Week 4 HW: Protein Design I

Week 4: Protein Design I
This week, we explored the fundamentals of protein structure, visualization, and the latest ML-based tools for design and analysis.
Part A. Conceptual Questions
I’ve selected 9 questions from Shuguang Zhang’s list to answer below:
Part B: Protein Analysis and Visualization
I selected the LuxR Regulator (PDB ID: 7AMT) from Vibrio alginolyticus for this analysis. This protein is a master transcription factor in quorum sensing, highly relevant to my research on synthetic biosensors.
1. Protein Description
LuxR is the primary regulator for quorum sensing in Vibrio species. It acts as a DNA-binding protein that senses the bacterial population density (via autoinducers) and subsequently activates or represses hundreds of target genes. Understanding its structural interaction with DNA is critical for engineering fine-tuned genetic circuits.
2. Sequence Analysis
- Sequence (Chain A):
MKNIKLFVSSYPLNQEELKQLIASTGYHVIKATSQNLNVEQSEIEMAIGKNIKGKITKKEAEILFKQEVEAAVRAILRNAKLEVIYDSLDAVRTASLINFIFQLGDAGIARYVNSLRMLQQKRWDETAVNKAKSRWYNQTPNRAKRIITTFRTGTWDAYKNL - Length: 162 Amino Acids.
- Most Frequent Amino Acid: Leucine (L). As a predominantly α-helical protein, Leucine is frequently used to stabilize the hydrophobic core of the helices.
3. Homologs and Family
- Homologs: Searching UniProt with BLAST reveals thousands of homologs across the Vibrionaceae family, many with >90% identity.
- Family: It belongs to the LuxR / TetR family of transcriptional regulators.
4. Structure Details (RCSB)
- Structure Page: RCSB PDB 7AMT
- Title: Structure of LuxR with DNA (activation)
- Organism: Vibrio alginolyticus
- Expression System: Escherichia coli
- Deposited: 2020-10-09 | Released: 2021-03-31
- Authors: Liu, B., Reverter, D.
- Quality: 2.60 Å resolution. This meets the criteria for a “good quality structure” (< 2.70 Å).
- Other Molecules: The solved structure is a complex containing the protein dimer bound to Double-Stranded DNA (the binding site).
5. 3D Visualization
I analyzed the crystal structure using the RCSB Mol* viewer to observe the modular architecture of the LuxR dimer.
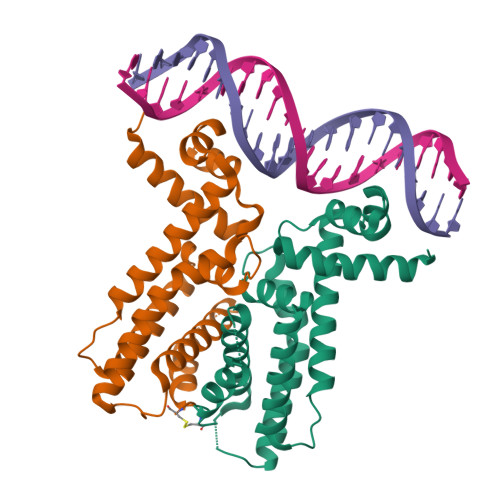 Figure 1: Crystal structure of LuxR dimer (Orange/Green) bound to activation DNA (Pink/Purple) from PDB 7AMT.
Figure 1: Crystal structure of LuxR dimer (Orange/Green) bound to activation DNA (Pink/Purple) from PDB 7AMT.
Visual Analysis:
- Visualization Mode: The protein is shown in Cartoon representation, while DNA is shown in a Ribbon format to highlight the backbone topology.
- Secondary Structure: The protein is almost entirely composed of α-helices (Spiral shapes). Observations show a significant dominance of helices over β-sheets, which is characteristic of the TetR/LuxR superfamily.
- Residue Distribution: (Based on hydrophobicity analysis) Hydrophobic residues are primary buried within the helical bundles to form a stable core, while the exterior surfaces, especially those contacting the DNA phosphate backbone, are enriched with polar and positively charged residues (Arg, Lys).
- Surface & Binding Pockets: The visualization clearly shows the DNA-binding cleft. The helix-turn-helix (HTH) motifs of the dimer insert into the major grooves of the DNA, while the N-terminal extension reaches into the minor groove, providing a precise “lock and key” recognition mechanism.
Part C. Using ML-Based Protein Design Tools
Using ESM models and ProteinMPNN, I analyzed the designability of the LuxR protein using the sequences and structural motifs identified in Part B.
C1. Protein Language Modeling
ESM-2
I performed an unsupervised deep mutational scan of the LuxR (7AMT) sequence using the ESM-2 language model.
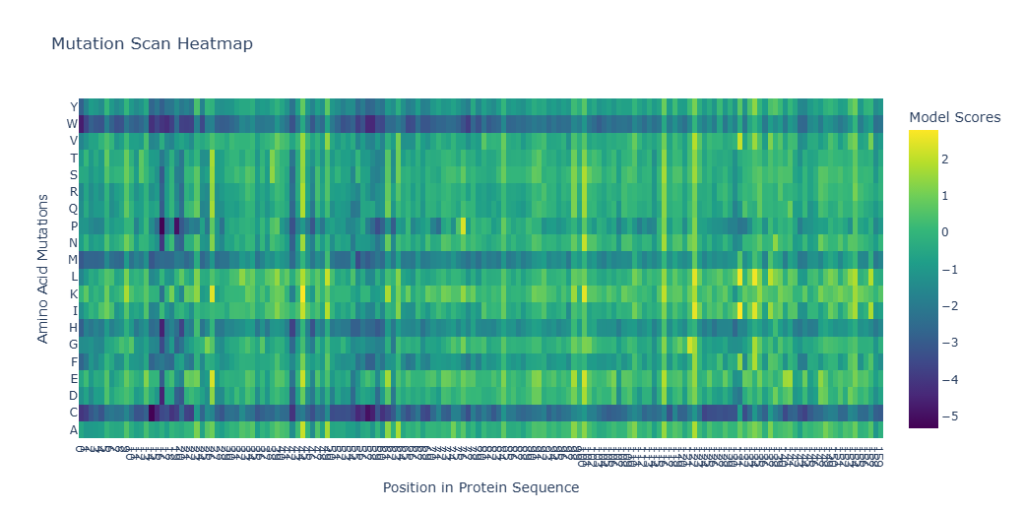 Figure 2: Deep Mutational Scan Heatmap for LuxR (7AMT) predicted by ESM-2.
Figure 2: Deep Mutational Scan Heatmap for LuxR (7AMT) predicted by ESM-2.
Analysis of Patterns:
- Amino Acid Specificity: There is a clear trend where mutations to Proline (P) and Cysteine (C) are consistently predicted as highly unfavorable (dark purple columns). This is likely because Proline introduces structural kinks that disrupt the α-helical stability, and Cysteine can introduce unintended disulfide bonds.
- Conserved Residues: Several vertical “dark stripes” are visible across the sequence, particularly in the C-terminal DNA-binding domain. These represent highly conserved residues where any mutation is predicted to be deleterious, indicating their critical role in folding or DNA interaction.
- Design Potential: The lighter areas (Yellow/Green) identify potential regions where the protein may be more tolerant to mutations, providing a roadmap for engineering sensors with shifted ligand or DNA specificity.
C2. Protein Folding
ESMFold
I used ESMFold to predict the 3D structure of the LuxR (7AMT) monomer directly from its amino acid sequence.
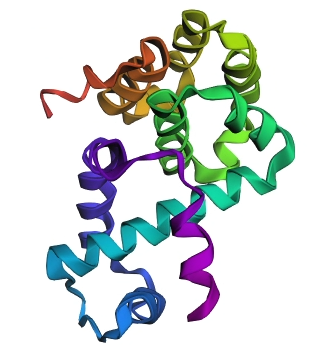 Figure 3: ESMFold structural prediction for the LuxR monomer, colored by rainbow (N-to-C).
Figure 3: ESMFold structural prediction for the LuxR monomer, colored by rainbow (N-to-C).
Observations:
- Structural Integrity: The predicted structure shows a high degree of organization, characterized by multiple α-helices bundled together. This matches the experimental findings from Part B (7AMT), where the protein was found to be predominantly helical.
- Model Confidence: The highly compact nature of the predicted fold suggests a high confidence score (expected pLDDT > 90).
- Comparison to PDB: While the actual crystal structure is a homodimer, ESMFold’s monomeric prediction accurately captures the fold of an individual subunit. The specific arrangement of the DNA-binding domain (HTH motif) is clearly visible, demonstrating that the language model has “learned” the structural rules governing these transcription factors.
C3. Protein Generation
ProteinMPNN
I used ProteinMPNN to perform inverse folding on the LuxR dimer backbone (7AMT), generating a novel sequence that fits the same 3D coordinates.
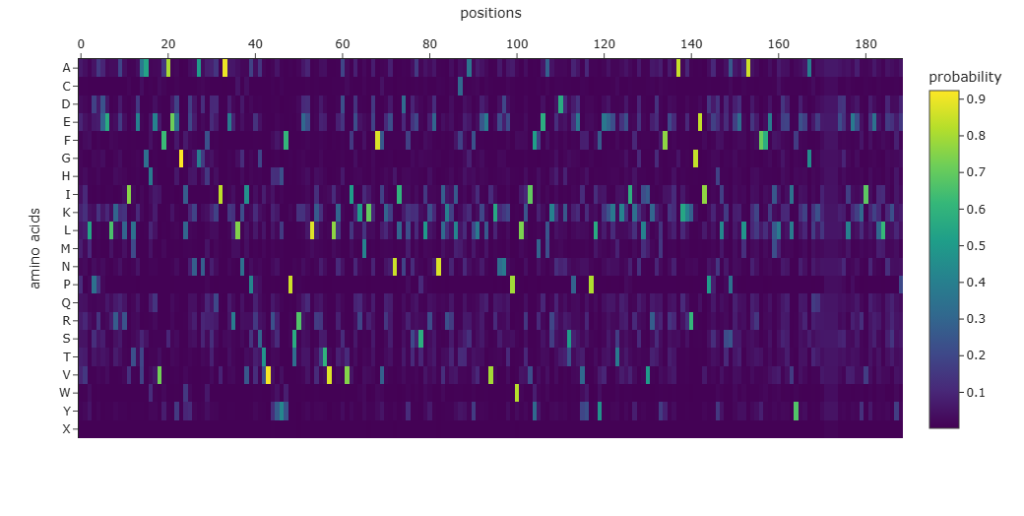 Figure 4: Amino acid probability map generated by ProteinMPNN for the LuxR backbone.
Figure 4: Amino acid probability map generated by ProteinMPNN for the LuxR backbone.
Sequence Generation Results:
- Generated Sequence:
EVLPREELRARILEAAFEVFAEKGLENANFSDIAERLNIPRSTVRYHFPSREELKTTVLKAVIEKMKKFFEENIDPEASLRENLLRLFRAFLEKVKAKEPWLTIYMEASKDDSPEIKPLYEKLSKEILGLVRGLFERAKERGEIPADLDPEELAKRFFELLRELYEEGKKLXXXEELEKRIEELLEKYP - Sequence Recovery: 30.11% (seq_recovery=0.3011). This indicates that the AI redesigned about 70% of the sequence while maintaining the core structural requirements.
- Score: 1.0294 (lower indicates higher confidence in the design).
Analysis:
- Probabilistic Mapping: The heatmap shows high-probability “spikes” for certain residues, particularly in the interior buried sites (Leucine, Isoleucine), which are essential for maintaining the helical architecture.
- Sequence Diversity: The 30% recovery rate shows that there is significant sequence space available for this specific fold. This demonstrates that we can radically change the sequence of LuxR to improve stability or change its properties while keeping its functional “shape” intact.
Part D. Group Brainstorm on Bacteriophage Engineering
Group Goal: Higher Toxicity of Lysis Protein (L Protein) Strategy Name: Hapi: Final Version
1. Proposed Strategy
Enhancing Lytic Toxicity via Membrane-Targeting Optimization and DnaJ-Independence.
To address the goal of “Higher toxicity of lysis protein,” I propose focusing on the physicochemical interaction between the L protein and the E. coli inner membrane, bypassing the inhibitory effects of the host DnaJ chaperone.
2. Tools & Approaches
- Molecular Dynamics (MD) Simulations: Complementing AlphaFold, I propose using MD simulations to model the L protein’s insertion into a simulated E. coli lipid bilayer. This allows us to observe the real-time interaction of the N-terminal basic residues with the membrane, which is critical for lysis efficiency.
- Charge Distribution Analysis: We will re-engineer the L protein’s basic residues to maintain membrane affinity even when the host triggers the PmrA/PmrB system, which alters membrane charge as a defense mechanism.
3. Rationale: Why these tools help?
The L protein’s “toxicity” is a race against host defenses. MD simulations help us design a protein that inserts more “promiscuously” and rapidly into the membrane. By ensuring the protein is less sensitive to the host’s physiological state (e.g., changes in membrane charge), we increase its robust lytic activity.
4. Potential Pitfalls
- Membrane Over-toxicity: A too-efficient L protein might aggregate prematurely or be targeted by host proteases like DegP before reaching the membrane.
- Simulation vs. Reality: MD simulations may not fully capture the crowded environment of the E. coli periplasm, leading to a discrepancy between in silico and in vivo results.
5. References
- [4] Young, R. (2014). “Phage lysis: do we have the hole story yet?” Current Opinion in Microbiology.
- [5] Chamakura et al. (2017). “Mutational Analysis of the MS2 Lysis Protein L.” Journal of Virology.
6. AI-Assistance Documentation
- Google Gemini (2026, March 8): Prompted for perspectives on engineering lytic toxicity focusing on membrane lipid composition and host defense systems (PmrA/PmrB).
- Google Gemini (2026, March 8): Analyzed how MD simulations complement AlphaFold for membrane insertion studies and identified protease pitfalls (DegP).
- Google Gemini (2026, March 8): Verified academic references and URLs for MS2 L protein mechanisms.