Aim 1.Construct Design: CBM27_RGD_MaSp1 Fusion Protein
Spider Silk-Snow Fungus Bioactive Biomaterial Optimised for Wound Care — CBM27_RGD_MaSp1_4x Fusion Protein · HTGAA Spring 2026

Documented (Update)
In Benchling:
- Full 8x construct (480 aa) fully annotated for in silico validation and Ginkgo pitch
- 4x construct (340 aa) fully annotated as synthesis-ready design
In Twist:
Saved draft order for CBM27_RGD_MaSp1_4x_Fusion as a Gene Fragment at $71.61 (~£56), 1,023 bp, complexity: Complex, orderable
Saved draft order for CBM27_RGD_MaSp1_4x_pET28a as a Clonal Gene in pET28a(+) at NdeI/XhoI insertion point at $145.45 (~£115), 1,005 bp insert, 6,297 bp total plasmid, complexity: Complex, orderable
Downloaded FASTA, GenBank and text files of the optimized gene fragment sequence
Downloaded GenBank file of the complete pET28a construct
Circular plasmid map generated and documented showing all functional elements
In Benchling: (Older)
Full 8x construct (480 aa) fully annotated 4x construct (340 aa) fully annotated as synthesis-ready design
In Twist:
Saved draft order for CBM27_RGD_MaSp1_4x_Fusion at $71.61 Downloaded FASTA, GenBank and text files of the optimized sequence
In IDT:
Documented complexity failure of 8x construct (score 57.3) Documented complexity failure of identical MaSp1 repeats (score 53)
The Four-Component Logic The fusion protein is built from four functional units arranged in a deliberate order from N-terminus to C-terminus:
The CBM27 domain, sourced from the Thermotoga maritima beta-mannanase Man5 (gene TM1227), physically anchors the silk network to the Tremella polysaccharide matrix by binding its mannan backbone. Without this anchor, the silk and polysaccharide components would phase-separate in wound exudate.
The flexible GGGGS x3 linker gives the CBM27 domain rotational freedom to engage the Tremella polysaccharide chains independently from the rest of the protein. This design follows validated precedent from CBM27 fusion protein literature. The RGD motif (GRGDS), derived from the minimal integrin-binding sequence of human fibronectin, actively recruits fibroblasts and keratinocytes to the material surface to accelerate tissue repair. This converts the dressing from a passive scaffold into an active wound-healing participant.
Eight MaSp1 consensus repeat units provide the mechanical backbone of the construct, self-assembling into beta-sheet nanofibrils that give the hydrogel its toughness and structural integrity. Construct Architecture The full fusion protein is 480 amino acids with the following domain map:
DomainPositionsFunctionCBM27 (buffered)1-176Tremella polysaccharide anchorGGGGS x3 Linker177-188Domain flexibilityRGD motif189-193Cell recruitmentMaSp1 x8 repeats194-474Mechanical structure6xHis tag475-480Ni-NTA purification Molecular weight: 44,409 Da. Isoelectric point: 7.29. Sequence Source and Verification

The CBM27 domain boundaries were verified against the UniProt entry for Thermotoga maritima Man5, which annotates the Carbohydrate Binding Module 27 at residues 495-664 of the full 669 aa protein. A five-residue buffer was added to each boundary to avoid clipping structurally important residues at the domain edge, giving a final CBM27 input of 177 amino acids.
The complete construct was assembled and annotated in Benchling as an AA sequence file named CBM27_RGD_MaSp1_Fusion within the Spider Silk - Tremella project.
Synthesis Complexity and the Codon Shuffling Requirement Initial synthesis feasibility analysis via the IDT Codon Optimization Tool confirmed that direct translation of 8 identical MaSp1 repeat units generates prohibitive sequence complexity, returning a total complexity score of 57.3 against a synthesis threshold of 24. The specific failure modes identified were:
Repeated DNA sequences exceeding 13 bases appearing at multiple locations across the insert 73.5% of the overall sequence composed of repeats longer than 8 bases A 100-base window at position 1165 with 85% GC content Hairpin structures forming between identical repeat regions

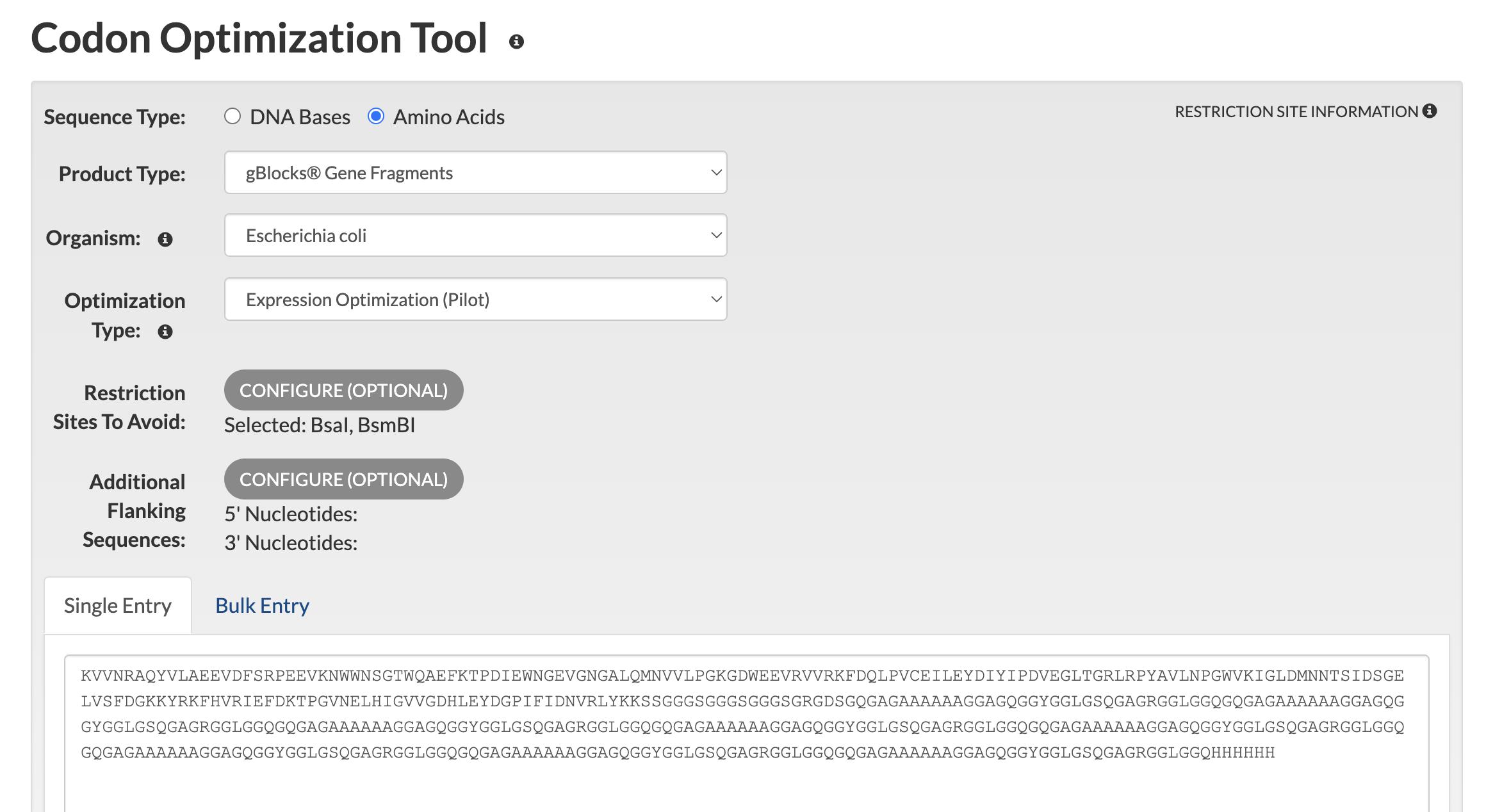


This result validates the codon shuffling strategy as a necessary design requirement rather than an optional refinement. The next step is to manually assign synonymous codons to each of the 8 MaSp1 repeats so that every repeat encodes an identical amino acid sequence but presents a distinct DNA sequence to the synthesis machinery.
1. Sequence Acquisition: CBM27 Domain
The carbohydrate binding module 27 domain was sourced from the Thermotoga maritima beta-mannanase Man5 (gene TM1227), a 669 amino acid protein. The CBM27 domain boundaries were verified against the UniProt entry, which annotates the domain at residues 495-664. A five-residue buffer was added to each boundary to avoid clipping structurally important residues at the domain edge, giving a final CBM27 input sequence of 177 amino acids spanning residues 490-669.
The extracted sequence was:
2. Construct Assembly in Benchling
The full fusion protein was assembled in Benchling as an AA sequence file named CBM27_RGD_MaSp1_Fusion within the Spider Silk - Tremella project folder. Each domain was added sequentially and annotated with a distinct colour:
| Domain | Positions | Colour | Function |
|---|---|---|---|
| CBM27 (buffered) | 1-176 | Blue | Tremella polysaccharide anchor |
| GGGGS x3 Linker | 177-188 | Grey | Domain flexibility |
| RGD motif | 189-193 | Pink | Cell recruitment |
| MaSp1 R1-R8 | 194-474 | Green | Mechanical structure |
| 6xHis tag | 475-480 | Yellow | Ni-NTA purification |
Total length: 480 amino acids. Molecular weight: 44,409 Da. Isoelectric point: 7.29.
The annotated sequence map confirmed all domains were correctly positioned and accounted for. The biochemical properties panel confirmed the sequence was 480 amino acids running from KVVN at the N-terminus to HHHH at the C-terminus.
3. Codon Optimization: CBM27 Domain
The CBM27 domain alone (177 aa) was submitted to the IDT Codon Optimization Tool with the following settings:
- Sequence type: Amino Acids
- Product type: gBlocks Gene Fragments
- Organism: Escherichia coli
- Restriction sites to avoid: BsaI (GGTCTC), BsmBI (CGTCTC)
The CBM27 domain passed initial screening with no complexity issues. The codon-optimized DNA output was 531 bp. No BsaI or BsmBI sites were introduced. Several other restriction sites were noted (PstI, SmaI, XmaI) but these are irrelevant to the Golden Gate assembly strategy and were not flagged as problems.
4. Synthesis Feasibility Testing: Full 8x Construct
IDT Test 1: Identical MaSp1 Repeats
The complete 480 amino acid sequence was submitted to IDT Codon Optimization with E. coli settings. IDT returned a complexity score of 57.3, well above the synthesis threshold of 24, with the following specific failure modes:
- A repeat sequence of 42 bases appearing at multiple locations
- 73.5% of the overall sequence composed of repeats longer than 8 bases
- A 100-base window at position 1165 with 85% GC content
- Hairpin structures forming between identical repeat regions
Result: Denied. Not synthesizable.
This confirmed the known problem with identical tandem silk repeats: even after codon optimization, the DNA homology between repeated units causes synthesis machinery slippage.
IDT Test 2: Codon-Shuffled MaSp1 Repeats
Eight synonymous codon-shuffled variants of the MaSp1 repeat were computationally generated, each encoding the identical amino acid sequence GQGAGAAAAAAGGAGQGGYGGLGSQGAGRGGLGGQ but using distinct codon assignments drawn from the E. coli K12 codon table. No two adjacent repeats shared more than 3 consecutive identical base pairs.
The full insert including CBM27, linker, RGD, all 8 shuffled repeats, 6xHis tag and stop codon was assembled as a 1,443 bp sequence and submitted to IDT gBlocks entry for complexity testing.
Result: Complexity score 53. Still Denied.
The codon shuffling reduced the score from 57.3 to 53 but was insufficient to bring it below the 24 threshold. The dominant remaining problem was a 42-base shared subsequence between two repeats in the alanine-rich region, where the limited synonymous codon options for alanine and glycine prevented sufficient DNA diversification across 8 repeats.
Twist Bioscience Test: Full 8x Construct
The same 480 amino acid sequence was submitted to Twist Bioscience’s gene ordering portal using their built-in amino acid import and codon optimization workflow with E. coli codon table, BsaI and BsmBI avoidance.
Result: Not Accepted.
Twist’s algorithm, which is more sophisticated than IDT’s and better handles repetitive sequences, also rejected the 8x construct. This was definitive confirmation that 8 identical MaSp1 repeats cannot be synthesized as a single gene fragment by any current commercial synthesis vendor.
5. Design Decision: 4x Repeat Construct
Based on the synthesis feasibility data, a second construct was designed using 4 MaSp1 repeat units instead of 8. This was created in Benchling by duplicating the original file and deleting repeats R5 through R8 (positions 334-473), producing a 340 amino acid construct named CBM27_RGD_MaSp1_4x_Fusion.
| Domain | Positions | Function |
|---|---|---|
| CBM27 (buffered) | 1-176 | Tremella polysaccharide anchor |
| GGGGS x3 Linker | 177-188 | Domain flexibility |
| RGD motif | 189-193 | Cell recruitment |
| MaSp1 R1-R4 | 194-333 | Mechanical structure |
| 6xHis tag | 334-340 | Ni-NTA purification |
6. Synthesis Feasibility Testing: 4x Construct on Twist
The 340 amino acid 4x construct was submitted to Twist Bioscience using the same amino acid import workflow. Twist performed codon optimization for E. coli with BsaI and BsmBI avoidance and returned the following result:
- Complexity: Complex
- Length: 1,023 bp
- Price: $71.61 (~£56)
- Status: Orderable
Complex in Twist terminology means the sequence is manufacturable but requires more careful synthesis handling than a standard sequence. It is not a rejection. The remaining complexity flags were minor repeat density warnings in the MaSp1 region, all categorised as warnings rather than errors.
Twist’s codon optimization was applied and the optimized sequence was downloaded in FASTA, GenBank and text formats. The order draft was saved to the Twist account.
7. Summary of Constructs and Status
| Construct | Repeats | Length | IDT Result | Twist Result | Purpose |
|---|---|---|---|---|---|
| CBM27_RGD_MaSp1_Fusion | 8x | 480 aa / 1,443 bp | Score 53, Denied | Not Accepted | In silico validation, Ginkgo pitch |
| CBM27_RGD_MaSp1_4x_Fusion | 4x | 340 aa / 1,023 bp | Not tested | Complex, $71.61, Orderable | Wet lab synthesis |
8. Backbone Vector Documentation

The insert was designed for expression in pET-28a(+) (Novagen, 5,365 bp), cloned at the NdeI/XhoI insertion points. This places the fusion protein under T7 promoter control with a C-terminal 6xHis tag provided by the vector for Ni-NTA purification. The vector carries kanamycin resistance for bacterial selection and a colE1 high copy origin of replication. The complete circular plasmid was designed directly in Twist Bioscience’s Clonal Gene ordering portal, which performs synthesis, cloning, transformation, colony picking and Sanger sequencing verification, delivering a ready-to-use sequence-verified plasmid. The full construct is 6,297 bp and was downloaded as a GenBank file and imported into Benchling as a circular DNA sequence for documentation. The plasmid is compatible with NEB PURExpress E6800 and Ginkgo Bioworks CFPS cell-free expression systems, both of which are T7 promoter driven E. coli based systems requiring no further cloning before expression.
9. Next Steps
In silico track: (see In silico page)
Submit the full 480 aa CBM27_RGD_MaSp1_Fusion sequence to AlphaFold3 to predict the 3D structure and confirm that the CBM27 domain and RGD motif are surface-exposed and not buried within the MaSp1 beta-sheet core.
Wet lab track:
The protein construct ready to be ordered from Twist. Preferred Ginkgo Bioworks (for remote lab experience) or LifeFabs assay as second option. Protocol draft started.