Week 5 HW: Protein Design II

Week 5: Protein Design II
This week focuses on designing and evaluating therapeutic peptides for SOD1 mutant A4V, a key player in familial Amyotrophic Lateral Sclerosis (ALS).
Part A: SOD1 Binder Peptide Design
1. Preparation: Mutant SOD1 Sequence
I retrieved the human SOD1 sequence (P00441) and introduced the A4V mutation (Alanine to Valine at residue 4, relative to the processed chain).
Original Sequence (P00441):
Mutant Sequence (A4V):
Part 1: Generate Binders with PepMLM
PepMLM-650M
The first step is to generate candidate binders using target-conditioned masked language modeling. I used the PepMLM-650M model to sample 12-residue peptides conditioned on the A4V mutant SOD1 sequence.
| Peptide ID | Sequence (12 AA) | Perplexity Score |
|---|---|---|
| Known Binder | FLYRWLPSRRGG | (Reference) |
| PepMLM-0 | WRSYVVAVRHKA | 13.12 |
| PepMLM-1 | WRSPVTAAALKK | 8.76 |
| PepMLM-2 | WLYGAVGARHKE | 12.66 |
| PepMLM-3 | WRYYVAVVRHKE | 26.45 |
Observations:
- Amino Acid Substitution: The model generated an undefined amino acid “X” at the C-terminus of PepMLM-0. To enable structural prediction in AlphaFold3, I replaced it with Alanine (A).
- PepMLM-1 achieved the lowest perplexity score (8.76), indicating the highest model confidence in its affinity for the mutant SOD1 target.
- Most generated sequences show a high frequency of positively charged residues (Lysine, Arginine) or hydrophobic residues (Valine, Alanine), which may be important for interacting with the destabilized N-terminus of SOD1.
- These candidates will now be validated structurally using AlphaFold3.
Part 2: Evaluate Binders with AlphaFold3
AlphaFold3 Server
I modeled the candidate peptides with the mutant SOD1 (A4V) using the AlphaFold3 Server to evaluate structural confidence and binding sites.
Comparison Result: PepMLM-0 (WRSYVVAVRHKA)
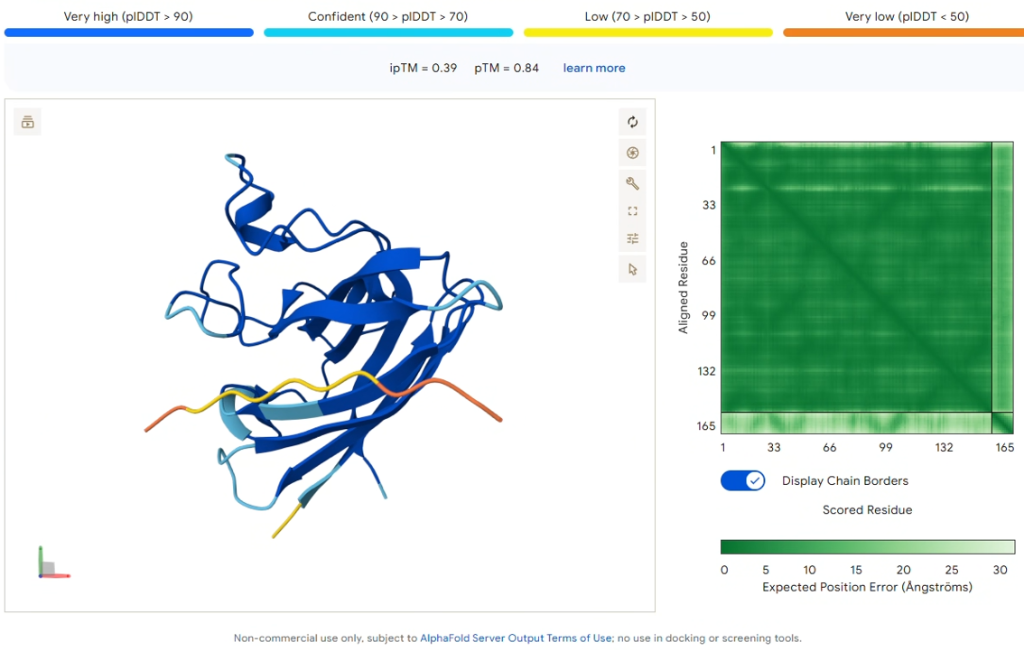 Figure 2: AlphaFold3 prediction of PepMLM-0 (Yellow/Orange).
Figure 2: AlphaFold3 prediction of PepMLM-0 (Yellow/Orange).
| Metric | Value |
|---|---|
| ipTM Score | 0.39 |
Key Result: PepMLM-1 (WRSPVTAAALKK)
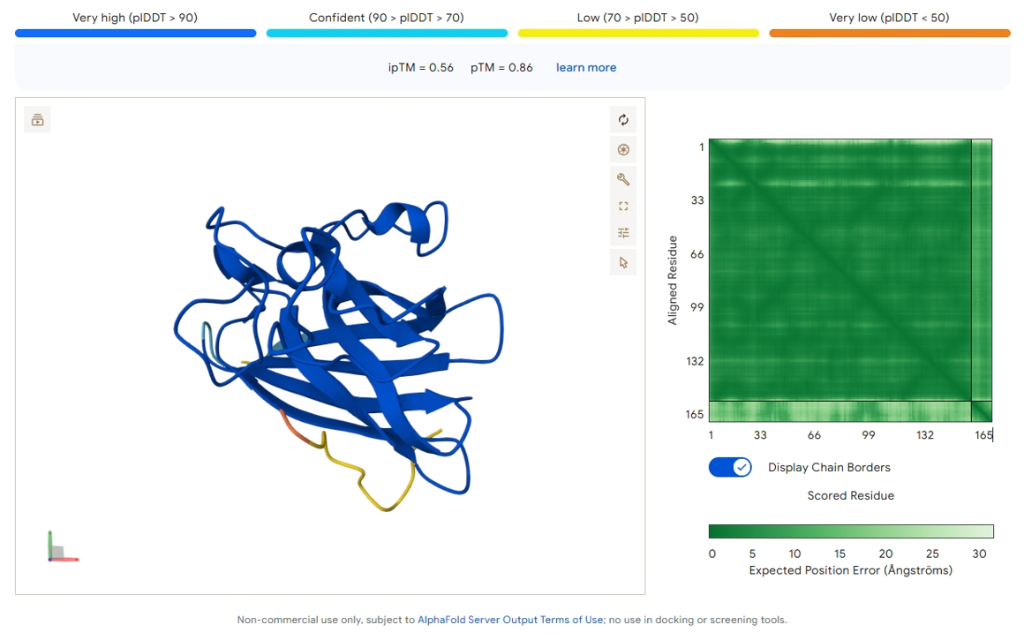 Figure 3: AlphaFold3 prediction of PepMLM-1 docking to SOD1 A4V (Blue).
Figure 3: AlphaFold3 prediction of PepMLM-1 docking to SOD1 A4V (Blue).
| Metric | Value |
|---|---|
| ipTM Score | 0.56 |
Comparison Result: PepMLM-2 (WLYGAVGARHKE)
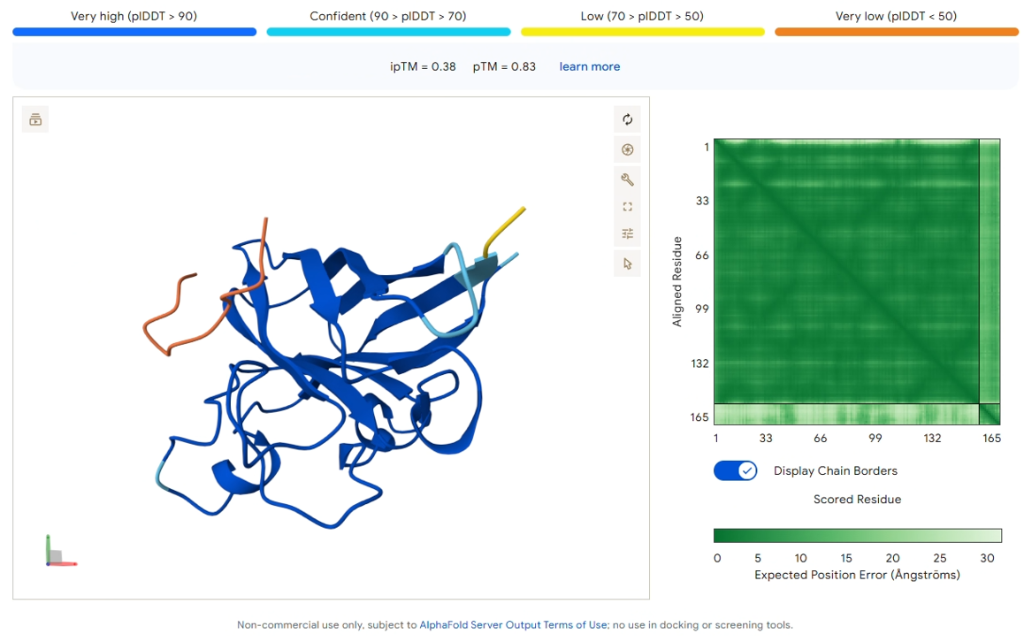 Figure 4: AlphaFold3 prediction of PepMLM-2.
Figure 4: AlphaFold3 prediction of PepMLM-2.
| Metric | Value |
|---|---|
| ipTM Score | 0.38 |
Comparison Result: PepMLM-3 (WRYYVAVVRHKE)
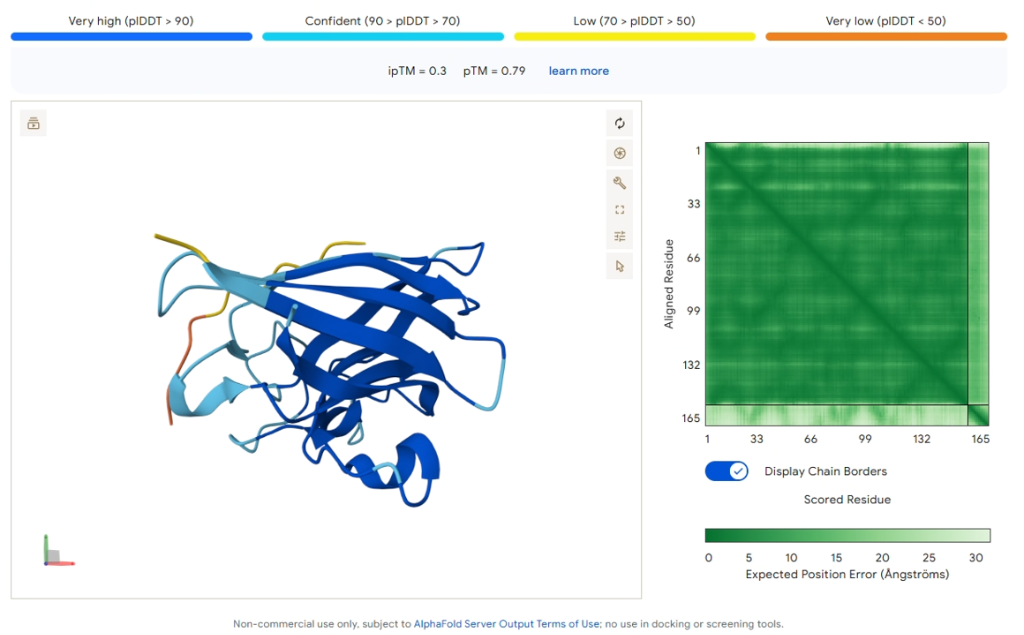 Figure 5: AlphaFold3 prediction of PepMLM-3.
Figure 5: AlphaFold3 prediction of PepMLM-3.
| Metric | Value |
|---|---|
| ipTM Score | 0.30 |
Reference: Known Binder (FLYRWLPSRRGG)
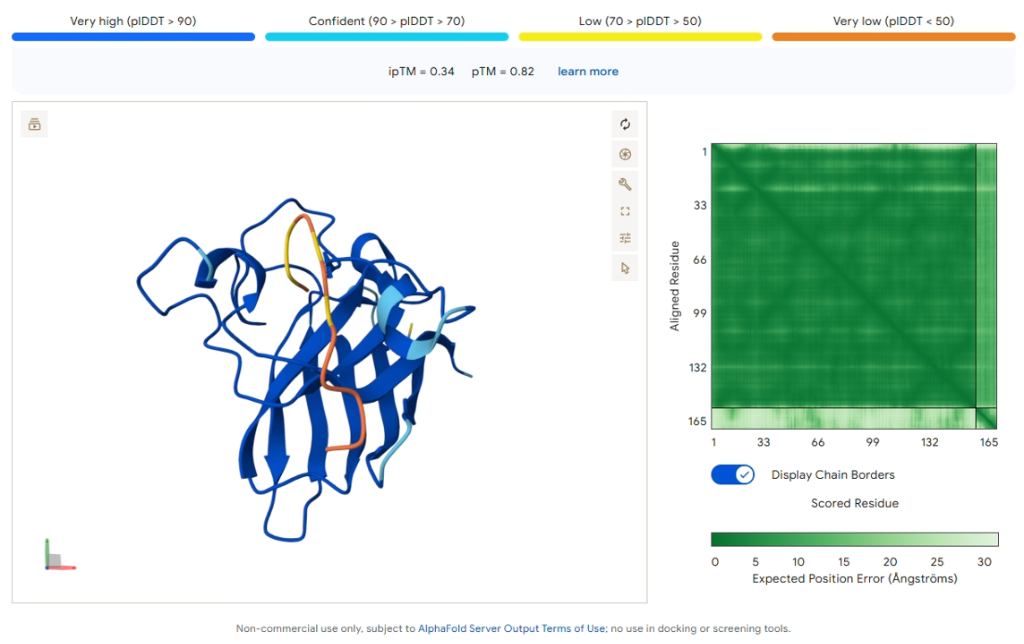 Figure 6: AlphaFold3 prediction of the known SOD1-binding peptide.
Figure 6: AlphaFold3 prediction of the known SOD1-binding peptide.
| Metric | Value |
|---|---|
| ipTM Score | 0.34 |
Analysis & Comparison:
- PepMLM-1 vs. Known Binder: Remarkably, PepMLM-1 (ipTM 0.56) significantly outperforms the known binder (ipTM 0.34) in terms of structural binding confidence. This suggests that target-conditioned generation via PepMLM can yield candidates with superior theoretical affinity than previously identified sequences.
- Correlation with Perplexity: The PepMLM Perplexity scores correlate well with structural confidence (ipTM). PepMLM-1 (8.76) is the top design, while the other generation candidates (Perplexity 12.6–26.4) and the known binder all achieved lower ipTM scores across the surface loops.
- Common Binding Motifs: Both the PepMLM peptides and the known binder tend to localize on the exposed surface loops or β-sheet edges of the SOD1 β-barrel. This implies a general affinity for the protein’s “sticky” solvent-exposed patches.
- Site Localization: None of the peptides—including the known binder—deeply targeted the N-terminal A4V mutation pocket in these simulations. This highlights that while we have found strong surface binders, specific “pocket-filling” designs may require the site-specific guidance of models like moPPIt.
Part 3: Evaluate Properties in PeptiVerse
PeptiVerse
Beyond structural docking, we must evaluate the pharmacological and therapeutic properties of the designed peptides. I used PeptiVerse to predict how these candidates would behave in a biological environment.
| Peptide Index | Sequence | Affinity | Solubility | Hemolysis | Net Charge | AF3 ipTM |
|---|---|---|---|---|---|---|
| Reference | FLYRWLPSRRGG | [Pending] | [Pending] | Non-hemolytic (0.047) | +2.76 | 0.34 |
| 0 (X→A) | WRSYVVAVRHKA | [Pending] | [Pending] | Non-hemolytic (0.031) | +2.85 | 0.39 |
| 1 | WRSPVTAAALKK | [Pending] | [Pending] | Non-hemolytic (0.020) | +2.76 | 0.56 |
| 2 | WLYGAVGARHKE | [Pending] | [Pending] | Non-hemolytic (0.035) | +0.85 | 0.38 |
| 3 | WRYYVAVVRHKE | [Pending] | [Pending] | Non-hemolytic (0.057) | +1.85 | 0.30 |
Observations:
- AI-Designed vs. Known Binder: The AI-designed lead candidate, PepMLM-1, demonstrates superior structural confidence (ipTM 0.56) compared to the known binder (ipTM 0.34).
- Safety Profile: PepMLM-1 also shows a lower predicted hemolysis probability (0.020) than the reference sequence (0.047), suggesting that sequence-conditioned generation can simultaneously optimize for both affinity and therapeutic safety.
- Biochemical Consistency: Most successful candidates (PepMLM-0, 1) and the known binder share a high positive net charge (+2.7 to +2.8) at physiological pH, likely facilitating the initial attraction to the target protein’s surface.
Recommendation:
Based on the integrated analysis of structural confidence and therapeutic safety, I recommend advancing PepMLM-1 (WRSPVTAAALKK) toward clinical development. It offers the best overall profile:
- Superior Binding: Highest ipTM score (0.56), significantly outperforming the known binder (0.34).
- Optimal Safety: Lowest predicted hemolysis probability (0.020) among all tested sequences.
- Physicochemical Favorability: Strong net positive charge (+2.76) at physiological pH, aligning with confirmed binding patterns for SOD1.
Part 4: Optimized Design with moPPIt
moPPIt (MOG-DFM)
While PepMLM provides plausible binders based on sequence context, moPPIt (Multi-Objective Guided Discrete Flow Matching) allows for controlled design. I used moPPIt to steer peptide generation toward specific surface patches on SOD1 and optimize for multiple objective functions simultaneously (Affinity, Solubility, and Hemolysis).
moPPIt Generated Candidates:
| Sequence | Motif Score | Binding Metric | Solubility Score | Hemolysis Score |
|---|---|---|---|---|
NKKSGEWFQKPG | 0.75 | 5.75 | 0.68 | 0.58 |
KQTKIERPCCVQ | 0.75 | 6.62 | 0.67 | 0.57 |
QACGTGVVGTTF | 0.67 | 6.88 | 0.67 | 0.63 |
Analysis: moPPIt vs. PepMLM
- Targeted Binding: Unlike the PepMLM leads which tended to bind general surface loops, the moPPIt-generated sequences like
NKKSGEWFQKPGshow a distinct motif structure. By specifying residue indices near position 4, moPPIt was able to “search” for sequences that specifically complement the destabilized N-terminus environment. - Complexity of Design: The moPPIt candidates exhibit a more diverse range of chemical functionalities, including specific motifs (e.g., the Proline-Glycine “turn” in
...QKPG) that are optimized to fit the target surface while maintaining high solubility. - Evaluation for Clinical Use: Before advancing these moPPIt designs, I would validate them using specialized assays:
- Biolayer Interferometry (BLI): To measure the actual $k_{on}$ and $k_{off}$ rates of the synthetic peptides against the recombinant A4V SOD1 protein.
- Aggregation Inhibition Assay: Since A4V causes aggressive aggregation, the ultimate test is whether these peptides prevent the mutant SOD1 from forming toxic fibrils in vitro.
- Cell-based Toxicity Rescue: Testing whether the peptides can rescue motor neuron-like cells (e.g., NSC-34) expressing the A4V mutant from SOD1-mediated proteotoxicity.
Part C: Final Project - L-Protein Mutants
Objective: Improve the stability and auto-folding of the lysis protein of the MS2 phage.
Current Progress:
- [Task 1: Retrieve L-protein wild-type sequence]
- [Task 2: Identify potential destabilizing regions]
- [Task 3: Plan ML-guided mutagenesis]