Hello! I’m Ian Sebastian Teran Garcia. I’m a third-year Biotechnology Engineering student in Cochabamba, Bolivia. I’m passionate about Synthetic Biology and Bioinformatics :)
I am also a co-founder of ReGlassia, a synthetic biology startup. You can know more about us here! : https://linktr.ee/re.glassia
This page includes Class Assigment and Week 2 Lecture Preparation Questions
Class Assignment
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
For HTGAA 2026, I’d like to propose the design and development of a synthetic biology based microbial system for the improvement of agricultural productivity in saline soils of the Bolivian Altiplano. This is because oil salinization is continuing to progress in the high-altitude areas of Bolivia as a consequence of climate change, water shortage and historical land use (Andrade, 2025). According to the Food and Agriculture Organization (n.d.), already a considerable fraction of irrigated and arid agricultural lands worldwide face the challenge of soil salinity. Scientific studies have shown that soil salinity significantly reduces crop yields, alters soil biological functions, and directly threatens food security, particularly in smallholder farming systems (Farooq et al., 2021). In the same way, the majority of smallholder farmers in the Altiplano rely on marginal soils, often where conventional fertilizers cannot be used effectively or are economically unaffordable and are a direct threat to local food security and livelihoods from salinization. This is why my proposed project aims to investigate the conceptual design for soil microorganisms that can sense such high salinity and improve soil structure and plant stress tolerance. However, beyond its technical feasibility, this application raises relevant ethical, environmental and governance issues surrounding environmental release and biosafety and also equitable access to biotechnology. Finally, as a Bolivian, I see this work as an opportunity to link cutting edge biological engineering with locally anchored solutions that address real challenges faced by vulnerable agricultural communities in my country.
Homework: Final Project 1. Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
I would like to measure multiple biological and functional aspects of the synthetic rhizosphere consortium composed of Pseudomonas fluorescens, Azospirillum brasilense, and Bacillus subtilis. Key variables include the production of osmoprotectants (such as proline or trehalose) under saline stress, nitrogen fixation efficiency, biofilm formation and exopolysaccharide (EPS) production, and the presence, sequence accuracy, and expression of engineered genetic constructs, including kill switch systems. At a higher level, the project will also assess microbial population dynamics and plant growth indicators such as root length and biomass, which serve as direct proxies for improved agricultural productivity under salt stress.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1. My contribution
Unfortunately, I was not able to contribute a pixel to the collective artwork, as I was in the middle of midterm exams at my university during that period, which limited my availability to participate.
2. What I liked about the project
I really liked the project because of its biological foundation and particularly its connection to cell-free fluorescent protein optimization and how it was used for a global pixel artwork designed by HTGAA students.
HOMEWORK 2
Part 1: Benchling & In-silico Gel Art
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs! HOMEWORK RESULTS :)
How many amino acid molecules do you take with 500 g of meat?
If we assume that meat is approximately 20% protein, then 500 grams of meat contains about 100 grams of protein. The average molecular weight of an amino acid is roughly 100 Daltons (100 g/mol). Dividing 100 grams by 100 g/mol gives approximately 1 mole of amino acids and one mole contains 6.02 × 10²³ molecules, the Avogadro’s number. Therefore, consuming 500 grams of meat means ingesting on the order of 10²³ amino acid molecules.
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several key components necessary for efficient and accurate DNA amplification. First, it includes Phusion DNA polymerase, a high-fidelity enzyme with proofreading activity (3’ → 5’ exonuclease), which reduces errors during DNA replication. It also contains dNTPs (deoxynucleotide triphosphates), which are the building blocks used to synthesize new DNA strands. The mix includes a reaction buffer, optimized with the correct pH and salt concentrations to ensure proper enzyme activity. Additionally, it contains Mg²⁺ ions, which act as essential cofactors for the polymerase. Some mixes may also include stabilizers to maintain enzyme activity during thermal cycling.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Continuous signal processing: Unlike Boolean circuits that operate in binary (On/Off), IANNs can process graded inputs and outputs, enabling more nuanced cellular responses.
Integration of multiple inputs: IANNs can combine many signals simultaneously and compute a weighted response, similar to an artificial neural network.
Instead of being limited to simple logic gates (and, or, not), IANNs can model nonlinear relationships between inputs and outputs.
Homework Part A: General and Lecturer-Specific Questions
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo expression systems, primarily due to its flexibility and precise control over experimental conditions. Because CFPS operates in an open environment without living cells, researchers can directly manipulate the concentrations of DNA templates, ions, cofactors, and other components in real time. This eliminates constraints associated with cellular viability, such as toxicity or metabolic burden. As a result, CFPS is particularly advantageous for the production of proteins that are toxic to host cells, such as antimicrobial peptides or pore-forming proteins. Additionally, CFPS enables rapid prototyping of genetic constructs, making it highly suitable for applications like synthetic biology circuit testing, where speed and iterative design are essential.
This is the first design I made, a green shiny bettle (really beautiful!):
The inspiration 🪲:
However, due to reagent limitations at my node, I decided to shift toward a much simpler idea that would be more feasible to implement. I wanted a design that could be cultured using the Opentrons OT-2 robot and later visualized under UV light once the colonies had grown. At the same time, I wanted to preserve an iconic and visually recognizable element, similar to the bright green beetle from my original design. Therefore, I designed a Smiling E. coli.!
This page includes Class Assigment and Week 2 Lecture Preparation Questions
Class Assignment
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
For HTGAA 2026, I’d like to propose the design and development of a synthetic biology based microbial system for the improvement of agricultural productivity in saline soils of the Bolivian Altiplano. This is because oil salinization is continuing to progress in the high-altitude areas of Bolivia as a consequence of climate change, water shortage and historical land use (Andrade, 2025). According to the Food and Agriculture Organization (n.d.), already a considerable fraction of irrigated and arid agricultural lands worldwide face the challenge of soil salinity. Scientific studies have shown that soil salinity significantly reduces crop yields, alters soil biological functions, and directly threatens food security, particularly in smallholder farming systems (Farooq et al., 2021). In the same way, the majority of smallholder farmers in the Altiplano rely on marginal soils, often where conventional fertilizers cannot be used effectively or are economically unaffordable and are a direct threat to local food security and livelihoods from salinization. This is why my proposed project aims to investigate the conceptual design for soil microorganisms that can sense such high salinity and improve soil structure and plant stress tolerance. However, beyond its technical feasibility, this application raises relevant ethical, environmental and governance issues surrounding environmental release and biosafety and also equitable access to biotechnology. Finally, as a Bolivian, I see this work as an opportunity to link cutting edge biological engineering with locally anchored solutions that address real challenges faced by vulnerable agricultural communities in my country.
Homework: Final Project 1. Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
I would like to measure multiple biological and functional aspects of the synthetic rhizosphere consortium composed of Pseudomonas fluorescens, Azospirillum brasilense, and Bacillus subtilis. Key variables include the production of osmoprotectants (such as proline or trehalose) under saline stress, nitrogen fixation efficiency, biofilm formation and exopolysaccharide (EPS) production, and the presence, sequence accuracy, and expression of engineered genetic constructs, including kill switch systems. At a higher level, the project will also assess microbial population dynamics and plant growth indicators such as root length and biomass, which serve as direct proxies for improved agricultural productivity under salt stress.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1. My contribution
Unfortunately, I was not able to contribute a pixel to the collective artwork, as I was in the middle of midterm exams at my university during that period, which limited my availability to participate.
What I liked about the project
I really liked the project because of its biological foundation and particularly its connection to cell-free fluorescent protein optimization and how it was used for a global pixel artwork designed by HTGAA students.
HOMEWORK 2
Part 1: Benchling & In-silico Gel Art
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs! HOMEWORK RESULTS :)
How many amino acid molecules do you take with 500 g of meat?
If we assume that meat is approximately 20% protein, then 500 grams of meat contains about 100 grams of protein. The average molecular weight of an amino acid is roughly 100 Daltons (100 g/mol). Dividing 100 grams by 100 g/mol gives approximately 1 mole of amino acids and one mole contains 6.02 × 10²³ molecules, the Avogadro’s number. Therefore, consuming 500 grams of meat means ingesting on the order of 10²³ amino acid molecules.
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc. Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several key components necessary for efficient and accurate DNA amplification. First, it includes Phusion DNA polymerase, a high-fidelity enzyme with proofreading activity (3’ → 5’ exonuclease), which reduces errors during DNA replication. It also contains dNTPs (deoxynucleotide triphosphates), which are the building blocks used to synthesize new DNA strands. The mix includes a reaction buffer, optimized with the correct pH and salt concentrations to ensure proper enzyme activity. Additionally, it contains Mg²⁺ ions, which act as essential cofactors for the polymerase. Some mixes may also include stabilizers to maintain enzyme activity during thermal cycling.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Continuous signal processing: Unlike Boolean circuits that operate in binary (On/Off), IANNs can process graded inputs and outputs, enabling more nuanced cellular responses.
Integration of multiple inputs: IANNs can combine many signals simultaneously and compute a weighted response, similar to an artificial neural network.
Instead of being limited to simple logic gates (and, or, not), IANNs can model nonlinear relationships between inputs and outputs.
Homework Part A: General and Lecturer-Specific Questions
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo expression systems, primarily due to its flexibility and precise control over experimental conditions. Because CFPS operates in an open environment without living cells, researchers can directly manipulate the concentrations of DNA templates, ions, cofactors, and other components in real time. This eliminates constraints associated with cellular viability, such as toxicity or metabolic burden. As a result, CFPS is particularly advantageous for the production of proteins that are toxic to host cells, such as antimicrobial peptides or pore-forming proteins. Additionally, CFPS enables rapid prototyping of genetic constructs, making it highly suitable for applications like synthetic biology circuit testing, where speed and iterative design are essential.
This is the first design I made, a green shiny bettle (really beautiful!):
The inspiration 🪲:
However, due to reagent limitations at my node, I decided to shift toward a much simpler idea that would be more feasible to implement. I wanted a design that could be cultured using the Opentrons OT-2 robot and later visualized under UV light once the colonies had grown. At the same time, I wanted to preserve an iconic and visually recognizable element, similar to the bright green beetle from my original design. Therefore, I designed a Smiling E. coli.!
Subsections of Homework
Week 1 HW: Principles and Practices
This page includes Class Assigment and Week 2 Lecture Preparation Questions
Class Assignment
1. First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
For HTGAA 2026, I’d like to propose the design and development of a synthetic biology based microbial system for the improvement of agricultural productivity in saline soils of the Bolivian Altiplano. This is because oil salinization is continuing to progress in the high-altitude areas of Bolivia as a consequence of climate change, water shortage and historical land use (Andrade, 2025). According to the Food and Agriculture Organization (n.d.), already a considerable fraction of irrigated and arid agricultural lands worldwide face the challenge of soil salinity. Scientific studies have shown that soil salinity significantly reduces crop yields, alters soil biological functions, and directly threatens food security, particularly in smallholder farming systems (Farooq et al., 2021).
In the same way, the majority of smallholder farmers in the Altiplano rely on marginal soils, often where conventional fertilizers cannot be used effectively or are economically unaffordable and are a direct threat to local food security and livelihoods from salinization. This is why my proposed project aims to investigate the conceptual design for soil microorganisms that can sense such high salinity and improve soil structure and plant stress tolerance. However, beyond its technical feasibility, this application raises relevant ethical, environmental and governance issues surrounding environmental release and biosafety and also equitable access to biotechnology. Finally, as a Bolivian, I see this work as an opportunity to link cutting edge biological engineering with locally anchored solutions that address real challenges faced by vulnerable agricultural communities in my country.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Main Goal: Ensuring Environmental Safety and Biosecurity
This goal focuses on preventing ecological harm and unintended consequences associated with the environmental use of engineered microorganisms.
-> Require environmental risk assessments prior to any field deployment.
Sub-goal 2: Reducing Ecological Uncertainty
-> Promote long-term monitoring of soil and microbial ecosystem impacts.
-> Establish protocols for detecting and responding to unintended ecological effects.
Main Goal: Promoting Equity and Responsible Use
This goal ensures that the benefits of the technology reach vulnerable communities without reinforcing existing inequalities.
Sub-goal 1: Supporting Smallholder Farmers
-> Ensure that the technology is affordable and adapted to local agricultural contexts.
-> Encourage community involvement in deployment decisions.
Sub-goal 2: Preventing Technological Exploitation
-> Avoid extractive research practices in developing regions.
-> Promote benefit-sharing and local capacity building.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
A) Biosafety by design through genetic containment.Purpose:
Current agricultural biotechnology often relies on external monitoring after deployment. This action proposes embedding biosafety mechanisms directly into the engineered organisms.
Design:
Implemented by academic researchers and biotech developers.
Reviewed by institutional biosafety committees and environmental regulators.
Assumptions:
Genetic containment systems function reliably in complex soil environments.
Risks of Failure & “Success”:
Failure: Evolutionary escape from containment mechanisms
Unintended Success: Reduced emphasis on ecological monitoring due to overconfidence in technical controls.
B) Regulatory frameworks for environmental synthetic biology.Purpose:
Environmental release regulations are often unclear or inconsistent. This action proposes clearer regulatory pathways specific to environmental synthetic biology.
Design:
National environmental and agricultural agencies conduct standardized risk assessments.
Assumptions:
Regulators have sufficient technical expertise.
Risks of Failure & “Success”:
Failure: Overregulation slows innovation
Unintended Success: Rapid approval without sufficient local adaptation
C) Community centered deployment and oversight.Purpose:
Agricultural technologies should align with the needs and values of affected communities.
Design:
Collaboration among researchers, NGOs, and local farming communities.
Participatory decision making processes.
Assumptions:
Community participation is meaningful and informed.
Risks of Failure & “Success”:
Failure: Delays due to conflicting priorities.
Unintended Success: Token participation without real influence.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
3
• By helping respond
2
2
3
Foster Lab Safety
• By preventing incident
1
2
N/A
• By helping respond
2
2
N/A
Protect the environment
• By preventing incidents
2
1
1
• By helping respond
2
2
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
2
• Feasibility?
1
2
2
• Not impede research
2
3
1
• Promote constructive applications
2
2
1
5. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Based on the comparative scoring of the governance options, the approach that I would prioritize is a combination of biosafety by design and community centered governance. This is because embedding safety mechanisms directly into engineered soil microorganisms is essential to prevent unintended ecological harm and to address biosecurity concerns at the earliest stage of development. This option performs strongly in preventing incidents and maintaining laboratory and environmental safety, making it a foundational requirement for any responsible application of environmental synthetic biology. At the same time, community centered governance is critical for ensuring that this technology is ethically deployed in the Bolivian Altiplano and engaging local farming communities helps align the technology with real agricultural needs, promotes trust and reduces the risk of inequitable or extractive use.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
A key ethical concern that stood out to me was the increasing use of artificial intelligence in synthetic biology because AI tools can greatly accelerate the design of engineered microorganisms, such as those proposed in my project to improve agricultural productivity in saline soils of the Bolivian Altiplano. However, a new ethical issue for me was the possibility that decisions driven by AI models may lack transparency or embed biases, potentially leading to unintended ecological consequences when organisms are applied in open environments. In consequence, to address these issues, I would suggest appropriate governance actions; for example, transparency in the use of AI for biological design, rigorous validation and risk assessment prior to environmental application. In addition, governance frameworks should encourage participatory approaches that involve local communities and ensure that resulting technologies are accessible, safe and aligned with local agricultural needs.
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson:
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
DNA polymerase copies DNA with high accuracy as the raw error rate of DNA polymerase is about 1 mistake per 10⁵ nucleotides copied. However, most DNA polymerases also have a proofreading function which corrects many of these mistakes, improving accuracy to about 1 error per 10⁷ - 10⁸ nucleotides and after replication, additional DNA repair systems fix remaining errors, bringing the final error rate to roughly 1 mistake per 10⁹ - 10¹⁰ nucleotides. On the other hand, the human genome is about 3 × 10⁹ base pairs long which means that without repair, thousands of errors would occur when a cell divides. For the last question, biology deals with this discrepancy through three layers of control which are polymerase proofreading, mismatch repair and other DNA repair pathways, keeping mutation rates low enough for genome stability while still allowing evolution.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Proteins are encoded using codons which are groups of three DNA nucleotides and there are 64 possible codons but only 20 amino acids plus stop signals. It is for this reason that most amino acids are encoded by multiple codons being this called degeneracy of the genetic code. On the other side, for an average human protein of about 400 amino acids, the number of possible DNA sequences that could encode the same protein is more than 10¹⁹ possible sequences. However, in practice, most of these sequences do not work well because some codons are translated more efficiently, certain sequences affect mRNA stability and others create unwanted secondary structures meanwhile some interfere with translation speed and protein folding. Moreover, regulatory elements, splicing signals, and GC content also limit which DNA sequences can successfully produce a functional protein in real cells.
Homework Questions from Dr. LeProust:
1.What’s the most commonly used method for oligo synthesis currently?
The most widely used nowadays is solid - phase phosphoramidite chemical synthesis and in this method DNA is built one nucleotide at a time on a solid support (controlled - pore glass). Also, each synthesis cycle adds one base through chemical reactions (deprotection, coupling, capping, oxidation) making this process fast and reliable for short DNA sequences being this the reason why it dominates both research and commercial oligo production.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because each synthesis step is not 100% efficient. As oligos get longer, small inefficiencies compound leading to incorrect sequences. For example, after 200 cycles the fraction of full - length, correct molecules drops sharply. In addition, longer oligos accumulate chemical side products, have higher error rates and are harder to purify.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
Because a 2000 bp gene would require 2000 consecutive chemical synthesis cycles that would result in a low yield of correct full - length DNA due to errors and the final product would be dominated by short fragments and mutated sequences, making purification not practical. Instead, long genes are made by assembling shorter, high - quality oligos through Gibson assembly or Golden Gate which improves accuracy and yield.
Homework Question from George Church:
1. [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids that all animals have are histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, valine, and arginine (HyperPhysics, n.d.). On the other hand my view of “lysine contingency” now makes me think that as all animals require lysine from their environment, synthetic biology could turn this constraint into a design principle in this area by engineering organisms that depend on externally supplied lysine and scientists would be able to control growth, improve biosafety and limit ecological spread. This would be very interesting for applications in agriculture, in my opinion.
References.
Andrade, D. (2025). Characterization, prediction, and remediation of salt-affected soils in the High Valley of Cochabamba - Bolivia (Doctoral thesis, Université de Liège - Gembloux Agro-Bio Tech). ORBi-University of Liège. https://orbi.uliege.be/handle/2268/325556
1. Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
I would like to measure multiple biological and functional aspects of the synthetic rhizosphere consortium composed of Pseudomonas fluorescens, Azospirillum brasilense, and Bacillus subtilis. Key variables include the production of osmoprotectants (such as proline or trehalose) under saline stress, nitrogen fixation efficiency, biofilm formation and exopolysaccharide (EPS) production, and the presence, sequence accuracy, and expression of engineered genetic constructs, including kill switch systems. At a higher level, the project will also assess microbial population dynamics and plant growth indicators such as root length and biomass, which serve as direct proxies for improved agricultural productivity under salt stress.
2. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Osmoprotectant levels will be quantified using high-performance liquid chromatography (HPLC) or mass spectrometry, which allow precise detection of small metabolites. Nitrogen fixation will be evaluated using the acetylene reduction assay (ARA) to measure nitrogenase activity, complemented by colorimetric assays for ammonia production. Biofilm formation will be quantified using crystal violet staining, while EPS production will be assessed using carbohydrate quantification assays. Gene expression levels associated with salt response and nitrogen fixation will be measured using quantitative PCR (qPCR), and reporter systems (fluorescence) may be used to monitor activation of engineered circuits such as salt-inducible promoters or kill switches. Plant performance will be evaluated through standard phenotyping methods, including biomass measurements and root morphology analysis.
3. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
I was thinking of DNA sequencing (Sanger or next-generation sequencing) will be used to confirm the accuracy of genetic constructs designed in Benchling, while gel electrophoresis will verify plasmid size and integrity. Mass spectrometry and HPLC will enable sensitive metabolite quantification, and qPCR will provide precise measurement of gene expression levels. Protein expression can be validated using Western blotting or fluorescence-based detection systems. Additionally, colony-forming unit (CFU) counts and live/dead staining assays will be used to evaluate kill switch functionality under different environmental conditions. Finally, 16S rRNA sequencing will allow monitoring of microbial community composition and stability within the consortium. Together, these technologies create a comprehensive and quantitative framework to validate the performance and safety of the designed system.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
According to the ExPASy Compute pI/Mw tool, the theoretical molecular weight of the eGFP construct is 28,006.60 Da (≈ 28.01 kDa), with a predicted pI of 5.90.
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
a) The formula provided expresses the charge state in terms of the ratios m/zₙ and m/zₙ₊₁, which represent two adjacent peaks in the mass spectrum.
Although it is written as m divided by z, these terms correspond directly to the experimentally measured m/z values of the peaks. Therefore, the equation can be simplified by replacing m/zₙ and m/zₙ₊₁ with the actual peak values. In this case, the peaks at 933.7148 and 965.9684 are adjacent.
Since m/z is inversely proportional to charge, the lower m/z value (933.7148) corresponds to the higher charge state (z+1), and the higher m/z value (965.9684) corresponds to the lower charge state (z).
z = (m / z(n+1)) / ( (m / z(n)) - (m / z(n+1)) )
z = (smaller peak) / (bigger peak − smaller peak)
z = 933.7148 / (965.9684 − 933.7148)
z = 933.7148 / 32.2536
z = 28.94 ≈ 29
b) The molecular weight (MW) was calculated using:
MW = z * (m/z) - z * mH
Where mH = 1.007276 Da (mass of a proton).
Substituting values:
MW = 30 * 933.7148 - 30 * 1.007276
MW = 28011.444 - 30.21828
MW = 27981.23 Da
The term mH represents the mass of a proton (H⁺), which is approximately 1.007276 Da. This value is used because, in electrospray ionization mass spectrometry (ESI-MS), proteins are ionized by gaining protons, forming positively charged ions of the form [M + zH]⁺ᶻ. As a result, the measured m/z value includes not only the mass of the protein but also the mass of the added protons. Each proton contributes both one unit of positive charge and an additional mass of about 1.007276 Da.
An error of approximately 0.0906% is considered very low in mass spectrometry, indicating that the experimentally calculated molecular weight is extremely close to the theoretical value.
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not.
Yes, the charge state can be observed because the small peaks in this region (1473.5333, 1473.7429, 1474.0481) correspond to the isotopic distribution of a single charge state. The spacing between adjacent isotopic peaks is approximately 0.3 m/z units.
Since isotopic spacing follows the relationship Δ(m/z) = 1/z, the charge state can be estimated as z = 1/0.3 = 3. Therefore, the peak corresponds to a charge state of approximately +3. While it is true that adjacent charge states in the full spectrum are separated by much larger differences in m/z, the charge state of an individual peak can still be determined from the isotopic spacing within the zoomed-in region.
Homework: Waters Part II — Secondary/Tertiary structure
1. Based on learnings in the lab, please explain the difference between native and denatured protein conformations.
What happens when a protein unfolds?
In its native state, a protein like eGFP is properly folded into a compact, globular structure stabilized by noncovalent interactions (hydrogen bonds, hydrophobic interactions, ionic interactions). Many basic residues that can accept protons are buried inside the structure.
When a protein becomes denatured, it unfolds into an extended conformation. This disrupts its tertiary structure and exposes previously buried residues, including basic amino acids (e.g., Lys, Arg, His), to the solvent
This is determined in a mass spectrometer by measuring the mass-to-charge ratio (m/z) of the protein ions produced during electrospray ionization on the Waters Xevo G3-QToF. As the protein enters the instrument, it picks up multiple protons, forming ions with different charge states. The instrument detects these ions as a series of peaks at different m/z values.
How is that determined with a mass spectrometer?
For a folded (native) protein, fewer protonation sites are accessible, so the protein carries fewer charges, and the detected peaks appear at higher m/z values with a narrow distribution. For a denatured protein, more sites are exposed, allowing more protons to attach, which produces ions with higher charge states that appear at lower m/z values and over a broader range. Thus, by analyzing the charge state distribution and the position of peaks in the spectrum, the mass spectrometer allows us to determine whether the protein is in a native or denatured conformation.
What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
The denatured protein (top spectrum) displays a broad distribution of many peaks at lower m/z values, indicating that the unfolded protein has acquired a higher number of charges. In contrast, the native protein (bottom spectrum) shows a narrower distribution with fewer peaks at higher m/z values, consistent with lower charge states. Overall, the denatured spectrum is more spread out and shifted to lower m/z, while the native spectrum is more compact and shifted to higher m/z.
2. Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800? What is the charge state? How can you tell?
The peak observed at approximately m/z ≈ 2800 in the native mass spectrum of eGFP corresponds to a specific charge state of the protein. In native mass spectrometry, proteins typically appear as a series of peaks rather than a single signal because they can carry multiple positive charges. Each peak in this series represents the same protein with a different number of charges (z), and determining this charge state is essential for interpreting the spectrum.
By zooming the region around m/z ≈ 2545, what initially appears to be a single peak is actually composed of multiple closely spaced isotopic peaks and their spacing provides direct information about the charge state. Specifically, the distance between adjacent isotopic peaks is equal to 1/z.
From the zoomed spectrum, the spacing between neighboring isotopic peaks is approximately 0.1 m/z units. Using the relationship Δ(m/z) = 1/z, the charge state can be calculated as z = 1/0.1 = 10. This indicates that the protein molecules contributing to this signal carry ten positive charges. Because the peaks in this region belong to the same charge envelope, the peak at m/z ≈ 2800 can therefore be assigned a charge state of +10.
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
Lysine (K): 20 residuesArginine (R): 6 residues
2. How many peptides will be generated from tryptic digestion of eGFP?
b) Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
c) Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
d) Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Using the ExPASy PeptideMass tool with trypsin digestion, a total of 19 peptides are predicted from the eGFP sequence.
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
23 chromatographic peaks are observed between 0.5 and 6 minutes with greater than 10% relative abundance: 0.61, 0.79, 1.20, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70, 4.30, 4.48, 4.64, 4.87, 5.06, 5.43.
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
The number of chromatographic peaks observed in the LC-MS peptide map (23 peaks) is slightly higher than the 19 peptides predicted from the tryptic digest using ExPASy.
This difference is expected because a single peptide can generate multiple signals in mass spectrometry. For example, peptides can appear with different charge states, form adducts (such as with sodium) or undergo minor modifications like oxidation, all of which produce additional peaks.
5. Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]^+) based on its m/z and z.
The peptide shown in Figure 5b has its most intense peak at m/z = 525.767, which corresponds to the most abundant charge state of the peptide.
To determine the charge (z), the isotopic peak spacing in the zoomed region is examined. The distance between adjacent isotopic peaks (for example, 525.767 to 526.259 to 526.768) is approximately 0.5 m/z units. Since isotopic spacing follows the relationship Δ(m/z) = 1/z, a spacing of about 0.5 indicates that z = 2. Therefore, the most abundant charge state of the peptide is +2.
The mass of the singly charged peptide ([M+H]+) can be calculated using the equation m/z = (M + zH)/z.
Rearranging gives M = z(m/z) − zH.
Substituting the values (with H ≈ 1 Da), M = 2 × 525.767 − 2 × 1 = 1049.534 Da.
Adding one proton gives the singly charged form: [M+H]+ = 1049.534 + 1 = 1050.534 Da.
Thus, the peptide has m/z ≈ 525.767, a charge state of +2, and a singly charged mass [M+H]+ of approximately 1050.53 Da.
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that Accuracy = (MWexperimental − MWtheoretical) / MWtheoretical)
FEGDTLVNR
Mass: 1050.5214
Position: 115-123
The experimental mass of the peptide was determined to be 1050.53 Da, while the theoretical mass from the ExPASy PeptideMass tool is 1050.5214 Da. The mass accuracy is calculated using the formula:
Accuracy = (1050.53 − 1050.5214) / 1050.5214 = 0.0086 / 1050.5214 = 0.00000819, which corresponds to 8.19 ppm.
This small error indicates excellent agreement between the experimental and theoretical masses.
7. What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
The percentage of the protein sequence confirmed by peptide mapping is 88%, as indicated by the sequence coverage shown in Figure 6.
8. Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence:
http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
The peptide sequence that best matches the fragmentation spectrum in Figure 5c is FEGDTLVNR. This sequence was identified by comparing the experimental peptide mass with the predicted tryptic peptides obtained from ExPASy and selecting the closest match. The predicted fragmentation pattern for this peptide shows a series of characteristic b-ions and y-ions, which correspond to fragmentation along the peptide backbone.
9. Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Yes, the peptide map data makes sense and supports that the protein is the eGFP standard. The results show 88% amino acid sequence coverage, which is considered excellent for protein identification by LC-MS.
Additionally, the high mass accuracy (under 10 ppm) indicates that the measured peptide masses closely match the theoretical values. The MS/MS fragmentation spectra further confirm the identity of the peptides, as the observed b-ion and y-ion patterns are consistent with the predicted sequences.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer
8FU Didecamer
8FU 3-Decamer
8FU 4-Decamer
Polypeptide Subunit Name
Subunit Mass
7FU
340 kDa
8FU
400 kDa
The 7FU decamer (340 kDa × 10) has a mass of 3.4 MDa and corresponds to the peak at 3.4 MDa.
The 8FU didecamer (400 kDa × 20) has a mass of 8.0 MDa and corresponds to the peak at 8.33 MDa.
The 8FU 3-decamer (400 kDa × 30) has a mass of 12.0 MDa and corresponds to the peak at ~12.67 MDa.
The 8FU 4-decamer (400 kDa × 40) has a mass of 16.0 MDa and corresponds to the signal around 16 MDa.
#Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical (kDa)
Observed/measured on the Intact LC-MS (kDa)
PPM Mass Error
28.01
27.98
906 ppm
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1. My contribution
Unfortunately, I was not able to contribute a pixel to the collective artwork, as I was in the middle of midterm exams at my university during that period, which limited my availability to participate.
2. What I liked about the project
I really liked the project because of its biological foundation and particularly its connection to cell-free fluorescent protein optimization and how it was used for a global pixel artwork designed by HTGAA students.
3. What could be improved for next year
For future versions, it could be interesting to include a live chat feature so participants can coordinate in real time and create more elaborate and intentional designs. Additionally, increasing the number of pixels beyond the 1,536 used in this edition could allow for more detailed and realistic compositions.
Update :) !:
There was a second part of the Pixel Artwork where I was able to contribute to the design of the bacterium shown on the right. Later, when I checked the design again, I noticed that other classmates had contributed to improving it as well, such as adding a smile and giving it an outline :D
(Also, haappy to be one of the main contributors this time :] )
Figure: Building genomes workflow and design overview.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
The lysate provides the essential molecular machinery required for transcription and translation, including ribosomes, metabolic enzymes, cofactors, and tRNAs. The presence of T7 RNA polymerase enables efficient transcription of target genes under T7 promoter control.
Salts / Buffer
Potassium Glutamate
Maintains ionic strength and mimics intracellular conditions, thereby stabilizing macromolecular interactions and supporting enzymatic activity.
HEPES-KOH pH 7.5¨
Serves as a buffering agent to maintain a stable pH, which is critical for optimal enzyme function during transcription and translation.
Magnesium Glutamate
Functions as an essential cofactor for ribosomes and polymerases, playing a key role in both transcriptional and translational processes.
Potassium Phosphate Monobasic / Dibasic
Contributes to buffering capacity and provides phosphate ions necessary for nucleotide metabolism and energy transfer reactions.
Energy / Nucleotide System
Ribose
Acts as a precursor for nucleotide biosynthesis, supporting sustained RNA production over extended reaction times.
Glucose
Serves as a metabolic energy source, enabling ATP regeneration through endogenous enzymatic pathways present in the lysate.
AMP, CMP, GMP, UMP
Provide nucleotide monophosphates that can be phosphorylated into their corresponding triphosphates, which are required substrates for RNA synthesis.
Guanine
Functions as a precursor in nucleotide salvage pathways, allowing for the biosynthesis of GMP and subsequently GTP for transcription.
Translation Mix (Amino Acids)
17 Amino Acid Mix
Supplies the majority of amino acids required for protein synthesis during translation.
Tyrosine
Provided separately due to solubility and stability constraints; essential for incorporation into nascent polypeptides.
Cysteine
Added separately due to its susceptibility to oxidation; plays a critical role in protein structure through disulfide bond formation.
Additives
Nicotinamide
Supports redox balance and enzymatic activity by contributing to NAD⁺-dependent metabolic processes within the reaction.
Backfill
Nuclease-Free Water
Used to adjust the final reaction volume while preventing nucleic acid degradation.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour PEP-NTP system relies on the direct addition of high-energy phosphate donors and nucleotide triphosphates, enabling rapid and high-yield protein synthesis within a short time frame. However, this approach is limited by the rapid depletion of energy substrates and accumulation of inhibitory byproducts.
In contrast, the 20-hour NMP-ribose-glucose system employs a metabolically sustained strategy in which substrates such as ribose and glucose support continuous nucleotide regeneration and ATP production. This results in prolonged reaction stability and sustained protein expression over extended periods.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur in the absence of externally supplied GMP because guanine can be converted into GMP through endogenous nucleotide salvage pathways present in the lysate. The resulting GMP can then be phosphorylated to GTP, which serves as the direct substrate for RNA polymerase during transcription.
References:
Carlson, E. D., Gan, R., Hodgman, C. E., & Jewett, M. C. (2012). Cell-free protein synthesis: Applications come of age. Biotechnology Advances, 30(5), 1185–1194.
https://doi.org/10.1016/j.biotechadv.2011.09.016
Swartz, J. R. (2012). Transforming biochemical engineering with cell-free biology. AIChE Journal, 58(1), 5–13.
https://doi.org/10.1002/aic.13701
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
1. sfGFP: Rapid Folding
sfGFP (superfolder GFP) can fold and form a functional chromophore in under 10 minutes, making it one of the fastest-maturing fluorescent proteins available. This is largely due to key stabilizing mutations like S30R and Y39N, which reinforce the β-barrel structure and improve global stability, meaning sfGFP begins producing fluorescence signal almost immediately after translation in a cell-free system, ideal for short incubations.
mRFP1 is reported to be a somewhat slowly-maturing monomer with low acid sensitivity; although it matures more than 10 times faster than its precursor DsRed, it still has a lower extinction coefficient, quantum yield, and photostability. In a cell-free context, this means a meaningful fraction of translated mRFP1 may remain non-fluorescent at any given time point, limiting the overall signal readout compared to what the protein concentration would predict.
3. mKO2: High Oxygen Dependence
mKO2 has a comparatively high dependence on oxygen tension for chromophore maturation, the oxygen tension at which 50% of fluorescence-positive cells is lost (pO₂·50) is ~0.9% for mKO2, and its kinetics of fluorescence recovery after reoxygenation are much slower than for greener variants like mAG. In a sealed or oxygen-limited cell-free reaction, this strong oxygen requirement means mKO2 may substantially underperform, as the chromophore oxidation step is rate-limiting and difficult to rescue once oxygen is depleted.
4. mTurquoise2: Exceptionally High Quantum Yield
mTurquoise2 has the highest quantum yield measured for a monomeric fluorescent protein, along with fast maturation, high photostability and a long mono-exponential fluorescence lifetime. It is also reported to be a rapidly-maturing monomer with very low acid sensitivity, making it one of the most reliable reporters in cell-free systems, its high quantum yield directly translates into more photons emitted per molecule, maximizing signal even at moderate expression levels.
5. mScarlet-I: Accelerated Maturation at the Cost of Quantum Yield
The single amino acid substitution T74I in mScarlet-I results in a marked maturation acceleration in cells, but at the cost of a moderate decrease in fluorescence quantum yield (0.54) and fluorescence lifetime (3.1 ns), though both values are still higher than those of all previously engineered bright mRFPs. This trade-off is particularly relevant in cell-free incubations: faster chromophore maturation is advantageous because it means fluorescence appears sooner, but the reduced quantum yield means the peak brightness will be lower than the parental mScarlet.
6. Electra2: Context-Dependent Brightness and Aggregation Risk
Electra2 is a monomeric blue fluorescent protein developed through hierarchical screening in bacterial and mammalian cells, optimized for intracellular brightness in a spectral range underserved by previous BFPs. However, its brightness performance varies significantly across expression systems — Electra2 outperforms mTagBFP2 in zebrafish but is dimmer in mouse brain under two-photon excitation, and there is a higher tendency for Electra1 (its close relative) to form puncta in neurons in vivo. In a cell-free system lacking the chaperone and quality-control machinery of living cells, Electra2’s tendency toward aggregation at higher concentrations could reduce functional fluorescent protein yield.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein: mKO2
Property to improve: Oxygen-dependent chromophore maturation
Hypothesis: Because mKO2 exhibits the strongest dependence on oxygen tension of all six proteins used, its fluorescence readout in a 36-hour cell-free incubation is likely limited by progressive oxygen depletion in the sealed reaction volume. As demonstrated in PURE system studies, oxygen is not only consumed during chromophore maturation of fluorescent proteins, but also by ATP regeneration pathways. For example, the pyruvate oxidase-based ATP regeneration system requires molecular oxygen to generate acetyl phosphate from pyruvate, creating direct competition for dissolved O₂.
Proposed adjustment: Supplement the mastermix with catalase (which regenerates O₂ from H₂O₂ produced during oxidative reactions) and use open or semi-permeable reaction vessels rather than sealed tubes to allow passive oxygen diffusion throughout the 36-hour incubation. This strategy is supported by studies in cell-free fluorescence fluctuation spectroscopy, where a droplet format was specifically designed to ensure sufficient oxygenation for chromophore maturation. The expected effect is a sustained supply of O₂ across the incubation window, enabling mKO2’s slower oxidation kinetics to run to completion and significantly increasing total fluorescence yield compared to an oxygen-depleted closed system.
Sources:
Pedelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C., & Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nature Biotechnology, 24(1), 79–88. https://www.nature.com/articles/nbt1172
Campbell, R. E., Tour, O., Palmer, A. E., Steinbach, P. A., Baird, G. S., Zacharias, D. A., & Tsien, R. Y. (2002). A monomeric red fluorescent protein. Proceedings of the National Academy of Sciences, 99(12), 7877–7882. https://www.pnas.org/doi/10.1073/pnas.082243699
Kagawa, W., Aida, T., Oguro, T., & Iida, R. (2012). Differential dependence on oxygen tension during the maturation process between monomeric Kusabira Orange 2 and monomeric Azami Green expressed in HeLa cells. Biochemical and Biophysical Research Communications, 422(2), 267–272. https://www.sciencedirect.com/science/article/abs/pii/S0006291X1200784X
Goedhart, J., von Stetten, D., Noirclerc-Savoye, M., Lelimousin, M., Joosen, L., Hink, M. A., van Weeren, L., Gadella, T. W. J., & Royant, A. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nature Methods, 9(3), 259–261. https://pubmed.ncbi.nlm.nih.gov/22434194/
Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., Aumonier, S., Gotthard, G., Royant, A., Hink, M. A., & Gadella, T. W. J. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nature Methods, 14(1), 53–56. https://www.nature.com/articles/nmeth.4074
Dunsing, V., Petrov, E. P., & Schwille, P. (2012). Chromophore maturation and fluorescence fluctuation spectroscopy of fluorescent proteins in a cell-free expression system. Biophysical Journal, 102(11), 2536–2545. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3367886/
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24)
Assigned protein: mKO2 (self-assigned, no email received)
Rationale
Based on the biophysical analysis in question 1 and the hypothesis in question 2, mKO2 was selected as the target protein for reagent optimization. Its primary limitation in a 36-hour cell-free incubation is its strong dependence on molecular oxygen for chromophore maturation, a resource that becomes progressively limiting in a closed 20 μL reaction as both the ATP regeneration machinery and the chromophore oxidation step compete for dissolved O₂.
The custom 2 μL supplement makes two small but targeted adjustments over the default preset, with the remaining volume completed with nuclease-free water:
1. Potassium Phosphate Dibasic (+25 nL): A modest increase above the preset 5.625 mM. Phosphate is a direct substrate in the oxidative ATP regeneration pathway; maintaining slightly higher phosphate availability over a 36-hour incubation helps sustain energy production and reduces metabolic competition for oxygen, leaving more dissolved O₂ available for mKO2 chromophore maturation.
2. Magnesium Glutamate (+25 nL): A small increase above the preset 6.975 mM. Mg²⁺ is an essential cofactor for RNA polymerase and ribosomes, and a modest bump is expected to improve translational yield, producing a larger pool of mKO2 polypeptide that can mature fluorescently as oxygen becomes available. The increase was kept minimal to avoid exceeding ~10 mM, above which excess Mg²⁺ can inhibit cell-free transcription-translation.
3. Nuclease-Free Water (1950 nL): Fills the remainder of the 2 μL supplement volume, keeping the total reaction at 20 μL without introducing unintended osmotic or ionic effects.
Expected Outcome: These conservative adjustments aim to sustain energy regeneration and translational output over the full 36-hour window, maximizing the total pool of matured mKO2 fluorophore. The hypothesis is that even small improvements in phosphate buffering and Mg²⁺ availability will shift the limiting factor away from energy depletion, allowing the reaction to run closer to its theoretical maximum fluorescence output given mKO2’s inherently slower oxidation kinetics.
(Total supplement volume: 2000 nL = 2 μL, within the assigned limit)
Additional Well: Electra2 - “Slow Burn” Strategy
Target Reporter:
Electra2
Operational Design:
Delayed Maturation / Late-Peak Pixel
Rationale
Electra2 was selected as a second experimental well to explore a dimension not emphasized in other designs within the collaborative painting: fluorescence persistence and delayed maturation dynamics rather than rapid early brightness.
Unlike fast-folding Aequorea-derived proteins such as sfGFP or mTurquoise2, Electra2 originates from a coral fluorescent protein lineage (eqFP611 via mRuby3-derived engineering), which undergoes comparatively slower and more complex chromophore maturation. This creates the possibility that fluorescence accumulation continues later into the 36-hour incubation window, even after other wells have plateaued.
To support this delayed maturation profile, the reaction environment is biased toward maximum pH stability using elevated HEPES buffering capacity. During long incubations, cell-free reactions gradually accumulate acidic metabolic byproducts that can reduce fluorescence intensity or prematurely quench partially matured chromophores. Increasing HEPES concentration helps preserve a near-optimal pH environment throughout the incubation period, extending the effective maturation window for Electra2.
Rather than maximizing instantaneous signal intensity, this design aims to produce a late-emerging fluorescence phenotype or a pixel that continues strengthening over time while faster systems stabilize or decay.
Reagents adjusted:
Reagent
Preset
Set to
HEPES-KOH pH 7.5
45.000 mM
Increased
Nuclease-Free Water
balance
balance
Expected Phenotype / Visual Outcome
A blue fluorescent pixel with delayed onset and prolonged signal persistence, serving as a temporal contrast element within the collaborative painting. The design demonstrates that fluorescence timing and maturation kinetics can be intentionally shaped in cell-free expression systems.
See this week’s lab protocol “Gel Art: Restriction Digests and Gel Electrophoresis” for details. Overview:
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
HOMEWORK RESULTS :)
1ST ATTEMPT
For my first attempt, I tried to form the phrase “Hi!”. It didn’t turn out as perfect as I imagined, but with practice, I hope to create more creative drawings.
2ND ATTEMPT
For my second attempt I tried to draw my own name in capital letters, “IAN”.
3RD ATTEMPT
For my third attempt I tried to draw the silhouette of an animal’s head.
Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
The protein I chose is …
Q68KI4 · NHX1_ARATH
Function:
Acts in low affinity electroneutral exchange of protons for cations such as Na+ or K+ across membranes. Can also exchange Li+ and Cs+ with a lower affinity. Involved in vacuolar ion compartmentalization necessary for cell volume regulation and cytoplasmic Na+ detoxification. Required during leaves expansion, probably to stimulate epidermal cell expansion. Confers competence to grow in high salinity conditions.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
[Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI]
Reverse Translation:
reverse translation of sp|Q68KI4|NHX1_ARATH Sodium/hydrogen exchanger 1 OS=Arabidopsis thaliana OX=3702 GN=NHX1 PE=1 SV=2 to a 1614 base sequence of most likely codons.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
[Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI]
Codon optimization is necessary because, although multiple codons can encode the same amino acid, each organism preferentially uses certain codons over others. For example, If a gene from one organism is expressed in a different host without optimization, rare codons may reduce translation efficiency, slow ribosome movement, decrease protein yield, or cause premature termination. The NHX1 coding sequence was optimized according to the codon usage preference of Escherichia coli, which was selected because it is one of the most widely used systems for recombinant protein expression due to its rapid growth, well - characterized genetics and availability of expression vectors and laboratory tools.
On the other hand, the codon optimization was performed using the IDT Codon Optimization Tool (Integrated DNA Technologies). During optimization, the tool adjusted synonymous codons to match E. coli codon bias while maintaining the original amino acid sequence. Additionally, to facilitate downstream cloning strategies, recognition sites for Type IIS restriction enzymes BsaI, BsmBI, and BbsI were avoided during the optimization process which ensures compatibility with Golden Gate assembly and prevents unwanted internal digestion of the gene sequence.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
My answer:
Cell-dependent protein expression
This option would clone the optimized NHX1 gene into an expression vector (plasmid) containing:
A strong promoter.
A ribosome binding site (RBS).
A selectable marker (antibiotic resistance gene)
A transcription terminator
The recombinant plasmid is then introduced into a host (Escherichia coli in this case), through transformation. Once inside the cell, the DNA sequence is transcribed, where RNA polymerase recognizes the promoter and synthesizes messenger RNA (mRNA) complementary to the coding strand of the DNA and translated where ribosomes bind to the mRNA and read the codons in triplets. Transfer RNAs (tRNAs) bring the corresponding amino acids, which are linked together through peptide bonds to form the NHX1 protein.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
In Benchling, select New DNA/RNA sequence
Give your insert sequence a name and select DNA with a Linear topology (this is a linear sequence that will be inserted into a circular backbone vector of our choosing).
The image above shows the Codon Optimized sequence of Q68KI4 · NHX1_ARATH.
Go through each piece of the given DNA sequences highlighted below (Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator) and paste the sequences into the Benchling file one after the other (replacing the coding sequence with your codon optimized DNA sequence of interest!). Each time you add a new piece of the sequence, make sure to annotate by right clicking over the sequence and creating an annotation that describes what each piece (e.g., Promoter, RBS, etc.) is.
Promoter (e.g. BBa_J23106):
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGC
RBS (e.g. BBa_B0034 with spacers for optimal expression):
CATTAAAGAGGAGAAAGGTACC
Start Codon:
ATG
Coding Sequence (your codon optimized DNA for a protein of interest, sfGFP for example):
Once you’ve completed this, click on Linear Map to preview the entire sequence. If you intend to have a TA review a sequence in the future, this is a good way to verify that all sections are annotated!
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.5. Import your sequence
You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
4.6. Choose Your Vector
Since we’re ordering a clonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle!
The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance.
Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Click into your sequence and select download construct (GenBank) to get the full plasmid sequence:
Go back to your Benchling account. Inside of a folder, click the import DNA/RNA sequence button and upload the GenBank file you just downloaded.
WOW! :)
Part 5: DNA Read/Write/Edit
5.1 DNA Read(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would sequence the cry4Ba gene from Bacillus thuringiensis isolates in the field, the promoter and regulatory regions controlling cry4Ba expression and comparable cry4 family homologs from different strains because I would like to understand the genetic diversity of cry4Ba which would be useful to explore new methods to improve efficacy against mosquito larvae, reveal natural sequence variation influencing toxicity and assist in environmental monitoring of Bt toxin dissemination.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?Also answer the following questions:
1. Is your method first-, second- or third-generation or other? How so?
The technology I would use is Illumina short-read sequencing which is a second generation sequencing method. It provides high accuracy, cost effectiveness and is well suited to bacterial genes.
2. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
DNA extraction
Fragmentation (to ~300 bp)
Adapter ligation
PCR enrichment
Library quantification & pooling
The input is Genomic DNA from Bacillus thuringiensis cultures.
3. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Sequencing-by-synthesis.
Each base is read by fluorescently labeled nucleotides incorporated one at a time.
Signals are captured and used for base calling.
4. What is the output of your chosen sequencing technology?
FASTQ files of read sequences and paired reads that can be aligned to reference genomes.
5.2 DNA Write(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would design and synthesize a codon-optimized cry4Ba gene for high toxin expression in a chosen bacterial host, variant versions with enhanced insecticidal activity and chimeric constructs combining parts of different Cry proteins to improve biological control of mosquitoes, increase production yield in recombinant strains and make toxin variants tailored to resistant insect populations.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use high throughput chemical DNA synthesis combined with assembly methods such as Gibson Assembly because chemical oligonucleotide synthesis (phosphoramidite chemistry) is the standard technology to produce short DNA fragments with controlled sequence and high purity and for this reason, they can be assembled into the full length cry4Ba gene using Gibson Assembly which joins overlapping oligonucleotides in a single reaction. I would choose this combination because it enables accurate synthesis of long genes, allows codon optimization for different expression hosts and supports easy modular design.
Also answer the following questions:
1. What are the essential steps of your chosen sequencing methods?
Oligo synthesis
Purification
Gene assembly
Cloning into an expression vector
2. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
Cost increases with length.
Errors may occur during oligo synthesis.
Requires verification.
5.3 DNA Edit(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I would edit the cry4Ba coding sequence to enhance toxicity or stability, regulatory elements to improve expression in non-Bt hosts and domains to broaden target specificity. The goal of this DNA Edit would be to develop improved insecticides or novel delivery systems.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR/Cas9 because it allows precise modification of DNA by using a guide RNA (gRNA) that directs the Cas9 nuclease to a specific sequence within the cry4Ba gene and it is highly specific, relatively easy to design, efficient in bacteria and also scalable for generating multiple toxin variants. On the other hand, If I wanted to introduce small point mutations to improve toxin activity or stability without creating double-strand breaks I would use CRISPR base editors enabling single nucleotide changes with greater precision and lower risk of unwanted insertions or deletions.
Also answer the following questions:
1. How does your technology of choice edit DNA? What are the essential steps?
CRISPR/Cas9 edits DNA by creating a targeted double strand break at a specific sequence defined by a designed guide RNA (gRNA) that would be complementary to the cry4Ba locus as it is computationally designed to match the desired target site and cloned or synthesized. Then, the Cas9 nuclease and gRNA are delivered into Bacillus cells via plasmid transformation. Once inside the cell, the gRNA directs Cas9 to the target sequence, where Cas9 introduces a precise cut in the DNA and the cell’s natural DNA repair mechanisms then repair the break either through non-homologous end joining or homologous recombination if a donor DNA template containing desired modifications is provided. Finally, edited colonies are screened and verified by PCR and sequencing to confirm the intended modification.
2. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Design guide RNA targeting cry4Ba.
Cas9 delivery vector or ribonucleoprotein.
Editing template with desired mutations.
Bacterial host cells.
3. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Off - target activity (needs careful design).
Editing efficiency depends on repair pathways.
Delivery methods vary in success.
Week 4 HW: Protein Design
Part A. Conceptual Questions
1. How many amino acid molecules do you take with 500 g of meat?
If we assume that meat is approximately 20% protein, then 500 grams of meat contains about 100 grams of protein. The average molecular weight of an amino acid is roughly 100 Daltons (100 g/mol). Dividing 100 grams by 100 g/mol gives approximately 1 mole of amino acids and one mole contains 6.02 × 10²³ molecules, the Avogadro’s number. Therefore, consuming 500 grams of meat means ingesting on the order of 10²³ amino acid molecules.
2. Why do humans eat beef but do not become a cow?
Humans do not become cows after eating beef because biological identity is not determined by the origin of consumed molecules but by the genetic information and regulatory networks. It is for this reason that proteins from beef are digested into individual amino acids, which are then absorbed and reused by our cells to synthesize human proteins according to our own DNA instructions.
3. Why are there only 20 natural amino acids?
There are only 20 canonical amino acids because they provide sufficient chemical diversity to build functional proteins while maintaining evolutionary stability. These amino acids cover a wide range of chemical properties: hydrophobic, polar, charged, aromatic, flexible, and rigid. Early in evolution, once the translation machinery became established, expanding the genetic code would have introduced significant risk of translational errors.
4. Can you make non - natural amino acids? Design some.
Yes, non - natural amino acids can be synthesized chemically or incorporated through expanded genetic code technologies. For example:
A fluorinated amino acid can be designed by replacing a methyl group with a trifluoromethyl group to increase hydrophobicity and protein stability.
Another possibility is a photo - switchable amino acid containing an azobenzene group, allowing protein conformation to be controlled with light.
A third design could include a redox - active moiety, such as a ferrocene group, enabling electron transfer within engineered proteins.
5. Where did amino acids come from before enzymes and life started?
Amino acids likely originated through prebiotic chemistry before the emergence of life and there are experiments such as the Miller - Urey experiment that demonstrated that amino acids can form spontaneously under simulated early Earth conditions involving simple gases and electrical discharge. Additionally, amino acids have been found in meteorites such as the Murchison meteorite, suggesting extraterrestrial delivery may have contributed to the prebiotic inventory.
6. If you make an α-helix using D - amino acids, what handedness would you expect?
Natural proteins are composed of L - amino acids and form predominantly right - handed α - helices. If a scientist constructs a helix entirely from D - amino acids, the chirality of each residue is inverted, which reverses the allowed backbone dihedral angles. As a result, the helix formed would be left - handed.
7. Why are most molecular helices right - handed?
Most molecular helices in biological systems are right - handed because life is based almost exclusively on L - amino acids because their stereochemistry constrains backbone geometry in a way that energetically favors right - handed helices, minimizing steric clashes and optimizing hydrogen bonding. If life had evolved using D - amino acids instead, left - handed helices would likely dominate.
8. Why do β - sheets tend to aggregate?
- What is the driving force?
β - sheets tend to aggregate because their backbone hydrogen bond donors and acceptors become exposed when proteins partially unfold. These exposed regions seek to form hydrogen bonds to stabilize themselves, often binding to similar β-strands from other molecules. The aggregation is driven by intermolecular hydrogen bonding, hydrophobic interactions and the entropic gain associated with releasing ordered water molecules from hydrophobic surfaces.
9. Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
Many amyloid diseases, including Alzheimer’s disease, involve the misfolding of proteins into stable cross - β - sheet fibril because they are particularly prone to forming highly ordered, self templating aggregates that grow through nucleation dependent polymerization. Although pathological in a biological context, these same structural properties make amyloid fibrils attractive as biomaterials and engineered amyloid-like peptides can form hydrogels, scaffolds for tissue engineering and nanostructured materials.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it
The protein I selected is the ribulose-1,5-bisphosphate carboxylase/oxygenase large subunit (RbcL) with UniProt accession Q8DIS5, entry name RBL_THEVB. This protein comes from the thermophilic cyanobacterium Thermosynechococcus vestitus BP-1. RuBisCO is the central enzyme of the Calvin cycle and is responsible for fixing atmospheric CO₂ into organic carbon during photosynthesis. I selected this protein because carbon fixation underlies all plant biomass production and therefore directly impacts agriculture, food security, and global carbon cycling. Studying RuBisCO at the structural level provides insight into how photosynthetic efficiency might be improved in crops.
2. Identify the amino acid sequence of your protein.
MAYTQSKSQKVGYQAGVKDYRLTYYTPDYTPKDTDILAAFRVTPQPGVPFEEAAAAVAAESSTGTWTTVWTDLLTDLDRYKGCCYDIEPLPGEDNQFIAYIAYPLDLFEEGSVTNMLTSIVGNVFGFKALKALRLEDLRIPVAYLKTFQGPPHGIQVERDKLNKYGRPLLGCTIKPKLGLSAKNYGRAVYECLRGGLDFTKDDENINSQPFQRWRDRFLFVADAIHKAQAETGEIKGHYLNVTAPTCEEMLKRAEFAKELEMPIIMHDFLTAGFTANTTLSKWCRDNGMLLHIHRAMHAVMDRQKNHGIHFRVLAKCLRMSGGDHIHTGTVVGKLEGDKAVTLGFVDLLRENYIEQDRSRGIYFTQDWASMPGVMAVASGGIHVWHMPALVDIFGDDAVLQFGGGTLGHPWGNAPGATANRVALEACIQARNEGRDLMREGGDIIREAARWSPELAAACELWKEIKFEFEAQDTI
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is: 475 aminoacids.
The most common amino acid is: A (alanine), which appears 47 times.
- How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
When the amino acid sequence of Q8DIS5 was analyzed using UniProt’s BLAST tool, the search returned 250 homologous protein sequences. These homologs correspond primarily to RuBisCO large subunits from cyanobacteria and photosynthetic organisms. The presence of numerous homologs indicates that this protein is highly conserved across species, reflecting its essential role in carbon fixation and global photosynthetic metabolism.
- Does your protein belong to any protein family?
Yes, the protein Q8DIS5 (RBL_THEVB) belongs to the RuBisCO superfamily, specifically Form I RuBisCO large subunits (RbcL family). This protein family includes the catalytic large chains of ribulose-1,5-bisphosphate carboxylase/oxygenase enzymes found in cyanobacteria, plants, and algae. Members of this family share highly conserved sequence motifs that are essential for carbon fixation, including residues involved in substrate binding and magnesium coordination at the active site.
3. Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure 2YBV was solved by X-ray crystallography, with:
Deposited: March 10, 2011
Released: March 28, 2012
Resolution: 2.30 Å
A resolution of 2.30 Å is considered high quality for structural analysis and visualization because at this resolution, the positions of amino acid side chains, cofactors, and active-site features are clearly defined.
- Are there any other molecules in the solved structure apart from protein?
Apart from the protein chains, crystallized RuBisCO structures typically include magnesium ions (Mg²⁺), which are essential cofactors for catalysis, as well as substrate analogs and water molecules that help stabilize the active site.
- Does your protein belong to any structure classification family?
Structurally, the RuBisCO large subunit belongs to the alpha/beta protein class with a conserved α/β barrel–like fold characteristic of the RuBisCO superfamily, illustrating both its functional role in catalysis and its classification within established structural families.
4. Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
When I visualized in cartoon representation, the protein displays a mixed α/β architecture. A central beta-sheet core is surrounded by multiple alpha helices, forming the characteristic α/β fold typical of RuBisCO large subunits. The beta sheets form the structural core of the enzyme, while the helices surround and stabilize this framework. Overall, the structure contains both helices and sheets, with a prominent beta-sheet catalytic core.
Moreover, when colored by secondary structure, the beta sheets appear concentrated in the central region, while alpha helices are distributed around the periphery. This confirms that the enzyme follows a classical α/β barrel-like organization commonly observed in metabolic enzymes.
On the other hand, coloring the structure by residue type reveals that hydrophobic residues are predominantly located in the interior of the protein, forming a stable hydrophobic core. In contrast, hydrophilic residues are mostly exposed on the surface, consistent with a soluble enzyme functioning in the chloroplast stroma. This distribution reflects proper protein folding and stability in aqueous environments.
Surface visualization of 2YBV reveals clear cavities and clefts between structural domains, indicating the presence of binding pockets. However, this specific crystal structure represents an apo form of the enzyme, as it does not contain a modeled Mg²⁺ ion or bound substrate. The only heteroatoms present in this structure are water molecules (HOH).
To visualize the catalytic metal ion, an additional RuBisCO structure (PDB ID 4RUB) was examined. In this structure, the Mg²⁺ ion appears as a green sphere located within a deep catalytic pocket and coordinated by nearby residues, confirming the structural location of the active site. Although Mg²⁺ is essential for enzymatic activity, it is not present in the 2YBV model itself.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
a) Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b) Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Distinct vertical bands of strongly negative scores are observed at several positions, indicating that most substitutions at these sites are predicted to be unfavorable. These residues appear to be highly constrained, suggesting structural or functional importance. For example, substitution of a hydrophobic wild-type residue with a chemically dissimilar charged residue at one of these constrained positions results in a pronounced decrease in log - likelihood. This pattern is consistent with disruption of structural stability, particularly if the residue contributes to hydrophobic packing or functional integrity. In contrast, other positions exhibit relatively mild score variations across multiple substitutions, indicating higher mutational tolerance. These sites likely correspond to surface-exposed or flexible regions of the protein.
Latent Space Analysis
a) Use the provided sequence dataset to embed proteins in reduced dimensionality.
b) Analyze the different formed neighborhoods: do they approximate similar proteins?
c) Place your protein in the resulting map and explain its position and similarity to its neighbors.
To evaluate the biological relevance of the learned embedding space, my protein of interest, RuBisCO (PDB ID: 2YBV), was embedded using the same ESM-2 pipeline applied to the SCOPe dataset. The resulting embedding was projected into the same reduced-dimensional space and analyzed using cosine similarity in the original high-dimensional representation.
The nearest neighbor to my protein (cosine similarity = 0.9904) corresponds to another ribulose-1,5-bisphosphate carboxylase-oxygenase from Oryza sativa. This extremely high similarity value indicates that the embedding space accurately captures functional and structural conservation. Given that RuBisCO is a highly conserved enzyme involved in carbon fixation, this result is biologically consistent and expected.
Beyond the closest homolog, additional neighboring proteins include large metabolic enzymes such as aconitase, nitrogenase, and ornithine decarboxylase. Many of these proteins belong to α/β structural classes in SCOPe, suggesting that the latent space organizes proteins not only by specific biochemical function but also by shared structural architecture.
C2. Protein Folding
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Total sequence length: 475
Running ESMFold inference for sequence with length 475…
Prediction complete. ptm: 0.919 plddt: 90.429
Results saved to test_d3e9a/
CPU times: user 1min 33s, sys: 8.76 s, total: 1min 41s
Wall time: 2min 15s
ExecutiveAlign: 3395 atoms aligned.
ExecutiveRMS: 132 atoms rejected during cycle 1 (RMSD=1.74).
ExecutiveRMS: 183 atoms rejected during cycle 2 (RMSD=1.04).
ExecutiveRMS: 97 atoms rejected during cycle 3 (RMSD=0.85).
ExecutiveRMS: 46 atoms rejected during cycle 4 (RMSD=0.79).
ExecutiveRMS: 22 atoms rejected during cycle 5 (RMSD=0.77).
Executive: RMSD = 0.761 (2868 to 2868 atoms)
The amino acid sequence of the protein (475 residues) was folded using ESMFold to predict its three-dimensional structure. The predicted model showed high confidence with a pTM score of 0.919 and a pLDDT value of approximately 90, indicating reliable structural prediction. The predicted structure was then aligned with the experimentally determined structure PDB 2YBV using PyMOL. The alignment resulted in an RMSD of 0.761 Å across 2868 atoms, indicating that the predicted coordinates match the original structure very closely.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To evaluate the effect of mutations on the protein structure, several point mutations were introduced into the amino acid sequence, including substitutions such as V->A, L->A, F->Y, G->A, and I-> A at different positions along the 475-residue protein. The modified sequence was then folded using ESMFold and compared with the experimental structure PDB 2YBV using PyMOL. Structural alignment produced an RMSD of 0.761 Å, indicating very high structural similarity between the mutated and original structures. These results suggest that the protein fold is highly conserved and resilient to sequence mutations, maintaining its overall three-dimensional structure despite several amino-acid substitutions.
Total sequence length: 475
Running ESMFold inference for sequence with length 475…
Prediction complete. ptm: 0.921 plddt: 90.388
Results saved to test_cdeb6/
CPU times: user 1min 45s, sys: 8.49 s, total: 1min 53s
Wall time: 2min 23s
Match: read scoring matrix.
Match: assigning 475 x 5591 pairwise scores.
MatchAlign: aligning residues (475 vs 5591)…
MatchAlign: score 2538.000
ExecutiveAlign: 3395 atoms aligned.
ExecutiveRMS: 132 atoms rejected during cycle 1 (RMSD=1.74).
ExecutiveRMS: 183 atoms rejected during cycle 2 (RMSD=1.04).
ExecutiveRMS: 97 atoms rejected during cycle 3 (RMSD=0.85).
ExecutiveRMS: 46 atoms rejected during cycle 4 (RMSD=0.79).
ExecutiveRMS: 22 atoms rejected during cycle 5 (RMSD=0.77).
Executive: RMSD = 0.761 (2868 to 2868 atoms)
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The amino acid probability heatmap generated by ProteinMPNN shows the likelihood of each amino acid at every position of the protein backbone. In the heatmap, several positions display strong probabilities for a single amino acid, which appear as bright signals for specific residues. These positions likely correspond to structurally constrained residues that are important for maintaining the stability of the protein fold. Other positions show more distributed probabilities across multiple amino acids, suggesting that these regions are more flexible and can tolerate substitutions.
When comparing the predicted sequence to the original sequence from PDB 2YBV, the sequence recovery was approximately 47.2%, meaning that about half of the residues match the native sequence while many positions were redesigned by the model. This observation is consistent with the heatmap, which indicates that while some residues are strongly constrained by the structure, other positions allow multiple amino acids. Overall, this result demonstrates that several different amino acid sequences can be compatible with the same protein backbone.
Input this sequence into ESMFold and compare the predicted structure to your original.
The sequence generated by ProteinMPNN was then folded using ESMFold to predict its three-dimensional structure. The predicted structure was subsequently compared to the original structure from PDB 2YBV.
Despite the differences between the designed sequence and the native sequence, the predicted structure maintained a similar overall fold. This suggests that the designed sequence is structurally compatible with the original backbone. These results illustrate an important principle of protein design: multiple distinct amino acid sequences can adopt very similar three-dimensional structures when the structural constraints of the protein backbone are preserved.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
PROJECT OBJECTIVE
Engineer the L protein of the MS2 phage to increase structural stability.
Disrupt or reduce its interaction with the bacterial chaperone DnaJ.
Preserve the C-terminal lysis domain to maintain lytic function.
Avoid mutations that interfere with structurally or evolutionarily coupled residues.
Phase 1: Mapping the DnaJ Interaction Interface
Since the exact binding interface between the L protein and DnaJ is unknown, the first step is to identify it computationally rather than introducing arbitrary mutations.
Use AlphaFold-Multimer to model the complex between L protein and DnaJ.
Generate multiple structural predictions and select the top-ranked models.
Identify consensus interface residues that consistently appear in the predicted binding interface.
Perform in silico alanine scanning of the N-terminal residues in the complex to determine which residues significantly contribute to binding energy (ΔΔG).
Analyze whether the N-terminal region resembles known DnaJ-binding motifs, typically hydrophobic residues flanked by basic amino acids.
This phase defines which residues are critical for interaction and should not be mutated randomly.
Phase 2: Targeted N-Terminal Redesign
Instead of deleting regions or performing extensive random substitutions, introduce controlled chemical modifications to disrupt interaction while preserving structural stability.
Focus on charge inversion strategies:
Basic residues (K, R) → Acidic residues (E, D)
Acidic residues (E, D) → Basic residues (K, R)
Disrupt hydrophobic interaction patches:
Hydrophobic residues (L, I, V, F) → Polar residues (S, T, N, Q)
Aromatic residues (F, Y, W) → Aliphatic or small residues
Generate a graded library of variants:
Minor charge modifications
Moderate interface perturbations
Strong hydrophobic disruption
This creates a Pareto front of variants balancing reduced DnaJ interaction and preserved protein stability.
Phase 3: Stability and Functional Filtering
To ensure that redesigned variants remain structurally viable and functionally relevant:
Use Rosetta or FoldX to calculate ΔΔG and verify that mutations do not destabilize the overall protein fold.
Confirm that mutations in the N-terminal region do not propagate structural stress toward the C-terminal lysis domain.
Identify residue pairs that co-evolved between the N-terminal and C-terminal regions.
Avoid mutating co-evolved residues independently to prevent functional disruption.
Evaluate aggregation propensity using tools such as Aggrescan3D to ensure that mutations do not create exposed hydrophobic patches leading to cytoplasmic aggregation.
Assess sequence plausibility using protein language models such as ESM to filter out unlikely or non-natural variants.
Key Limitations:
The DnaJ binding mode may be transient or dynamic, reducing AlphaFold-Multimer accuracy.
Protein language model scores do not guarantee in vivo functionality.
Intrinsically disordered regions may not be accurately modeled.
Computational predictions must ultimately be validated experimentally.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Generated sequences:
Binder
Pseudo Perplexity
WRYGVTALAHWX
10.28 ⭐
KRYPVVGLEWKX
14.16
KHYPPVVVAHKK
14.86
WRYYAAVVRHKK
19.84
Known Binder: FLYRWLPSRRGG
Four candidate peptides of length 12 were generated using PepMLM conditioned on the mutant SOD1 sequence. The model assigned pseudo-perplexity scores to each peptide, which reflect the likelihood of the sequence under the model. Lower perplexity values indicate higher confidence. Among the generated peptides, WRYGVTALAHWX had the lowest pseudo-perplexity (10.28), suggesting it is the most plausible binder candidate. For comparison, the known SOD1-binding peptide FLYRWLPSRRGG was also included.
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
a) ipTM = 0.45pTM = 0.78
b) ipTM = 0.38pTM = 0.87
c) ipTM = 0.24pTM = 0.87
d) ipTM = 0.27pTM = 0.71
e) Known Binder ipTM = 0.35pTM = 0.83
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Model 1
The peptide sits along the side of the β-sheet barrel.
It contacts surface loops near the barrel edge.
Surface-bound, not buried.
Not near the extreme N-terminus (A4V region).
Model 2
The peptide is positioned above the β-barrel core, interacting mainly with loop regions.
Still surface-exposed.
No clear contact with the dimer interface.
Model 3
The peptide extends toward a flexible loop projecting from the barrel.
Appears loosely associated, with much of the peptide exposed.
Again not near the N-terminal A4V site.
Model 4
-The peptide approaches between β-strands and adjacent loops, slightly closer to the barrel surface.
Part of the peptide appears partially tucked against the protein, but still largely surface-bound.
Model 5
The peptide lies along the β-sheet surface, contacting residues on the outer barrel face.
The orientation is consistent with surface docking rather than deep insertion.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
AlphaFold3 was used to model complexes between mutant SOD1 and each candidate peptide. The predicted interface TM-scores (ipTM) ranged from 0.24 to 0.45, indicating generally weak but plausible protein–peptide interactions. Most peptides appeared surface-bound along the β-barrel region of SOD1, interacting primarily with exposed loop regions rather than the N-terminal region where the A4V mutation occurs. The known SOD1-binding peptide FLYRWLPSRRGG produced an ipTM score of 0.35. Notably, one PepMLM-generated peptide (WRYGVTALAHWA) showed a higher ipTM score of 0.45, suggesting a potentially stronger interaction than the reference peptide. These results indicate that some generated peptides may represent promising candidates for further optimization and evaluation.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
PEPTIDE #1:
PEPTIDE #2:
PEPTIDE #3:
PEPTIDE #4:
PEPTIDE #5:
Peptide
ipTM (AF3)
Binding Affinity (pKd)
Solubility
Hemolysis (Prob)
Net Charge
WRYGVTALAHWA
0.45
6.223
Soluble (1.00)
0.048
+1.85
KRYPVVGLEWKA
0.38
5.542
Soluble (1.00)
0.033
+1.76
KHYPPVVVAHKK
0.24
4.835
Soluble (1.00)
0.018
+2.93
WRYYAAVVRHKK
0.27
6.045
Soluble (1.00)
0.032
+3.84
FLYRWLPSRRGG
0.31
5.968
Soluble (1.00)
0.047
+2.76
PeptiVerse predictions were used to evaluate the therapeutic properties of the generated peptides, including binding affinity, solubility, hemolysis probability, and net charge. The peptide WRYGVTALAHWA showed the highest predicted binding affinity (pKd = 6.223) and the highest AlphaFold3 interface score (ipTM = 0.45), suggesting a relatively stronger interaction with SOD1 compared to the other candidates and the reference peptide FLYRWLPSRRGG. All peptides were predicted to be highly soluble with low hemolysis probabilities, indicating generally favorable therapeutic properties. Although some peptides displayed higher net charges, WRYGVTALAHWA maintained a moderate charge and low toxicity risk. Based on the combined structural and therapeutic predictions, WRYGVTALAHWA appears to provide the best balance of binding strength and developability and I selected it as the peptide to advance for further optimization.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Peptides generated:
a) STCKYKKIGGTL
b) GRYKCYCRDSRY
c) DDTITCKKKQCT
In this step, peptides were generated using moPPIt, which applies Multi-Objective Guided Discrete Flow Matching to design binders toward specific residues on the target protein while simultaneously optimizing multiple objectives such as binding affinity, solubility, and toxicity. The mutant sequence of Superoxide Dismutase 1 was provided as the target, and residues 4-7 near the A4V mutation were selected to guide peptide binding toward the N-terminal region of the protein. Compared to the peptides generated with PepMLM, the moPPIt peptides displayed more structured residue patterns, including higher frequencies of positively charged residues such as lysine and arginine and the presence of cysteine residues that may contribute to stabilizing protein–peptide interactions. This suggests that moPPIt performs directed optimization of binding motifs rather than broadly sampling plausible sequences.
Before advancing these peptides toward clinical studies, several validation steps would be required. First, computational structural modeling using AlphaFold3 or molecular docking could confirm whether the peptides bind near the intended SOD1 residues. Property prediction tools such as PeptiVerse could further evaluate binding affinity, solubility and toxicity risks. Finally, experimental validation would be necessary, including in vitro binding assays, aggregation inhibition assays for mutant SOD1, and toxicity testing in relevant cellular systems.
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
L-Protein Engineering | Option 1: Mutagenesis
STEP 1:
A multiple sequence alignment of homologous L-protein sequences was performed using Clustal Omega to identify conserved and variable regions across related bacteriophages. The alignment revealed that the transmembrane region, located in the C-terminal portion of the protein, is highly conserved, particularly in residues forming a hydrophobic helix (LVLIFLAIFLSKFTNQLLLSLL). This high level of conservation suggests a critical functional role in membrane insertion and pore formation during bacterial lysis. In contrast, the N-terminal soluble region displayed greater sequence variability, indicating a higher tolerance to mutations. Based on these observations, conserved residues were avoided during mutational design, while more variable positions, especially in the soluble domain, were prioritized as potential targets for mutation.
STEP 2:
To evaluate the effect of mutations across the L-protein sequence, a protein language model (ESM-2) was used to compute log-likelihood ratio (LLR) scores for all possible amino acid substitutions at each position. This approach estimates how favorable a mutation is relative to the wild-type residue based on learned sequence patterns from large protein datasets. Positive LLR scores indicate mutations that are more likely to be tolerated or beneficial for protein stability, while negative scores suggest deleterious effects. The results were compiled into a ranked list of candidate mutations, allowing the identification of positions and substitutions with the highest predicted improvement. These scores were then used as a primary filter to guide mutation selection, in combination with conservation analysis from the multiple sequence alignment.
The protein language model identified several mutations with high positive LLR scores, indicating potentially favorable substitutions. The top-ranked mutations included K50L (LLR = 2.56), C29R (LLR = 2.39), Y39L (LLR = 2.24), C29S (LLR = 2.04), and S9Q (LLR = 2.01). Additional high-scoring mutations were observed at positions within both the soluble and transmembrane regions, such as T52L (LLR = 1.81), N53L (LLR = 1.86), and A45L (LLR = 1.54), particularly favoring substitutions to hydrophobic residues in the transmembrane domain. These results suggest that increasing hydrophobicity in the membrane region and selecting tolerated substitutions in variable regions may improve protein stability and folding.
STEP 3:
To assess how well the model predictions reflect real functional outcomes, the LLR scores were compared with available experimental lysis data for L-protein mutants. While some overlap between high-scoring mutations and experimentally tested variants was observed, many of the top-ranked mutations identified by the model were not present in the experimental dataset. Therefore, the experimental data was used when available, but for many candidate mutations, selection relied primarily on LLR scores in combination with conservation analysis.
STEP 4:
Based on the combined analysis of LLR scores, sequence conservation, and structural considerations, five mutations were selected as potential candidates for improving the L-protein. In the soluble region, the mutations S9Q and K23R were chosen due to their high LLR scores and location in more variable regions, suggesting a higher tolerance for substitutions that may improve folding stability. In the transmembrane region, K50L and T52L were selected, as both mutations introduce more hydrophobic residues, which is consistent with the conserved nature of this domain and may enhance membrane insertion and pore formation. Additionally, a combined mutant (S9Q + K50L) was designed to explore potential additive effects between improved folding in the soluble region and enhanced hydrophobicity in the transmembrane domain.
AlphaFold predictions were used to assess the structural impact of the selected mutations. The wild-type protein showed a pTM score of 0.44, while most mutants exhibited similar values around 0.43, indicating no significant structural disruption. Notably, the T52L mutant showed a slightly higher pTM score of 0.46, suggesting a modest improvement in structural stability. This result is consistent with the introduction of a more hydrophobic residue in the transmembrane region, which may favor membrane insertion. Overall, these findings indicate that the proposed mutations are structurally tolerated and may contribute to improved protein stability.
Week 6 HW: Genetic Circuits Part I
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab:
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several key components necessary for efficient and accurate DNA amplification. First, it includes Phusion DNA polymerase, a high-fidelity enzyme with proofreading activity (3’ → 5’ exonuclease), which reduces errors during DNA replication. It also contains dNTPs (deoxynucleotide triphosphates), which are the building blocks used to synthesize new DNA strands. The mix includes a reaction buffer, optimized with the correct pH and salt concentrations to ensure proper enzyme activity. Additionally, it contains Mg²⁺ ions, which act as essential cofactors for the polymerase. Some mixes may also include stabilizers to maintain enzyme activity during thermal cycling.
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature depends mainly on the melting temperature (Tm) of the primers. Tm is influenced by primer length, GC content (since G-C pairs have stronger bonding than A-T), and sequence composition. Typically, the annealing temperature is set about 5°C below the Tm.
Moreover, other factors include primer specificity, as mismatches lower effective binding, and salt concentration, which affects DNA duplex stability. If the temperature is too low, nonspecific binding may occur; if too high, primers may not bind efficiently.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR amplifies a specific DNA region using primers and DNA polymerase, allowing you to generate large amounts of a defined fragment and even introducing mutations or overlaps. It is highly flexible and does not require specific restriction sites. In contrast, restriction enzyme digestion cuts DNA at specific recognition sequences using restriction enzymes. This method is precise but limited by the presence of those recognition sites in the DNA.
PCR is preferable when you need to amplify DNA, modify sequences or add overlaps for cloning. On the other hand, restriction digestion is preferable when working with existing plasmids and known restriction sites, especially for traditional cloning methods.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
DNA fragments must have overlapping homologous regions (typically 20–40 base pairs) at their ends. These overlaps can be designed into PCR primers or generated through careful restriction digestion. It is also important to verify that sequences are correct (no mutations) and in the proper orientation. DNA fragments should be clean and free of contamination. Finally, checking sequences using software (like Benchling) ensures that overlaps align correctly for seamless assembly.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli induced by chemical treatment or electroporation. In chemical transformation, cells are treated with calcium chloride, which neutralizes the negative charges on DNA and the cell membrane. A heat shock step creates a temporary imbalance in the membrane, allowing DNA to enter the cell. Alternatively, in electroporation, an electrical pulse creates transient pores in the membrane, through which DNA can pass. Once inside, the plasmid replicates independently if it has an origin of replication.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
- Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly relies on Type IIS restriction enzymes that cut DNA outside their recognition sites, generating unique overhangs. These overhangs are designed to be complementary between adjacent fragments, ensuring correct assembly order. During the reaction, the enzyme cuts the DNA and DNA ligase joins the fragments together. Because the recognition sites are removed after cutting, the assembled DNA cannot be re-cut, making the process highly efficient. Multiple fragments can be assembled in a single reaction tube in a predefined sequence.
Diagram:
- Model this assembly method with Benchling or Asimov Kernel!
The Golden Gate Assembly was modeled in Benchling by inserting a pre-designed genetic circuit I previously had, into the pXTK058 backbone. The circuit consisted of a constitutive promoter, a ribosome binding site (RBS), the coding sequence for butyryl-CoA dehydrogenase and a terminator, forming a complete expression cassette.
Type IIS restriction enzyme sites (BbsI) were used to generate compatible overhangs for directional assembly. The Assembly Wizard was used to simulate the process by treating these overhangs as overlaps. The final construct was verified to ensure correct insertion, orientation, and sequence integrity.
Assignment: Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository4
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
HTGAA WEEK #6: IAN SEBASTIAN TERAN GARCIA’S HOMEWORK
1. Exploring the devices in the Bacterial Demos Repo:
Found a construct called “Circuit 3” in the Bacterial Demos Repore and I could observe it corresponds to a plasmid backbone containing the AmeR resistance gene, which allows bacterial selection under antibiotic conditions. The promoter pAmeR drives the expression of this resistance gene.
2. Recreating the Repressilator:
After recreating the repressilator, no noticeable differences were observed between the original circuit obtained from the repository and the one recreated in the simulation environment. Both produced nearly identical graphical outputs, indicating that the reconstruction was accurate and functionally equivalent.
Graphs interpretation:
The graph of RNA concentrations over time shows clear periodic oscillations for the three transcripts. Each gene’s mRNA level rises and falls in a regular pattern, with a noticeable phase shift between them. When one gene is highly expressed, it represses the next gene, causing its expression to decrease, while the third gene begins to increase. This cyclical pattern confirms that the circuit is functioning as an oscillator, with coordinated and repeating changes in gene expression.
Similarly, the protein concentration graph also displays oscillatory behavior, although the fluctuations are smoother and slightly delayed compared to the RNA levels. This delay occurs because protein production depends on the prior synthesis of mRNA, and proteins generally have longer degradation times. Therefore, protein dynamics tend to be more stable and less abrupt than RNA dynamics, which is consistent with biological expectations.
The RNAP flux graph represents the transcriptional activity of each gene at a specific moment in time. Higher values indicate stronger promoter activity, meaning that more RNA polymerase is actively transcribing that gene. In contrast, lower values suggest that the gene is being repressed. This snapshot reflects the regulatory interactions within the circuit at that particular time point.
Finally, the ribosome flux graph shows the rate of protein synthesis for each gene. Similar to the RNAP flux, higher values correspond to increased translation activity. The patterns observed here are consistent with the RNA levels but may show slight delays due to the time required for translation. Overall, these flux measurements provide additional confirmation of the dynamic regulation and oscillatory behavior of the repressilator system.
2.2. Repressilator simulation recreated by me:
3. My constructs:
3.1. Construct 1.
The first genetic circuit consists of the TetR gene under the control of the inducible pBad promoter. This represents a simple gene expression system without regulatory feedback. The RNA concentration graph shows a rapid increase followed by a stable plateau, indicating that transcription is activated and reaches a steady state where production and degradation are balanced.
The protein concentration shows a delayed increase compared to RNA levels, which is expected due to the time required for translation. Eventually, protein levels stabilize, indicating equilibrium.
The RNAP and ribosome flux graphs show relatively constant activity, suggesting sustained transcription and translation under the simulated conditions. Overall, this circuit behaves as a simple inducible expression system, producing a stable amount of TetR without dynamic regulation.
3.2. Construct 2.
For the second construct, the genetic circuit expresses QacR under the control of the pSrpR promoter. Similar to the first construct, this system lacks explicit feedback regulation and behaves as a simple expression module. The RNA concentration rapidly increases and stabilizes, indicating steady transcriptional activity. Compared to Construct 1, the higher RNA levels suggest that the pSrpR promoter is stronger under the simulation conditions.
Moreover, the protein concentration follows the expected delayed increase and reaches a higher steady-state level. RNAP and ribosome fluxes confirm sustained transcription and translation activity. Overall, this construct demonstrates stable gene expression and highlights how promoter strength affects system output.
3.3. Construct 3.
The third construct includes two genes which are QacR, under the inducible pBad promoter and LitR under the pLitR promoter, introducing regulatory interactions and feedback into the system as the simulation results show a strong dominance of LitR expression, while QacR remains near zero. This suggests that the inducible promoter pBad is not sufficiently activated under the simulation conditions, resulting in minimal QacR production.
As a consequence, repression of pLitR by QacR is ineffective, allowing LitR to accumulate. Additionally, LitR negatively regulates its own promoter, creating a feedback loop that stabilizes its expression level.
The protein concentration reflects this behavior, with LitR reaching a high steady-state level and QacR remaining negligible. RNAP and ribosome fluxes confirm strong transcription and translation for LitR and minimal activity for QacR.
Week 7 HW: Genetic Circuits Part II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Continuous signal processing: Unlike Boolean circuits that operate in binary (On/Off), IANNs can process graded inputs and outputs, enabling more nuanced cellular responses.
Integration of multiple inputs: IANNs can combine many signals simultaneously and compute a weighted response, similar to an artificial neural network.
Instead of being limited to simple logic gates (and, or, not), IANNs can model nonlinear relationships between inputs and outputs.
Parameters like the promoter strength, binding affinity and degradation rates can be tuned to adjust how strongly each input influences the output.
Cells can make context - dependent decisions rather than rigid binary responses.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful application of an intracellular artificial neural network (IANN) is the classification of cancer cells based on microRNA expression profiles.
In this system, the inputs are the intracellular concentrations of specific microRNAs (for example, miR-21 or miR-34), which are differentially expressed in cancerous cells. These microRNAs regulate gene expression by repressing or permitting translation of target mRNAs, effectively acting as weighted inputs in the network.
The output is the expression of a reporter or therapeutic gene, such as a fluorescent protein or an apoptosis-inducing factor. The IANN integrates the multiple microRNA inputs and produces a response only when the combined signal exceeds a threshold, similar to a perceptron.
However, this approach faces several limitations because biological noise in gene expression can reduce accuracy and unintended interactions may interfere with circuit behavior. Additionally, there are constraints on the number of inputs that can be reliably implemented. Finally, delivering such engineered systems into patients remains a significant practical limitation.
Reference:
Xie, Z., Wroblewska, L., Prochazka, L., Weiss, R., & Benenson, Y. (2011). Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science (New York, N.Y.), 333(6047), 1307–1311. https://doi.org/10.1126/science.1205527
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
This diagram I made on Canva represents an intracellular artificial neural network composed of two layers. In Layer 1, the inputs X₁ and X₂ are transcribed (Tx) and translated (Tl) to produce an endoribonuclease (E), which acts as an intermediate regulatory signal. This enzyme then interacts with Layer 2, where inputs X₃ and X₄ are transcribed into mRNA. The endoribonuclease E negatively regulates this layer by cleaving the mRNA, thereby reducing its availability for translation. As a result, the production of the fluorescent protein Y is modulated at the translational level. This design mimics a multilayer perceptron, where the first layer processes inputs to generate a hidden signal (E), and the second layer integrates both direct inputs and regulatory signals to determine the final output.
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are primarily based on mycelium, the filamentous network of fungi, which can bind organic matter into solid structures. One of the most well-known examples is mycelium-based packaging, used as an alternative to polystyrene foam for protecting goods during shipping. These materials are lightweight, biodegradable, and can be molded into custom shapes. Another example is mycelium leather, a sustainable alternative to animal leather, used in fashion and upholstery. Additionally, fungal materials are used in construction, such as insulation panels and biodegradable bricks, due to their thermal resistance and low density. Some applications also include acoustic panels and biocomposites for furniture.
Fungal materials are biodegradable, renewable, and can be grown using agricultural waste, which significantly reduces environmental impact compared to plastics or synthetic foams. Their production typically requires less energy and generates fewer emissions. Furthermore, they exhibit useful properties such as thermal insulation, fire resistance, and lightweight structure.
However, there are also limitations as fungal materials generally have lower mechanical strength compared to plastics or metals, which restricts their use in load-bearing applications. They can be sensitive to moisture and environmental conditions if not properly treated. Additionally, scaling production while maintaining consistency can be challenging, and their durability over long periods may be lower than that of conventional materials.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Category
Description
Why it matters
Stronger materials
Engineer fungi to produce enhanced structural proteins or denser mycelium networks
Improves mechanical strength for construction, packaging, and durable biomaterials
Water resistance
Modify fungi to synthesize hydrophobic compounds
Increases durability in humid environments and expands real-world applications
Self-healing materials
Program fungi to regrow and repair damaged structures
Extends lifespan of materials and reduces maintenance costs
Antimicrobial properties
Engineer production of antimicrobial compounds
Prevents contamination and increases safety in medical or packaging uses
Responsive (smart) materials
Enable fungi to respond to stimuli (light, temperature, chemicals)
Allows development of adaptive or sensing materials
Fungi vs Bacteria in Synthetic Biology
Feature
Fungi
Bacteria
Cell structure
Multicellular, filamentous (mycelium)
Unicellular
Material formation
Naturally forms 3D structures
Cannot form structures without scaffolds
Protein secretion
High secretion capacity
Limited secretion
Substrate use
Can degrade complex biomass (e.g., agricultural waste)
Prefer simpler substrates
Growth speed
Slower
Faster
Genetic manipulation
More complex
Easier
Best use case
Living materials, biomaterials, structure-based applications
Fast production of molecules, simple genetic circuits
Shin, H.-J., Ro, H.-S., Kawauchi, M., & Honda, Y. (2025). Review on mushroom mycelium-based products and their production process: From upstream to downstream. Bioresources and Bioprocessing, 12, 3. https://doi.org/10.1186/s40643-024-00836-7
Week 9 HW: Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo expression systems, primarily due to its flexibility and precise control over experimental conditions. Because CFPS operates in an open environment without living cells, researchers can directly manipulate the concentrations of DNA templates, ions, cofactors, and other components in real time. This eliminates constraints associated with cellular viability, such as toxicity or metabolic burden. As a result, CFPS is particularly advantageous for the production of proteins that are toxic to host cells, such as antimicrobial peptides or pore-forming proteins. Additionally, CFPS enables rapid prototyping of genetic constructs, making it highly suitable for applications like synthetic biology circuit testing, where speed and iterative design are essential.
2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system consists of several essential components that collectively replicate the molecular machinery of protein synthesis. The core component is the cell extract (lysate), which contains ribosomes, transfer RNAs (tRNAs), aminoacyl-tRNA synthetases, and various translation factors required for protein assembly. A DNA or messenger RNA (mRNA) template provides the genetic instructions encoding the target protein. Amino acids serve as the building blocks for protein synthesis, while an energy system—typically composed of ATP, GTP, and associated regeneration pathways—fuels transcription and translation processes. Additionally, salts and cofactors such as magnesium and potassium ions are necessary to maintain proper structural and functional conditions for enzymatic activity. When DNA is used as a template, transcriptional enzymes such as T7 RNA polymerase are also included to generate mRNA.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in CFPS because protein synthesis is an energy-intensive process that rapidly consumes ATP and GTP. Without a continuous supply of energy, translation halts prematurely, leading to low protein yields. To address this limitation, CFPS systems incorporate energy regeneration mechanisms that recycle ADP into ATP.
One commonly used method that I could use involves phosphoenolpyruvate (PEP) in combination with pyruvate kinase, which efficiently regenerates ATP during the reaction. Alternative systems, such as creatine phosphate with creatine kinase or glucose-based metabolic pathways, can also be employed depending on the desired duration and efficiency of protein production. These strategies extend reaction lifetimes and significantly improve overall protein yield.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic and eukaryotic CFPS systems differ in complexity, cost, and functional capabilities. Prokaryotic systems, such as those derived from Escherichia coli, are widely used due to their simplicity, high protein yield, and cost-effectiveness. However, they lack the machinery required for many post-translational modifications. These systems are well suited for expressing proteins that do not require complex folding or modifications, such as fluorescent reporters like GFP or metabolic enzymes.
In contrast, eukaryotic CFPS systems, including wheat germ or rabbit reticulocyte extracts, provide a more physiologically relevant environment that supports proper folding, disulfide bond formation, and certain post-translational modifications. Consequently, they are more appropriate for producing complex proteins such as human hormones or antibodies, where structural accuracy is critical for functionality.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The expression of membrane proteins in CFPS systems presents unique challenges due to their hydrophobic nature and dependence on lipid environments for proper folding and stability. These proteins are prone to aggregation when synthesized in aqueous conditions. To overcome these challenges, CFPS reactions can be supplemented with membrane-mimicking systems such as liposomes, nanodiscs, or mild detergents that facilitate proper insertion and stabilization of the protein.
Additionally, molecular chaperones may be included to assist in correct folding. Careful optimization of ionic conditions, particularly magnesium and potassium concentrations, as well as modulation of expression rates, can further enhance protein quality. These strategies collectively create a suitable environment for functional membrane protein production.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low protein yield in CFPS systems can arise from several factors and one common issue is inefficient transcription or translation, which may result from weak promoters, suboptimal ribosome binding sites, or degraded DNA templates. This can be addressed by optimizing genetic elements, increasing template concentration, or ensuring DNA integrity.
A second factor is insufficient energy supply; rapid depletion of ATP can prematurely terminate protein synthesis. Implementing or optimizing an energy regeneration system can significantly improve yields. A third potential cause is protein misfolding or degradation, often due to the absence of proper folding conditions or the presence of proteases in the extract. This can be mitigated by adding molecular chaperones, reducing reaction temperature, or incorporating protease inhibitors. Systematic optimization of these parameters is essential to achieve efficient and reliable protein production.
Homework Question from Kate Adamala
1. Pick a function and describe it.
a) What would your synthetic cell do? What is the input and what is the output?
The synthetic minimal cell is designed to function as a biosensor and detoxification system for mercury contamination in aqueous environments. The input is the presence of mercury ions (Hg²⁺), which are detected by a mercury-responsive regulatory element inside the synthetic cell. In response, the system activates gene expression. The output consists of two components: (i) the production of a fluorescent reporter protein (GFP), which enables detection, and (ii) the enzymatic conversion of Hg²⁺ into elemental mercury (Hg⁰), a less toxic and more diffusible form.
b) Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. Without encapsulation, the system would lack spatial organization and controlled interaction with the environment. The components responsible for sensing and response would diffuse freely, reducing efficiency and eliminating the ability to function as a defined, cell-like unit. Encapsulation is essential to maintain compartmentalization and regulate the exchange of molecules.
c) Could this function be realized by genetically modified natural cell?
Yes, this function could be implemented in genetically modified bacteria carrying mercury-resistance operons. However, such approaches involve the use of living genetically modified organisms, which raises biosafety and regulatory concerns. In contrast, synthetic minimal cells provide a non-living, modular alternative that allows precise control over system components and avoids environmental risks associated with engineered cells.
d) Describe the desired outcome of your synthetic cell operation.
The desired outcome is that, in the presence of mercury, the synthetic minimal cell simultaneously detects, reports, and detoxifies the contaminant. This results in both a measurable fluorescent signal and a reduction in mercury toxicity, enabling combined environmental sensing and remediation.
2. Design all components that would need to be part of your synthetic cell.
a) What would be the membrane made of?
The membrane would consist of a lipid bilayer composed of phospholipids such as POPC combined with cholesterol. This composition provides structural stability, appropriate fluidity, and controlled permeability, mimicking natural biological membranes.
b) What would you encapsulate inside? Enzymes, small molecules.
The synthetic cell would encapsulate a complete cell-free transcription/translation (Tx/Tl) system, including ribosomes, tRNAs, enzymes, amino acids, nucleotides, and cofactors. Additionally, it would contain the DNA encoding the mercury-responsive genetic circuit, an energy regeneration system (e.g., phosphoenolpyruvate-based), and all necessary components for protein synthesis, including the fluorescent reporter and detoxification enzymes.
c) Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
The Tx/Tl system would be derived from bacterial extracts (Escherichia coli), as this system is efficient, cost-effective and compatible with the mercury-responsive regulatory elements used in the design.
Since the system does not require complex post-translational modifications, a prokaryotic expression system is sufficient.
d) How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Communication with the environment will be achieved through a combination of membrane permeability and specific transport mechanisms. Mercury ions (Hg²⁺), which are not readily permeable through lipid membranes, will enter the synthetic cell via membrane transport proteins such as MerT or MerP. Once inside, they activate the regulatory system. The detoxified product (Hg⁰) is more hydrophobic and can diffuse out of the membrane. The fluorescent signal remains inside the vesicle and can be detected externally using appropriate instrumentation.
3. Experimental details
a) List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids:
POPC (phosphatidylcholine)
Cholesterol
Genes:
merR (mercury-responsive transcriptional regulator)
merT and merP (mercury transport proteins)
merA (mercury reductase enzyme)
gfp (fluorescent reporter under mercury-inducible promoter)
Additional components:
Bacterial cell-free Tx/Tl system (E. coli extract)
Energy regeneration system (e.g., PEP + pyruvate kinase)
b) How will you measure the function of your system?
The function of the system will be evaluated using two complementary methods. First, fluorescence measurements will be used to quantify GFP expression as an indicator of mercury detection, using techniques such as plate readers or fluorescence microscopy. Second, chemical analysis of mercury transformation will be performed to confirm detoxification, using analytical methods such as atomic absorption spectroscopy to measure the conversion of Hg²⁺ to Hg⁰.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Application field: Textiles in fashion.
Write a one-sentence summary pitch sentence describing your concept.
A smart textile incorporating freeze-dried cell-free systems that detects environmental pollutants and responds by producing visible color changes and neutralizing harmful compounds.
How will the idea work, in more detail? Write 3-4 sentences or more.
The proposed system consists of fabrics embedded with freeze-dried cell-free transcription/translation (Tx/Tl) reactions distributed within microcapsules integrated into the textile fibers. Upon exposure to environmental stimuli such as air pollutants (e.g., nitrogen oxides or volatile organic compounds), the system is activated by ambient moisture (humidity or sweat), which rehydrates the cell-free components. The embedded genetic circuits are designed to sense specific chemical signatures and trigger the expression of reporter proteins that produce visible color changes, allowing real-time detection. In addition, the system can express enzymes capable of partially degrading or neutralizing harmful compounds in the immediate surroundings. This creates a dual-function material that acts both as a biosensor and a localized remediation system.
What societal challenge or market need will this address?
Current monitoring systems are often centralized and do not provide individuals with real-time, localized information about their exposure. This smart textile addresses the need for personal, wearable environmental monitoring, empowering users to make informed decisions about their surroundings. Furthermore, integrating a remediation function adds value by not only detecting pollutants but also contributing to their reduction at a microenvironmental level. This concept is particularly relevant for urban populations, industrial workers and populations exposed to poor air quality.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
One key limitation of cell-free systems is their dependence on hydration for activation. This can be addressed by designing the textile to utilize ambient humidity, sweat, or embedded hydrogel layers that retain moisture and enable controlled activation. Stability during storage can be improved through freeze-drying (lyophilization) combined with protective matrices such as sugars (trehalose), which preserve biological activity over extended periods. To address the one-time-use limitation, the textile can be engineered with replaceable or rechargeable patches containing the cell-free components.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Sustainable agriculture is essential for long-duration space missions, where food must be produced in controlled and resource-limited environments. Beneficial soil bacteria play a critical role in plant growth by promoting nutrient availability and stress resistance. However, microgravity and space radiation may alter bacterial gene expression and reduce their effectiveness. Understanding how plant growth–promoting bacteria respond to space conditions is therefore essential for developing reliable bioregenerative life-support systems. This topic is significant for enabling food production beyond Earth and scientifically interesting for studying microbial adaptation to extreme environments.
(It is a topic I always wanted to explore more about)
2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Stress-response and plant-growth–related genes in Bacillus subtilis (spo0A, sigB and auxin-related pathways).
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Bacillus subtilis is a model plant growth–promoting bacterium known for its resilience and ability to enhance plant health.
In space, environmental stressors such as microgravity and radiation may disrupt its gene expression, affecting its capacity to support plant growth. By analyzing stress-response and growth-related genes, whether beneficial bacterial functions are maintained under space conditions could be evaluated. This directly addresses the challenge of ensuring reliable microbial support systems for extraterrestrial agriculture.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
The hypothesis is that space conditions, including microgravity and radiation, alter the expression of key stress-response and plant growth–promoting genes in Bacillus subtilis. Specifically, it is expected that stress-response genes such as sigB will be upregulated, while genes associated with plant growth promotion may be downregulated or dysregulated.
The research goal is to determine whether these changes can be detected using the BioBits® cell-free system as a rapid, portable diagnostic tool, and by linking gene expression outputs to fluorescent reporters, this system could enable real-time monitoring of microbial health and functionality in space. This approach supports the development of robust microbial systems for sustainable agriculture beyond Earth.
5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Samples of Bacillus subtilis grown under simulated microgravity conditions will be compared to Earth controls. DNA or RNA will be extracted and amplified using the miniPCR. Target sequences will be introduced into the BioBits cell-free system with reporter constructs to measure gene expression via fluorescence. The P51 Molecular Fluorescence Viewer will be used to quantify signal intensity. Controls will include non-stressed bacteria and no-template reactions. Data will consist of fluorescence levels corresponding to gene activity, allowing comparison of stress-response and growth-related gene expression.
Morrison, M. D., Fajardo-Cavazos, P., & Nicholson, W. L. (2017). Cultivation in space flight produces minimal alterations in Bacillus subtilis physiology and spore formation. NPJ Microgravity, 3(1).
https://pubmed.ncbi.nlm.nih.gov/28821547/
Week 3 HW: Lab Automation
This is the first design I made, a green shiny bettle (really beautiful!):
The inspiration 🪲:
However, due to reagent limitations at my node, I decided to shift toward a much simpler idea that would be more feasible to implement. I wanted a design that could be cultured using the Opentrons OT-2 robot and later visualized under UV light once the colonies had grown. At the same time, I wanted to preserve an iconic and visually recognizable element, similar to the bright green beetle from my original design. Therefore, I designed a Smiling E. coli.!
importmath################################GREENSECTION(Body+Flagella)################################pipette_20ul.pick_up_tip()center=center_location**Ovalbody:**a=16b=8points=40foriinrange(points):ifi%8==0:pipette_20ul.aspirate(8,location_of_color('Green'))angle=2*math.pi*i/pointsx=a*math.cos(angle)y=b*math.sin(angle)loc=center.move(types.Point(x=x,y=y,z=0))dispense_and_detach(pipette_20ul,1,loc)**Flagella:**flagella_points=6foriinrange(flagella_points):pipette_20ul.aspirate(6,location_of_color('Green'))angle=2*math.pi*i/flagella_pointsstart_x=(a+1)*math.cos(angle)start_y=(b+1)*math.sin(angle)fortinrange(5):fx=start_x+t*2*math.cos(angle)fy=start_y+t*2*math.sin(angle)loc=center.move(types.Point(x=fx,y=fy,z=0))dispense_and_detach(pipette_20ul,1,loc)pipette_20ul.drop_tip()################################REDSECTION(Eyes+Smile)################################pipette_20ul.pick_up_tip()**Eyes:**pipette_20ul.aspirate(4,location_of_color('Red'))left_eye=center.move(types.Point(x=-5,y=2,z=0))right_eye=center.move(types.Point(x=5,y=2,z=0))dispense_and_detach(pipette_20ul,2,left_eye)dispense_and_detach(pipette_20ul,2,right_eye)**Smile:**smile_points=15pipette_20ul.aspirate(15,location_of_color('Red'))foriinrange(smile_points):angle=math.pi*i/smile_pointsx=6*math.cos(angle)y=-3*math.sin(angle)-2loc=center.move(types.Point(x=x,y=y,z=0))dispense_and_detach(pipette_20ul,1,loc)pipette_20ul.drop_tip()**Don't forget to end with a drop_tip()**
RESULT :) :
At the end of it all, the design was successfully brought to life on the agar plate using the Opentrons OT-2 and seeing it finally appear felt incredibly exciting :,D.
AI tools (ChatGPT and Gemini) assisted in suggesting mathematical approaches for generating an oval body and radial flagella. I reviewed, modified and finalized the code to ensure correct simulation behavior and compliance with lab constraints (volume limits).
Post-Lab Questions:
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
DeRoo, J. B., Jones, A. A., Slaughter, C. K., Ahr, W. T., Stroup, S. M., Thompson, G. B., & Snow, C. D. (2025). Automation of protein crystallization scaleup via Opentrons-2 liquid handling. SLAS Technology, 32, Article 100268. https://doi.org/10.1016/j.slast.2025.100268
This paper describes how an Opentrons - 2 liquid handling robot can be used to automate and scale up protein crystallization experiments which is a foundational step in structural biology that is traditionally manual intensive. The robot was programmed via Python scripts to prepare 24 well sitting drop crystallization trials with precise reagent mixing and drop deposition. By comparing results against standard manual setup, the study showed that automation:
Reduce hands-on labor and variability, improving reproducibility.
Produce consistent crystal growth for both model proteins (hen egg white lysozyme) and a periplasmic protein from Campylobacter jejuni.
Scale preparation in a way that could benefit labs doing structural studies or materials research requiring uniform crystal batches.
I find this application novel because protein crystallization is a critical but laborious step in X - ray crystallography and related structural methods. Most labs still perform it manually or with high-cost automation. This work shows that a relatively low - cost, open-programmable robot like Opentrons - 2 can reliably handle complex setup steps, lowering the barrier to high-throughput crystallization workflows and enabling new scale and reproducibility in structural biology applications
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet.
What I Would Automate?
A) Automated DNA Assembly & Construct Library Design
Using a liquid handling robot such as the Opentrons OT-2, I would automate Golden Gate or Gibson assembly reactions to generate a small library of salt-inducible constructs:
To test salt responsiveness the Opentrons OT-2 liquid handling robot would:
Prepare a 96-well plate with increasing NaCl concentrations (0–400 mM).
Inoculate engineered strains.
If I would use a cloud lab platform such as Ginkgo Bioworks’s Ginkgo Nebula (conceptually), the workflow would include:
-Automated liquid handling for salt gradients.
-Plate sealing and incubation.
-Automated plate reader measurements.
-Data export for downstream analysis.
SECTION 1: ABSTRACT Provide a concise, self-contained summary of your project (minimum 150 words). The abstract should allow a reader to understand the purpose, approach, and expected outcomes of the work without referring to other sections.
Your abstract should briefly address the following elements:
a. Significance: What problem or question does the project address, and why is it important?
PROJECT OBJECTIVE
Engineer the L protein of the MS2 phage to increase structural stability. Disrupt or reduce its interaction with the bacterial chaperone DnaJ. Preserve the C-terminal lysis domain to maintain lytic function. Avoid mutations that interfere with structurally or evolutionarily coupled residues. Phase 1: Mapping the DnaJ Interaction Interface
Since the exact binding interface between the L protein and DnaJ is unknown, the first step is to identify it computationally rather than introducing arbitrary mutations.
Subsections of Projects
Individual Final Project
SECTION 1: ABSTRACT
Provide a concise, self-contained summary of your project (minimum 150 words). The abstract should allow a reader to understand the purpose, approach, and expected outcomes of the work without referring to other sections.
1. Your abstract should briefly address the following elements:
a. Significance: What problem or question does the project address, and why is it
important?
b. Broad Objective: What is the overall goal of the project?
c. Hypothesis: What prediction or principle is the project testing or demonstrating?
d. Specific Aims: What key steps or milestones will be completed to achieve the
objective?
e. Methods: What experimental or technical approaches will be used?
Abstract
Soil salinity is an increasing constraint on agricultural productivity, particularly in arid and high-altitude regions such as the Bolivian Altiplano, where environmental conditions promote the accumulation of salts and severely limit crop growth. This problem disproportionately affects smallholder farming communities whose livelihoods depend on crops such as quinua (Chenopodium quinoa), making the development of accessible, sustainable biological solutions an urgent priority.
The overall objective of this project is to design a synthetic rhizosphere consortium capable of enhancing crop resilience under saline stress by integrating three complementary microbial functions: osmoprotection, nitrogen fixation, and soil stabilization through biofilm formation. The central hypothesis is that a functionally coordinated microbial consortium, comprising Pseudomonas fluorescens, Azospirillum brasilense, and Bacillus subtilis, can improve plant tolerance to salinity more effectively than any single organism alone, by simultaneously addressing multiple dimensions of salt stress at the root-soil interface.
The specific aims include: (1) designing salt-responsive genetic circuits for each organism in Benchling, (2) validating metabolic feasibility using Flux Balance Analysis (COBRApy), (3) simulating consortium interactions using BacArena, and (4) establishing a framework for future experimental validation using Arabidopsis thaliana as a model plant, with long-term translation to Altiplano native crops.
Methods include in silico genetic circuit design in Benchling using sequences from NCBI and the iGEM Parts Registry, Gibson Assembly for construct integration, codon optimization via IDT, genome-scale metabolic modeling with COBRApy (model iJN476), and consortium interaction simulation with BacArena. All three constructs incorporate auxotrophic kill switches for biosafety and environmental containment.
The first aim is to design a synthetic rhizosphere consortium in silico and establish the experimental foundation for its laboratory validation. Using Benchling as the primary platform, we will construct modular, salt-responsive genetic circuits for three complementary organisms: Pseudomonas fluorescens (osmoprotection), Azospirillum brasilense (nitrogen fixation), and Bacillus subtilis (biofilm formation). A conditional kill switch will be incorporated into each construct to ensure biosafety and environmental containment.
Phase 1A - In Silico Genetic Circuit Design
Design salt-responsive genetic constructs for each organism in Benchling:
P. fluorescens: P_algU → betA → betB (glycine betaine production)
A. brasilense: P_nifH → nifHDK (nitrogen fixation under salinity stress)
B. subtilis: P_epsA → epsA → tapA (biofilm matrix formation)
Incorporate auxotrophic kill switches in all three constructs for biosafety.
Perform codon optimization of heterologous genes (betA, betB) for P. fluorescens using the IDT Codon Optimization Tool.
Validate metabolic feasibility of glycine betaine production in P. fluorescens via Flux Balance Analysis (COBRApy, model iJN1411).
Simulate consortium interactions between P. fluorescens and B. subtilis using BacArena to assess metabolic compatibility and predict inter-species metabolite exchange.
Synthesize DNA constructs via Twist Bioscience and obtain expression plasmids from Addgene.
Transform each bacterial strain:
P. fluorescens: electroporation
A. brasilense: electroporation
B. subtilis: competent cell transformation
Confirm successful transformation via colony PCR.
Validate individual strain performance under saline conditions:
P. fluorescens: glycine betaine quantification via HPLC
A. brasilense: nitrate production via colorimetric assay
B. subtilis: exopolysaccharide production via crystal violet staining
Validate kill switch functionality: CFU assay with and without DAP (P. fluorescens) and tryptophan (B. subtilis).
Validate consortium performance: culture all three strains together under saline stress and measure combined outputs.
Aim 2: Development Aim - Plant Validation Under Salinity Stress
The second aim extends the engineered consortium into a plant-based validation system, evaluating its capacity to improve growth and stress tolerance under saline conditions. Arabidopsis thaliana due to its well-annotated genome, short life cycle and availability of salt-responsive mutant lines (Chu et al., 2019). Results obtained with A. thaliana will inform future translation to native Bolivian Altiplano crops.
Comparison: Consortium-inoculated plants vs non-inoculated controls under saline vs normal conditions.
Microbial community monitoring: 16S rRNA sequencing to confirm consortium stability in the rhizosphere.
Optimize microbial ratios between the three strains for maximum plant consortium.
Delivery: Two delivery strategies will be evaluated in parallel:
Seed coating: lyophilized consortium applied directly to seeds prior to germination.
Soil inoculant: liquid consortium applied to soil at planting.
Lyophilization is the preferred long-term delivery format as it eliminates cold chain requirements and extends shelf life — critical for deployment in remote regions such as the Bolivian Altiplano.
Aim 3: Visionary Aim - Directed Evolution for the Bolivian Altiplano
The Core Problem: Ecological Fragility of Synthetic Consortia
In natural soil ecosystems, plants actively recruit microorganisms through root exudates, generating selective rhizosphere microbiomes adapted to local environmental pressures. In the Bolivian Altiplano, native crops such as quinua (Chenopodium quinoa) coexist with microbial communities shaped by chronic salinity, drought, nutrient limitation and high UV exposure. Consequently, a purely synthetic consortium composed of engineered bacterial strains may exhibit limited ecological persistence once introduced into real agricultural soils.
Synthetic microbial communities are particularly vulnerable to competitive exclusion by native halotolerant microorganisms, instability across successive plant generations and incompatibility with host-specific rhizosphere signaling (Chang et al., 2021). Additionally, microbial communities are inherently dynamic systems whose composition changes continuously through ecological succession, potentially causing loss of engineered functionality over time (Sánchez et al., 2021).
Previous studies on artificial microbiome selection demonstrated that engineered communities frequently experience functional drift, reduced heritability, and declining performance after repeated propagation cycles (Blouin et al., 2015; Chang et al., 2021). These limitations are especially relevant in saline soils, where environmental fluctuations strongly influence microbial community composition and plant–microbe interactions.
To address these ecological limitations, we propose a rational directed iterative evolution strategy inspired by top-down microbiome engineering frameworks (Chang et al., 2021; Sánchez et al., 2021). Rather than relying exclusively on bottom-up rational assembly, this approach integrates ecological selection, enrichment, perturbation, and adaptive stabilization to evolve a field-compatible microbial consortium.
Directed evolution of microbial communities can be conceptualized as a guided exploration of an “ecological structure–function landscape,” in which microbial composition and collective functions co-evolve under selective environmental pressures (Sánchez et al., 2021).
Iteration
Action
Expected Outcome
1-Enrichment
Co-culture synthetic consortium with native Altiplano soil microbiota and quinua rhizosphere samples
Identification of compatible native strains and ecological partners
2-Selection
Select for enhanced osmoprotection, nitrogen fixation, oxidative stress tolerance, and biofilm stability under saline conditions
Emergence of an adaptive hybrid consortium
3-Validation
Test evolved communities on quinua under simulated Altiplano conditions
Quantification of plant growth promotion and rhizosphere persistence
4-Iteration
Repeat enrichment and selection under progressively higher salinity stress
Development of a field-resilient consortium
This iterative framework mimics natural ecological adaptation while preserving the desired engineered traits. Environmental perturbations such as increasing salinity, nutrient limitation, and osmotic stress can shift community composition toward more resilient stable states (Sánchez et al., 2021). Furthermore, migration from native microbial pools may replenish functional diversity and prevent evolutionary stagnation during selection cycles (Chang et al., 2021).
Ecological and Evolutionary Rationale:
Traditional bottom-up synthetic biology approaches assume that microbial interactions can be fully predicted from individual strain behavior. However, microbial communities frequently exhibit higher-order interactions, emergent properties, and multistable ecological states that cannot be anticipated from pairwise interactions alone (Guo & Boedicker, 2016; Sánchez-Gorostiaga et al., 2019).
Directed evolution bypasses this limitation by allowing beneficial ecological interactions to emerge naturally through selection. According to Sánchez et al. (2021), effective community-level evolution requires:
Phenotypic variation between microbial communities
Heritable transmission of community-level functions
Stabilization of ecological succession dynamics
Recurrent perturbation and selection cycles
Importantly, generational stability is critical because microbial communities continuously change through ecological succession. Communities must therefore reach stable equilibrium states before their functions can be reliably inherited across generations. Several ecological engineering strategies described by Chang et al. (2021) and Sánchez et al. (2021) directly support our proposed workflow, including:
Migration from native microbial pools
Dilution-to-extinction bottlenecking
Community coalescence
Environmental pulse perturbations
Selective enrichment under stress conditions
Functional Targets for Directed Evolution:
Salt Stress Adaptation
Salinity imposes osmotic imbalance, ion toxicity, oxidative stress, and water limitation on both plants and microorganisms. Therefore, evolved consortia will be selected for enhanced halotolerance through:
Increased biosynthesis of osmoprotectants:
-trehalose
-glycine betaine
-ectoine
Enhanced Na⁺ extrusion and ion homeostasis systems
Exopolysaccharide (EPS) production for soil water retention
Reduction of reactive oxygen species (ROS) accumulation
Improved membrane stability under osmotic stress
Nitrogen Fixation Under Salinity
Nitrogen fixation efficiency is highly sensitive to oxidative and osmotic stress because nitrogenase enzymes are easily inhibited under saline conditions. Consequently, selection will favor microbial communities capable of maintaining diazotrophic activity despite environmental stress.
Selection targets include:
Nitrogenase activity under high salinity
Stable conversion of atmospheric N₂ into ammonium
Improved nitrogen assimilation by quinua plants
Maintenance of metabolic cooperation between consortium members
Increased nutrient availability in nutrient-poor Altiplano soils
Artificial microbiome selection has previously been used to enhance plant-associated functions under abiotic stress, including drought and salinity tolerance (Mueller et al., 2016; Jochum et al., 2019).
Biofilm Stability and Rhizosphere Persistence
Long-term rhizosphere persistence represents one of the principal challenges for engineered microbiomes. Biofilms provide structural stability, facilitate nutrient exchange, and protect microbial communities against environmental fluctuations.
The consortium will therefore be evolved for:
Increased extracellular matrix production
Enhanced root adhesion and colonization
Cooperative metabolic interactions
Improved protection against desiccation and salinity
Resistance to invasion by competing microorganisms
Stable persistence across successive plant generations
Chang et al. (2021) demonstrated that communities evolved through iterative perturbation and selection exhibit greater resistance to ecological invasion compared to rationally assembled synthetic consortia. This property is essential for field deployment in highly competitive native soils.
Expected Impact
This Aim seeks to integrate ecological adaptation with synthetic biology to create a next-generation agricultural microbiome optimized for saline soils of the Bolivian Altiplano. Instead of producing a static engineered consortium, this strategy aims to generate an ecologically stabilized and evolutionarily conditioned rhizosphere microbiome capable of long-term persistence under extreme environmental stress.
By combining rational engineering with directed community evolution, the final consortium is expected to provide:
Sustained nitrogen fixation under salinity
Enhanced plant osmotic stress tolerance
Greater rhizosphere colonization efficiency
Long-term ecological resilience
Improved quinua productivity in saline soils
The ultimate vision of this project is a modular, open-source microbial platform for climate-resilient agriculture that can be:
Adapted to different saline environments beyond the Bolivian Altiplano.
Scaled through low-cost fermentation and lyophilization infrastructure.
Deployed directly by farming communities without dependence on cold chain logistics or chemical fertilizers.
Iteratively improved through continued directed evolution as environmental conditions change with climate.
In the long term, this approach represents a replicable model for engineering resilient agricultural microbiomes in response to the growing global challenge of soil salinization driven by climate change and unsustainable irrigation practices.
SECTION 3: BACKGROUND
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap in knowledge or capability that your project addresses.
1. Briefly summarize two peer-reviewed research citations relevant to your research
(minimum four sentences).
Citation 1
Hiernaux, P., et al. (2024). Complementarity of Sentinel-1 and Sentinel-2 data for soil salinity monitoring to support sustainable agriculture practices in the Central Bolivian Altiplano. Sustainability, 16(14), 6200. https://www.mdpi.com/2071-1050/16/14/6200
The Bolivian Altiplano is a remote endorheic region that suffers from the major problem of soil salinization, threatening the sustainability of agriculture activity. Located at an average elevation of 3,700 meters above sea level, the region around Lake Poopó in the central Altiplano faces severe and progressive salt accumulation driven by high evaporation rates and the endorheic nature of the basin. This study monitored soil salinity across a five-year period using satellite imagery and machine learning, revealing the extent and seasonal dynamics of salinization in the region. The findings highlight an urgent need for mitigation strategies that are accessible to remote farming communities, precisely the gap this project addresses through the development of a deployable microbial inoculant. Globally, soil salinization poses a critical threat to agricultural productivity, ecosystem resilience, and regional resource sustainability, with primary and secondary salinization processes intensifying under climate change and unsustainable land-use practices. Together, these findings establish the Bolivian Altiplano as one of the most vulnerable agricultural regions in South America and justify the urgent development of low-cost, biologically based salinity mitigation strategies.
Citation 2
Bukhat, S., et al. (2021). Potential of plant growth promoting bacterial consortium for improving the growth and yield of wheat under saline conditions. Frontiers in Plant Science. PMC9557047. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9557047/
Research on multi-strain bacterial consortia demonstrated that co-inoculation of PGPR strains, including Pseudomonas fluorescens, caused significant positive impacts on shoot length, root length, fresh weight, and biochemical traits of wheat at the highest salinity levels tested, outperforming any single-strain inoculation. The multi-strain consortium significantly improved chlorophyll content, relative water content and K/Na ratio, which are key indicators of salinity tolerance in plants. These results provide direct experimental evidence that combining PGPR strains with complementary growth-promoting traits produces synergistic effects that cannot be achieved by a single organism alone. This finding is foundational to the design rationale of this project, which combines three functionally distinct organisms: Pseudomonas fluorescens for osmoprotection, Azospirillum brasilense for nitrogen fixation and Bacillus subtilis for biofilm formation, each addressing a different dimension of salinity stress simultaneously. The study also tested bacterial compatibility prior to consortium development, a methodological consideration directly reflected in the BacArena simulation component of this project.
Citation 3
Gerbore, J., et al. (2024). A “love match” score to compare root exudate attraction and feeding of the plant growth-promoting rhizobacteria Bacillus subtilis, Pseudomonas fluorescens, and Azospirillum brasilense. Frontiers in Microbiology. PMC11456545. https://pmc.ncbi.nlm.nih.gov/articles/PMC11456545/
A study evaluating the exact three organisms used in this project, Bacillus subtilis, Pseudomonas fluorescens and Azospirillum brasilense, found that P. fluorescens and A. brasilense responded more efficiently to plant root exudates than B. subtilis and proposed a quantitative “love match” score to evaluate plant-PGPR pair compatibility based on chemotaxis and bacterial growth responses. This research is particularly significant for this project because it directly evaluates the three chosen organisms together, confirming that they are well-established biofertilizers with documented responses to plant signals. The study found that all three organisms responded positively to root exudates from multiple plant species, suggesting that the consortium could be compatible with a range of host plants, including Arabidopsis thaliana. Furthermore, the quantitative framework proposed by this study could be directly applied in future experimental work to predict and optimize the rhizosphere performance of this consortium.
Citation 4
Ilyas, N., et al. (2024). Plant growth-promoting bacteria (PGPB)-induced plant adaptations to stresses: an updated review. PeerJ. https://peerj.com/articles/17882/
A comprehensive review documented that microbial consortia of Bacillus subtilis and Azospirillum brasilense upregulate root and shoot development, carbon dioxide uptake, transpiration, and leaf chlorophyll index under stress conditions, with inoculation improving grain yield and nitrogen accumulation in wheat. Additionally, Bacillus subtilis, Azospirillum brasilense and Pseudomonas fluorescens have been shown to increase the concentration of stress-protective compounds in plants under abiotic stress. This review consolidates a growing body of literature confirming that the three organisms selected for this project have individually and collectively demonstrated plant growth-promoting properties under stress conditions. The review also highlights that PGPR function through multiple complementary mechanisms, including osmoprotectant production, nitrogen fixation, exopolysaccharide synthesis and phytohormone modulation, underscoring why a multi-organism consortium targeting each of these mechanisms simultaneously represents a more robust strategy than single-strain inoculants. This mechanistic diversity is the biological foundation of the consortium design presented in this project.
2. Explain how your project is novel or innovative. (Minimum 3 sentences.)
a. Examples of topics to discuss:
i. New applications or uses of existing biological tools or concepts.
ii. Development of new approaches, methodologies, or technologies.
iii. Ways the project challenges existing paradigms or assumptions.
iv. How the work expands the boundaries of synthetic biology.
This project represents a significant departure from conventional approaches to agricultural salinity management, which have historically relied on either chemical soil amendments, such as gypsum application and leaching or the development of genetically modified salt-tolerant crop varieties. Both approaches face critical limitations: chemical amendments are costly, require specialized equipment and are inaccessible to smallholder farmers in remote regions, while transgenic crop development involves lengthy regulatory processes and raises socioeconomic concerns about seed sovereignty.
This project proposes a different paradigm because instead of modifying the plant or the soil chemistry, it engineers the invisible microbial community at the root-soil interface, the rhizosphere, to actively buffer the plant against salt stress from the outside in.
The novelty of this project lies first in its systems-level design philosophy. Rather than introducing a single organism with a single function, this project engineers three phylogenetically distinct bacteria, a Pseudomonad, a Spirillum and a Firmicute, each assigned a specific, non-redundant functional role within the consortium: osmoprotection (P. fluorescens), nitrogen fixation (A. brasilense), and soil stabilization through biofilm (B. subtilis). This division of labor mirrors the functional redundancy and specialization observed in natural soil ecosystems but imposes synthetic genetic control over when and how each function is expressed. The use of salt-responsive promoters, P_algU in P. fluorescens and P_epsA in B. subtilis, ensures that the consortium activates its protective functions precisely when the plant needs them most.
Second, the integration of synthetic biology tools, including Gibson Assembly, codon optimization and auxotrophic kill switches into a multi-organism agricultural consortium represents an application of tools primarily developed for single-organism chassis systems. While Gibson Assembly, auxotrophic containment and salt-responsive promoters have each been used individually in synthetic biology contexts, their combined application across three different host organisms in a coordinated consortium designed for open-field agriculture is unprecedented. The auxotrophic kill switch design in particular addresses one of the most persistent barriers to the field deployment of engineered microorganisms which are biosafety and ecological containment. By requiring an external metabolite supplement (DAP, tryptophan or aromatic amino acids) for survival, each organism is intrinsically confined to controlled environments with a robust containment strategy that does not depend on active gene expression or environmental triggers.
Third, its long-term vision includes the use of controlled adaptive evolution under contained or semi-contained conditions to improve the environmental robustness of the synthetic consortium while maintaining biosafety safeguards. By integrating engineered functions with insights from native Altiplano microbiota, the project aims to develop biologically resilient systems that are both effective and ecologically responsible. In doing so, it expands synthetic biology beyond static engineered organisms toward more adaptive and environmentally integrated biotechnological platforms.
3. Explain why your project matters and what impact it could have. (Minimum 5 sentences.)
a. Examples of topics to discuss:
i. The problem addressed: What pressing real-world problem does your project attempt to solve?
ii. Importance of the problem: Why is this problem significant, or what critical barrier to progress in the field does it represent?
iii. Broader societal contribution: How could the outcomes of your project benefit society beyond the immediate research context?
iv. Advancement of knowledge or capability: How might the project improve scientific understanding, technical capability, or clinical practice within one or more fields?
v. Field-level change: If your aims are achieved, how could the concepts, methods, technologies, treatments, services, or preventative approaches used in this field of research change?
Soil salinization is one of the most pressing and underaddressed threats to global food security. Secondary salinization now threatens 20% of irrigated lands worldwide, with projections suggesting 50% of croplands may be affected by 2050 — driven by climate change, decreased precipitation, and poor irrigation practices (Butcher et al., 2016). The FAO’s first major global assessment of salt-affected soils in 50 years estimates that over 1,381 million hectares, 10.7% of total global land area, are currently affected with models indicating that increasing aridity could expand this to between 24 and 32% of total land surface, with the vast majority of aridification expected to occur in developing countries (FAO, 2024). In the Bolivian Altiplano specifically, this problem is not a future projection but a present reality. This remote endorheic region that suffers from the major problem of soil salinization, threatening the sustainability of agriculture activity (Hiernaux et al., 2024). The farming communities of the Altiplano depend on crops like quinua, fava bean, barley, cañahua, etc; as their primary food and income sources. This project directly targets this gap, not with a solution designed for industrial agriculture but with one designed for resource-limited, remote and extreme environments.
The critical barrier this project addresses is salinity itself and the absence of accessible, biological and deployable solutions for smallholder farmers in saline environments. Soil salinity stress is considered highly detrimental for agriculture because of its devastating effects on productivity and food security, in addition to having important ecological and socioeconomic repercussions, with salinity negatively affecting microbial diversity in the plant rhizosphere and limiting water conductance, soil porosity, and aeration (Ilyas et al., 2021).
Existing biofertilizer products on the market typically contain a single PGPR strain and are designed for temperate, well-irrigated agricultural systems and not for the extreme conditions of high-altitude endorheic basins like the Altiplano. A lyophilized, multi-functional microbial consortium that can be applied as a seed coating, without cold chain, without specialized equipment, represents a solution designed from the ground up for accessibility, scalability and ecological compatibility.
Beyond its agricultural application, this project has the potential to contribute to broader societal goals in several dimensions. Nitrogen fertilizer production and use account for approximately 5% of global greenhouse gas emissions and food security’s reliance on synthetic nitrogen fertilizers represents one of the most significant environmental costs of modern agriculture (Cassman & Dobermann, 2022). By providing biological nitrogen fixation through A. brasilense, this consortium could meaningfully reduce dependence on synthetic nitrogen fertilizers in Altiplano farming systems. A biological alternative that delivers fixed nitrogen directly to the rhizosphere could dramatically improve this efficiency. Furthermore, the lyophilized delivery format envisions a product that farming communities could store and apply themselves, potentially creating local biotechnology capacity in regions historically excluded from the benefits of the bioeconomy. PubMed CentralPubMed
In soil microbiology, it provides a framework for integrating genome-scale metabolic modeling (COBRApy, model iJN1411) and agent-based consortium simulation (BacArena) to predict the behavior of engineered microbial communities before committing to costly wet lab experiments. The use of multi-strain co-inoculation strategies has been shown to cause significant positive impacts on shoot length, root length, fresh weight and biochemical traits. Moreover, the compatibility of strains must be assessed prior to consortium development, a methodological consideration directly reflected in the BacArena simulation component of this project (Bukhat et al., 2021). The directed evolution component of the long-term vision further pushes the boundary of what is currently possible in agricultural biotechnology, proposing a rational, iterative approach to adapting synthetic consortia to specific ecological contexts.
Finally, research evaluating the compatibility of B. subtilis, P. fluorescens, and A. brasilense together has already proposed quantitative frameworks for predicting plant-PGPR pair performance, establishing a scientific foundation for the rational design of multi-organism consortia (Gerbore et al., 2024), and this project builds on that foundation by adding synthetic genetic control, environmental responsiveness and biosafety containment, moving PGPR science from empirical strain selection toward rational consortium engineering. Moreover, if a lyophilized synthetic consortium can be shown to improve crop resilience in one of the world’s most extreme agricultural environments, it opens the door to a new generation of biological products designed specifically for the frontlines of climate change, arid regions, saline soils and degraded lands, where the need is greatest and current solutions are most inadequate.
4. Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility). (Minimum 2 paragraphs.)
a. First paragraph: Include what ethical implications are involved in your project. Try to suggest ethical principle(s) you may apply (e.g. non-maleficence, justice)?
b. Second paragraph: Describe the measures that should be taken to ensure that your project is ethical (both in how the research is conducted and in its broader implications for society). You may wish to answer the following questions:
i. What action(s) do you propose?
ii. What are potential unintended consequences of your proposed actions?
iii. What could you have been wrong (e.g., incorrect assumptions and uncertainties)?
iv. What are alternatives to your proposed actions?
v. Note: in an NIH proposal, an ethics statement is used to describe the relevance of this research to public health.
This project sits at the intersection of synthetic biology, food security and environmental justice, each of these dimensions carries significant ethical weight. The deliberate engineering of microorganisms for open-environment deployment raises immediate concerns related to the principle of non-maleficence: the obligation to avoid causing harm.
Introducing genetically engineered bacteria into the rhizosphere of the Bolivian Altiplano, a complex, poorly characterized and ecologically fragile ecosystem carries the risk of unintended consequences, including competitive displacement of native soil microbiota, horizontal gene transfer of engineered constructs to non-target organisms and disruption of existing nitrogen cycling and soil microbiome dynamics that local crops have co-evolved with over centuries. The principle of justice is equally central to this project: the Bolivian Altiplano is home to Indigenous Quechua and Aymara farming communities whose agricultural systems, cultural identities and food sovereignty are deeply intertwined. Any biotechnology deployed in this context must be developed with, not for, these communities, ensuring that the benefits of the technology are equitably distributed and that local knowledge and consent are respected throughout the research process. Finally, the principle of responsibility demands transparency about what this technology can and cannot do: this project is an in silico design and its real-world efficacy remains unvalidated. Overstating its potential could mislead policymakers, funders or communities into premature adoption of an unproven system.
On the other hand, the measures proposed to ensure this project is conducted ethically operate at multiple levels. At the biosafety level, all three engineered organisms incorporate auxotrophic kill switches, requiring external metabolite supplementation for survival, providing a passive, robust containment mechanism that does not depend on active monitoring or enforcement. This directly addresses the risk of uncontrolled environmental persistence. At the research conduct level, any future experimental validation should be preceded by contained greenhouse studies, followed by small-scale field trials with continuous monitoring of soil microbial community composition using 16S rRNA sequencing, before any open-field deployment is considered. At the community engagement level, the project envisions the development of the lyophilized inoculant as an open-source, non-proprietary product, explicitly designed to prevent corporate capture of a technology developed for marginalized communities.
Potential unintended consequences that must be acknowledged include: the possibility that the synthetic consortium outcompetes native PGPR strains that local crops depend on; that the kill switch fails under unexpected environmental conditions; or that the directed evolution component introduces unforeseen traits into the hybrid consortium. Critically, this project assumes that salinity is the primary limiting factor for Altiplano agriculture, an assumption that may be incorrect, as water availability, frost, UV radiation and socioeconomic factors also constrain productivity. Alternative approaches that avoid genetic engineering entirely, such as metagenomics-guided enrichment of native salt-tolerant consortia or the development of biochar-based soil amendments should be evaluated in parallel as safer, more ecologically conservative options. Ultimately, the ethical imperative of this project is not just to do no harm but to actively advance the well-being of communities on the front lines of climate change, making beneficence not merely a principle but a design requirement.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
1. Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take). (min. 15 lines/sentences—a numbered list is acceptable)
a. Include specific methods/tools/technologies/biological concepts for each part of the final project and analysis.
b. This section will be used to determine whether the experiments are well designed, feasible, and likely to succeed in testing your hypothesis.
c. Often this section is broken into discrete tasks/sub-aims.
d. For each experiment and/or analysis, include a description of your expected results.
e. If possible, include figure(s) that visually show a broad workflow of your project or a specific aspect of your experimental plan.
f. Reminder: All HTGAA projects must include some DNA design!
1. In silico genetic circuit design - P. fluorescens (Days 1-2, ~5 hours)
Design of the osmoprotection circuit in Benchling using a synthetic AlgU-responsive promoter (P_algU, based on Firoved & Deretic 2003 consensus: -35 box GAACTT, 16 nt spacer, -10 box TCTGA, 31 bp total), betA (choline dehydrogenase, 1,671 bp) and betB (betaine aldehyde dehydrogenase, 1,425 bp) coding sequences from E. coli K-12 (NCBI accession X52905), RBS BBa_B0034 (12 bp, iGEM Parts Registry), and terminator BBa_B0015 (129 bp, iGEM Parts Registry), assembled into the broad host-range backbone pBBR1MCS2 (Addgene #26702, KanR, 5,148 bp) via Gibson Assembly with 40 bp overlaps.
Expected result: complete annotated plasmid map in Benchling with status “Ready to assemble.”
2. Codon optimization of betA and betB (Day 2)
Codon optimization of both heterologous genes for P. fluorescens using the IDT Codon Optimization Tool, with P. aeruginosa as reference organism due to shared GC content (~60-63%) and codon usage preferences. Both genes were re-uploaded to Benchling replacing the original E. coli sequences.
Expected result: optimized sequences replacing rare codons with Pseudomonas-preferred synonymous alternatives, improving translation efficiency without altering the amino acid sequence or enzymatic function of BetA and BetB.
3. Auxotrophic kill switch - P. fluorescens (Day 2)
Design of biosafety containment for P. fluorescens through deletion of dapA (4-hydroxy-tetrahydrodipicolinate synthase, 465 bp, locus tag CPH89_RS26560), an essential gene for diaminopimelate (DAP) biosynthesis and cell wall formation. Without DAP, P. fluorescens cannot synthesize peptidoglycan and undergoes cell lysis. Documented in Benchling with dual annotations: CDS (wildtype) and Misc Feature (DELETION TARGET). In wet lab, deletion would be performed using lambda Red recombination or CRISPR/Cas9.
Expected result: passive containment because bacteria cannot survive in natural Altiplano soil where free DAP is unavailable.
4. In silico genetic circuit design - B. subtilis (Days 2-3, ~5 hours)
Design of the biofilm circuit in Benchling using the native P_epsA promoter (NCBI NC_000964, complement region 3529856-3530056, 201 bp), which responds to salt stress through the Spo0A/SinR regulatory cascade, epsA (BSU_34370, 705 bp, modulator of EpsB kinase) and tapA (BSU_34400, 762 bp, TasA anchoring protein essential for biofilm matrix assembly) from B. subtilis 168, assembled into pBP_Pveg backbone (Addgene #112776, CmR, 2,204 bp) via Gibson Assembly with 40 bp overlaps. Both genes were located on the complementary strand of NC_000964 and reverse complemented using a custom Python script to obtain correct 5’ to 3’ coding sequences.
Expected result: complete B. subtilis plasmid map with status “Assembled.”
5. Auxotrophic kill switch - B. subtilis (Day 3)
Design of biosafety containment for B. subtilis through deletion of trpC (indole-3-glycerol phosphate synthase, 750 bp, locus tag BSU_24310) — an essential gene for tryptophan biosynthesis. Without tryptophan, B. subtilis cannot synthesize essential proteins and dies. Documented in Benchling with dual annotations: CDS (wildtype) and Misc Feature (DELETION TARGET).
Expected result: passive containment because bacteria cannot survive in natural Altiplano soil where free tryptophan is unavailable in sufficient concentrations.
6. In silico genetic circuit design - A. brasilense (Day 4, ~5 hours)
Design of the nitrogen fixation circuit using the native P_nifH promoter (~200 bp upstream of nifH, sigma-54 and NifA-dependent) and nifHDK operon from A. brasilense Sp7 (NCBI GCF_008274965.1): nifH (nitrogenase reductase, 882 bp), nifD (nitrogenase alpha subunit, 1,440 bp), and nifK (nitrogenase beta subunit). All three genes form a polycistronic operon controlled by a single P_nifH promoter. Since A. brasilense is a native diazotroph, FeMo cofactor biosynthesis genes (nifB, nifE, nifN) are already present in the chromosome and there is no need to include them in the plasmid. Assembled into pBBR1MCS2 via Gibson Assembly.
Expected result: complete A. brasilense plasmid map with status “Assembled.”
7. Auxotrophic kill switch - A. brasilense (Day 4, integrated)
Design of biosafety containment for A. brasilense through deletion of aroA (3-phosphoshikimate 1-carboxyvinyltransferase), an essential gene for biosynthesis of all three aromatic amino acids: phenylalanine, tyrosine, and tryptophan. Without aromatic amino acids, A. brasilense cannot synthesize essential proteins and structural components. Documented in Benchling with dual annotations: CDS (wildtype) and Misc Feature (DELETION TARGET).
Expected result: passive containment because bacteria cannot survive in natural Altiplano soil where free aromatic amino acids are unavailable.
Flux Balance Analysis (FBA) performed using COBRApy (Ebrahim et al., 2013) in Google Colab, with the P. putida KT2440 model iJN746 (Nogales et al., 2008) as a metabolic proxy for P. fluorescens, justified by shared Pseudomonas genus and conserved central metabolic pathways. Choline uptake (EX_chol_e) was set to -10 mmol/gDW/h as carbon substrate. CHOLD (betA) and BETALDHx (betB) were assigned minimum flux constraints of 1.0 mmol/gDW/h to simulate constitutive P_algU-driven expression under salt stress. Salinity stress was modeled by restricting the biomass reaction (BiomassKT_TEMP) to 100%, 70%, and 40% of maximum growth rate, corresponding to 0, 75, and 150 mM NaCl respectively. Glycine betaine export (EX_glyb_e) and transport reactions (GLYBtex, GLYBabcpp) were opened to allow secretion.
Expected result: glycine betaine production sustained at 10 mmol/gDW/h across all salinity conditions, with growth rate decreasing from 2.64 h⁻¹ (normal) to 1.47 h⁻¹ (severe stress), confirming that the betA/betB pathway is metabolically feasible in Pseudomonas without causing metabolic collapse.
Agent-based metabolic simulation of P. fluorescens and B. subtilis co-culture using BacArena in R, with genome-scale metabolic models iJN746 (P. putida proxy) and iBB1018 (B. subtilis 168) respectively. A. brasilense was excluded due to the absence of a validated genome-scale metabolic model in any public database (BiGG, BioModels). Simulation will assess inter-species metabolite exchange, competitive exclusion risk and consortium stability under saline conditions.
Expected result: confirmation of metabolic compatibility between P. fluorescens and B. subtilis with no predicted competitive inhibition, supporting the feasibility of the three-organism consortium.
10. Future wet lab - DNA synthesis and transformation (Weeks 1-2)
Order codon-optimized constructs from Twist Bioscience. Transform P. fluorescens and A. brasilense by electroporation (2.5 kV, 25 µF, 200 Ω) and B. subtilis by competent cell transformation using the standard two-step starvation protocol. Select transformants on LB agar with appropriate antibiotics: kanamycin 50 µg/mL (P. fluorescens and A. brasilense) and chloramphenicol 5 µg/mL (B. subtilis). Confirm transformation by colony PCR using construct-specific primers, visualized by 1% agarose gel electrophoresis.
Expected result: antibiotic-resistant colonies confirmed by band of expected size.
Grow each transformed strain in liquid media supplemented with 0, 75, and 150 mM NaCl. Measure functional outputs: glycine betaine production by HPLC or colorimetric assay (P. fluorescens), nitrogenase activity by Acetylene Reduction Assay (ARA) — measuring ethylene production as a proxy for N₂ fixation (A. brasilense), and biofilm formation by crystal violet staining quantified at OD₅₉₀ (B. subtilis). Each condition performed in biological triplicates.
Expected result: statistically significant increase (p < 0.05, Student’s t-test) in each functional output under salt stress compared to non-transformed wild-type controls.
CFU assays for each strain grown in media with and without supplementation of DAP (0.3 mM, P. fluorescens), tryptophan (0.5 mM, B. subtilis), and aromatic amino acids (0.5 mM each, A. brasilense). Plate on LB agar after 24h growth and count colonies. Perform live/dead staining (SYTO 9 / propidium iodide) as secondary validation.
Expected result: complete growth inhibition (0 CFU/mL) without metabolite supplementation in all three strains, confirming kill switch functionality and biosafety containment.
Co-culture the three engineered strains at optimized ratios (1:1:1 initial, then optimized based on individual strain data). Lyophilize consortium and apply as seed coating to Arabidopsis thaliana Col-0 seeds. Grow on 0.5x MS agar plates with 0 and 150 mM NaCl for 14 days, then transfer to soil for 21 days. Measure germination rate, rosette diameter, number of leaves, fresh and dry biomass, and chlorophyll fluorescence (Fv/Fm ratio by pulse amplitude modulation fluorometry). Compare consortium-inoculated plants vs non-inoculated controls.
Expected result: statistically significant improvement (p < 0.05) in all plant performance metrics under saline conditions in inoculated plants.
15. Future wet lab - Microbial community monitoring (Weeks 6-8)
Extract DNA from rhizosphere soil of inoculated and non-inoculated A. thaliana plants. Perform 16S rRNA amplicon sequencing (V3-V4 region) using Illumina MiSeq platform. Analyze community composition using QIIME2 pipeline.
Expected result: all three engineered strains detectable and stable in rhizosphere of inoculated plants throughout the experiment, with no significant displacement of native microbiota composition.
Translate optimal microbial ratios and NaCl screening conditions identified in plant inoculation experiments into Opentrons OT-2 Python liquid handling protocols for high-throughput combinatorial screening of consortium performance in 96-well plates. Measure OD₆₀₀ and functional outputs automatically.
Expected result: automated, reproducible preparation of consortium inoculants at multiple microbial ratios, eliminating pipetting error and enabling rapid optimization of consortium composition.
fromopentronsimportprotocol_apimetadata={'protocolName':'HTGAA2026 - Rhizosphere Consortium Screening','author':'Ian Sebastian Teran Garcia','description':'''Combinatorial screening of synthetic rhizosphere consortium
(P. fluorescens + A. brasilense + B. subtilis) at multiple
microbial ratios under NaCl salinity stress conditions.
Bolivian Altiplano Project - HTGAA 2026''','apiLevel':'2.13'}defrun(protocol:protocol_api.ProtocolContext):# ── LABWARE ──────────────────────────────────────────────────tiprack_1=protocol.load_labware('opentrons_96_tiprack_300ul',1)tiprack_2=protocol.load_labware('opentrons_96_tiprack_300ul',2)# Bacterial stocks — each organism in separate tubetube_rack=protocol.load_labware('opentrons_24_tuberack_eppendorf_2ml_safelock_snapcap',3)# NaCl solutions — different concentrationsreservoir=protocol.load_labware('nest_12_reservoir_15ml',4)# 96-well plate for screeningplate=protocol.load_labware('nest_96_wellplate_200ul_flat',5)# Pipettesp300_multi=protocol.load_instrument('p300_multi_gen2','left',tip_racks=[tiprack_1])p300_single=protocol.load_instrument('p300_single_gen2','right',tip_racks=[tiprack_2])# ── REAGENTS ─────────────────────────────────────────────────# Tube rack positionsPf_stock=tube_rack['A1']# P. fluorescens — OD600 = 1.0Ab_stock=tube_rack['A2']# A. brasilense — OD600 = 1.0Bs_stock=tube_rack['A3']# B. subtilis — OD600 = 1.0media=tube_rack['A4']# LB media base# Reservoir — NaCl solutionsNaCl_0mM=reservoir['A1']# 0 mM NaCl — normal conditionsNaCl_75mM=reservoir['A2']# 75 mM NaCl — moderate stressNaCl_150mM=reservoir['A3']# 150 mM NaCl — severe stress# ── PLATE LAYOUT ─────────────────────────────────────────────# Columns 1-4: 0 mM NaCl# Columns 5-8: 75 mM NaCl# Columns 9-12: 150 mM NaCl# # Row A: Pf only (100% Pf, 0% Ab, 0% Bs)# Row B: Ab only (0% Pf, 100% Ab, 0% Bs)# Row C: Bs only (0% Pf, 0% Ab, 100% Bs)# Row D: Pf + Ab (50:50)# Row E: Pf + Bs (50:50)# Row F: Ab + Bs (50:50)# Row G: Full consortium 1:1:1# Row H: Full consortium 2:1:1 (Pf dominant)# ── BACTERIAL RATIOS (µL per well, total volume = 200 µL) ────ratios={'A':{'Pf':100,'Ab':0,'Bs':0},# Pf only'B':{'Pf':0,'Ab':100,'Bs':0},# Ab only'C':{'Pf':0,'Ab':0,'Bs':100},# Bs only'D':{'Pf':50,'Ab':50,'Bs':0},# Pf + Ab'E':{'Pf':50,'Ab':0,'Bs':50},# Pf + Bs'F':{'Pf':0,'Ab':50,'Bs':50},# Ab + Bs'G':{'Pf':33,'Ab':33,'Bs':34},# Full 1:1:1'H':{'Pf':67,'Ab':17,'Bs':16},# Full 2:1:1}nacl_cols={'0mM':[1,2,3,4],'75mM':[5,6,7,8],'150mM':[9,10,11,12]}nacl_sources={'0mM':NaCl_0mM,'75mM':NaCl_75mM,'150mM':NaCl_150mM}# ── STEP 1: ADD NaCl MEDIA TO ALL WELLS ──────────────────────protocol.comment("Step 1: Dispensing NaCl media to all wells")forcondition,colsinnacl_cols.items():source=nacl_sources[condition]forcolincols:p300_multi.pick_up_tip()p300_multi.aspirate(100,source)p300_multi.dispense(100,plate.columns()[col-1][0])p300_multi.drop_tip()# ── STEP 2: ADD BACTERIA AT SPECIFIED RATIOS ─────────────────protocol.comment("Step 2: Adding bacterial strains at specified ratios")rows=['A','B','C','D','E','F','G','H']bacteria_sources={'Pf':Pf_stock,'Ab':Ab_stock,'Bs':Bs_stock}forrowinrows:ratio=ratios[row]forbacteria,volumeinratio.items():ifvolume>0:source=bacteria_sources[bacteria]forcolinrange(1,13):well=plate.wells_by_name()[f'{row}{col}']p300_single.pick_up_tip()p300_single.aspirate(volume,source)p300_single.dispense(volume,well)p300_single.mix(3,50,well)# mix 3x after dispensingp300_single.drop_tip()# ── STEP 3: SEAL AND INCUBATE ─────────────────────────────────protocol.comment("""
Step 3: Protocol complete.
Seal plate with breathable membrane.
Incubate at 28°C for 48 hours with shaking at 200 rpm.
Measure OD600 at 0h, 24h, and 48h.
After incubation, perform:
- Crystal violet staining for biofilm (B. subtilis)
- Colorimetric assay for glycine betaine (P. fluorescens)
- Acetylene Reduction Assay for N2 fixation (A. brasilense)
""")
Opentrons OT-2 - Plate Design
The 96-well plate is organized along two axes, rows and columns, creating a fully factorial experimental design that simultaneously tests all microbial combinations across all salinity conditions in a single automated run.
Columns - Salinity Conditions (the environmental variable)
The 12 columns are divided into three groups of four replicates each, representing the three salinity conditions used throughout this project:
Columns 1-4: 0 mM NaCl - normal growth conditions, used as baseline control
Columns 5-8: 75 mM NaCl - moderate salinity stress, equivalent to early-stage soil salinization
Columns 9-12: 150 mM NaCl - severe salinity stress, representative of Bolivian Altiplano conditions
Having four replicates per condition ensures statistical robustness and allows detection of well-to-well variability.
Rows - Microbial Combinations (the biological variable)
The 8 rows test every possible combination of the three organisms, from individual strains to the full consortium, allowing direct comparison of single-strain vs multi-strain performance.
Row A:P. fluorescens only (100%) - isolates osmoprotection contribution
Row B:A. brasilense only (100%) - isolates nitrogen fixation contribution
Row C:B. subtilis only (100%) - isolates biofilm formation contribution
Collect rhizosphere soil samples from native quinua (Chenopodium quinoa) fields across multiple sites in the Bolivian Altiplano, specifically targeting soils with documented high salinity (>150 mM NaCl equivalent). Create a native microbiome library by culturing samples under progressive saline stress conditions (75, 150, 300 mM NaCl) to enrich for naturally salt-tolerant PGPR. Co-culture this native microbiome library with the synthetic consortium in iterative selection cycles of 4-6 weeks each — selecting combinations that maximize quinua germination rate, root biomass, and nitrogen content under saline stress. After each iteration, characterize the winning microbial combinations by 16S rRNA amplicon sequencing and whole genome sequencing to identify which native strains integrated successfully with the engineered consortium. Repeat for a minimum of 4 iterations until consortium performance stabilizes.
Expected result: a hybrid consortium combining the precision of the engineered strains with the ecological robustness of naturally Altiplano-adapted microorganisms, outperforming the synthetic-only consortium in quinua growth metrics.
18. Future long-term - Field translation and lyophilized product development (Year 3)
Test the optimized hybrid consortium on quinua under greenhouse conditions mimicking the Bolivian Altiplano environment: altitude equivalent pressure, high UV radiation, temperature fluctuations between -5°C and 20°C, and saline soil substrate collected directly from the Altiplano. Optimize lyophilization protocol, including cryoprotectant selection (trehalose, skim milk) and storage conditions to maximize consortium viability after freeze-drying. Formulate final product as a lyophilized seed coating applicable by hand without specialized equipment. Conduct small-scale open field trials in collaboration with local Altiplano farming communities, with continuous monitoring of soil microbial composition (16S rRNA), plant performance, and consortium persistence over one full growing season.
Expected result: field-ready lyophilized microbial inoculant with demonstrated efficacy under real Altiplano conditions, shelf-stable for at least 12 months without refrigeration and deployable directly by local farmers as a seed coating.
2. We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
DNA Sequencing (future validation — Sanger sequencing of constructs)
DNA Editing (kill switch deletions via CRISPR or recombination)
Databases (NCBI, GenBank, iGEM Parts Registry, BiGG, BioModels)
Lab Automation (Opentrons OT-2 protocol design)
Creating Code for Laboratory Automation (Opentrons Python protocol)
Using Liquid Handling Robots (Opentrons OT-2 - 96-well consortium screening)
Designing a Twist Order (codon-optimized constructs for future synthesis)
Use of Benchling (primary platform for all circuit design)
Models and Notebooks (COBRApy FBA + BacArena consortium simulation)
Chassis Selection (P. fluorescens, A. brasilense, B. subtilis)
Registry of Standard Biological Parts (BBa_B0034, BBa_B0015)
Plasmid Preparation (future - pBBR1MCS2 and pBP_Pveg)
Bacterial Culturing (future - transformation and validation)
Gibson Assembly (all three constructs assembled via Gibson)
CRISPR/Cas9 (future - kill switch gene deletions)
Gel Electrophoresis (future - colony PCR verification)
Primer Design or Selection (future - construct verification primers)
PCR Reactions (future - colony PCR confirmation)
1. Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project. (min. 4 sentences)
2. Identify any How To Grow (Almost) Anything Industry Council companies which are associated with your final project (optional).
Techniques - Expanded Description
1. Use of Benchling
Benchling served as the primary platform for all genetic circuit design in this project. For each of the three organisms, Pseudomonas fluorescens, Azospirillum brasilense and Bacillus subtilis, individual genetic parts were imported from NCBI and the iGEM Parts Registry, annotated with their functional roles (promoter, RBS, CDS, terminator) and assembled into complete circular plasmid constructs using the Gibson Assembly workflow. The plasmid map visualization tool was used to verify part order, topology, overlap regions, and annotation colors, creating a complete and reproducible digital record of the consortium’s genetic architecture.
Additionally, Benchling Notebook was used to document all design decisions, part sources, circuit logic, kill switch rationale, and measurable outcomes for each organism, providing a structured experimental record that mirrors the documentation standards of professional biotechnology research :))
2. Gibson Assembly
Gibson Assembly was selected as the assembly method for all three constructs due to its flexibility with multi-fragment assemblies, compatibility with any DNA sequence regardless of restriction sites and high efficiency for constructs in the 5-12 kb range. For each construct, 40 bp overlaps were designed between adjacent fragments, P_algU promoter, RBS, coding sequences, terminator and backbone. They were verified using the Benchling Assembly Wizard, which automatically calculated overlap regions and confirmed construct integrity with “Ready to assemble” or “Assembled” status. In future wet lab validation, Gibson Assembly reactions will be performed using the NEB HiFi Assembly Master Mix at 50°C for 60 minutes, followed by transformation into competent cells and antibiotic selection. Constructs will be confirmed by Sanger sequencing prior to transformation into the final host organisms, P. fluorescens, A. brasilense and B. subtilis, ensuring sequence accuracy before any functional validation experiments.
Industry Council Connections
Addgene: Source of expression backbones used in all three constructs: pBBR1MCS2 (#26702) for P. fluorescens and A. brasilense, and pBP_Pveg (#112776) for B. subtilis
Twist Biosciences: Planned DNA synthesis partner for codon-optimized betA and betB constructs for future wet lab validation
Opentrons: Opentrons OT-2 liquid handling robot protocols were designed for high-throughput combinatorial screening of consortium performance under multiple NaCl concentrations in 96-well plates.
Ginkgo Bioworks: Potential partner for scale-up fermentation, consortium production, and autonomous lab validation of engineered strains
Biome Consortia: Direct alignment with the project’s core concept of engineering microbial consortia for agricultural and environmental applications
New England Biolabs: Planned supplier of NEB HiFi Assembly Master Mix for future Gibson Assembly reactions and Q5 polymerase for colony PCR verification
Thermo Fisher Scientific: Planned supplier of reagents for HPLC glycine betaine quantification, crystal violet biofilm staining, and live/dead staining for kill switch validation
SecureDNA: Relevant to the biosafety and environmental containment framework of this project, particularly the kill switch design and GMO release protocols
Waters Corporation: HPLC instrumentation for future quantification of glycine betaine production by engineered P. fluorescens.
SECTION 5: Results & Quantitative Expectations
1. You are required to validate at least one aspect of your final project aims. This is to ensure that you are able to successfully apply a relevant synthetic biology technique to your project.
Include figures if you have them—accuracy is critical in figures, tables, and graphs
Here is a non-exhaustive list of acceptable validations:
1. Designing DNA relevant to your final project.
2. Performing a PCR reaction using primers relevant to your final project.
3. Performing a Gibson assembly relevant to your final project.
4. Creating and performing a cell-free assay related to your final project.
5. Creating and running code to validate an aspect of your final project.
6. Developing a model or completing a computational analysis relevant to your project.
7. Designing DNA construct(s) that can express at least one gene of interest, ordering it (via Twist), and testing expression of the construct(s) (potentially using an Opentrons robot).
1. What aspect of your final project did you choose to validate? (min. 2 sentences)
To anyone, feel free to check the genetics constructs on Benchling :D:
This project validated two complementary aspects of the synthetic rhizosphere consortium design. First, the complete in silico genetic circuit design for all three organisms, Pseudomonas fluorescens, Azospirillum brasilense and Bacillus subtilis, was validated through Gibson Assembly simulation in Benchling, confirming that all genetic parts are correctly ordered, properly annotated, and have sufficient 40 bp overlap regions for successful assembly.
Second, the metabolic feasibility of glycine betaine biosynthesis in Pseudomonas was computationally validated using Flux Balance Analysis (FBA) in COBRApy, demonstrating that the engineered betA/betB pathway can sustain production of 10 mmol/gDW/h of glycine betaine across all salinity conditions tested (0, 75, and 150 mM NaCl) without causing metabolic collapse, even as growth rate decreased by 44% under severe saline stress. Together,
These two validations confirm both the structural integrity of the genetic constructs and the metabolic viability of the osmoprotection strategy, providing a strong computational foundation for future experimental validation.
Genetic Constructs
Construct
Size (bp)
Backbone
Kill Switch
Gibson_Pf_OsmoProtect_KillSwitch_v1
9328
pBBR1MCS2
ΔdapA
Gibson_Bs_Biofilm_KillSwitch_v1
4788
pBP_Pveg
ΔtrpC
Gibson_Ab_NitrogenFixation_KillSwitch_v1
10735
pBBR1MCS2
ΔaroA
Genetic Parts Summary
Part
Organism
Function
Source
Size
P_algU synthetic
P. fluorescens
Salt-inducible promoter
Synthetic (Firoved & Deretic 2003)
31 bp
betA
P. fluorescens
Choline dehydrogenase
NCBI E. coli K-12, X52905
1,671 bp
betB
P. fluorescens
Betaine aldehyde dehydrogenase
NCBI E. coli K-12, X52905
1,425 bp
ΔdapA
P. fluorescens
Kill switch-DAP auxotrophy
NCBI P. fluorescens ATCC13525
465 bp
P_epsA native
B. subtilis
Salt-responsive promoter
NCBI NC_000964
201 bp
epsA
B. subtilis
EpsB kinase modulator
NCBI NC_000964, BSU_34370
705 bp
tapA
B. subtilis
Biofilm matrix protein
NCBI NC_000964, BSU_34400
762 bp
ΔtrpC
B. subtilis
Kill switch-Trp auxotrophy
NCBI NC_000964, BSU_24310
750 bp
P_nifH native
A. brasilense
Nitrogen-responsive promoter
NCBI GCF_008274965.1
200 bp
nifH
A. brasilense
Nitrogenase reductase
NCBI GCF_008274965.1
882 bp
nifD
A. brasilense
Nitrogenase alpha subunit
NCBI GCF_008274965.1
1,440 bp
ΔaroA
A. brasilense
Kill switch-aromatic aa auxotrophy
NCBI GCF_008274965.1
1338 bp
BBa_B0034
All
Ribosome binding site
iGEM Parts Registry
12 bp
BBa_B0015
All
Transcriptional terminator
iGEM Parts Registry
129 bp
Pseudomonas fluorescens:
Bacillus subtilis:
Azospirillum brasilense:
2. Write down a detailed protocol of how you validated this aspect of your final project. (Numbered list or paragraph is fine)
Validation Protocol
Part 1 - Genetic Circuit Design and Gibson Assembly Validation in Benchling
Created a new project in Benchling named HTGAA2026_RhizosphereConsortium with eight folders: 01_Pf_Osmoprotectant, 02_Pf_KillSwitch, 03_Registry_Parts, 04_Notebook, 05_Bs_Biofilm, 06_Bs_KillSwitch, 07_Ab_NitrogenFixation and 08_Ab_KillSwitch.
Imported all genetic parts into the Registry folder as individual DNA sequences:
BBa_B0034 (RBS, 12 bp) and BBa_B0015 (terminator, 129 bp) from the iGEM Parts Registry
betA (1,671 bp) and betB (1,425 bp) from NCBI accession X52905 (E. coli K-12)
epsA (705 bp) and tapA (762 bp) from NCBI NC_000964 (B. subtilis 168)
nifH (882 bp), nifD (1,440 bp), and nifK from NCBI GCF_008274965.1 (A. brasilense Sp7)
Backbone pBBR1MCS2 (5,148 bp) from Addgene #26702
Backbone pBP_Pveg (2,204 bp) from Addgene #112776
For all genes located on the complementary strand of their respective genomes (epsA, tapA, nifH, nifD, nifK), performed reverse complementation using a custom Python script to obtain the correct 5’ to 3’ coding sequences before importing into Benchling.
Designed a synthetic AlgU-responsive promoter for P. fluorescens (31 bp) based on the published consensus sequence from Firoved & Deretic (2003): -35 box GAACTT, 16 nt spacer, -10 box TCTGA. Extracted the native P_epsA promoter (201 bp) from the region complement(3529856..3530056) of NC_000964, and the native P_nifH promoter (~200 bp upstream of nifH) from GCF_008274965.1.
Performed codon optimization of betA and betB for Pseudomonas using the IDT Codon Optimization Tool with P. aeruginosa as reference organism. Updated sequences in Benchling replacing original E. coli sequences.
Assembled each construct as a new DNA sequence in its respective folder, concatenating parts in the correct order:
Annotated each part within the construct with its functional type (Promoter, RBS, CDS, Terminator) and color-coded for visualization.
Designed kill switch modules for each organism:
P. fluorescens: ΔdapA (465 bp, CPH89_RS26560) - DAP auxotrophy
B. subtilis: ΔtrpC (750 bp, BSU_24310) - tryptophan auxotrophy
A. brasilense: ΔaroA - aromatic amino acid auxotrophy
Each documented with dual annotations: CDS (wildtype) and Misc Feature (DELETION TARGET)
Configured Gibson Assembly for each construct in Benchling using the Assembly Wizard:
Added backbone and insert fragments
Set overlap length to 40 bp
Clicked Autopopulate to calculate overlaps automatically
Verified “Ready to assemble” or “Assembled” status for all three constructs
Verified final plasmid maps in circular view, confirming correct part order, topology, and annotation for all three constructs.
Part 2 - Metabolic Feasibility Validation using COBRApy
Opened Google Colab and created a new notebook named HTGAA2026_COBRApy_Pf_GlycineBetaine.
Installed COBRApy using !pip install cobra -q and imported required libraries: cobra, requests, pandas, and matplotlib.
Downloaded the P. putida KT2440 genome-scale metabolic model iJN746 from BioModels (MODEL1507180068) and loaded it using cobra.io.read_sbml_model(). Used iJN746 as a metabolic proxy for P. fluorescens due to the absence of a validated P. fluorescens model in public databases, justified by shared Pseudomonas genus and conserved central metabolic pathways.
Explored the model to identify glycine betaine-related reactions, confirming the presence of:
CHOLD - choline dehydrogenase (equivalent to betA)
BETALDHx - betaine aldehyde dehydrogenase (equivalent to betB)
EX_glyb_e - glycine betaine export reaction
GLYBtex and GLYBabcpp - glycine betaine transport reactions
Configured the model for FBA simulation:
Set choline uptake EX_chol_e lower bound to -10 mmol/gDW/h
Set EX_chol_e upper bound to 0 (uptake only)
Opened glycine betaine export and transport reactions
Set minimum flux constraints of 1.0 mmol/gDW/h on CHOLD and BETALDHx to simulate P_algU-driven expression
Set objective function to maximize EX_glyb_e (glycine betaine export)
Ran FBA under three salinity conditions by restricting the biomass reaction (BiomassKT_TEMP) to simulate growth inhibition:
0 mM NaCl: 100% maximum growth rate (3.6794 h⁻¹)
75 mM NaCl: 70% maximum growth rate
150 mM NaCl: 40% maximum growth rate
Recorded glycine betaine production, growth rate, and solver status for each condition.
Generated a two-panel bar chart using matplotlib showing glycine betaine production and growth rate across all three salinity conditions, saved as FBA_glycine_betaine_salinity.png.
Interpreted results and documented findings in Google Colab notebook, linking metabolic feasibility to the Benchling circuit design.
3. What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8. (min. 4 sentences)
The primary technique utilized in this project was DNA construct design in Benchling, the industry-standard platform for genetic circuit design used by leading synthetic biology companies including Ginkgo Bioworks and Twist Bioscience. All three genetic circuits were designed from scratch using sequences retrieved from databases, specifically NCBI GenBank for coding sequences (betA, betB, epsA, tapA, nifH, nifD, nifK) and the Registry of Standard Biological Parts (iGEM) for standardized RBS BBa_B0034 and terminator BBa_B0015, demonstrating the ability to navigate and extract relevant biological information from multiple public repositories simultaneously.
Gibson Assembly was used as the assembly strategy for all three constructs, with 40 bp overlaps calculated and verified through the Benchling Assembly Wizard, a technique that requires understanding of DNA homology, exonuclease activity and fragment design principles that were directly applied in the design of each construct. Models and notebooks were central to the computational validation component: COBRApy was used to perform Flux Balance Analysis (FBA) on a genome-scale metabolic model, requiring knowledge of constraint-based metabolic modeling, objective function definition and the interpretation of flux distributions, skills that bridge synthetic biology with systems biology and metabolic engineering.
Additionally, bioethical considerations were integrated throughout the entire validation process, from the design of auxotrophic kill switches as biosafety containment mechanisms, to the explicit acknowledgment of the limitations of in silico validation and the need for staged experimental testing before any field deployment of the engineered consortium.
4. You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data (e.g., using the Asimov Kernel or another simulation method). (min.2 sentences)
The primary data presented in this project was generated computationally using Flux Balance Analysis (FBA) in COBRApy, a well-established simulation method in systems biology and metabolic engineering. The analysis produced quantitative flux data for glycine betaine production and growth rate across three salinity conditions (0, 75, and 150 mM NaCl), revealing that the engineered betA/betB pathway sustained maximum glycine betaine production of 10 mmol/gDW/h under all conditions tested, while growth rate decreased progressively from 2.64 h⁻¹ under normal conditions to 1.47 h⁻¹ under severe saline stress, a 44% reduction confirming the metabolic cost of salinity stress on Pseudomonas central metabolism.
These results were visualized as a two-panel bar chart comparing glycine betaine production and growth rate across all three conditions, providing clear graphical evidence that the betA/betB pathway is metabolically feasible without causing cellular collapse, directly validating the core design rationale of the P. fluorescens osmoprotection circuit designed in Benchling. The solver status remained “optimal” across all three conditions, which in FBA terms means the model successfully found a feasible solution satisfying all metabolic constraints simultaneously, confirming that glycine betaine biosynthesis can be sustained alongside core cellular functions including growth, energy generation and biosynthesis of essential metabolites.
Computational Validation: Can Pseudomonas Produce Glycine Betaine Under Salt Stress?
Objective:
The goal of this analysis was to determine whether Pseudomonas fluorescens has sufficient metabolic capacity to produce glycine betaine through the engineered betA/betB pathway under saline stress conditions, without compromising its own growth and survival.
Methods:
Flux Balance Analysis (FBA) was performed using COBRApy (Ebrahim et al., 2013) in Google Colab. Due to the absence of a validated genome-scale metabolic model for P. fluorescens in public databases, the closely related P. putida KT2440 model iJN746 was used as a metabolic proxy. Both organisms belong to the Pseudomonas genus and share highly conserved central metabolic pathways, making this a valid approximation for feasibility analysis.
Key modeling decisions:
Choline uptake was set to -10 mmol/gDW/h as the carbon substrate for glycine betaine biosynthesis.
CHOLD (betA) and BETALDHx (betB) were assigned minimum flux constraints to simulate constitutive expression driven by the synthetic P_algU promoter.
BHMT, the reaction that internally consumes glycine betaine was constrained to simulate osmoprotectant accumulation under stress.
Salinity stress was modeled by restricting the biomass reaction to 100%, 70%, and 40% of maximum growth rate, corresponding to 0, 75, and 150 mM NaCl respectively.
Results
Condition
Glycine Betaine (mmol/gDW/h)
Growth Rate (h⁻¹)
Status
0 mM NaCl (normal)
10.00
2.64
Optimal
75 mM NaCl (moderate)
10.00
2.58
Optimal
150 mM NaCl (severe)
10.00
1.47
Optimal
Limitations
FBA is a static optimization model and does not simulate gene regulation or dynamic stress responses. It cannot predict whether P_algU will activate under salt stress, that requires experimental validation.
iJN746 is a P. putida model, not P. fluorescens. While the central metabolic pathways are conserved, strain-specific differences may exist.
The minimum flux constraints on CHOLD and BETALDHx simulate promoter activation. This is a modeling assumption, not a direct measurement of gene expression.
Interpretation
The betA/betB pathway is metabolically feasible in Pseudomonas. The model remained optimal under all salinity conditions tested, meaning the bacterium can simultaneously grow and produce glycine betaine without metabolic collapse. This confirms that the heterologous expression of betA and betB from E. coli K-12 is compatible with Pseudomonas central metabolism.
Even under 150 mM NaCl, where growth rate dropped by 44% (from 2.64 to 1.47 h⁻¹), glycine betaine production remained at maximum capacity (10 mmol/gDW/h). This is consistent with the biological rationale of the circuit: P_algU activates betA/betB precisely when salt stress is highest.
The iJN746 model contains native CHOLD and BETALDHx reactions, confirming that Pseudomonas has the required cofactors, substrates, and thermodynamic conditions to support the betA/betB pathway, validating the circuit design in Benchling.
Step 2: Download and load the iJN746 metabolic model
This step downloads the iJN746 genome-scale metabolic model of Pseudomonas putida from BioModels and loads it into COBRApy using SBML format.
fromgoogle.colabimportfiles# Upload the SBML file downloaded manually from BioModelsuploaded=files.upload()# Replace with the exact uploaded filename if differentmodel_file="MODEL1507180068_urn.xml"# Load the SBML model into COBRApymodel=cobra.io.read_sbml_model(model_file)print("Model loaded successfully")print(f"Reactions: {len(model.reactions)}")print(f"Metabolites: {len(model.metabolites)}")print(f"Genes: {len(model.genes)}")
Step 3: Explore the baseline metabolic model
This step evaluates the basal growth rate of the model and identifies reactions associated with glycine betaine biosynthesis and osmoprotection pathways.
This step searches for the biomass reaction used by the model as the growth objective during Flux Balance Analysis.
# Search biomass reactionprint("Searching for biomass reaction:\n")forrxninmodel.reactions:if"biomass"inrxn.id.lower()or"biomass"inrxn.name.lower():print(f"ID: {rxn.id}")print(f"Name: {rxn.name}")print("---")
Step 5: Verify choline uptake and betA pathway activity
This step verifies whether choline can enter the metabolic network and evaluates the activity of the betA-associated reaction involved in glycine betaine biosynthesis.
withmodel:# Enable choline uptakemodel.reactions.get_by_id("EX_chol_e").lower_bound=-10model.reactions.get_by_id("EX_chol_e").upper_bound=0# Set biomass as objective functionmodel.objective="BiomassKT_TEMP"# Run optimizationsolution=model.optimize()print(f"Status: {solution.status}")print(f"Growth rate: {solution.fluxes['BiomassKT_TEMP']:.4f}")print(f"Flux EX_chol_e: {solution.fluxes['EX_chol_e']:.4f}")print(f"Flux CHOLD (betA): {solution.fluxes['CHOLD']:.4f}")
Step 6: Perform Flux Balance Analysis under salinity stress
This step simulates glycine betaine production under increasing salinity stress conditions by constraining biomass formation and activating the betA/betB pathway.
results=[]withmodel:# Base configurationmodel.reactions.get_by_id("EX_chol_e").lower_bound=-10model.reactions.get_by_id("EX_chol_e").upper_bound=0model.reactions.get_by_id("EX_glyb_e").lower_bound=0model.reactions.get_by_id("EX_glyb_e").upper_bound=999999model.reactions.get_by_id("GLYBtex").lower_bound=-999999model.reactions.get_by_id("GLYBabcpp").lower_bound=-999999model.reactions.get_by_id("BHMT").upper_bound=0# Simulate activation of betA and betB under P_algUmodel.reactions.get_by_id("CHOLD").lower_bound=1.0model.reactions.get_by_id("BETALDHx").lower_bound=1.0# Set glycine betaine export as objectivemodel.objective="EX_glyb_e"# Salinity conditionssalt_conditions=[("0 mM NaCl (normal)",1.0),("75 mM NaCl (moderate)",0.7),("150 mM NaCl (severe)",0.4),]base_growth=3.6794forcondition,fractioninsalt_conditions:# Constrain growth according to stress intensitymodel.reactions.get_by_id("BiomassKT_TEMP").upper_bound=base_growth*fraction# Optimize modelsolution=model.optimize()# Store resultsresults.append({"Condition":condition,"Glycine betaine (mmol/gDW/h)":round(solution.objective_value,4),"Growth rate (h⁻¹)":round(solution.fluxes.get("BiomassKT_TEMP",0),4),"betA flux":round(solution.fluxes.get("CHOLD",0),4),"betB flux":round(solution.fluxes.get("BETALDHx",0),4),"Status":solution.status})# Convert results into DataFramedf=pd.DataFrame(results)# Display resultsprint(df.to_string(index=False))
Step 7: Visualize FBA results under salinity stress
This step generates comparative plots showing glycine betaine production and growth rate under different NaCl stress conditions.
fig,(ax1,ax2)=plt.subplots(1,2,figsize=(12,5))fig.suptitle("FBA Analysis: Glycine Betaine Production in Pseudomonas\nunder Salinity Stress",fontsize=13,fontweight="bold")# Extract valuesconditions=[r["Condition"]forrinresults]glyb_vals=[r["Glycine betaine (mmol/gDW/h)"]forrinresults]growth_vals=[r["Growth rate (h⁻¹)"]forrinresults]colors=["#2ecc71","#f39c12","#e74c3c"]# Plot 1 — Glycine betaine productionbars1=ax1.bar(conditions,glyb_vals,color=colors,edgecolor="white",linewidth=1.5)ax1.set_title("Glycine Betaine Production\n(betA/betB pathway)",fontweight="bold")ax1.set_ylabel("mmol/gDW/h")ax1.set_ylim(0,max(glyb_vals)*1.3)forbar,valinzip(bars1,glyb_vals):ax1.text(bar.get_x()+bar.get_width()/2,bar.get_height()+0.1,f"{val:.2f}",ha="center",fontweight="bold")ax1.tick_params(axis="x",labelsize=8)# Plot 2 — Growth ratebars2=ax2.bar(conditions,growth_vals,color=colors,edgecolor="white",linewidth=1.5)ax2.set_title("Growth Rate under Salinity Stress",fontweight="bold")ax2.set_ylabel("h⁻¹")ax2.set_ylim(0,max(growth_vals)*1.3)forbar,valinzip(bars2,growth_vals):ax2.text(bar.get_x()+bar.get_width()/2,bar.get_height()+0.02,f"{val:.2f}",ha="center",fontweight="bold")ax2.tick_params(axis="x",labelsize=8)# Save figureplt.tight_layout()plt.savefig("FBA_glycine_betaine_salinity.png",dpi=150,bbox_inches="tight")plt.show()print("Figure saved")
2. Did you encounter any unexpected challenge(s) when performing your validation? If so, describe the challenge(s) and strategies to overcome it. If not, discuss potential problems, difficulties, limitations, and/or alternative strategies to overcome challenges in your final project. (min. 4 sentences).
1. Incorrect promoter selection for P. fluorescens - resolved
The initially proposed salt-responsive promoter for P. fluorescens was the E. coli osmY promoter (P_osmY), which was later identified as sigma-S dependent, a sigma factor not conserved in Pseudomonas. This was resolved by designing a synthetic AlgU-responsive promoter based on the published consensus sequence from Firoved & Deretic (2003), which uses the native Pseudomonas salt-stress sigma factor AlgU. This challenge highlighted the importance of verifying promoter-sigma factor compatibility across species before committing to a design.
2. Taxonomic reclassification of A. brasilense Sp245 - resolved
During genome retrieval, the initially targeted strain A. brasilense Sp245 was found to have been reclassified as Azospirillum baldaniorum sp, causing confusion in NCBI database searches. This was resolved by switching to A. brasilense Sp7 (NCBI GCF_008274965.1), which retains the original A. brasilense designation and has a well-characterized nif gene cluster. This is documented in the notebook as a design note to maintain transparency.
3. Complementary strand sequences - resolved
Multiple target genes, including epsA, tapA, nifH, nifD and nifK, were located on the complementary strand of their respective genomes, meaning the sequences retrieved from NCBI were in the reverse orientation. This was resolved by writing a custom Python script to perform reverse complementation of each sequence before importing into Benchling, ensuring all coding sequences were in the correct 5’ to 3’ orientation for expression.
4. Absence of P. fluorescens genome-scale metabolic model - partially resolved
No validated genome-scale metabolic model exists for P. fluorescens in any public database (BiGG, BioModels), which prevented direct FBA analysis of the target organism. This was partially resolved by using the P. putida KT2440 model iJN746 as a metabolic proxy, justified by shared Pseudomonas genus and conserved central metabolic pathways. However, this remains a limitation of the analysis, as strain-specific metabolic differences between P. putida and P. fluorescens cannot be fully excluded.
5. BiGG database server unavailability - resolved
During the COBRApy analysis, the BiGG database server (bigg.ucsd.edu) was unavailable and refused all connections, preventing direct model download. This was resolved by retrieving the iJN746 model from BioModels (MODEL1507180068) as an alternative source, which provided the same model in SBML format compatible with COBRApy.
6. Glycine betaine production initially returning zero flux - resolved
Initial FBA runs returned zero glycine betaine production despite choline being available as a substrate. Investigation revealed two issues: first, the choline exchange reaction (EX_chol_e) had incorrect bounds that only allowed export, not uptake; and second, the internal BHMT reaction was consuming all produced glycine betaine before it could be exported. These were resolved by correcting the EX_chol_e bounds to allow uptake, opening glycine betaine transport reactions, and setting minimum flux constraints on CHOLD and BETALDHx to simulate P_algU-driven expression — ultimately achieving the expected production of 10 mmol/gDW/h.
7. Backbone availability on Addgene - resolved
The initially planned backbone for B. subtilis (pHT01, Addgene #26861) was not available on Addgene at the time of design. This was resolved by identifying an alternative backbone from the BacilloFlex toolkit, pBP_Pveg (Addgene #112776), which is specifically designed for B. subtilis expression and is part of a well-validated modular assembly system.
SECTION 6: ADDITIONAL INFORMATION
12. List all references cited in this assignment (bullet-point list)
Aim 3
Chu, T. N., Tran, B. T. H., Van Bui, L., & Hoang, M. T. T. (2019). Plant growth-promoting rhizobacterium Pseudomonas PS01 induces salt tolerance in Arabidopsis thaliana. BMC Research Notes, 12, 1. https://doi.org/10.1186/s13104-019-4046-1
Chang, C.-Y., Osborne, M. L., Bajic, D., & Sanchez, A. (2020). Artificially selecting microbial communities using propagule strategies. Evolution, 74(11), 2392–2403. https://doi.org/10.1111/evo.14094
Chang, C.-Y., Vila, J. C. C., Bender, M., Li, R., Mankowski, M. C., Bassette, M., Borden, J., Golfier, S., Sanchez, P. G. L., Waymack, R., Zhu, X., Diaz-Colunga, J., Estrela, S., Rebolleda-Gomez, M., & Sanchez, A. (2021). Engineering complex communities by directed evolution. Nature Ecology & Evolution, 5(7), 1011–1023. https://doi.org/10.1038/s41559-021-01457-5 (NCBI) (PubMed)
Blouin, M., Karimi, B., Mathieu, J., & Lerch, T. Z. (2015). Levels and limits in artificial selection of communities. Ecology Letters, 18(10), 1040–1048. https://doi.org/10.1111/ele.12482
Guo, X., & Boedicker, J. Q. (2016). The contribution of high-order metabolic interactions to the global activity of a four-species microbial community. PLoS Computational Biology, 12(9), e1005079. https://doi.org/10.1371/journal.pcbi.1005079
Jochum, M. D., McWilliams, K. L., Pierson, E. A., & Jo, Y.-K. (2019). Host-mediated microbiome engineering of drought tolerance in the wheat rhizosphere. PLoS ONE, 14(12), e0225933. https://doi.org/10.1371/journal.pone.0225933
Mueller, U. G., Juenger, T. E., Kardish, M. R., Carlson, A. L., Burns, K. M., Edwards, J. A., Smith, C. C., Fang, C.-C., & Des Marais, D. L. (2021). Artificial selection on microbiomes to breed microbiomes that confer salt tolerance to plants. mSystems, 6(6), e01125-21. https://doi.org/10.1128/mSystems.01125-21 (nih) (PubMed)
Sánchez, A., Chang, C.-Y., Díaz-Colunga, J., Estrela, S., Rebolleda-Gómez, M., & Vila, J. C. C. (2021). Directed evolution of microbial communities. Annual Review of Biophysics, 50, 323–341. https://doi.org/10.1146/annurev-biophys-101220-072829
Sánchez-Gorostiaga, A., Bajic, D., Osborne, M. L., Poyatos, J. F., & Sanchez, A. (2019). High-order interactions distort the functional landscape of microbial consortia. PLoS Biology, 17(12), e3000550. https://doi.org/10.1371/journal.pbio.3000550
Salinity & Bolivian Altiplano
Hiernaux, P., et al. (2024). Complementarity of Sentinel-1 and Sentinel-2 data for soil salinity monitoring to support sustainable agriculture practices in the Central Bolivian Altiplano. Sustainability, 16(14), 6200. https://www.mdpi.com/2071-1050/16/14/6200
Bukhat, S., et al. (2021). Potential of plant growth promoting bacterial consortium for improving the growth and yield of wheat under saline conditions. Frontiers in Plant Science. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9557047/
Gerbore, J., et al. (2024). A “love match” score to compare root exudate attraction and feeding of Bacillus subtilis, Pseudomonas fluorescens, and - Azospirillum brasilense. Frontiers in Microbiology. https://pmc.ncbi.nlm.nih.gov/articles/PMC11456545/
Ilyas, N., et al. (2024). Plant growth-promoting bacteria (PGPB)-induced plant adaptations to stresses: an updated review. PeerJ. https://peerj.com/articles/17882/
Chu, T. N., Tran, B. T. H., Van Bui, L., & Hoang, M. T. T. (2019). Plant growth-promoting rhizobacterium Pseudomonas PS01 induces salt tolerance in Arabidopsis thaliana. BMC Research Notes, 12, 1. https://doi.org/10.1186/s13104-019-4046-1
Azospirillum brasilense
Tropaldi, L., et al. (2025). Azospirillum brasilense as a bioinoculant to alleviate the effects of salinity on quinoa seed germination. Plants, 14(24), 3829. https://www.mdpi.com/2223-7747/14/24/3829
Wisniewski-Dye, F., et al. (2011). Azospirillum genomes reveal transition of bacteria from aquatic to terrestrial environments. PLoS Genetics. https://doi.org/10.1371/journal.pgen.1002430
Prikryl, Z., et al. (2002). pBBR1-based vectors for monitoring Azospirillum-wheat interactions. PubMed 12084480.
Lamark, T., et al. (1991). DNA sequence of bet genes encoding enzymes for the osmoregulatory choline-glycine betaine pathway in Escherichia coli. Molecular Microbiology. https://doi.org/10.1111/j.1365-2958.1991.tb00748.x
Menegat, S., Ledo, A., & Tirado, R. (2022). Greenhouse gas emissions from global production and use of nitrogen synthetic fertilisers in agriculture. Scientific Reports. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9411506/
Ebrahim, A., Lerman, J. A., Palsson, B. Ø., & Hyduke, D. R. (2013). COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Systems Biology, 7, 74. https://doi.org/10.1186/1752-0509-7-74
Nogales, J., Palsson, B. Ø., & Thiele, I. (2008). A genome-scale metabolic reconstruction of Pseudomonas putida KT2440: iJN746 as a cell factory. BMC Systems Biology, 2, 79. https://doi.org/10.1186/1752-0509-2-79
Freilich, S., et al. (2011). BacArena: individual-based metabolic modelling of heterogeneous microbes in complex communities. PLoS Computational Biology. https://doi.org/10.1371/journal.pcbi.1002020
16S rRNA amplicon sequencing — microbial community monitoring
Genomics core facility
$400
Google Colab Pro - COBRApy + BacArena analysis
Google
$10/month
Equipment (if not available at host institution)
Item
Supplier
Estimated Cost
Opentrons OT-2 liquid handling robot
Opentrons
$10,000
Electroporator - bacterial transformation
Bio-Rad Gene Pulser
$3,000
Plate reader - OD600 + fluorescence
Thermo Fisher
$8,000
Benchtop centrifuge
Eppendorf
$2,500
-80°C freezer - strain storage
Thermo Fisher
$5,000
Budget Summary
Category
Estimated Cost
DNA Design & Synthesis
$660-960
Plasmids & Backbones
$150
Enzymes & Assembly
$235
Bacterial Strains
$720
Antibiotics & Supplements
$190
Culture Media & Consumables
$445
Validation Assays
$1,030
Plant Experiments
$565
Sequencing & Bioinformatics
$510
Total (reagents + consumables)
~$4,505-4,805 USD
Equipment (if needed)
~$28,500 USD
Total (with equipment)
~$33,000-33,300 USD
Note: Equipment costs assume no access to shared core facility infrastructure. Most academic institutions provide access to electroporators, plate readers, centrifuges, and growth chambers, reducing the total budget to approximately $4,500-4,800 USD for reagents and consumables only.
Thank you for reading my project! :)
To close, thank you to my HTGAA 2026 node in Quito, Ecuador and to all the TAs who helped make this happen. Thank you for opening the doors of synthetic biology to Latin America. We have so much to contribute and projects like this one are just the beginning.
XXOO Ian Teran
Group Final Project
PROJECT OBJECTIVE
Engineer the L protein of the MS2 phage to increase structural stability.
Disrupt or reduce its interaction with the bacterial chaperone DnaJ.
Preserve the C-terminal lysis domain to maintain lytic function.
Avoid mutations that interfere with structurally or evolutionarily coupled residues.
Phase 1: Mapping the DnaJ Interaction Interface
Since the exact binding interface between the L protein and DnaJ is unknown, the first step is to identify it computationally rather than introducing arbitrary mutations.
Use AlphaFold-Multimer to model the complex between L protein and DnaJ.
Generate multiple structural predictions and select the top-ranked models.
Identify consensus interface residues that consistently appear in the predicted binding interface.
Perform in silico alanine scanning of the N-terminal residues in the complex to determine which residues significantly contribute to binding energy (ΔΔG).
Analyze whether the N-terminal region resembles known DnaJ-binding motifs, typically hydrophobic residues flanked by basic amino acids.
This phase defines which residues are critical for interaction and should not be mutated randomly.
Phase 2: Targeted N-Terminal Redesign
Instead of deleting regions or performing extensive random substitutions, introduce controlled chemical modifications to disrupt interaction while preserving structural stability.
Focus on charge inversion strategies:
Basic residues (K, R) → Acidic residues (E, D)
Acidic residues (E, D) → Basic residues (K, R)
Disrupt hydrophobic interaction patches:
Hydrophobic residues (L, I, V, F) → Polar residues (S, T, N, Q)
Aromatic residues (F, Y, W) → Aliphatic or small residues
Generate a graded library of variants:
Minor charge modifications
Moderate interface perturbations
Strong hydrophobic disruption
This creates a Pareto front of variants balancing reduced DnaJ interaction and preserved protein stability.
Phase 3: Stability and Functional Filtering
To ensure that redesigned variants remain structurally viable and functionally relevant:
Use Rosetta or FoldX to calculate ΔΔG and verify that mutations do not destabilize the overall protein fold.
Confirm that mutations in the N-terminal region do not propagate structural stress toward the C-terminal lysis domain.
Identify residue pairs that co-evolved between the N-terminal and C-terminal regions.
Avoid mutating co-evolved residues independently to prevent functional disruption.
Evaluate aggregation propensity using tools such as Aggrescan3D to ensure that mutations do not create exposed hydrophobic patches leading to cytoplasmic aggregation.
Assess sequence plausibility using protein language models such as ESM to filter out unlikely or non-natural variants.
Key Limitations:
The DnaJ binding mode may be transient or dynamic, reducing AlphaFold-Multimer accuracy.
Protein language model scores do not guarantee in vivo functionality.
Intrinsically disordered regions may not be accurately modeled.
Computational predictions must ultimately be validated experimentally.
From WEEK 5 HW:
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
L-Protein Engineering | Option 1: Mutagenesis
STEP 1:
A multiple sequence alignment of homologous L-protein sequences was performed using Clustal Omega to identify conserved and variable regions across related bacteriophages. The alignment revealed that the transmembrane region, located in the C-terminal portion of the protein, is highly conserved, particularly in residues forming a hydrophobic helix (LVLIFLAIFLSKFTNQLLLSLL). This high level of conservation suggests a critical functional role in membrane insertion and pore formation during bacterial lysis. In contrast, the N-terminal soluble region displayed greater sequence variability, indicating a higher tolerance to mutations. Based on these observations, conserved residues were avoided during mutational design, while more variable positions, especially in the soluble domain, were prioritized as potential targets for mutation.
STEP 2:
To evaluate the effect of mutations across the L-protein sequence, a protein language model (ESM-2) was used to compute log-likelihood ratio (LLR) scores for all possible amino acid substitutions at each position. This approach estimates how favorable a mutation is relative to the wild-type residue based on learned sequence patterns from large protein datasets. Positive LLR scores indicate mutations that are more likely to be tolerated or beneficial for protein stability, while negative scores suggest deleterious effects. The results were compiled into a ranked list of candidate mutations, allowing the identification of positions and substitutions with the highest predicted improvement. These scores were then used as a primary filter to guide mutation selection, in combination with conservation analysis from the multiple sequence alignment.
The protein language model identified several mutations with high positive LLR scores, indicating potentially favorable substitutions. The top-ranked mutations included K50L (LLR = 2.56), C29R (LLR = 2.39), Y39L (LLR = 2.24), C29S (LLR = 2.04), and S9Q (LLR = 2.01). Additional high-scoring mutations were observed at positions within both the soluble and transmembrane regions, such as T52L (LLR = 1.81), N53L (LLR = 1.86), and A45L (LLR = 1.54), particularly favoring substitutions to hydrophobic residues in the transmembrane domain. These results suggest that increasing hydrophobicity in the membrane region and selecting tolerated substitutions in variable regions may improve protein stability and folding.
STEP 3:
To assess how well the model predictions reflect real functional outcomes, the LLR scores were compared with available experimental lysis data for L-protein mutants. While some overlap between high-scoring mutations and experimentally tested variants was observed, many of the top-ranked mutations identified by the model were not present in the experimental dataset. Therefore, the experimental data was used when available, but for many candidate mutations, selection relied primarily on LLR scores in combination with conservation analysis.
STEP 4:
Based on the combined analysis of LLR scores, sequence conservation, and structural considerations, five mutations were selected as potential candidates for improving the L-protein. In the soluble region, the mutations S9Q and K23R were chosen due to their high LLR scores and location in more variable regions, suggesting a higher tolerance for substitutions that may improve folding stability. In the transmembrane region, K50L and T52L were selected, as both mutations introduce more hydrophobic residues, which is consistent with the conserved nature of this domain and may enhance membrane insertion and pore formation. Additionally, a combined mutant (S9Q + K50L) was designed to explore potential additive effects between improved folding in the soluble region and enhanced hydrophobicity in the transmembrane domain.
AlphaFold predictions were used to assess the structural impact of the selected mutations. The wild-type protein showed a pTM score of 0.44, while most mutants exhibited similar values around 0.43, indicating no significant structural disruption. Notably, the T52L mutant showed a slightly higher pTM score of 0.46, suggesting a modest improvement in structural stability. This result is consistent with the introduction of a more hydrophobic residue in the transmembrane region, which may favor membrane insertion. Overall, these findings indicate that the proposed mutations are structurally tolerated and may contribute to improved protein stability.












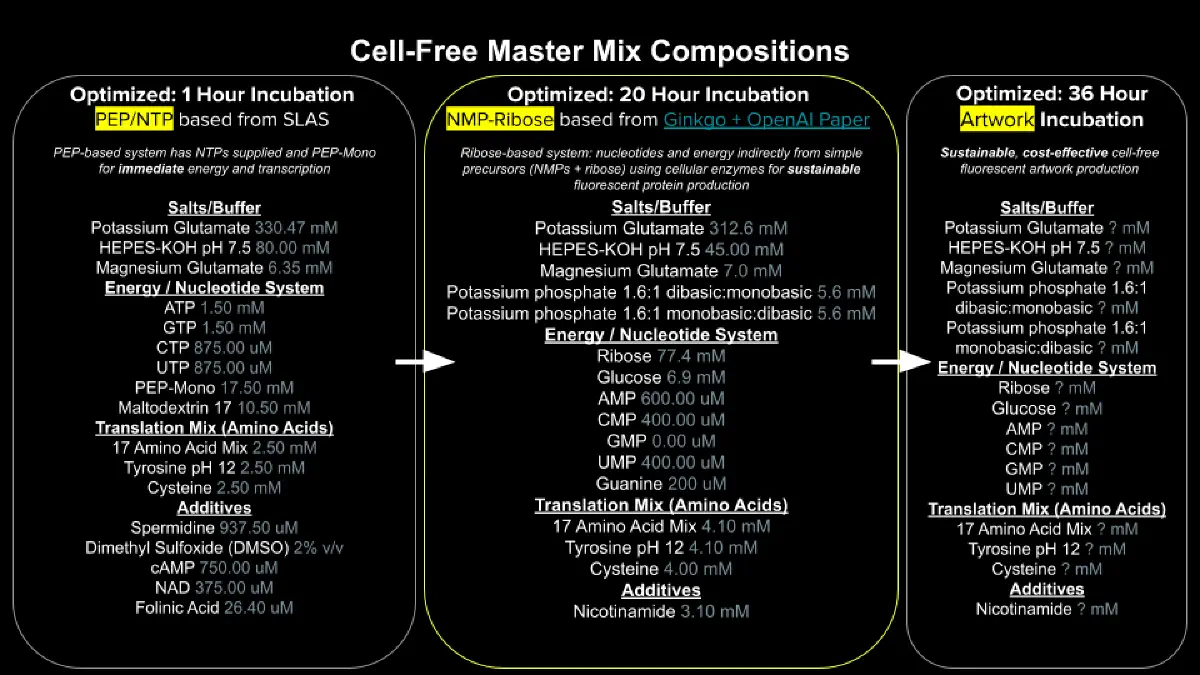

%20(2).png)
%20(5).png)
%20(1).png)
%20(1)%20(1).png)
%20(4).png)