Week 2 HW: DNA Read, Write, and Edit

Week 2: DNA Read, Write, and Edit
Part 1 & 2: Gel Art
Describe your process of creating Gel Art using Benchling and restriction digests.
1.1 In-silico Design (Benchling)
Enzymes used: HindIII, SacI, KpnI
Design Concept: For this project, I designed a “Gel Art” pattern inspired by a European circular arch bridge. I wanted to use the different migration speeds of DNA fragments to recreate the structural elegance of an ancient stone bridge.
Inspiration:

Simulated Gel Pattern:

1.2 Lab Execution
- Protocol followed: “Gel Art: Restriction Digests and Gel Electrophoresis”
- Results: (Awaiting wet-lab execution results)
Part 3: DNA Design Challenge
3.1 Protein Choice
- Chosen Protein: LuxR (Aliivibrio fischeri)
- Why: I chose LuxR because it is a fundamental component for building genetic circuits. LuxR/LuxI system is one of the most well-characterized Quorum Sensing systems, used to create sophisticated biological logic gates and population-controlled behaviors in synthetic biology.
- Mechanism (LuxR/LuxI Quorum Sensing):
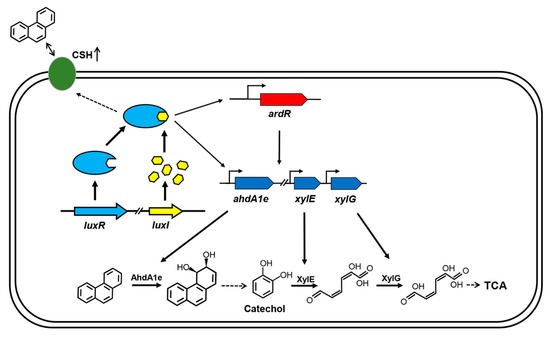 Image source: MDPI - International Journal of Molecular Sciences, 2020
Image source: MDPI - International Journal of Molecular Sciences, 2020
- Protein Sequence:
3.2 Reverse Translation
- Reverse Translation Process: Starting from the LuxR protein sequence, I determined the original nucleotide sequence of the luxR gene from Aliivibrio fischeri. I used the Gene Corner Reverse Translate Tool for this process. In synthetic biology, this “reverse” process allows us to understand how nature codes for the protein, providing a baseline for synthesis.
- Wild-type DNA Sequence (luxR):
3.3 Codon Optimization
Optimized for Organism: Escherichia coli (K-12)
Optimization Tool: I utilized the IDT Codon Optimization Tool to adapt the sequence for high expression in E. coli.
Why Optimize? Codon optimization is crucial because different organisms prefer different “synonymous” codons to represent the same amino acid. This is known as codon usage bias. In E. coli, rare codons (like those found in A. fischeri) can lead to:
- Low Expression: Ribosomes stalling at rare codons, reducing protein yield.
- Truncated Proteins: Ribosomes falling off the mRNA before finishing.
- Misfolding: The timing of translation speed affects how the protein folds. Additionally, optimization helps remove internal restriction sites (like BsaI, BbsI) and strong secondary structures that might interfere with DNA synthesis or translation.
Codon-Optimized DNA Sequence for E. coli:
3.4 Production Technology
Process (Cell-Free Protein Synthesis - CFPS): I am particularly interested in producing this protein using cell-free methods, such as the PURE (Protein synthesis Using Recombinant Elements) system or TX-TL (Transcription-Translation) cell extracts (e.g., from E. coli).
How it works: In a cell-free system, instead of transforming the DNA into a living cell, we simply mix the DNA template with a cocktail of “biological machinery” in a test tube. This cocktail includes:
- RNA Polymerase: To transcribe the DNA into mRNA.
- Ribosomes: To translate the mRNA into a protein.
- tRNAs & Amino Acids: The building blocks and adapters for protein assembly.
- Energy Sources: ATP and regeneration systems to fuel the process.
Why Cell-Free?
- Speed: Protein can be produced in hours rather than days, as there is no need for cell growth or transformation.
- Direct Prototyping: We can use linear DNA (like the codon-optimized sequence I designed) directly without cloning it into a plasmid.
- Safety & Control: Since there are no living cells, it is easier to study proteins that might be toxic to a host cell, and we have precise control over the reaction environment.
Dual-Method Compatibility: Ultimately, this design is highly versatile: it is possible to use both cell-free (in vitro) and cell-dependent (in vivo) methods. While cell-free systems offer rapid prototyping and a controlled environment, the same optimized sequence can be cloned into a plasmid and transformed into E. coli for stable, large-scale production. This flexibility allows us to choose the most appropriate method depending on the experimental goals.
3.5 [Optional] Central Dogma in Nature
- Transcription and Translation: In nature, a single gene can sometimes code for multiple proteins through mechanisms like alternative splicing (in eukaryotes) or overlapping open reading frames (ORFs) (in viruses and some bacteria like our lux operon). For example, in the lux system, the organization of genes allows for coordinated expression of the entire bioluminescence machinery from a single promoter.
Part 4: Twist DNA Synthesis Order
4.1 DNA Insert Construction
Name: LuxR_Cassette_v1
Full Insert Sequence (FASTA):
Components Breakdown:
- Promoter (J23106):
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC(Constitutive promoter) - RBS (B0034):
CATTAAAGAGGAGAAAGGTACC(Strong RBS with spacers) - Start Codon:
ATG - Coding Sequence: Optimized luxR (Stop codon removed for C-terminal tagging)
- 7x His Tag:
CATCACCATCACCATCATCAC(For protein purification) - Stop Codon:
TAA - Terminator (B0015):
CCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
- Promoter (J23106):
4.2 Vector Selection
Cloning Vector: pTwist Amp High Copy
Strategy: This vector was chosen for its high copy number in E. coli, ensuring high yields of the plasmid for downstream applications. The cassette is flanked by cloning sites for easy extraction if needed. You can view the full annotated sequence on Benchling here.
Final Plasmid Map:
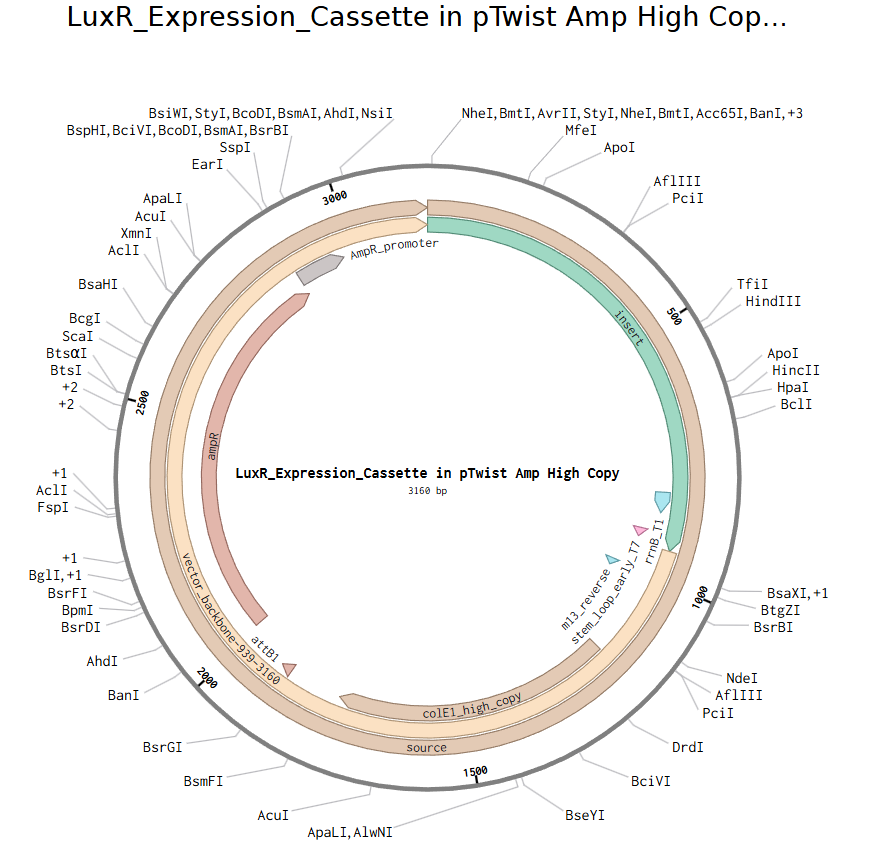 Visual representation of the LuxR_Expression_Cassette inserted into the pTwist Amp High Copy vector.
Visual representation of the LuxR_Expression_Cassette inserted into the pTwist Amp High Copy vector.

Part 5: DNA Read/Write/Edit
5.1 DNA Read (Sequencing)
- (i) Target: I want to sequence environmental DNA and synthesized constructs in real-time at the “bench” or in the field. This is crucial for early detection of unintended mutations or environmental contamination.
- (ii) Technology: Oxford Nanopore Technologies (ONT)
- Generation: 3rd Generation (Single-molecule sequencing).
- Input & Preparation: Long-read DNA. Minimal preparation is key; using ONT’s Rapid Sequencing Kits, we can perform transposase-based fragmentation and adapter ligation in under 10 minutes.
- Essential Steps: DNA molecules pass through a protein nanopore embedded in a membrane. As they pass, they disrupt the electrical current.
- Base Calling: The changes in current are decoded using neural networks (base-callers like Guppy or Dorado) into a sequence of A, T, C, and G.
- Output: FASTQ files of “long reads,” allowing me to see entire genetic circuits in a single piece.
- Why ONT? It is highly portable (MinION) and provides data in real-time, which is essential for the “Biosecurity by Design” concept below.
5.2 DNA Write (Synthesis)
- (i) Target: Synthetic genetic circuits based on quorum sensing regulators like LuxR (as designed in Part 3 and 4). These would be distributed as part of “safe-to-use” biological toolkits for decentralized research.
- (ii) Technology: Phosphoramidite Synthesis (Modern Array-based)
- Essential Steps: Controlled coupling of A, T, C, and G nucleotides onto a substrate (like a silicon chip for Twist Bioscience), followed by deprotection and oxidation cycles.
- Limitations: While accurate and scalable for fragments, constructing multi-kb circuits still requires hierarchical assembly (Gibson or Golden Gate). Errors increase with length, requiring the biosecurity measures described in Part 5.3.
5.3 DNA Edit (Biosecurity Kill Switch)
- (i) Target: I want to edit the Host Genome (E. coli or cell-free chassis) to include an autonomously triggered “Biosecurity Kill Switch.”
- (ii) Technology: CRISPR-Cas9 System
The Concept: If the ONT sequencer (Read) detects a specific error, unintended mutation, or the presence of a hazardous sequence, it triggers the expression of a specialized CRISPR-Cas system.
Mechanism: I would design the system where a guide RNA (gRNA) targets essential genomic sequences or the synthetic circuit itself. Once triggered, Cas9 creates double-strand breaks in the genome, effectively “self-destructing” the cell or the DNA pool to prevent the spread of a dangerous or dysfunctional agent.
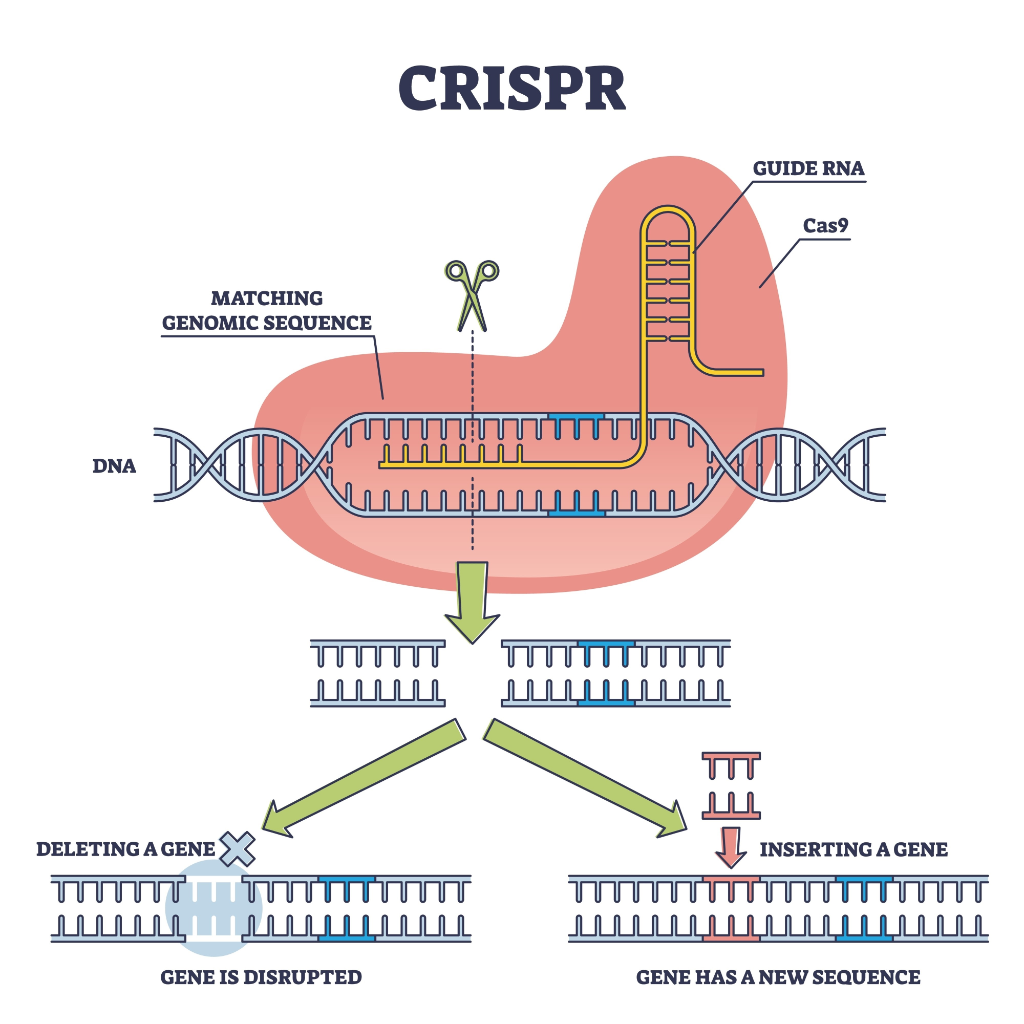
Preparation & Input: Requires a plasmid carrying the
Cas9gene under a conditional promoter and specializedgRNAsdesigned for high precision.Limitations: The primary limitation is “escaper” mutants—cells that survive by mutating the CRISPR target site. To mitigate this, multiple essential sites must be targeted simultaneously (multiplexing).