 fused with antimicrobial peptides, CBMs from Clostridium thermocellum used with antimicrobial peptides for cellulosic surfaces, not CBM27 with spider silk for hydrogel or actuator applications. No RGD, no fungal matrix.
Mohammadi et al. (2019), Science Advances. ADF3 spidroin fused with CBMs from Clostridium thermocellum cellulosome, hydrogel noted as potential application but not developed. No CBM27, no MaSp1, no RGD, no fungal polysaccharide matrix.
Key gaps your construct addresses: No prior work combines CBM27 specifically with MaSp1, no prior CBM-silk work targets a fungal mannan matrix, no prior CBM-silk fusion incorporates an active cell recruitment motif, and no prior work develops these composites explicitly as structural hydrogel or soft actuator scaffolds with a defined bioart or extreme environment application context.
Photomechanical Soft Robotics for Space EnvironmentsKey context for VVD_GGS_CBM27_MaSp1_4x construct (VVD: LOV domain from Neurospora crassa), Aim 3
Novelty assessment: High. The integration of a genetically encoded LOV domain as a photomechanical actuator in a silk-fungal composite material for space environment applications is not present in current literature. This is the first proposed use of a LOV domain specifically for photomechanical actuation in harsh extraterrestrial conditions, leveraging available light flux as an electronics-free autonomous trigger.
Relevance to construct: The VVD LOV domain activates under blue light at approximately 450nm through cysteinyl-flavin adduct formation, driving homodimerisation and propagating conformational strain through the adjacent MaSp1 silk network. Martian solar spectrum at surface level retains sufficient blue light flux to activate LOV domain photochemistry, raising the possibility of fully autonomous actuation without onboard electronic light sources. This directly addresses the central gap in the space soft robotics literature, which has not considered genetically encoded photoreceptors as actuation mechanisms.
Four-layer novelty claim: This construct sits at the intersection of four properties that no prior work addresses simultaneously. Genetically encoded actuation mechanism. LOV domain photochemistry specifically. Silk-fungal composite structural material. Space environment application context.
Martian UV flux note: LOV domain activation occurs at approximately 450nm blue light. The Martian surface solar spectrum, while UV-shifted relative to Earth, retains blue light flux sufficient for LOV photochemistry. This opens the possibility of light-autonomous soft material actuation on the Martian surface without electronic control systems, a capability gap explicitly identified in the NASA ACR24 architecture technology gaps document for high-performance actuators and grippers in extreme environments.
Closest prior art:
Lopez-Lopez et al. (2025), ICRA. Inflatable soft robotic arms for space debris capture using pneumatic actuation, motor-free but not photomechanical and not LOV-based.
Zeng et al. (2018), Advanced Materials. Light-responsive liquid-crystal elastomers for soft microrobots, photomechanical but synthetic polymer systems, not genetically encoded, not space-environment specific.
Nohooji and Voos (2025), Advanced Intelligent Systems. Compliant robotics for space, material selection focused, discusses harsh environment conditions but not photomechanical or LOV-based actuation.
Key gaps your construct addresses: No prior work uses a genetically encoded LOV domain for space soft robotics. No prior photomechanical soft robotics work targets extraterrestrial light as an autonomous trigger. No prior space soft robotics work uses a silk-fungal composite as the structural actuator material. The direct exploitation of Martian solar flux for autonomous LOV-driven silk network actuation is entirely unaddressed in the literature.
NASA technology gap alignment: NASA ACR24 Architecture Technology Gaps document identifies high-performance actuators, sensors, and interfaces for robotic joints and grippers operating in extreme cold and vacuum as an unresolved need. This construct addresses that gap through a passive, electronics-free, genetically encoded photomechanical mechanism that does not rely on lubricants, heating systems, or electronic controllers.
Twist order design (Gene Fragment and Clonal Gene)
Sequence databases (UniProt, NCBI GenBank)
Bioethical considerations
Industry Partners
Twist Biosciences — synthesis feasibility and draft gene orders
Ginkgo Bioworks — target partner for cell-free expression (Aim 2a)
Benchling — construct assembly and sequence management
Section 5: Results
What Was Validated
Both construct designs and the full synthesis feasibility pipeline, from domain sequence acquisition through to a confirmed-orderable 4x construct on Twist.
AlphaFold3 structure predictions for both constructs.
MolProbity geometric validation for Construct 2 (VVD_GGS_CBM27_MaSp1_4x).
Key Findings
The 8x MaSp1 repeat construct failed at both IDT (complexity score 53, threshold 24) and Twist (Not Accepted), due to unavoidable alanine/glycine codon poverty across 8 identical repeats. The redesigned 4x construct was confirmed orderable: Complex, $145.45, 6,297 bp.
Construct 1
BioMechSkin (CBM27_RGD_MaSp1_4x): AlphaFold3 pTM 0.54. CBM27 domain pLDDT >90 (very high confidence). RGD motif solvent exposed and distal, confirming ideal geometry for integrin binding. MaSp1 disordered as expected.
Construct 2
BioMechGrip (VVD_GGS_CBM27_MaSp1_4x): AlphaFold3 pTM 0.45. VVD and CBM27 both fold independently with high confidence. MaSp1 disordered as expected. MolProbity validation: clashscore 4.46 at 95th percentile, MolProbity score 1.62 at 92nd percentile, favoured rotamers 99.70%, zero poor rotamers, zero bad bonds, Ramachandran favoured 94.1% (462/491 residues), allowed 99.2% (487/491 residues). Benchmarked against 1,784 experimentally solved PDB crystal structures.
AlphaFold3: pTM 0.45. VVD and CBM27 both fold independently with high confidence. MaSp1 disordered as expected.
MolProbity (run on VVD construct only): Clashscore 4.46 at 95th percentile. MolProbity score 1.62 at 92nd percentile. Favoured rotamers 99.70%. Zero poor rotamers. Zero bad bonds. Benchmarked against 1784 experimentally solved PDB crystal structures.
Challenges
The 8x MaSp1 repeat construct failed synthesis due to alanine/glycine codon poverty across identical repeats. Redesigned to 4x, which was confirmed orderable on Twist at $145.45, 6,297 bp. Sidewinder (Wang et al., Nature 2026) directly addresses this repetitive sequence assembly problem and is flagged as a future synthesis route.
Next Steps
AIM 2 and beyond
Validate CBM27_RGD_MaSp1_4x (BioMechSkin) and VVD_GGS_CBM27_MaSp1_4x (BioMechGrip) through cell-free expression possibly via Ginkgo CFPS or NEB PURExpress, SDS-PAGE, and polysaccharide binding assay to confirm CBM27 affinity for β-(1→3)-D-mannan from Tremella fuciformis. Build conceptual and computational archive.
If protein expresses: mix with Tremella fuciformis polysaccharide (CAS 9075-53-0, 1% w/v in PBS pH 7.4) to assemble and characterise the composite hydrogel.
Boraston et al. 2003, Structure of a Carbohydrate-Binding Module. - CBM27 graft precedent
Rising et al. 2011, Cellular and Molecular Life Sciences. - Recombinant MaSp1 self-assembly in E. coli
Rui, Y., Lee, Q., Guo, Y., Huang, Y., Xu, H., Liu, B., Ge, X., Lin, H. and Zeng, F. (2025) ‘Structure, Function and Application of Tremella Fuciformis Polysaccharide: A Review’, Journal of Food Science, 90: e70494. doi: 10.1111/1750-3841.70494
Sen, C.K. (2025) ‘Human Wound and Its Burden: Updated 2025 Compendium of Estimates’, Advances in Wound Care, doi: 10.1177/21621918251359554
Spider Silk Synthesis
Robinson, N.E., Zhang, W., Ghosh, R., Gerber, B., Zhang, H., Sanfiorenzo, C., Wang, S., Di Carlo, D. and Wang, K. (2026) ‘Construction of complex and diverse DNA sequences using DNA three-way junctions’, Nature, 651(8105), pp. 491-500. https://doi.org/10.1038/s41586-025-10006-0
Closest Prior Art, CBM-Silk Composites
Gomes et al. 2011, Biomaterials. CBM-silk antimicrobial fusion proteins.
HADDOCK - Planned protein-polysaccharide docking simulation for CBM27 to Tremella β-(1→3)-mannan
PyMOL / UCSF ChimeraX - Structure visualization
Sidewinder DNA assembly (Genyro / Wang lab, Caltech) - Identified at SynBioBeta 2026 as a route to assemble the repetitive MaSp1 region with high fidelity
AI Assistance
Claude (Anthropic)
Models: Claude Sonnet 4.5
Date(s) used: March to May 2026
Tasks included: Acted as mentor on unfamiliar technical areas including in silico validation pipeline planning, budget tradeoffs (PURExpress, Twist gBlock vs Clonal Gene), code for project website and final documentation. Also used for sanity-checking sequences and stepwise Benchling workflows.
Industry Partners & External Resources
Twist Bioscience - Synthesis feasibility and draft gene orders
Ginkgo Bioworks - Target partner for cell-free expression (Aim 2a)
Genyro / Sidewinder - SynBioBeta 2026, May 6 2026; identified as future assembly route
SynBioBeta 2026 conference - Sidewinder presentation by Adrian Woolfson and Prof. Kaihang Wang
Acknowledgments
HTGAA 2026 course instructors, TAs, and guest lecturers (MIT Media Lab)
Node leads, TAs and cohort
All homework design contributors
The HTGAA 2026 cohort as a whole
Subsections of Individual Final Project-Co-RSynBioR
Aim 1.Construct Design: CBM27_RGD_MaSp1 Fusion Protein
Spider Silk-Snow Fungus Bioactive Biomaterial Optimised for Wound Care — CBM27_RGD_MaSp1_4x Fusion Protein · HTGAA Spring 2026
Documented (Update)
In Benchling:
Full 8x construct (480 aa) fully annotated for in silico validation and Ginkgo pitch
4x construct (340 aa) fully annotated as synthesis-ready design
In Twist:
Saved draft order for CBM27_RGD_MaSp1_4x_Fusion as a Gene Fragment at $71.61 (~£56), 1,023 bp, complexity: Complex, orderable
Saved draft order for CBM27_RGD_MaSp1_4x_pET28a as a Clonal Gene in pET28a(+) at NdeI/XhoI insertion point at $145.45 (~£115), 1,005 bp insert, 6,297 bp total plasmid, complexity: Complex, orderable
Downloaded FASTA, GenBank and text files of the optimized gene fragment sequence
Downloaded GenBank file of the complete pET28a construct
Circular plasmid map generated and documented showing all functional elements
In Benchling: (Older)
Full 8x construct (480 aa) fully annotated
4x construct (340 aa) fully annotated as synthesis-ready design
In Twist:
Saved draft order for CBM27_RGD_MaSp1_4x_Fusion at $71.61
Downloaded FASTA, GenBank and text files of the optimized sequence
In IDT:
Documented complexity failure of 8x construct (score 57.3)
Documented complexity failure of identical MaSp1 repeats (score 53)
The Four-Component Logic
The fusion protein is built from four functional units arranged in a deliberate order from N-terminus to C-terminus:
The CBM27 domain, sourced from the Thermotoga maritima beta-mannanase Man5 (gene TM1227), physically anchors the silk network to the Tremella polysaccharide matrix by binding its mannan backbone. Without this anchor, the silk and polysaccharide components would phase-separate in wound exudate.
The flexible GGGGS x3 linker gives the CBM27 domain rotational freedom to engage the Tremella polysaccharide chains independently from the rest of the protein. This design follows validated precedent from CBM27 fusion protein literature.
The RGD motif (GRGDS), derived from the minimal integrin-binding sequence of human fibronectin, actively recruits fibroblasts and keratinocytes to the material surface to accelerate tissue repair. This converts the dressing from a passive scaffold into an active wound-healing participant.
Eight MaSp1 consensus repeat units provide the mechanical backbone of the construct, self-assembling into beta-sheet nanofibrils that give the hydrogel its toughness and structural integrity.
Construct Architecture
The full fusion protein is 480 amino acids with the following domain map:
The CBM27 domain boundaries were verified against the UniProt entry for Thermotoga maritima Man5, which annotates the Carbohydrate Binding Module 27 at residues 495-664 of the full 669 aa protein. A five-residue buffer was added to each boundary to avoid clipping structurally important residues at the domain edge, giving a final CBM27 input of 177 amino acids.
The complete construct was assembled and annotated in Benchling as an AA sequence file named CBM27_RGD_MaSp1_Fusion within the Spider Silk - Tremella project.
Synthesis Complexity and the Codon Shuffling Requirement
Initial synthesis feasibility analysis via the IDT Codon Optimization Tool confirmed that direct translation of 8 identical MaSp1 repeat units generates prohibitive sequence complexity, returning a total complexity score of 57.3 against a synthesis threshold of 24. The specific failure modes identified were:
Repeated DNA sequences exceeding 13 bases appearing at multiple locations across the insert
73.5% of the overall sequence composed of repeats longer than 8 bases
A 100-base window at position 1165 with 85% GC content
Hairpin structures forming between identical repeat regions
This result validates the codon shuffling strategy as a necessary design requirement rather than an optional refinement. The next step is to manually assign synonymous codons to each of the 8 MaSp1 repeats so that every repeat encodes an identical amino acid sequence but presents a distinct DNA sequence to the synthesis machinery.
1. Sequence Acquisition: CBM27 Domain
The carbohydrate binding module 27 domain was sourced from the Thermotoga maritima beta-mannanase Man5 (gene TM1227), a 669 amino acid protein. The CBM27 domain boundaries were verified against the UniProt entry, which annotates the domain at residues 495-664. A five-residue buffer was added to each boundary to avoid clipping structurally important residues at the domain edge, giving a final CBM27 input sequence of 177 amino acids spanning residues 490-669.
The full fusion protein was assembled in Benchling as an AA sequence file named CBM27_RGD_MaSp1_Fusion within the Spider Silk - Tremella project folder. Each domain was added sequentially and annotated with a distinct colour:
Domain
Positions
Colour
Function
CBM27 (buffered)
1-176
Blue
Tremella polysaccharide anchor
GGGGS x3 Linker
177-188
Grey
Domain flexibility
RGD motif
189-193
Pink
Cell recruitment
MaSp1 R1-R8
194-474
Green
Mechanical structure
6xHis tag
475-480
Yellow
Ni-NTA purification
Total length: 480 amino acids. Molecular weight: 44,409 Da. Isoelectric point: 7.29.
The annotated sequence map confirmed all domains were correctly positioned and accounted for. The biochemical properties panel confirmed the sequence was 480 amino acids running from KVVN at the N-terminus to HHHH at the C-terminus.
3. Codon Optimization: CBM27 Domain
The CBM27 domain alone (177 aa) was submitted to the IDT Codon Optimization Tool with the following settings:
Sequence type: Amino Acids
Product type: gBlocks Gene Fragments
Organism: Escherichia coli
Restriction sites to avoid: BsaI (GGTCTC), BsmBI (CGTCTC)
The CBM27 domain passed initial screening with no complexity issues. The codon-optimized DNA output was 531 bp. No BsaI or BsmBI sites were introduced. Several other restriction sites were noted (PstI, SmaI, XmaI) but these are irrelevant to the Golden Gate assembly strategy and were not flagged as problems.
4. Synthesis Feasibility Testing: Full 8x Construct
IDT Test 1: Identical MaSp1 Repeats
The complete 480 amino acid sequence was submitted to IDT Codon Optimization with E. coli settings. IDT returned a complexity score of 57.3, well above the synthesis threshold of 24, with the following specific failure modes:
A repeat sequence of 42 bases appearing at multiple locations
73.5% of the overall sequence composed of repeats longer than 8 bases
A 100-base window at position 1165 with 85% GC content
Hairpin structures forming between identical repeat regions
Result: Denied. Not synthesizable.
This confirmed the known problem with identical tandem silk repeats: even after codon optimization, the DNA homology between repeated units causes synthesis machinery slippage.
IDT Test 2: Codon-Shuffled MaSp1 Repeats
Eight synonymous codon-shuffled variants of the MaSp1 repeat were computationally generated, each encoding the identical amino acid sequence GQGAGAAAAAAGGAGQGGYGGLGSQGAGRGGLGGQ but using distinct codon assignments drawn from the E. coli K12 codon table. No two adjacent repeats shared more than 3 consecutive identical base pairs.
The full insert including CBM27, linker, RGD, all 8 shuffled repeats, 6xHis tag and stop codon was assembled as a 1,443 bp sequence and submitted to IDT gBlocks entry for complexity testing.
Result: Complexity score 53. Still Denied.
The codon shuffling reduced the score from 57.3 to 53 but was insufficient to bring it below the 24 threshold. The dominant remaining problem was a 42-base shared subsequence between two repeats in the alanine-rich region, where the limited synonymous codon options for alanine and glycine prevented sufficient DNA diversification across 8 repeats.
Twist Bioscience Test: Full 8x Construct
The same 480 amino acid sequence was submitted to Twist Bioscience’s gene ordering portal using their built-in amino acid import and codon optimization workflow with E. coli codon table, BsaI and BsmBI avoidance.
Result: Not Accepted.
Twist’s algorithm, which is more sophisticated than IDT’s and better handles repetitive sequences, also rejected the 8x construct. This was definitive confirmation that 8 identical MaSp1 repeats cannot be synthesized as a single gene fragment by any current commercial synthesis vendor.
5. Design Decision: 4x Repeat Construct
Based on the synthesis feasibility data, a second construct was designed using 4 MaSp1 repeat units instead of 8. This was created in Benchling by duplicating the original file and deleting repeats R5 through R8 (positions 334-473), producing a 340 amino acid construct named CBM27_RGD_MaSp1_4x_Fusion.
Domain
Positions
Function
CBM27 (buffered)
1-176
Tremella polysaccharide anchor
GGGGS x3 Linker
177-188
Domain flexibility
RGD motif
189-193
Cell recruitment
MaSp1 R1-R4
194-333
Mechanical structure
6xHis tag
334-340
Ni-NTA purification
6. Synthesis Feasibility Testing: 4x Construct on Twist
The 340 amino acid 4x construct was submitted to Twist Bioscience using the same amino acid import workflow. Twist performed codon optimization for E. coli with BsaI and BsmBI avoidance and returned the following result:
Complexity: Complex
Length: 1,023 bp
Price: $71.61 (~£56)
Status: Orderable
Complex in Twist terminology means the sequence is manufacturable but requires more careful synthesis handling than a standard sequence. It is not a rejection. The remaining complexity flags were minor repeat density warnings in the MaSp1 region, all categorised as warnings rather than errors.
Twist’s codon optimization was applied and the optimized sequence was downloaded in FASTA, GenBank and text formats. The order draft was saved to the Twist account.
7. Summary of Constructs and Status
Construct
Repeats
Length
IDT Result
Twist Result
Purpose
CBM27_RGD_MaSp1_Fusion
8x
480 aa / 1,443 bp
Score 53, Denied
Not Accepted
In silico validation, Ginkgo pitch
CBM27_RGD_MaSp1_4x_Fusion
4x
340 aa / 1,023 bp
Not tested
Complex, $71.61, Orderable
Wet lab synthesis
8. Backbone Vector Documentation
The insert was designed for expression in pET-28a(+) (Novagen, 5,365 bp), cloned at the NdeI/XhoI insertion points. This places the fusion protein under T7 promoter control with a C-terminal 6xHis tag provided by the vector for Ni-NTA purification. The vector carries kanamycin resistance for bacterial selection and a colE1 high copy origin of replication. The complete circular plasmid was designed directly in Twist Bioscience’s Clonal Gene ordering portal, which performs synthesis, cloning, transformation, colony picking and Sanger sequencing verification, delivering a ready-to-use sequence-verified plasmid. The full construct is 6,297 bp and was downloaded as a GenBank file and imported into Benchling as a circular DNA sequence for documentation. The plasmid is compatible with NEB PURExpress E6800 and Ginkgo Bioworks CFPS cell-free expression systems, both of which are T7 promoter driven E. coli based systems requiring no further cloning before expression.
9. Next Steps
In silico track: (see In silico page)
Submit the full 480 aa CBM27_RGD_MaSp1_Fusion sequence to AlphaFold3 to predict the 3D structure and confirm that the CBM27 domain and RGD motif are surface-exposed and not buried within the MaSp1 beta-sheet core.
Wet lab track:
The protein construct ready to be ordered from Twist. Preferred Ginkgo Bioworks (for remote lab experience) or LifeFabs assay as second option. Protocol draft started.
Protocol for Assay
Protocol for Assay
Draft v 2
Experimental Protocol: CBM27_RGD_MaSp1_4x Fusion Expression and Tremella Composite Formation
Construct: CBM27_RGD_MaSp1_4x_Fusion Vector: pET28a (NdeI/XhoI, C-terminal 6xHis tag) Protein MW: ~37 kDa (340 aa) Expression system: Cell-free protein synthesis (CFPS) — see Aim 2 context below
Aim 2 Context: Why This Assay Exists
This protocol is the experimental component of Aim 2a of the Exoskin project. Aim 1 established the computational and synthesis-feasibility foundation: the CBM27_RGD_MaSp1_4x construct has been designed in Benchling, codon-optimised for E. coli K12, validated structurally by AlphaFold3 (CBM27 pLDDT >90, RGD solvent-exposed, MaSp1 disordered as expected), and confirmed orderable by Twist Biosciences at 6,297 bp in pET28a ($145.45, Clonal Gene). HADDOCK docking of CBM27 against Tremella β-(1→3)-D-mannan is pending due to a documented gap in carbohydrate force field databases for fungal mannans; this makes wet lab confirmation of polysaccharide binding the critical validation step.
The protocol below is designed for submission to the Ginkgo Bioworks (via HTGAA Twist aim 1 order submission to be confrimed) automated CFPS platform. Steps are written to be compatible with liquid-handling automation and standard Ginkgo reagent sets. Where a step requires upfront specification (e.g. chaperone supplementation), this is flagged explicitly, because automated cloud lab runs cannot be modified mid-protocol after submission.
Alternative execution route: If Ginkgo partnership is not confirmed in time, this protocol can be run manually using NEB PURExpress E6800 in any standard molecular biology lab. All volumes and conditions are identical between routes.
Step 1: Cell-Free Protein Synthesis
Materials needed:
PURExpress Solution A
PURExpress Solution B
Murine RNase Inhibitor
Template DNA: pET28a_CBM27_RGD_MaSp1_4x plasmid (circular, 250 ng) or linear PCR product
DnaK chaperone mix (supplementary, see note below)
Nuclease-free water
Ice
Chaperone specification note (required for Ginkgo submission): MaSp1 repeat regions are prone to aggregation and beta-sheet stacking due to their glycine/alanine-rich repetitive sequence. DnaK/DnaJ/GrpE chaperone supplementation should be specified upfront in the Ginkgo protocol submission. Typical addition: 2 µM DnaK, 0.4 µM DnaJ, 0.1 µM GrpE in the final reaction volume. If using manual PURExpress, add chaperones after assembling the base reaction and before incubation.
Protocol:
Thaw Solutions A and B on ice. Do not vortex.
Assemble the following reaction on ice in this exact order in a 1.5 ml microcentrifuge tube:
10 µl Solution A
7.5 µl Solution B
0.5 µl RNase Inhibitor (20 units)
2 µl template DNA (250 ng)
0.5 µl DnaK chaperone mix (see note above)
4.5 µl nuclease-free water
Total volume: 25 µl
Mix gently by pipetting up and down 5 times. Do not vortex.
Incubate at 37°C for 2 hours.
Place on ice immediately after incubation.
Run alongside: one negative control reaction with no template DNA, identical volumes and conditions.
Step 2: His-Tag Purification (Mandatory Before Step 3)
This step is required before proceeding to Tremella composite formation. The raw PURExpress reaction contains many E. coli ribosomal and accessory proteins. Mixing unpurified reaction with Tremella polysaccharide risks false-positive gel formation from non-specific protein-polysaccharide interactions. Ni-NTA purification isolates only the 6xHis-tagged fusion protein.
Materials needed:
Ni-NTA agarose resin (e.g. Qiagen, cat. 30210) or magnetic Ni-NTA beads
Binding buffer: 50 mM NaH₂PO₄, 300 mM NaCl, 10 mM imidazole, pH 8.0
Wash buffer: 50 mM NaH₂PO₄, 300 mM NaCl, 20 mM imidazole, pH 8.0
Elution buffer: 50 mM NaH₂PO₄, 300 mM NaCl, 250 mM imidazole, pH 8.0
Spin columns or magnetic rack
PBS pH 7.4 for buffer exchange
Protocol:
Dilute the 25 µl CFPS reaction with 225 µl binding buffer (1:10 dilution).
Add 25 µl pre-equilibrated Ni-NTA resin slurry. Mix on rotary mixer for 30 minutes at 4°C.
Wash resin twice with 200 µl wash buffer. Centrifuge 700 × g, 2 minutes each wash.
Elute with 50 µl elution buffer. Incubate 5 minutes at room temperature, then centrifuge 700 × g, 2 minutes.
Buffer exchange into PBS pH 7.4 using a 10 kDa MWCO spin concentrator to remove imidazole.
Recover purified protein in approximately 20–25 µl PBS.
Reserve 2.5 µl for SDS-PAGE (Step 3). The remaining ~18–22 µl proceeds to Step 4.
Step 3: SDS-PAGE Confirmation
Materials needed:
10-20% Tris-glycine precast gel
SDS loading buffer (4x)
Protein ladder (10-250 kDa range)
Running buffer (Tris-glycine-SDS)
Coomassie Blue stain (or silver stain for higher sensitivity)
Protocol:
Take 2.5 µl of the purified eluate and add 2.5 µl of 2x SDS loading buffer (or 0.83 µl of 4x buffer + 1.67 µl nuclease-free water to reach 1x final concentration).
Heat at 95°C for 5 minutes to denature proteins.
Load onto gel alongside protein ladder and a lane with the same volume of purified negative control eluate.
Run at 200V for 35 minutes.
Stain with Coomassie Blue for 1 hour. Destain with water overnight, or use methanol/acetic acid destain for faster results.
Look for a band at approximately 37 kDa.
Success criterion: A visible band at 37 kDa in the expression lane that is absent in the negative control lane. Proceed to Step 4 only if this criterion is met.
If no band is observed: Do not proceed to composite formation. Troubleshoot expression first: check template DNA integrity by nanodrop and gel, verify T7 promoter orientation in the construct, and consider a second run with increased template (up to 500 ng) or extended incubation (4 hours at 37°C). If aggregation is suspected (smear or high-MW band), confirm DnaK chaperone supplementation was included.
Step 4: Tremella Composite Formation (only if Step 3 successful and not confirmed if can be run after above if at Ginkgo Bio)
Materials needed:
Tremella fuciformis polysaccharide: minimum 95% purity, fungal-derived, CAS 9083-80-1 (note: use a supplier that specifies β-(1→3)-linked mannan as the primary backbone structure; Sigma-Aldrich or Funakoshi are appropriate sources — confirm catalogue specification before ordering)
PBS buffer pH 7.4
Purified fusion protein from Step 2 (~18–22 µl)
Microcentrifuge tubes
Rotary mixer
Positive control: PURExpress negative control eluate (same volume as fusion protein sample)
Protocol:
Prepare a 1% w/v Tremella polysaccharide solution by dissolving 10 mg dried Tremella polysaccharide in 1 ml PBS pH 7.4. Stir gently at room temperature for 2 hours until fully dissolved. The solution should be visibly viscous. If it does not dissolve fully, warm to 37°C for 30 minutes before cooling back to room temperature.
Take the purified fusion protein from Step 2 (~18–22 µl). Record exact volume.
Add an equal volume of the 1% Tremella polysaccharide solution to the protein. Mix gently by pipetting 10 times. Do not vortex.
Prepare a parallel control by mixing the same volume of the purified negative control eluate with the same volume of 1% Tremella solution.
Incubate both tubes at room temperature for 30 minutes on a rotary mixer at low speed, to allow CBM27 domain binding to the Tremella β-(1→3)-D-mannan backbone.
Assess viscosity using the tube inversion test: invert each tube 180° and observe flow behaviour over 10 seconds. A gel or high-viscosity hydrogel will not flow freely; a low-viscosity solution will run to the cap immediately.
As a semi-quantitative measure, attempt to aspirate 10 µl from each tube using a standard 20 µl pipette tip. Note resistance to aspiration on a simple 0/1/2 scale (0 = no resistance, 1 = moderate resistance, 2 = cannot aspirate). Record for both the fusion protein tube and the control tube.
Success criterion: Visible increase in viscosity or gel formation in the fusion protein tube that is absent or substantially lower in the negative control tube. A tube inversion score of 1 or 2 in the fusion protein tube versus 0 in the control tube constitutes a positive result.
If no gel formation is observed: CBM27 may not be folding correctly under cell-free conditions, or the Tremella preparation may not contain sufficient β-(1→3)-mannan backbone to support CBM27 binding. Troubleshooting options: (1) repeat with a different Tremella polysaccharide source or lot; (2) run a second CFPS reaction with extended incubation and additional DnaK; (3) attempt CD spectroscopy on the purified protein to assess CBM27 secondary structure if equipment is available.
What Each Result Means
Result
Interpretation
Band at 37 kDa on gel, absent in control
Protein expressed successfully. Proceed to Step 4.
No band on gel
Expression failed. Check DNA template integrity, T7 promoter orientation, and DnaK supplementation. Do not proceed.
Band present but at wrong MW
Possible truncation or degradation. Check RNase inhibitor and RNA integrity.
Gel formation / high viscosity in Step 4
CBM27 is anchoring the silk to the Tremella matrix. Composite hydrogel formation confirmed. Aim 2a and 2b success.
No gel formation in Step 4, band confirmed in Step 3
Protein expressed but CBM27 may not be binding. Check Tremella source specification; consider docking simulation to revisit binding geometry.
No gel formation in Step 4, no band in Step 3
Expression failed. Step 4 result is uninterpretable. Restart from expression troubleshooting.
Ginkgo Submission Checklist
If submitting to Ginkgo Bioworks cloud lab, confirm the following are specified in your protocol submission before sending:
Plasmid sequence file (Benchling export, .gb or .fa) for CBM27_RGD_MaSp1_4x in pET28a
Expected band size stated as 37 kDa (for SDS-PAGE automated imaging parameters)
Ni-NTA purification step requested prior to composite assembly
Tremella polysaccharide source and CAS number specified (CAS 9083-80-1, minimum 95% purity, β-(1→3)-mannan backbone confirmed)
Negative control (no-template CFPS) requested in parallel
Group Final Project
L-Protein Mutants
Problem: How to improve the stability and auto-folding of the lysis protein of an MS2-phage? This mechanism is key to understanding how phages may help address antibiotic resistance.
After going through the readings, including the group final project document a Plan A would be:
(This stays within scope, MurJ and multi-target approaches seem intersting though…)
Use computational tools like AlphaFold2 or ProteinMPNN to identify mutations that improve intrinsic stability and auto-folding of the lysis protein
Target mutations that strengthen the hydrophobic core, eliminate aggregation-prone regions, or introduce stabilising interactions like salt bridges
Engineer the lysis protein to fold correctly without requiring DnaJ or any other bacterial chaperone
Design mutations that also accelerate oligomerisation or enhance membrane pore-forming activity for faster lysis
Synthesise the mutant gene via Twist, clone into plasmid using Gibson Assembly, validate structural integrity with Nuclera, then test in E. coli.



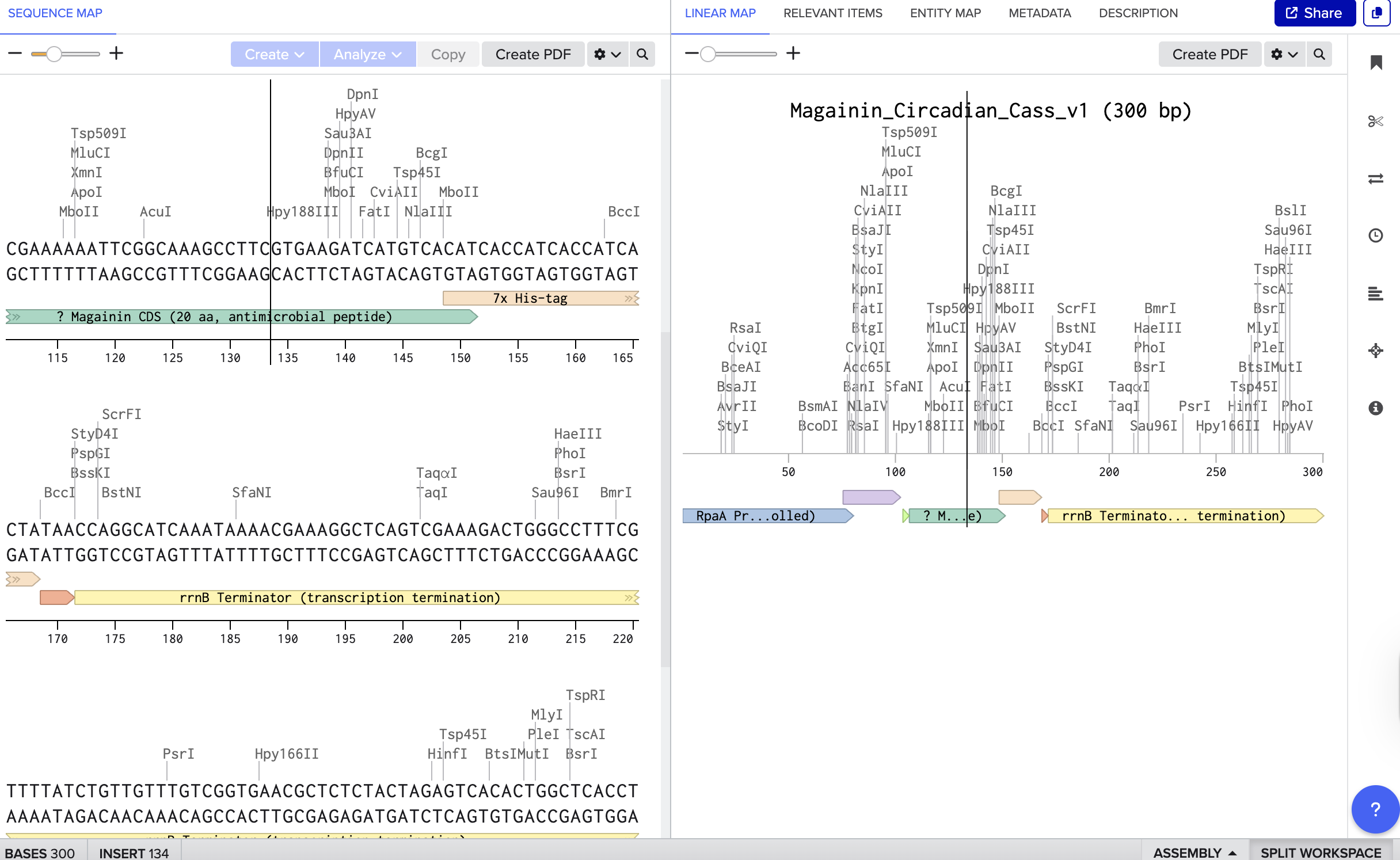
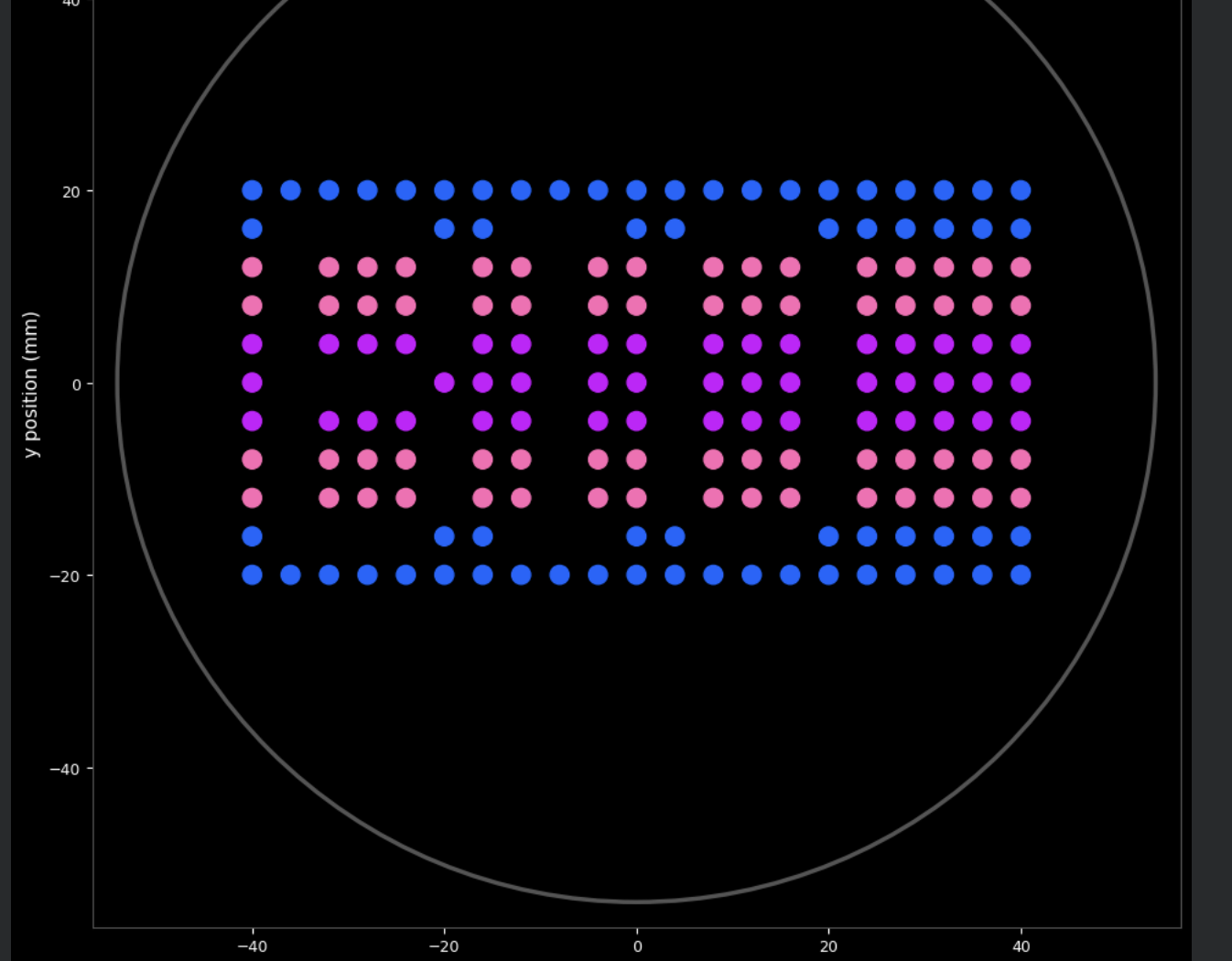
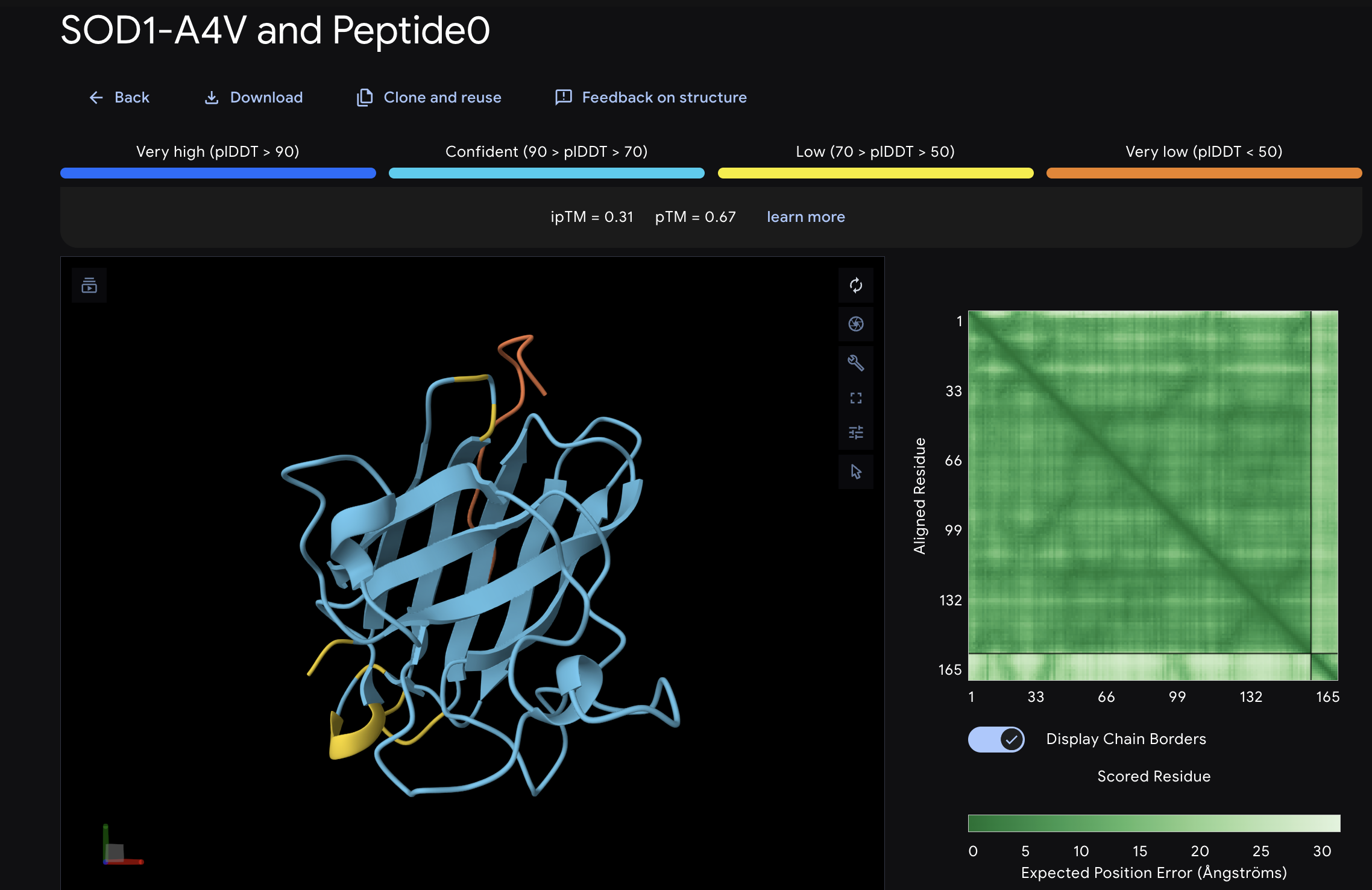

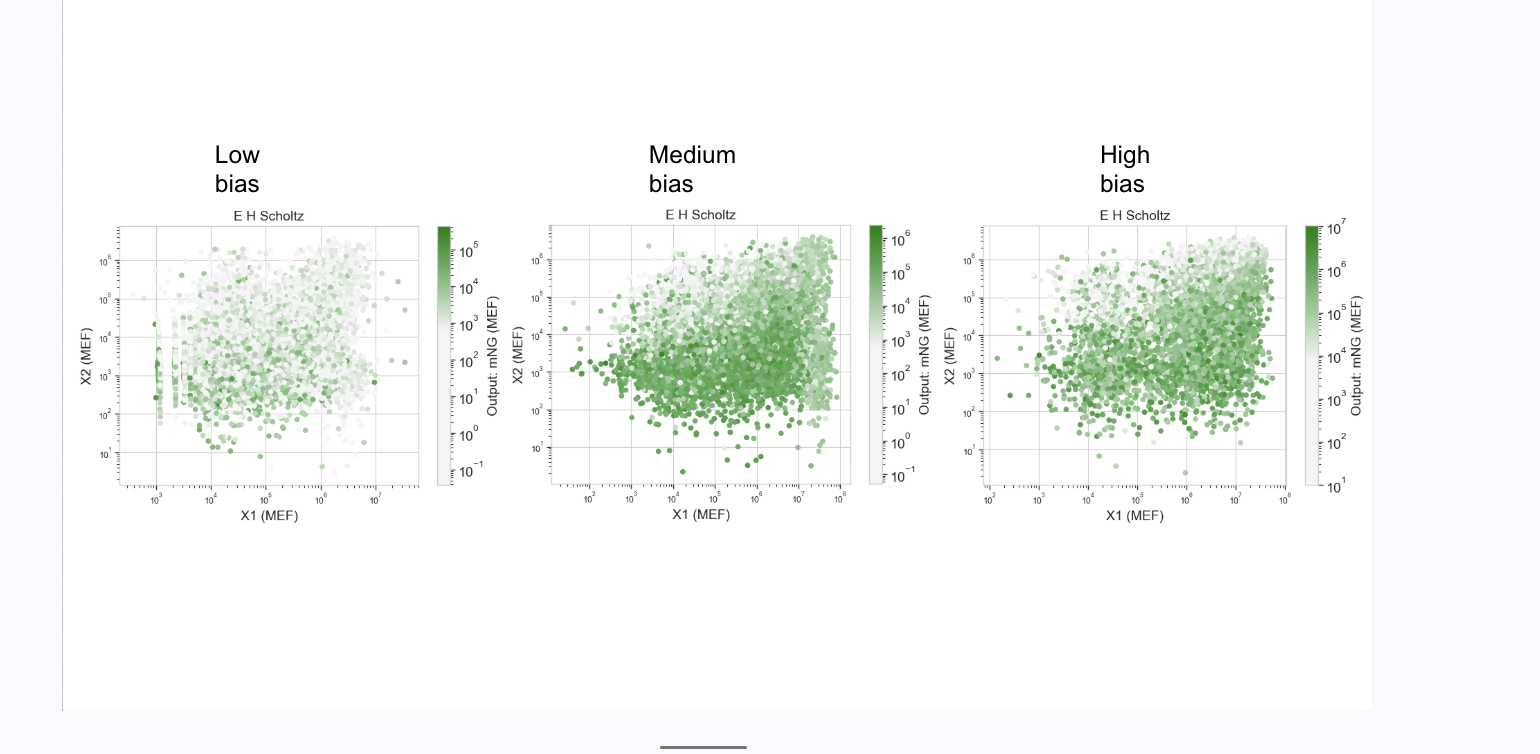





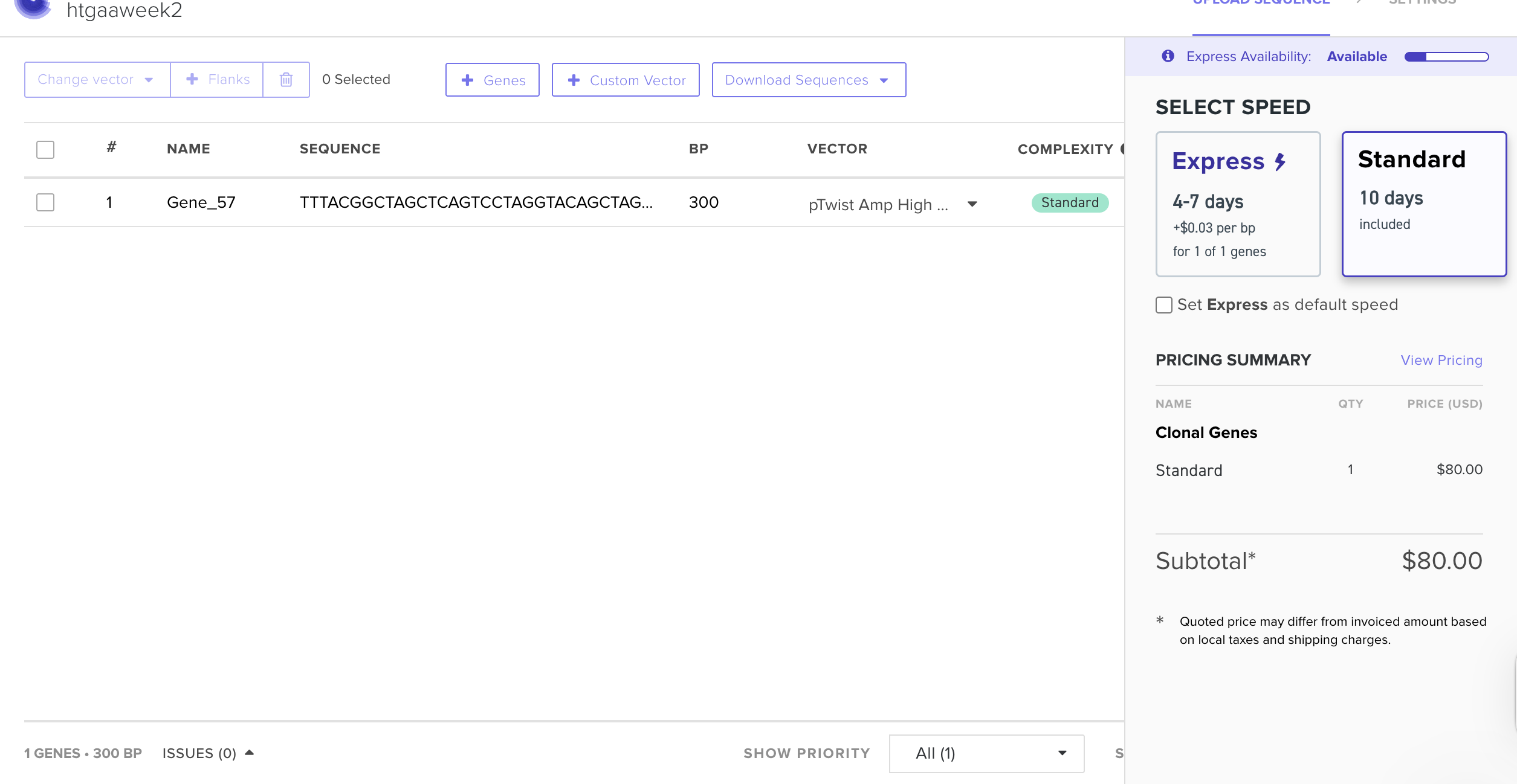
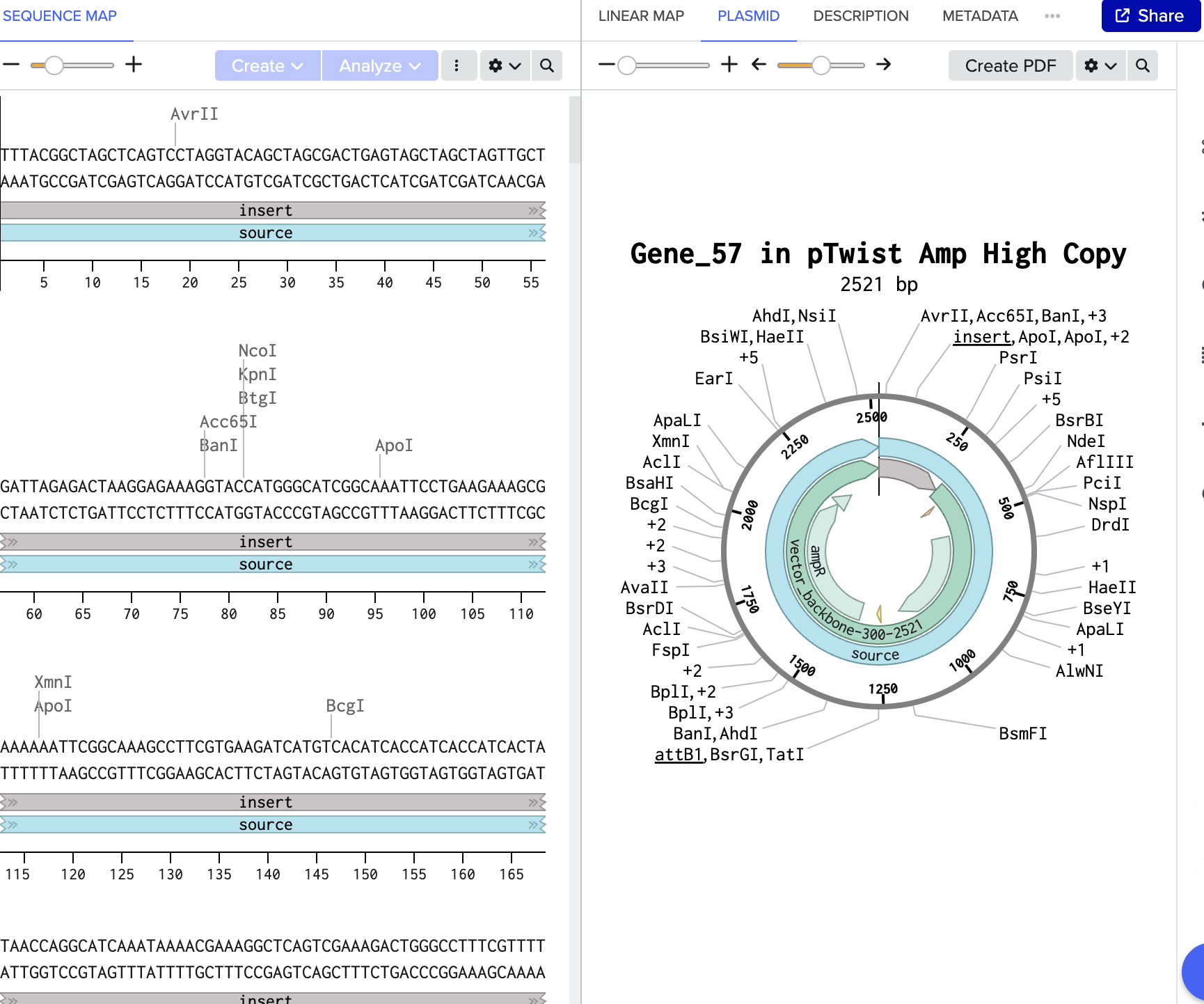


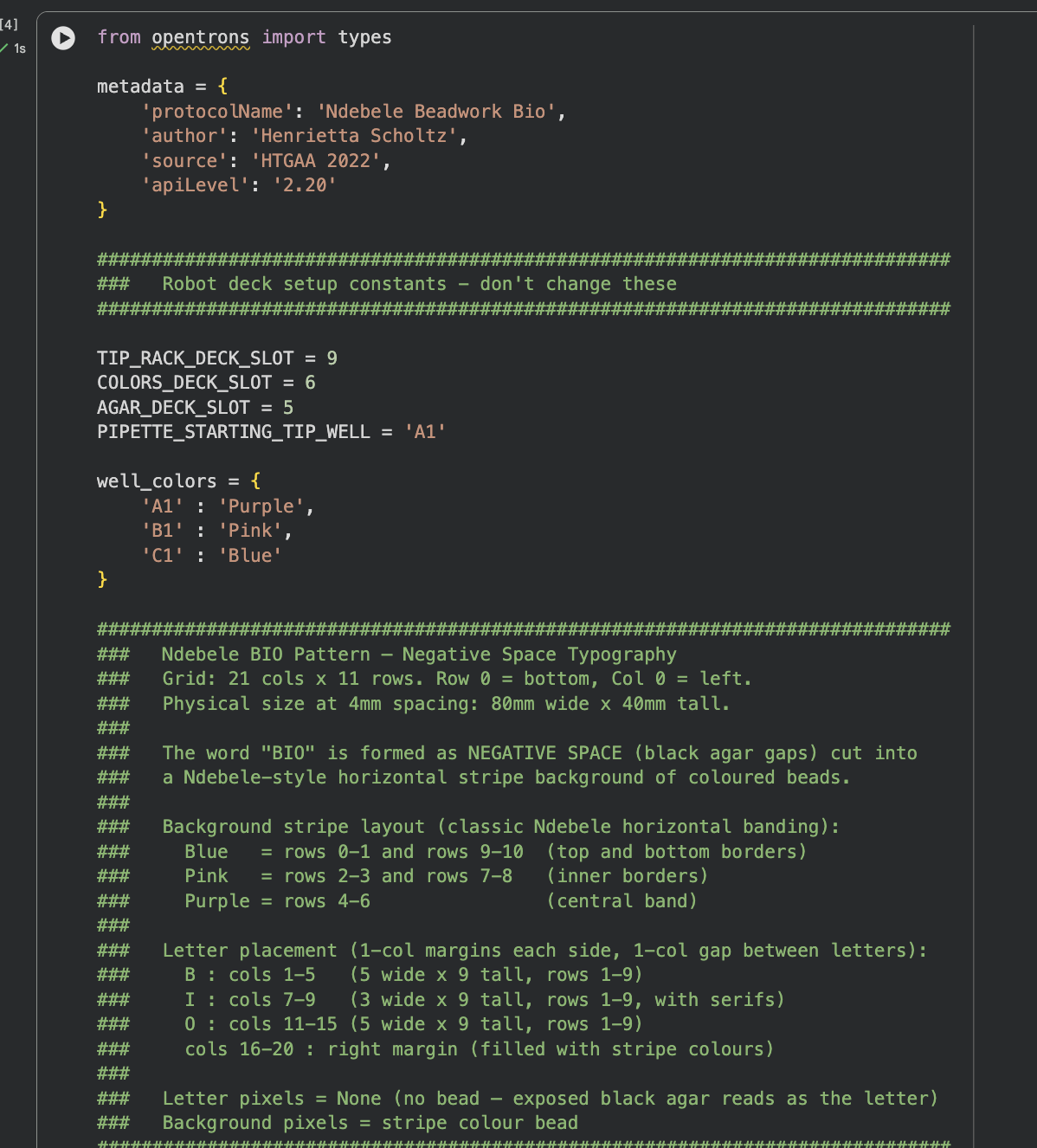



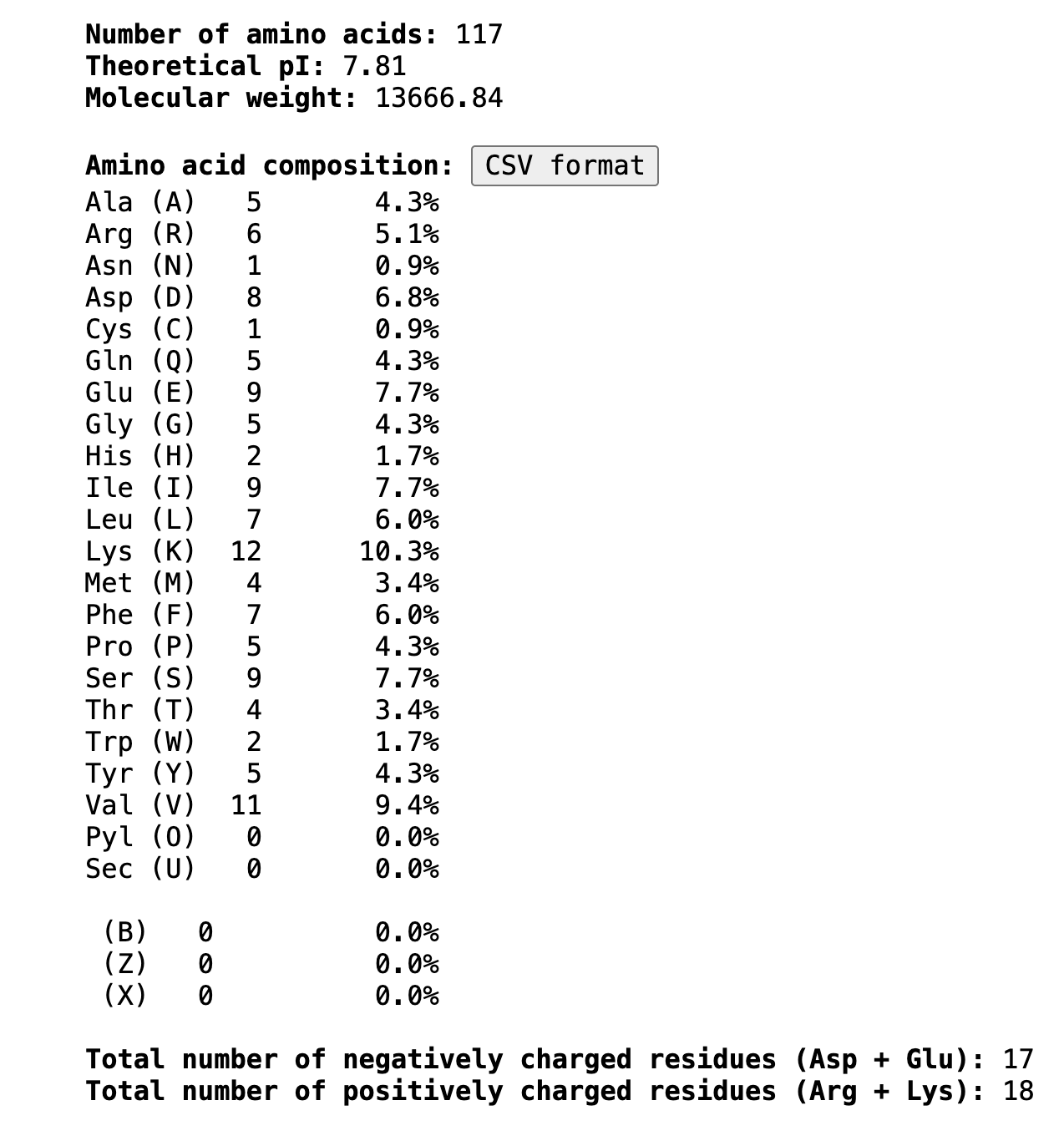
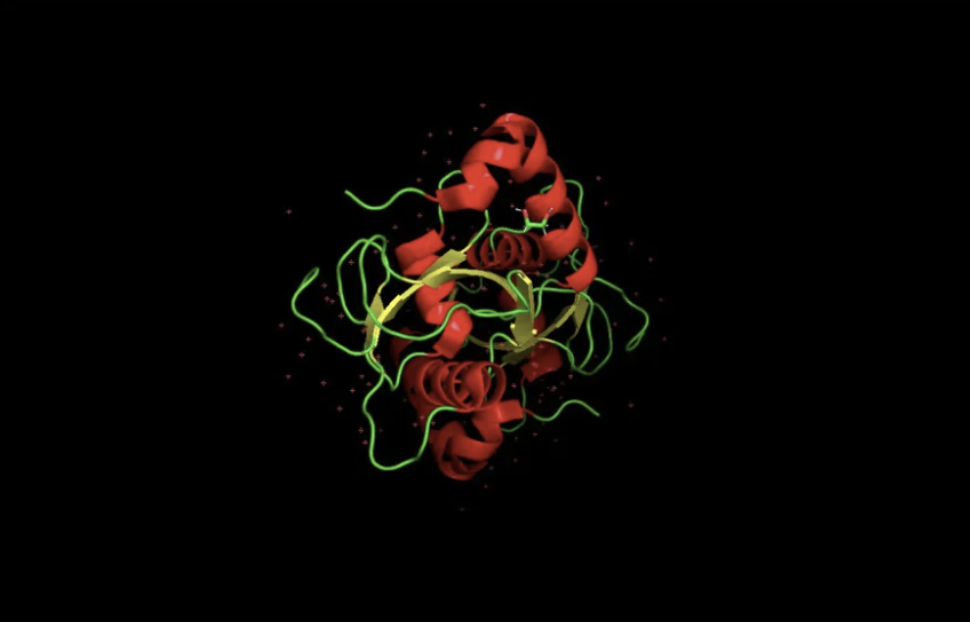 When colored by secondary structure, GABARAPL2 shows a clear dominance of red (helices) over yellow (beta sheets). Green loops connect these elements throughout the structure.
When colored by secondary structure, GABARAPL2 shows a clear dominance of red (helices) over yellow (beta sheets). Green loops connect these elements throughout the structure.






 ___
___