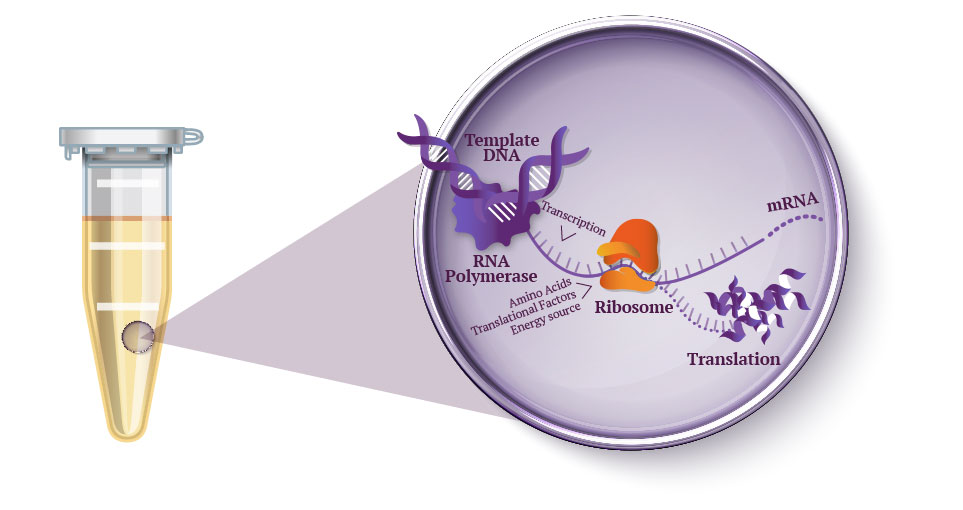
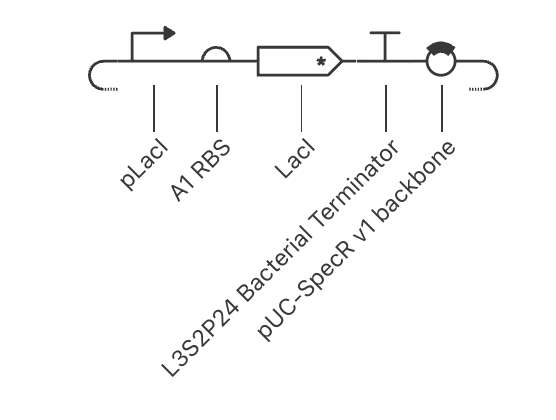
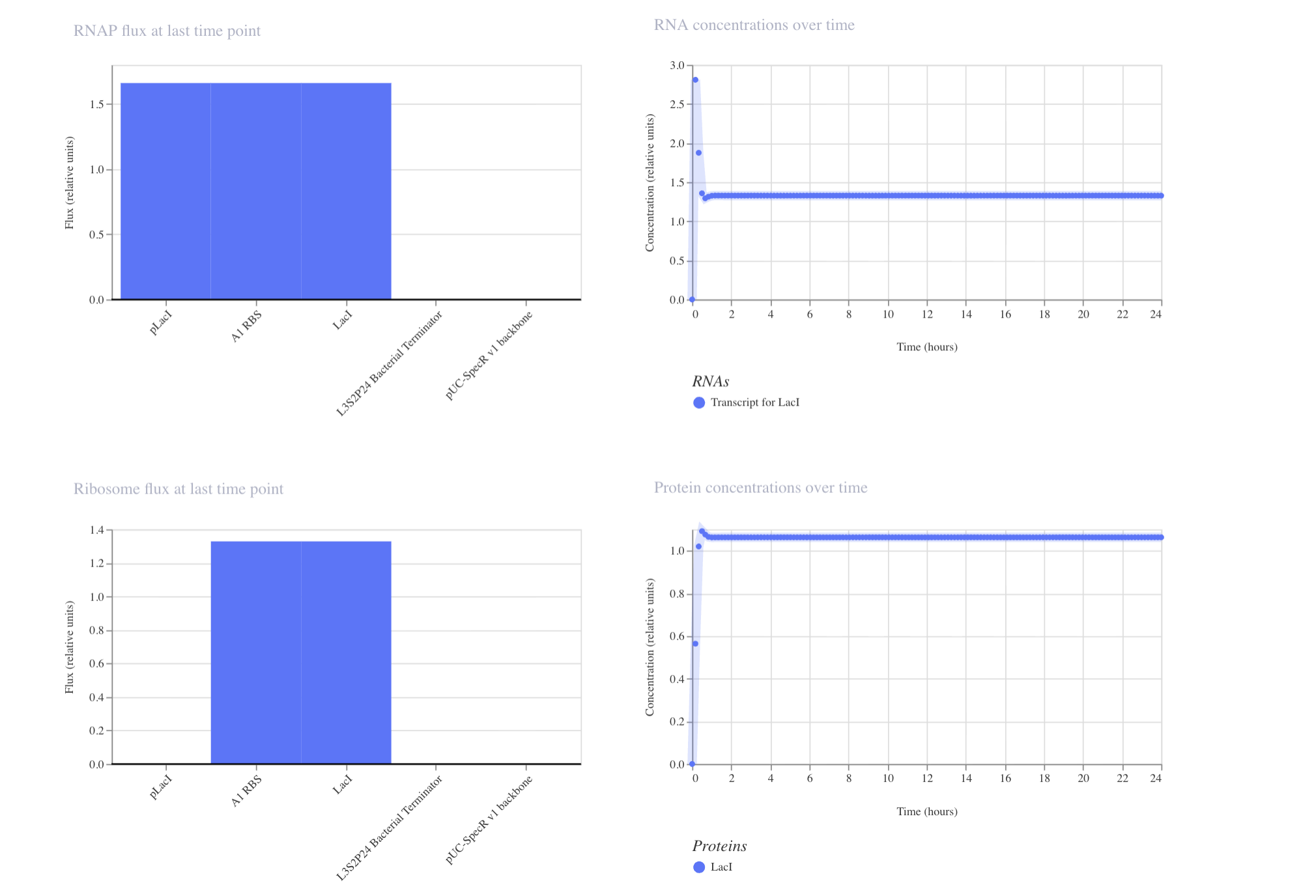
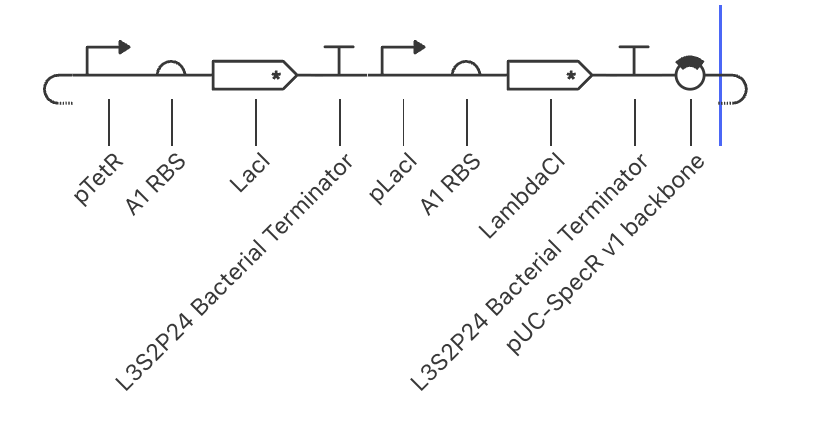
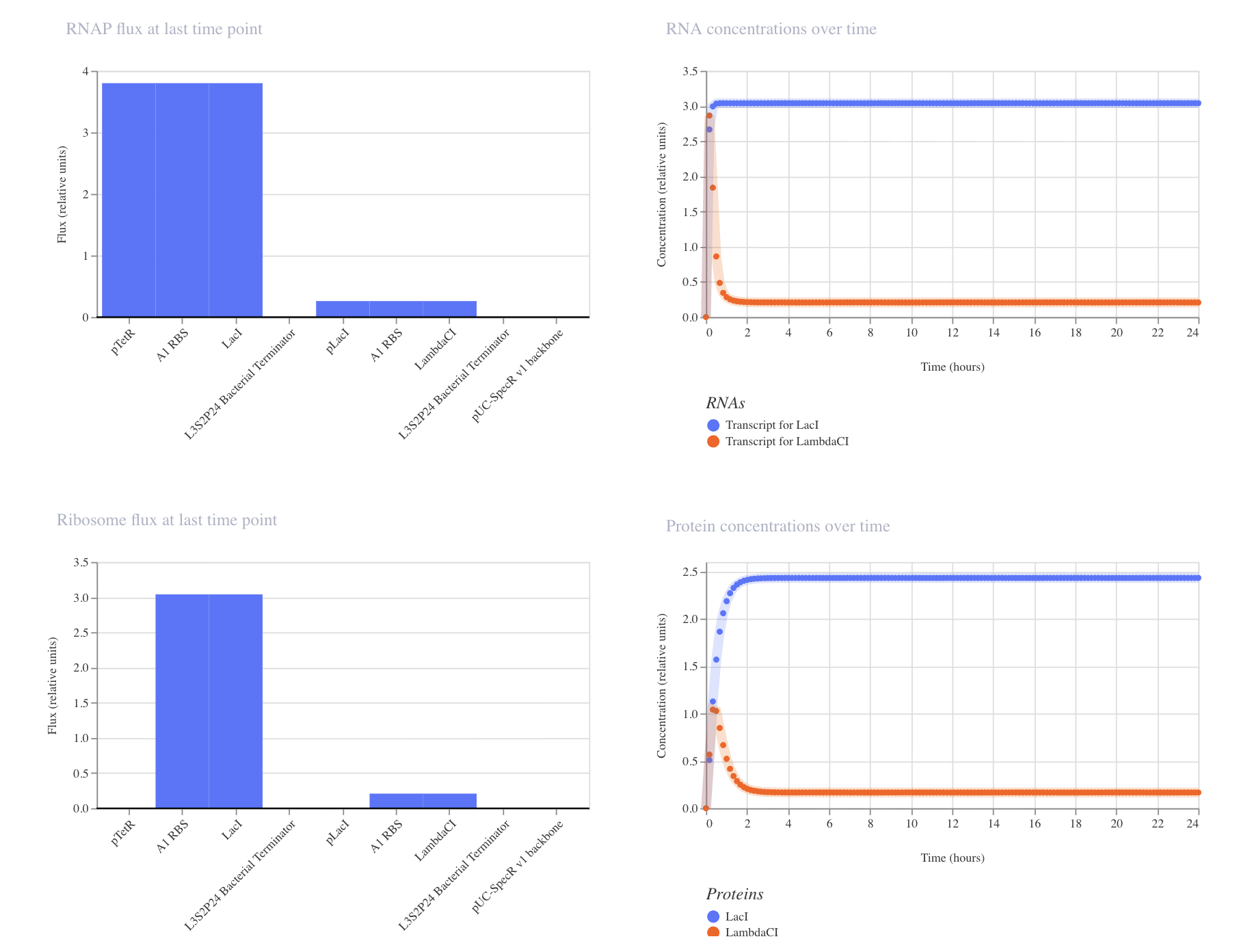
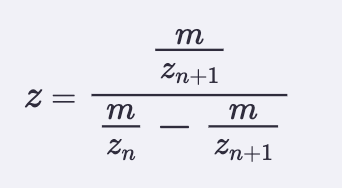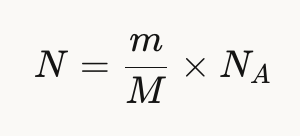Question 1 In the 1980’s, Keith Wood became the first to make a tobacco plant glow using firefly luciferase. However, he faced a critical limitation: they could not synthesize their own luciferin, requiring an external luciferin spray to emit light. 1 As Wood pivoted to fungal pathways, the firefly route has been largely abandoned despite its superior light efficiency. However, recent breakthrough have reopened this door, including the discovery of spontaneous benzoquinone + cysteine L-luciferin formation, and the identification of ACOT1 in D-luciferin transformation. My project aims to put these breakthroughs together, engineering plants capable of bioluminscence through firefly pathways.
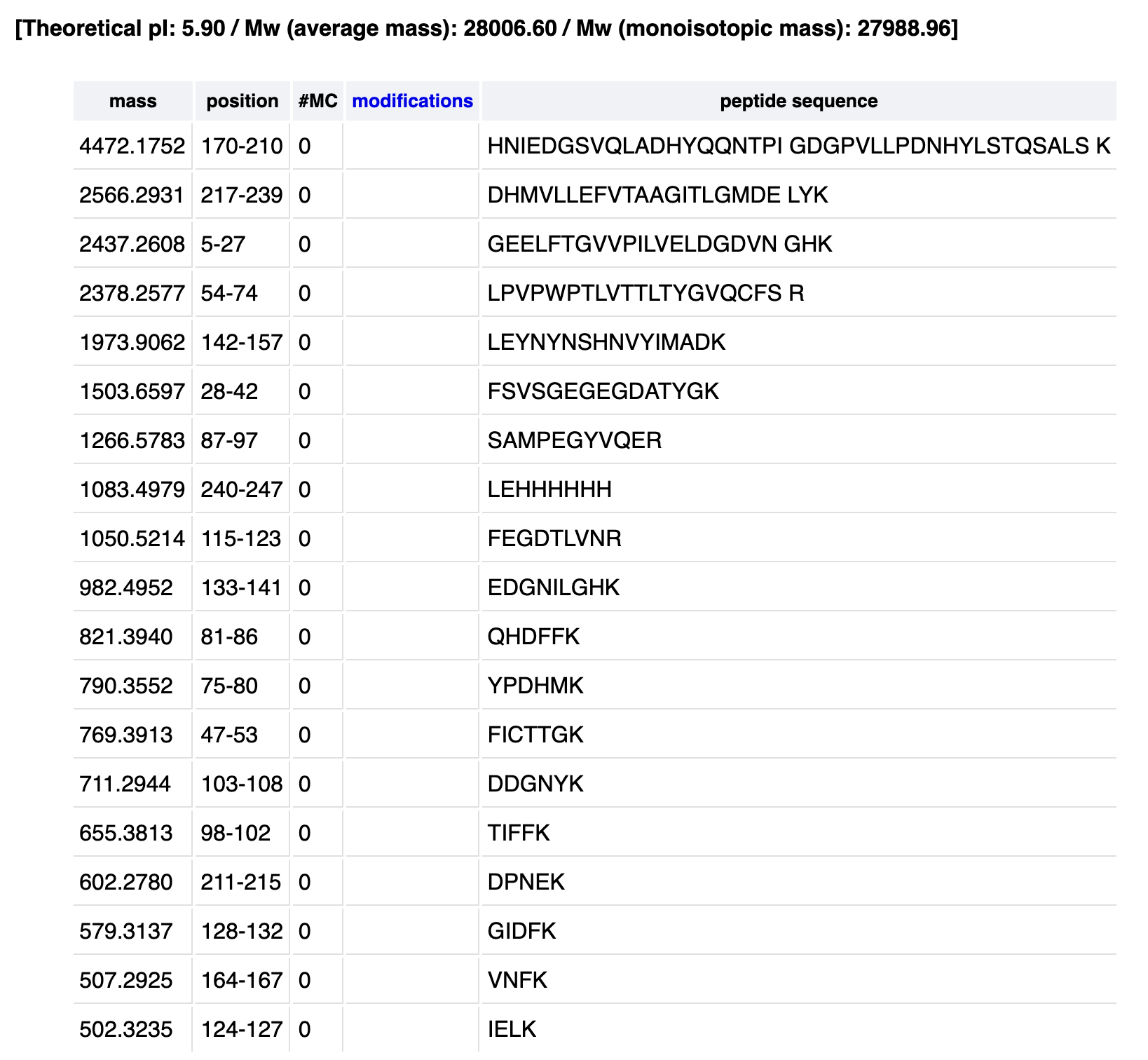
Homework: Waters Part I — Molecular Weight eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
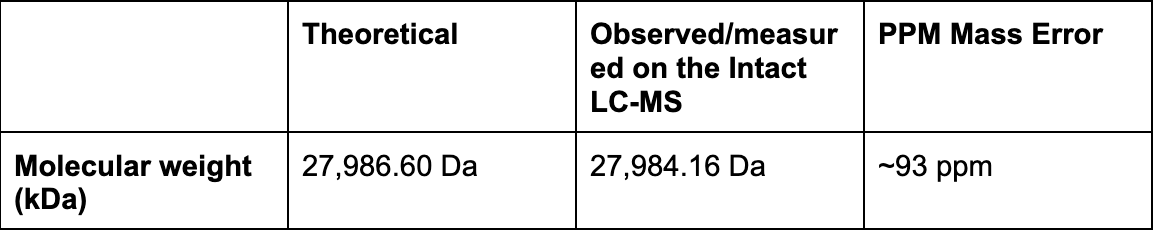
Using the online calculator, the theoretical mass is 28006.60 Da. After subtracting the chromophore maturation (-20 Da), my predicted molecular weight comes out to 27,986.60 Da.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I’m so sorry, I didn’t meet the deadline to submit but I will definitely be applying to be a TA this fall. :)
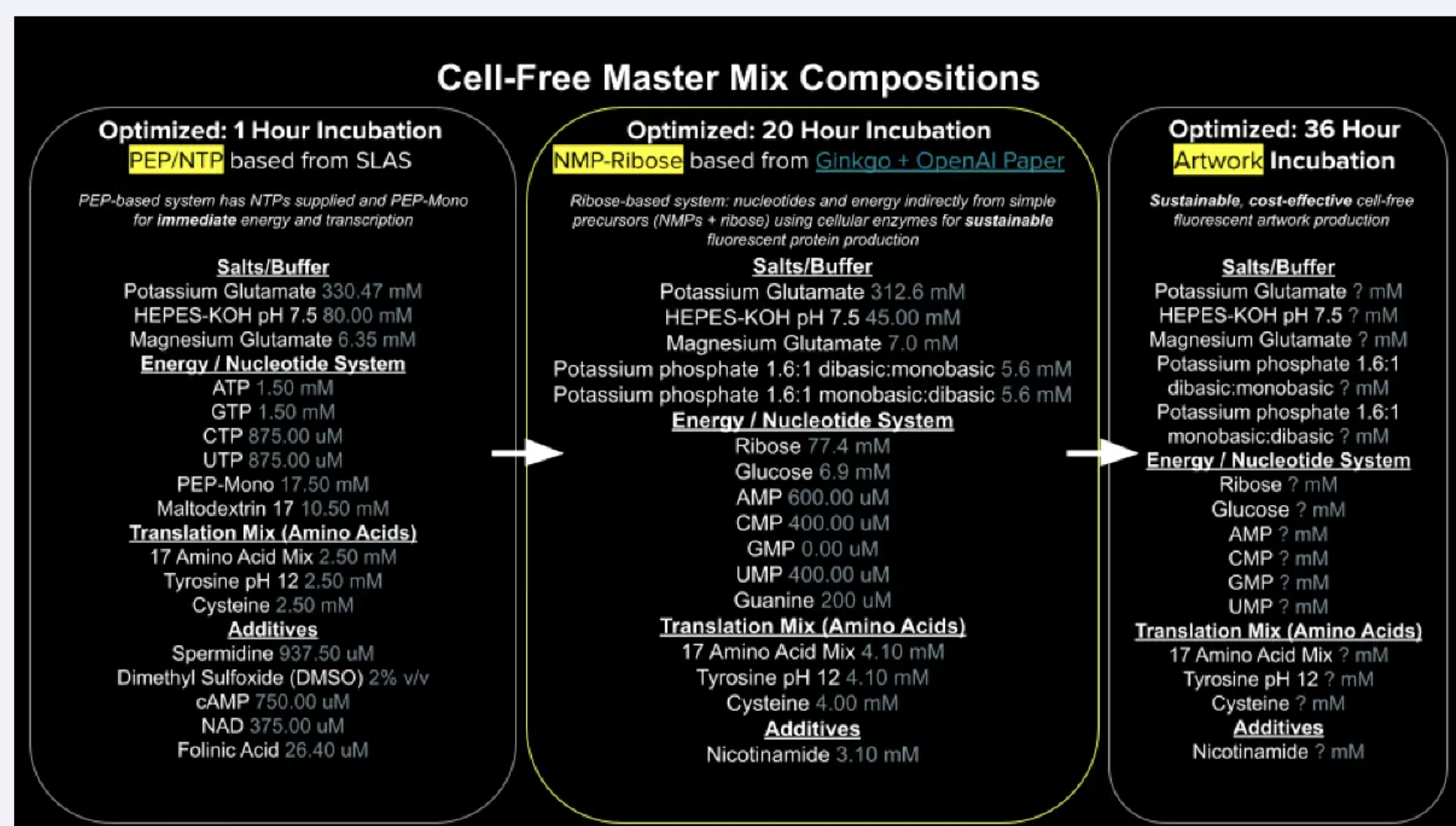
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Part 1: Benchling & In-silico Gel Art If ignoring ladder and lane 1, there is the design! I tried to recreate No Face from Spirited Away.
link to benchling: https://benchling.com/s/seq-8pB9vY3uTYXRrqsIKJS6?m=slm-FP5NlW0BAaqfbSmaoUg0
DNA Design Challenge For my homework, I decided to pick the enzyme that is the strongest candidate for facilitating luciferin synthesis. I picked my enzyme from my independent research. First I downloaded Fallon’s paper data from Fallon et al. 2018, “Firefly genomes illuminate parallel origins of bioluminescence in beetles” in eLife (DOI: 10.7554/eLife.36495. This experiment compared gene expression between the fat body (a firefly’s liver) versus the lantern (the organ that makes light) to find which genes are highly expressed in the lantern. I ran a filter in the file PPYR_OGS1.1_fatbody-vs…_test.txt texts, keeping only the statistically significant genes of TPM ≥ 50 and sleuth b ≥ 3. The TPM measures how actively a gene is being expressed in the lantern tissue, a higher TMP signaling a higher likelihood of luciferin expression. The sleuth is the statistical software Fallon used, that estimates log2 fold change. The higher the b sleuth, the more expression a gene has specifically in the lantern than fat body. I also ran qval ≤ 1e-10 to adjust for random noise.
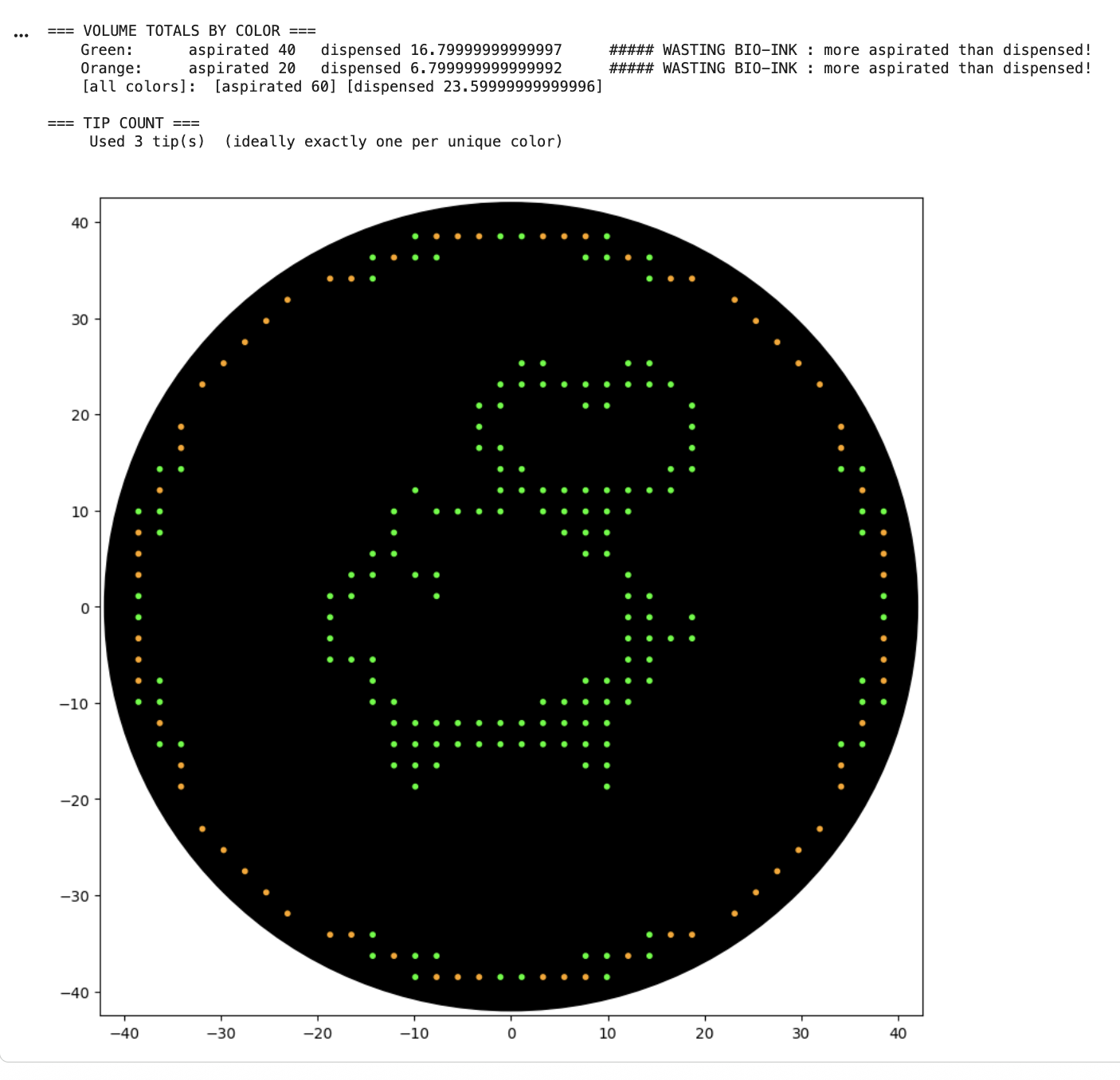
Python Script for Opentrons Artwork After designing in http://opentrons-art.rcdonovan.com/ (a pig with a heart in the centre), I chose to run the Collab notebook for simplicity. I used Claude Code to help generate my coordinates into proper code, before running all.
After my first simulation I noticed “WASTING BIO-INK : more aspirated than dispensed” warnings, so I edited my code. The next simulation still seemed to be wasting ink, but Claude informed me it’s an issue with floating point rounding.
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) A dalton is a unit of mass defined as 1/12th the mass of an unbound neutral Carbon-12 atom at rest, equivalent to 1.66X10-24 grams. It is commonly used in molecular biology to calculate the mass of large molecules into grams. Meat protein is roughly 25% protein by mass, therefore 500g x 0.25 = 125g of protein since almost all amino acids are concentrated in the protein of meat.
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
SOD1 A4V mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Notestranslation begins with Methionine in eukaryotes, therefore the position of translation is technically 5 (A -> V). Protein language models train on amino acid sequences. Masked language modelling trains sequences by randomly masking some positions, and training the model to fill in the masked positions based on context. PepMLM does the same thing but to peptide binder design, where it masks all the peptides in the proteins you give, iteratively fills in those masked positions from most to least confident. Low perplexity = more confident, high perplexity = model uncertain.
Assignment: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Components included are Phusion DNA Polymerase, deoxynucleotides (dNTPs), and HF Reaction Buffer containing MgCl₂. Physion DNA polymerase is a Pyrococcus-like enzyme. The polymerase domain reads the strand and synthesizes it by adding dNTPs one at a time (through phosphodeister bond formation). This requires a primer to already be present in the template. The enhancing domain fused onto the polymerase keeps the enzyme attached to the DNA template, speeding up the process. The exonuclease domain reads this newly synthesized stand and corrects any mistakes. Deoxynucleotides (dNTPs) are the building blocks of DNA at optimized concentrations. The HF reaction buffer helps control the pH for optimal activity, while the MgCl₂ positions the dNTPs correctly and stabilizes the transition state during bond formation.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs (Intracellular Artificial Neural Networks) have several advantages over traditional genetic circuits. Booleans inherently work in a binary fashion: there is either a statement ment that makes it true/false, and therefore, either on/off. IANNs have the ability to process continuous signals, allowing for inputs to be weighted against each other before an output. This makes the system more sensitive to differences, not just whether something is true or false. This is great for cells as molecular concentrations exist on a spectrum. Another reason is its reconfigurability: you can reprogram the IANN circuit without needling to completely rebuild it. IANNs also make multi-input easier to configure, as an arbitrary number of inputs can be collapsed into one step, instead of building onto each other in booleans. Lastly, boolean circuits are static, with inherently fixed behaviour after it is built. IANNs are defined by weights, allowing for greater flexibility if you were to modify the weight thresholds.
Homework Part A: General and Lecturer-Specific Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis provides multiple advantages over traditional methods due to its lack of membrane. You don’t have to cross the membrane, and the cell’s homeostasis controls don’t resist any experimentations. Furthermore, since everything is now a controllable variable, you have better control over composition (such as concentrations) of amino acids, substrates, etc. You also maintain better folding control and template flexibility (since you don’t have to clone into plasmids). Cases where this is beneficial include space travel (since cell-free synthesis allows for freeze-drying, which is more shelf-stable in storage), and anywhere else resources may be limited to run a fermenter (war zones, rural clinics, etc.) Another case is the expression of “unhostable” proteins, which would in other cases, kill its living cell. The cell-free environment means you bypass the actual cell host.
Subsections of Homework
Week 1 HW: Principles and Practices
Question 1
In the 1980’s, Keith Wood became the first to make a tobacco plant glow using firefly luciferase. However, he faced a critical limitation: they could not synthesize their own luciferin, requiring an external luciferin spray to emit light. 1 As Wood pivoted to fungal pathways, the firefly route has been largely abandoned despite its superior light efficiency. However, recent breakthrough have reopened this door, including the discovery of spontaneous benzoquinone + cysteine L-luciferin formation, and the identification of ACOT1 in D-luciferin transformation. My project aims to put these breakthroughs together, engineering plants capable of bioluminscence through firefly pathways.
As we know:
Fireflies create an enzyme called luciferace that can oxidize D-luciferin.
Luciferase can be put synthetically engineered into plants, but must be sprayed with luciferin to synthesize luciferin.
Kanie et al. (2016) discovered arbutin can spontaneously react with L-cysteine (naturally found in plants) to create L-luciferin.
Zhang (2020) discovered ACOT1 as the enzyme responsible for catalyzing the reaction from L-luciferin to D-luciferin.
In theory, these reactions could make a plant glow with many foreseen and unforeseen modifications.
My proposed reaction:
Arbutin =
GLUCOSE + HYDROQUINONE
Glucose -0 -Hydroquinone gets cut with BGL (β-glucosidas).
Hydroquinone ->
Benzoquinone
We inject our first gene (lactase) to remove two -0H groups, creating benzoquinone. Important to make sure the benzoquinone is trapped where it meets cysteine immediately or its proteins can damage the cytoplasm of the plant
Benzoquinone + Cysteine
=
L-LUCIFERINE
As we have discovered, benzoquinone spontaneously reacts with cysteine under certain conditions, producing L-Luciferin
L-Luciferin + ACOT1 =
D-luciferin AND L-LUCIFERIN
We inject ACOT1 (naturally occurring in the firefly) to catalyze the reaction into D-luciferin (the form that glows)
D-luciferin + ATP = LUCIFERYL-ADENYLATE + PPI
LUCIFERYL-ADENYLATE + 02 = OXYLUCIFERIN + CO2 + AMP
OXYLUCIFERIN ->
OXYLUCIFERIN + PHOTON
(Well understood steps of how d-Lucefirin is created into light)
Beyond the ethical application of synthetic genomics, an important goal would be to ensure the plant will not be detrimental or disrupt natural ecosystems. (Example: GloFish goldfish being an invasive species after someone released them in Brazil.) Goals pertaining to this specific issue will be broken down below.
Option 1: Lab-work will be contained and carefully scrutinized to ensure plants are not accidentally released. This includes but is not limited to: proper disposal, proper containment, proper sanitization.
Option 2: Plants will eventually be tested in greenhouse settings, or other settings replicating natural ecosystems to verify no immediate or obvious harm and gather data in case of contamination.
Option 3: Plants that are modified will be non-invasive and common (ex. Petunias), and will be modified only to the extent needed for autonomous self-bioluminescence.
Option 4: Research in later stages will be done with the aim of open-environment release, where doing so will be safe for natural ecosystems.
Does the option:
Option 1
Option 2
Option 3
Option 4
Protect the environment?
• By preventing harm to natural ecosystems?
ME
RE
ME
ME
• By helping respond
n/a
ME
n/a
SE
Enhance Biosecurity
• By preventing incidents
ME
SE
SE
ME
• By helping respond
n/a
SE
n/a
SE
Foster Lab Safety
• By preventing incident
ME
n/a
n/a
MIN
• By helping respond
n/a
MOD
n/a
MIN
Other considerations
• Minimizing costs and burdens to stakeholders
SE
MIN
MOD
ME
• Feasibility?
RE
MIN
RE
MOD
• Not impede research
n/a
SE
n/a
SE
• Is net good for the world?
RE
RE
SE
RE
ME = Most Effective
MOD = Moderately Effective
RE = Relatively Effective
SE = Somewhat Effective
MIN = Minimally Effective
n/a = n/a
A second goal is to remain responsible with tools and research. As a newcomer to synthetic biology, I want to ensure proper attribution to the work of the reseachers who make this possible.
Option 1: Prevent misuse of plasmids/enzymes/resources from previous academic papers.
Option 2: Be respectful to my lab space and peers, maintaining proper safety and etiquette.
Does the option:
Option 1
Option 2
Protect the environment?
• By preventing harm to natural ecosystems?
SE
RE
• By helping respond
n/a
RE
Enhance Biosecurity
• By preventing incidents
SE
RE
• By helping respond
SE
MIN
Foster Lab Safety
• By preventing incident
ME
ME
• By helping respond
SE
RE
Other considerations
• Minimizing costs and burdens to stakeholders
ME
MOD
• Feasibility?
ME
RE
• Not impede research
ME
RE
• Is net good for the world?
ME
RE
ME = Most Effective
MOD = Moderately Effective
RE = Relatively Effective
SE = Somewhat Effective
MIN = Minimally Effective
n/a = n/a
Question 3
Action 1
Purpose: As this is such a new field, these is little regulation on the genetic modification of plants for commercial/art purposes. I propose a more thorough, environmental review before the release of plants.
Design: Researchers would need to submit their own opinions and data, while federal regulators would need to propose their own processes.
Assumptions: That everyone will be acting in good faith, not unnecessarily over or under regulating, and that regulators have the knowledge required to make educated decisions.
Risks: Over-regulation would cause extreme bureaucracy in the the bio-plant space, and cause resentment in the scientific community. Under-regulation might cause detrimental harm to natural ecosystems in Canada.
Action 2
Purpose: People lack incentive or the freedom to purse such projects. Companies can make it more attractive for workers to purse bioengineering for commercial or art purposes.
Design: Corporations or the government of Canada can create programs that incentivize bio-plant discoveries, making it more accessible and attractive for individuals.
Assumptions: Corporations or government programs would be executed well-enough for progress, and enough attention would be directed at students/workers to ensure some level of success.
Risks: Corporations may over-rely on potential profits, pushing individuals into projects that may not be beneficial for society. This is where Action 1 regulations should ideally be applicable. Programs may not be executed in good faith or passion, leaving people uninspired and destitute.
Action 3
Purpose: People lack access to labs, and most places do not have community bio-labs. Autonomous labs will still take time to be implemented on a wider scale. People should be able to get certificates for level-1 biohazard for at-home labs from the government.
Design: A certain certification process should undergo with organizations representing the government. Once you prove you can meet certain standards, a visit should be granted to confirm. If granted, you are certified for a certain amount of time until you re-certify.
Assumptions: Individuals engaging in home labs are responsible, and strive to truly meet these standards at all time. Federal regulators act in good faith, and are responsible with their certifications.
Risks: Individuals running home labs accidentally contaminate or cause injury to themselves or others.
Week 2 Lecture Prep
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Error rate of polymerase in nature is 1 x 10−6 or 1:106
Human genome: 3.2 Gbp (billion base pairs) or 3.2×109 bases
So if error rate is 1 × 10−6 per base: (1×10−6)×(3.2×109) = 3.2×103 = 3,200 errors
Biology deals with this through the MutS Repair System, where a DNA repair protein scans the replicated DNA, and binds where there are errors in the pairing. Once the MutS is binded, that part of the strand is cut, and the exonuclease removes the error. Lastly, the DNA polymerase resynthesizes the correct pairing, ligase seals it, and the error is removed. This system improves fidelity by 100-1000x, effectively lowering the error number in human genomes to 0.3-1 per copy.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The average human protein is estimated to be around 400 amino acids ( 1036 bp). There are an average 3 numbers of different codons that encode the same amino acid, therefore the number of possible sequencing is ≈3400, which is an absurdly high number. However, this does not translate in practice. Firstly, different organisms prefer different codons, which is why we optimize our codons depending on our project. Biology still cares about the nucleotide sequence, despite identical codon sequences. Other reasons include the mRNA behaving differently depending on the structure, certain codes negatively affecting how the protein folds, RNA instability, among other practical hurdles. Slide 62 has an interesting argument as to why optimal alphabet size falls in the tens, not hundreds.
Homework Questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method is phosphoramidite solid-phase DNA synthesis, of which Twist is the main commercial provider for. While phosphoramidite has been optimized since its founding in 1981, the overall method has remained the same. The DNA is built one base at a time using a repeating cycle. First a DNA strand is attached to a solid surface, and a DMT protecting group blocks the 5’ end. Once a chemical acid removes the DMT and exposes the 5’OH, after which a phosphoramidite nucleotide is added to the free 5’-OH (~99–99.5% per cycle efficiency). Since not every strand couples successfully, Twist chemically caps the failed strand. Then the new linkage is chemically oxidized to form a stable bond. Then the cycle repeats.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because with every additional nucleotide, the yield drops since efficiency is not at 100%, but 99.5%. Therefore, for an 100 nucleotide length strand, 0.995100 ≈ 60%, where only 60% of molecules will reach their full length, while the rest are truncated somewhere else.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Because at 2000bp, the percentage of successful molecules will essentially be at 0. (0.9952000 ≈ 0.005%) There would be almost no full length molecules. This is a hard, biological wall we are yet to solve.
Homework Question from George Church
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any. What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in animals are Arginine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, and Valine. These are obtained through an animal’s diet, considered essential as they generally cannot be synthesized by animals. The “lysine contingency” is an idea that was popularized by the franchise Jurassic Park. In the movies, dinosaurs were engineered to be unable to make the lysine amino acid without a human-supply, so that in theory, they would not be able to survive without human guidance. This was done to prevent escape, and safety regulations. However, this wouldn’t seem to work in real life. Animals are already “lysine contingent”; we are unable to synthesize lysine. That’s why lysine is found in a wide variety of animal diets, and can be easily supplemented by food. Virtually all food sources and ecosystems contain lysine, scavenging would already easily supply it. Furthermore, there are many other amino acids we require. Signalling one out is arbitrary. Effective containment would rely on dependencies that are rare in nature.
Other References
Iwano, S., Sugiyama, M., Hama, H., Watakabe, A., Hasegawa, N., Kuchimaru, T., … Miyawaki, A. (2018). Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science, 359(6378), 935–939. https://doi.org/10.1126/science.aaq1067
Kanie, S., Abe, K., Hirano, T., & Niwa, H. (2016). One-pot non-enzymatic formation of firefly luciferin in a neutral buffer from p-benzoquinone and cysteine. Scientific Reports, 6, 24794. https://doi.org/10.1038/srep24794
Zhang, R., Chen, L., Jiao, J., Zhou, Y., Liu, Y., & Wang, Y. (2020). Genomic and experimental data provide new insights into luciferin biosynthesis and bioluminescence evolution in fireflies. Scientific Reports, 10, 15882. https://doi.org/10.1038/s41598-020-72900-z
Applicable AI Promots:
Summarize Keith Wood’s lifework and research in firefly related plant bioluminescence.
(Image of example table) explain what they mean by options, and how the graph works.
Explain the MutS mismatch repair system in detail.
How many different ways are there to code (DNA nucleotide code) for an average human protein? Why doesn’t it translate in practice?
Explain how phosphoramidite solid-phase DNA synthesis works in detail.
Using the online calculator, the theoretical mass is 28006.60 Da. After subtracting the chromophore maturation (-20 Da), my predicted molecular weight comes out to 27,986.60 Da.
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine z for each adjacent pair of peaks (n, n+1) using:
I picked peaks 903.7148 and 875.4421
Z = 875.4421/903.7148 - 875.4421
Z = 875.4421/28.2727
Z = 30.96 rounded to 31
Charge is +31 for peak 875.4421
Therefore charge must be +32 for 903.7148
Determine the MW of the protein using the relationship between m/Zn, MW and z:
Taking the formula from recitation: MW = (n x m/Zn) - n
MW = (31 x 875.4421) - 31
MW = 28,015.16 − 31
MW = 27,984.16 Da
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Accuracy = ∣27,986 - 27,986.60∣ / 27,986.60
Accuracy = 2.60 / 27,986.60
Accuracy = 0.0000929
Accuracy = 0.0000929 x 100
Accuracy = 0.0093% (≈ 93 ppm)
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Yes (although it may be inaccurate because I had to guess some of the numbers form the blurry image). We know the isotope peaks with an ion of charge z are spaced apart at z = 1. Looking at the isotope peaks in the zoomed photo:
The consistent spacing is around 0.052m/z. Therefore:
Z = 1.0.052
Z = 19.2
The charge state is +19.
Homework: Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking the biochemical properties tab will show you a count for each amino acid).
There are 20 lysines and 6 arganines, totaling 26 sites:
How many peptides will be generated from tryptic digestion of eGFP?
The total number of peptides generated is 19.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
The peaks are:
0.61
0.79
1.20 (decided to count this one)
1.43
1.80
1.85
1.93
2.17
2.26
2.54
2.78
3.27
3.53
3.59
3.70
4.30
4.48
4.64
4.87
5.06
5.43
There are 21 chromatographic peaks.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
No, there are slightly more peaks in the chromatogram (21) than predicted by Peptide Mass (19). This is likely due to human error (me counting peaks that are below relative abundance), missed cleavages, or missed small peptides that still produced peaks.
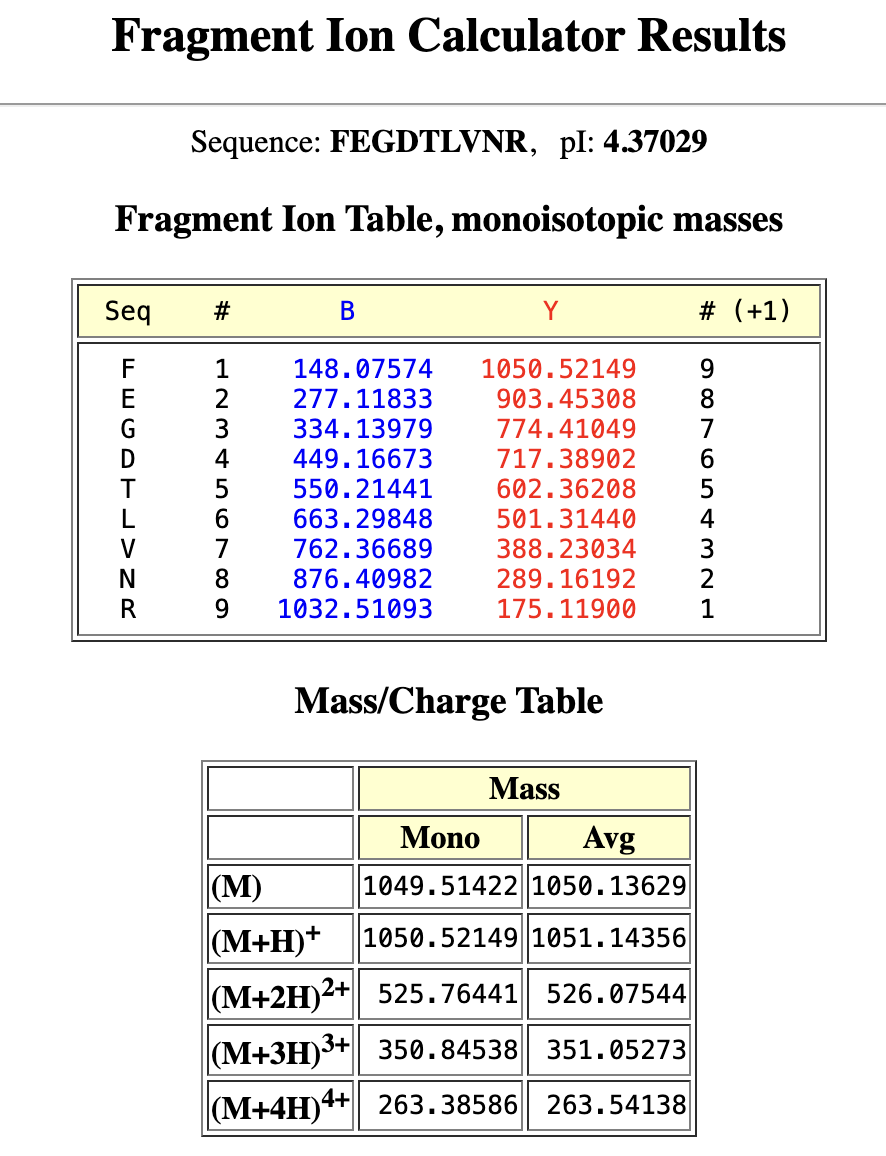
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ( M+H⁺ ) based on its m/z and z
The most abundant m/z is the tallest peak of 525.76712. Using the zoomed in image:
525.76712 → 526.25918: Δ = 0.49206
526.25918 → 526.76845: Δ = 0.50927
526.76845 → 527.26098: Δ = 0.49253
Average isotope spacing ≈ 0.50 m/z
Z = 1/0.50 = +2
The most abundant charge state is doubly charged.
For the [M+H]⁺ (singly charged) mass:
M = 2 x (525.76712) - 2 (equation from earlier)
M = 1051.53424−2.01456
M = 1049.5197 Da
M+H+ = M+1.00728
M+H+ = 1049.5197+1.00728
M+H+ = 1050.527 Da
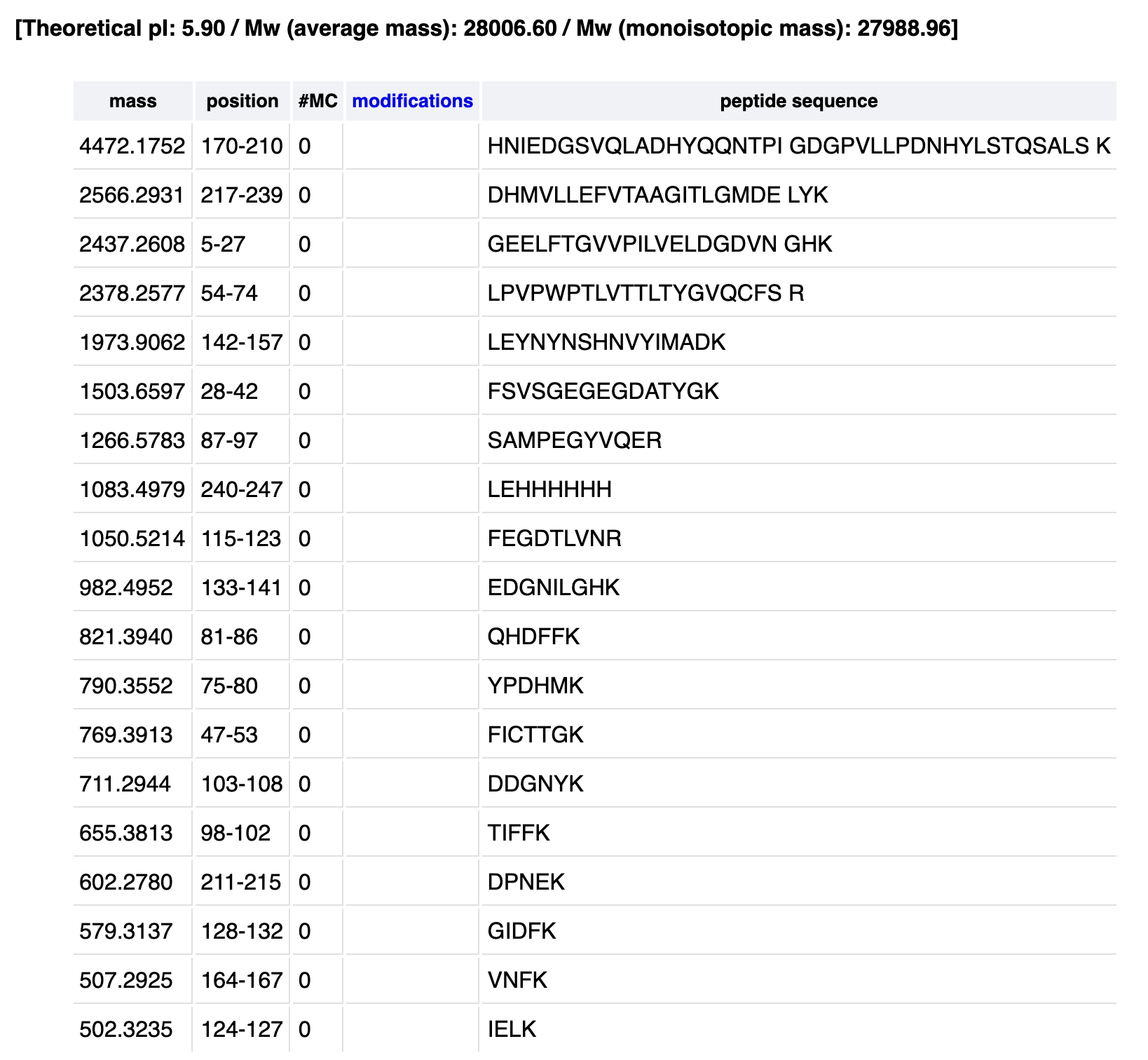
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
From my PeptideMass table1050.527 matches the predicted 1050.5214 :
1050.5214 Da → FEGDTLVNR (positions 115–123)
Accuracy = ∣1050.5244 - 1050.5214∣ / 1050.5214
Accuracy = 0.003 / 1050.55214
Accuracy = 2.86×10^−6
Error = 2.86 ppm
What is the percentage of the sequence that is confirmed by peptide mapping?
The percentage confirmed by peptide mapping is 88% of the eGFP sequence. The unidentified regions are in white and probably correspond to small peptides below detection threshold.
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Yes, as mentioned earlier it’s FEGDTLVNR (115-123).
M+H⁺ = 1050.52149 → matches Figure 5b peak at 1050.52438
M+2H²⁺ = 525.76441 → matches Figure 5b peak at 525.76712
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Yes, the peptide map data confirm the protein in eGFP standard. Firstly, there is a high sequence coverage (88%). Secondly, there is accurate peptide mass matches, where the peptide identified FEGDTLVNR with [M+H]⁺ = 1050.524 Da matches the PeptideMass prediction of 1050.521 Da to within 3 ppm. Thirdly, the fragmentation pattern from figure 5c confirms this sequence, matching the predicted series for FEGDTLVNR, confirming the actual amino acid sequence of the peptide.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Calculating each ones mass:
7FU Decamer: 10 × 340 kDa = 3.4 MDa
8FU Didecamer: 20 × 400 kDa = 8.0 MDa
8FU 3-Decamer: 30 × 400 kDa = 12.0 MDa
8FU 4-Decamer: 40 × 400 kDa = 16.0 MDa
Matching to Figure 7:
3.4 -> yes, there is a peak for 7FU Decamer
8.33 -> yes there is a peak for 8FU Didecamer
12.67 -> yes there is a peak for 8FU 3-Decamer
16 -> yes there is a small peak for 8FU 4-Decamer
All four oligomeric species are on the spectrum in Figure 7, albeit at different intensities.
Since KLH exists natively as a didecamers, 8.33MDa is the tallest peak.
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I’m so sorry, I didn’t meet the deadline to submit but I will definitely be applying to be a TA this fall. :)
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): E-Coli cells that have been opened, where their inside machinery provides the mechanisms for transcription and translation of your proteins. The BL21 has a modification to help your protein not get degraded, the DE3 carries an inserted polymerase gene for fast transcription from t7 promoters, and Star has a modification so that the mRNA isn’t chewed up before translation.
Potassium Glutamate: Provides K+ which is integral for ribosome assembly, tRNA folding and translation fidelity. Glutamate specifically gives K+ without the downsides of KCI.
HEPES-KOH pH 7.5: A sort of pH sponge that soaks up the acidic waste produced during the reaction that burns sugar for energy. This helps preserve enzymes and ribosome activity by holding the pH near 7.5 (optimal operating point mimicking E.Coli’s native pH).
Magnesium Glutamate: Magnesium is an integral concentration-sensitive component in cell-free systems that allows for ribosome stabilization, codon pairing, and ATP reactions. Too little levels cause the ribosomes to dissociate, and too much causes translation to be error prone.
Potassium phosphate monobasic: Supplies inorganic phosphates that lysate kinases attach onto NMPs so that they can become NTPs for transcription and translation.
Potassium phosphate dibasic: Supplies the same inorganic phosphate pool as above that kinases use to charge NMPs into NTPs and ADP into ATP. It’s paired with the monobasic at 1.6:1 to help the reaction pH stable at 7.5 over long incubation.
Ribose: A sustainable and readily available form of carbon/sugar that the cell uses as raw material to build its nucleotides from scratch, and that gets burned through during ATP generation. This slow release of sugar and salvage strategy was the core innovation that lets the reaction run for 20 hours.
Glucose: Another cheap sugar that feeds into glycolysis to generate ATP. It complements ribose by providing a steady metabolic fuel to the lysate, and allowing ribose to be freely used for building nucleotides.
AMP: cheap, low-energy form of adenosine.The lysate’s kinases boost it up the energy ladder, allowing it to be used for transcription and translation.
CMP: cheap, low-energy form of cytidine. The lysate’s kinases boost it up the energy ladder, allowing it to be used for transcription and translation.
GMP: cheap, low-energy form of guanosine. It’s supplied as free guanine, letting the lysate enzymes build GMP from scratch.
UMP: cheap, low-energy form of uridine. The lysate’s kinases boost it up the energy ladder, allowing it to be used for transcription and translation.
Guanine: Free base form of G, the nitrogenous base with no sugar and no phosphate, a cheap precursor of G. The lysate’s salvage enzymes attach with other material to build GMP, which kinases then up to GTP for use in transcription.
17 Amino Acid Mix: A premixed solution of 17 of the standard amino acid, which are building blocks the lysate’s load onto tRNAs, and then ribosomes string together into proteins during translation. The other 3 amino acids are added separately because of solubility and stability issues that would otherwise cause them to degrade.
Tyrosine: One of the 20 amino acid building blocks added separately as it has poor solubility at neutral pH. Instead it is prepared as a separate stock dissolved in highly basic water. The small amount of alkalinity added with it is absorbed by the HEPES buffer mentioned earlier.
Cysteine: One of the 20 amino acid building blocks added separately because its free thiol group oxidizes rapidly in storage. It’s kept as a separate stock to ensure the cysteine in the reaction is usable when needed during translation.
Nicotinamide: Vitamin B3, which is the precursor that the lysate’s salvage enzymes use to build NAD+, an electron shuffling molecule that glycolysis needs to produce ATP. It’s topped up to replenish the NAD+/NADH pool as it gets continuously consumed over the 20 hour incubation.
Backfill
Nuclease Free Water: Ultra-pure water that is treated prior to ensure it is free of RNases and Dnases (enzymes that can destroy RNA and DNA or contaminate the reactions).
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above.
The main difference comes down to supply: the 1-hour systems supplies are already pre-charged and ready to use NTPs (ATP, GTP, CTP, UTP) and PEP-Mono supplies a phosphate donor for fast ATP regeneration. This gives immediate but short-lived transcription and translation, burning out within an hour as the free phosphate accumulates magnesium. The 20-hour system supplies cheap precursor NMPs (AMP, CMP, UMP), free guanine, ribose and glucose, relying on the lysate to slowly build the pathways for NTPs and ATP regeneration. Paired up with the NAD+ replenishment, the system produces over a much longer incubation time, at a lower cost but also slower speed.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
The lysate has a salvage enzyme called HGRPT that takes free guanine and attaches it to PRPP (an activated form of ribose) to produce GMP in a single enzymatic step. The lusate’s kinases then boost GMP up the energy ladder into GDP, and then GTP, which RNA polymerase then incorporates as the G letter during transcription. Therefore, GMP is not missing from the reaction, but is being built on demand from cheaper precursors.
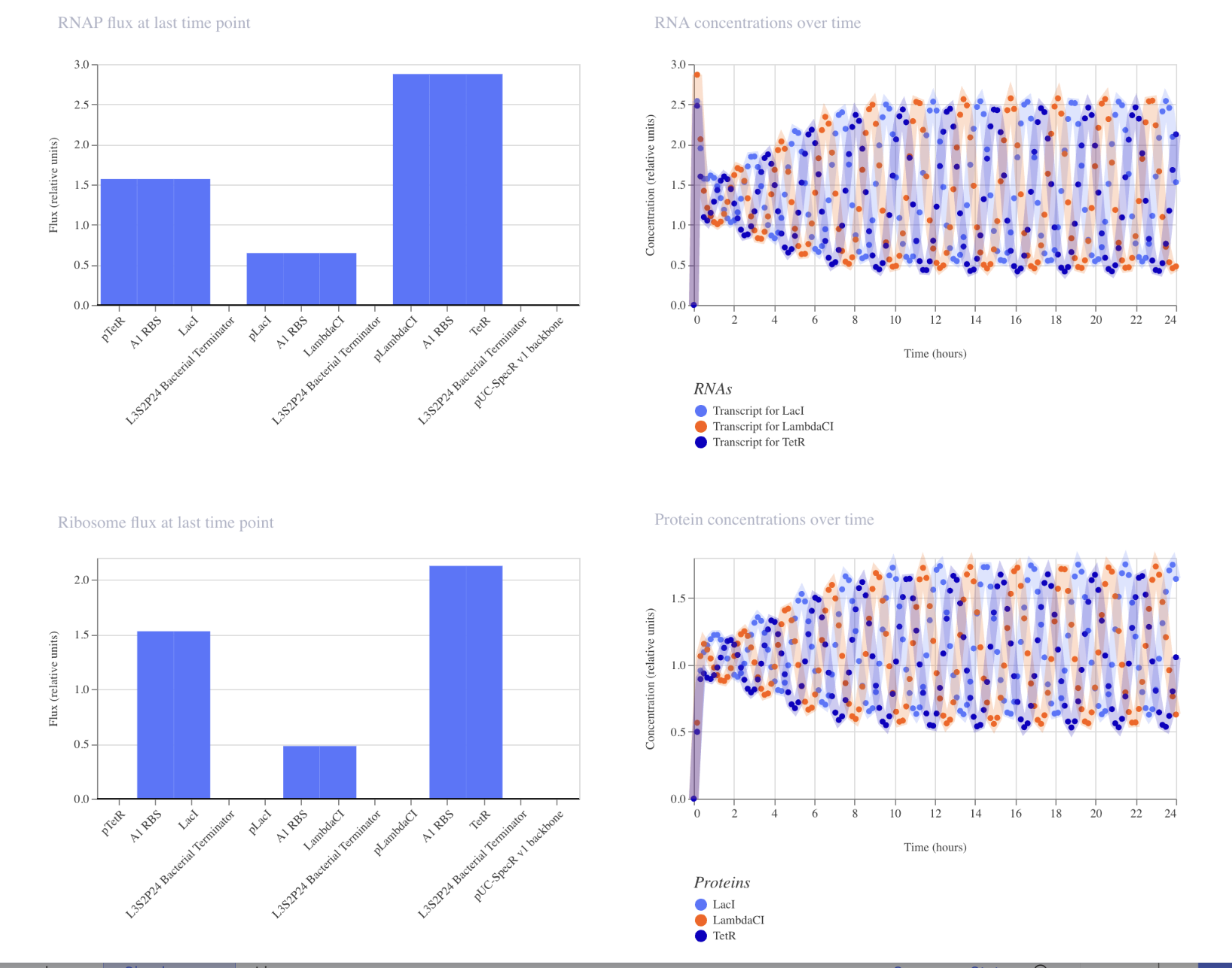
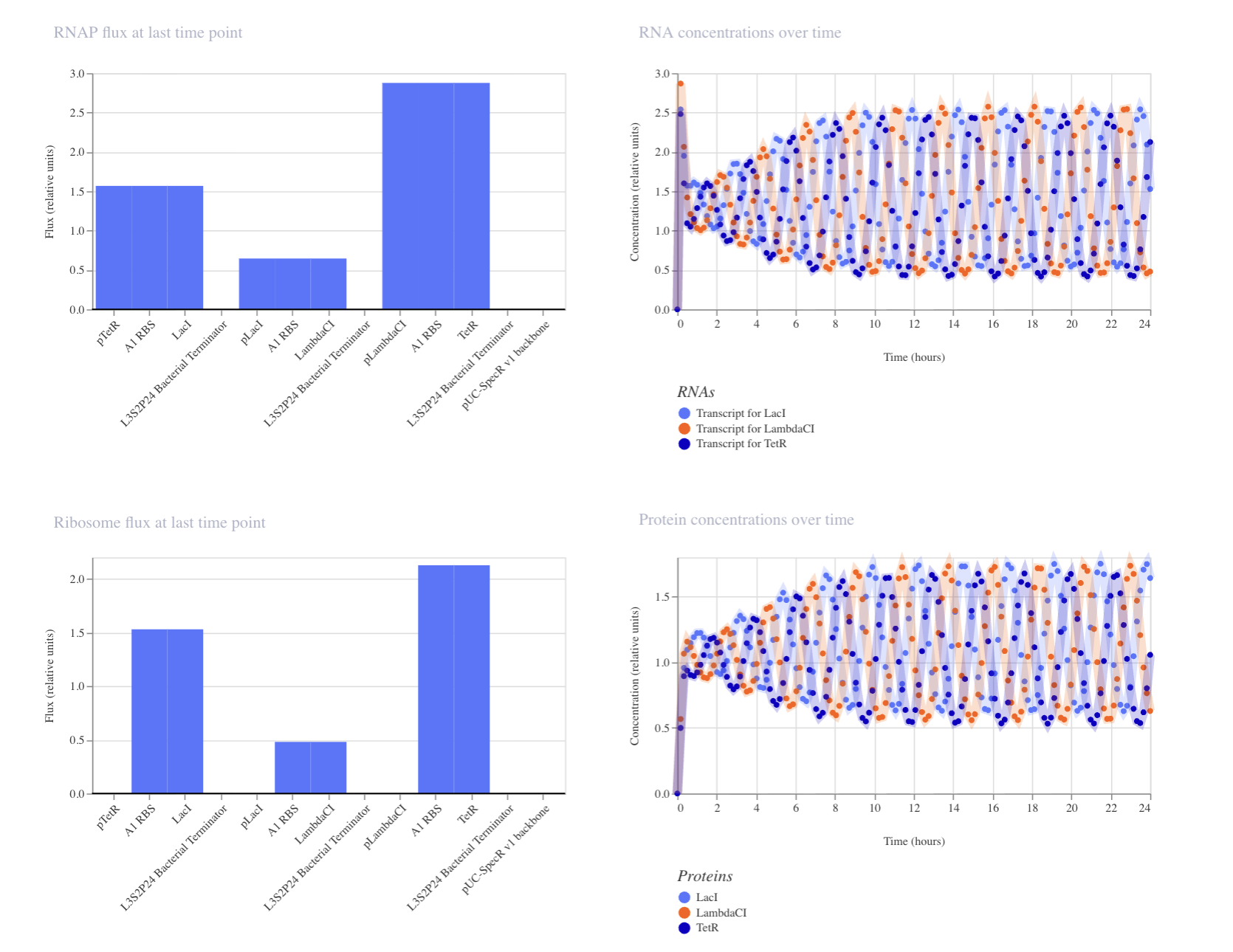
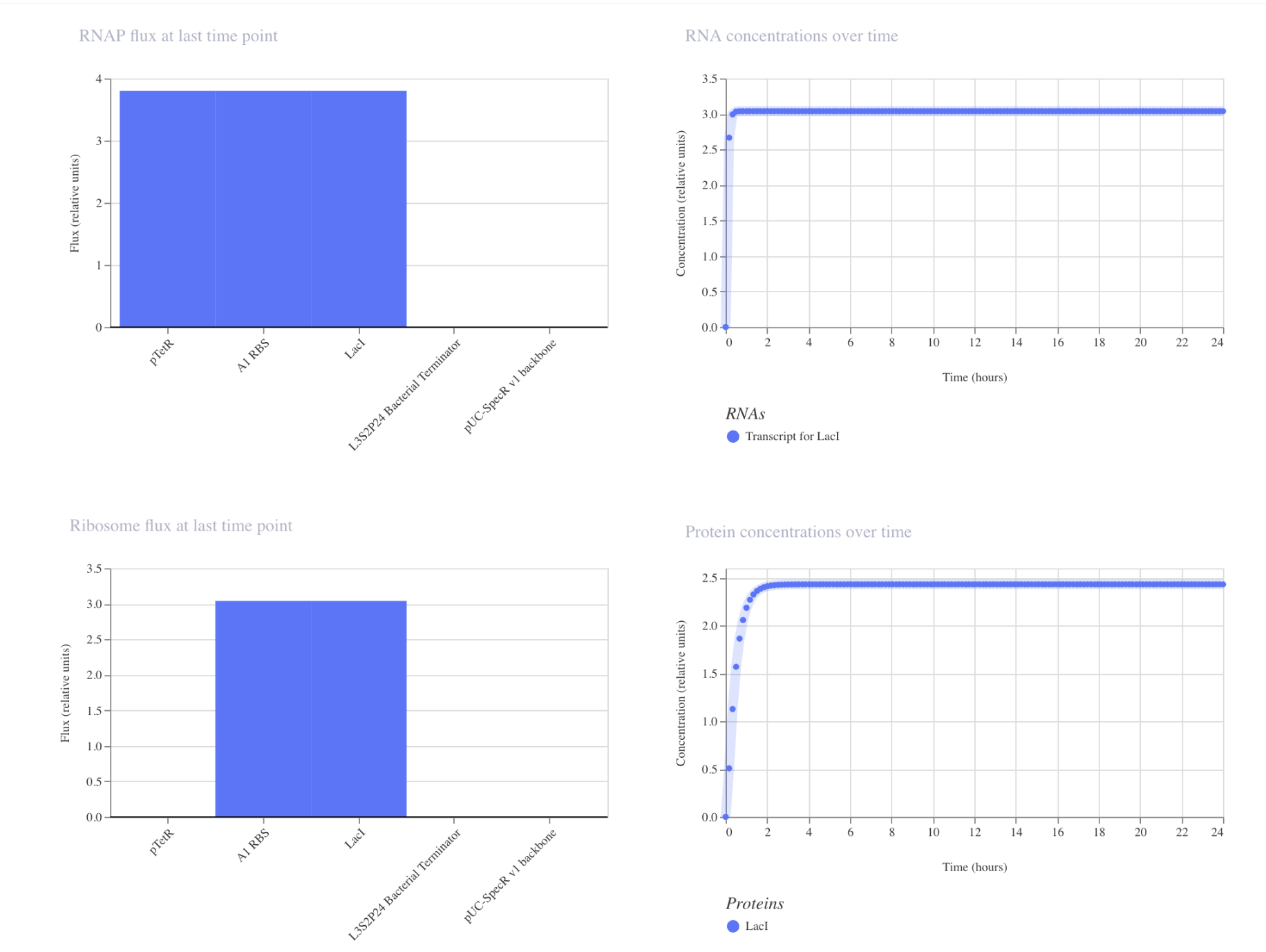
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc)
sfGFP: A very rapidly-maturing weak dimer with a maturation time of 13.6 minutes, allowing for the chromophore to reach its fluorescence state quickly enough to give a strong readout within the CFPS reaction timeframe. It was engineered to fold robustly even when fused to poorly-soluble partners, crucial in cell-free systems.
mRFP1: A slowly-maturing monomer with a maturation time of 60 minutes and a pKA of 4.5, so its chromophore needs significantly more oxidation and time to reach the red-emitting state. In a cell-free system reaction, a lot of the protein produced may still be immature non-fluorescence at readout, underestimating its actual yield. However low pKA means once mature the signal stays stable as the reaction acidifies.
mKO2: Has a maturation time of 108 minutes because of its three-ring chromophore requiring extra oxidative cyclization steps beyond standard chromophore formation. This makes it sensitive to oxygen-limited environments of a close cell-free system tube, where the chromophore maturation may stall or remain incomplete.
mTurquoise2: Strong quantum yield of 0.93 (93% of the absorbed photons are re-emitted as fluorescence) giving very bright readout per molecule even at modest CFPS yields. The kPA is very low at 3.1 making it strong against pH drift from glycolytic acid waste during long incubations, so signal stables stable even as the pH drops.
mScarlet_I: The link to this one gives mScarlet instead of mScarlet_I, so I typed in mScarlet_I in the search, and used that link’s info instead. It’s a rapidly maturing monomer at 36 minutes engineered from its parent’s (mScarlet) mutation. The quantum yield is still pretty solid. The faster maturation allows more synthesized proteins to reach fluorescence state within the finite oxygen timeframe, even if it glows a bit more dim than its parents.
Electra2: A monomeric blue fluorescence protein engineered from mRuby3, achieving a pretty high quantum yield of 0.76. The maturation time is not reported in the link provided.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
You can’t add oxygen to the master mix the same way you’d add nicotinamide. However, you can still engineer oxygen availability to be more readily available for mKO2, since it’s sensitive to oxygen-limited environments. It’s limited due to three main reasons:
Standard chromophore oxidation (every GFP-family FP requires molecular oxygen to oxidize its chromophore into its fluorescence state)
Extra thazoline ring (this requires an additional oxidative cyclization step)
Reaction-tube hypoxia (closed CFPS tubes go hypoxic as the lysate is burning sugar through glycolysis, eating the available O2.
Ways you can add more oxygen:
Use a larger headspace above the tube (run it in a tube where it’s mostly air-volume above the liquid)
Increase surface area, running it in a flat-bottom plate instead of a tube, allowing for better oxygenation.
Put it in a shaking incubator, allowing for more oxygen infusion. You can further improve on this by using breathable seals.
Reduce glucose, meaning less glycolysis and less oxygen consumption.
Add some sort of O2 carrying additives to extend oxygenation.
You could also hypothetically run the reaction in an O2 enriched chamber, or set up some sort of continuous-exchange CFPS with an oxygen permeable membrane. There are many ways you can supplement more oxygen into the reaction, allowing for more efficient mKO2.
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
If ignoring ladder and lane 1, there is the design! I tried to recreate No Face from Spirited Away.
For my homework, I decided to pick the enzyme that is the strongest candidate for facilitating luciferin synthesis. I picked my enzyme from my independent research. First I downloaded Fallon’s paper data from Fallon et al. 2018, “Firefly genomes illuminate parallel origins of bioluminescence in beetles” in eLife (DOI: 10.7554/eLife.36495. This experiment compared gene expression between the fat body (a firefly’s liver) versus the lantern (the organ that makes light) to find which genes are highly expressed in the lantern. I ran a filter in the file PPYR_OGS1.1_fatbody-vs…_test.txt texts, keeping only the statistically significant genes of TPM ≥ 50 and sleuth b ≥ 3. The TPM measures how actively a gene is being expressed in the lantern tissue, a higher TMP signaling a higher likelihood of luciferin expression. The sleuth is the statistical software Fallon used, that estimates log2 fold change. The higher the b sleuth, the more expression a gene has specifically in the lantern than fat body. I also ran qval ≤ 1e-10 to adjust for random noise.
Afterwards, I ran my candidates through Fallon’s enzyme annotation file (PPYR_OGS1.1.enzyme.ids.txt) which lists the predicted enzymes based on Fallon’s work in InterProScan. Although both filters have arbitrary thresholds with likely blind spots, it produced a good starting point.
I then ran my candidates through HMMsearch for function, and BLAST to check if the enzyme is expressed only in fireflies, or all insects (which would point to different, non-applicable functions such as exoskeleton hardening). One candidate was already discussed in Zhang (2020), but has not yet been experimentally validated. The rest are potentially novel candidates not yet found in literature.
I then did reverse BLAST to see if the top-hit organisms point back to their assigned enzyme. This was done to check if they’re true orthologs, or if BLAST was matching generic similarities. This narrowed down my search to four candidates.
I decided to go along with PPYR_02911 as my homework protein for several reasons. Firstly, it’s an oxidative enzyme (P450), which is the class you’d expect in the bottleneck. We know that firefly luciferin requires a benzoquinone precursor, therefore requiring an oxidative step facilitated by an oxidative enzyme. Secondly, it’s a tandem duplicate of one of my other candidates (PPYR_02910), being only 4kb apart. They are 87% identical, pointing to possible gene neofunctionalization. Thirdly, its BLAST hits were almost all bioluminescent species, and its Lantern TPM was the highest of the group. Lastly, the candidate proposed by Zhang (2020) likely facilitates in storage, pointing to a support function. I’m more interested in finding the missing catalytic step.
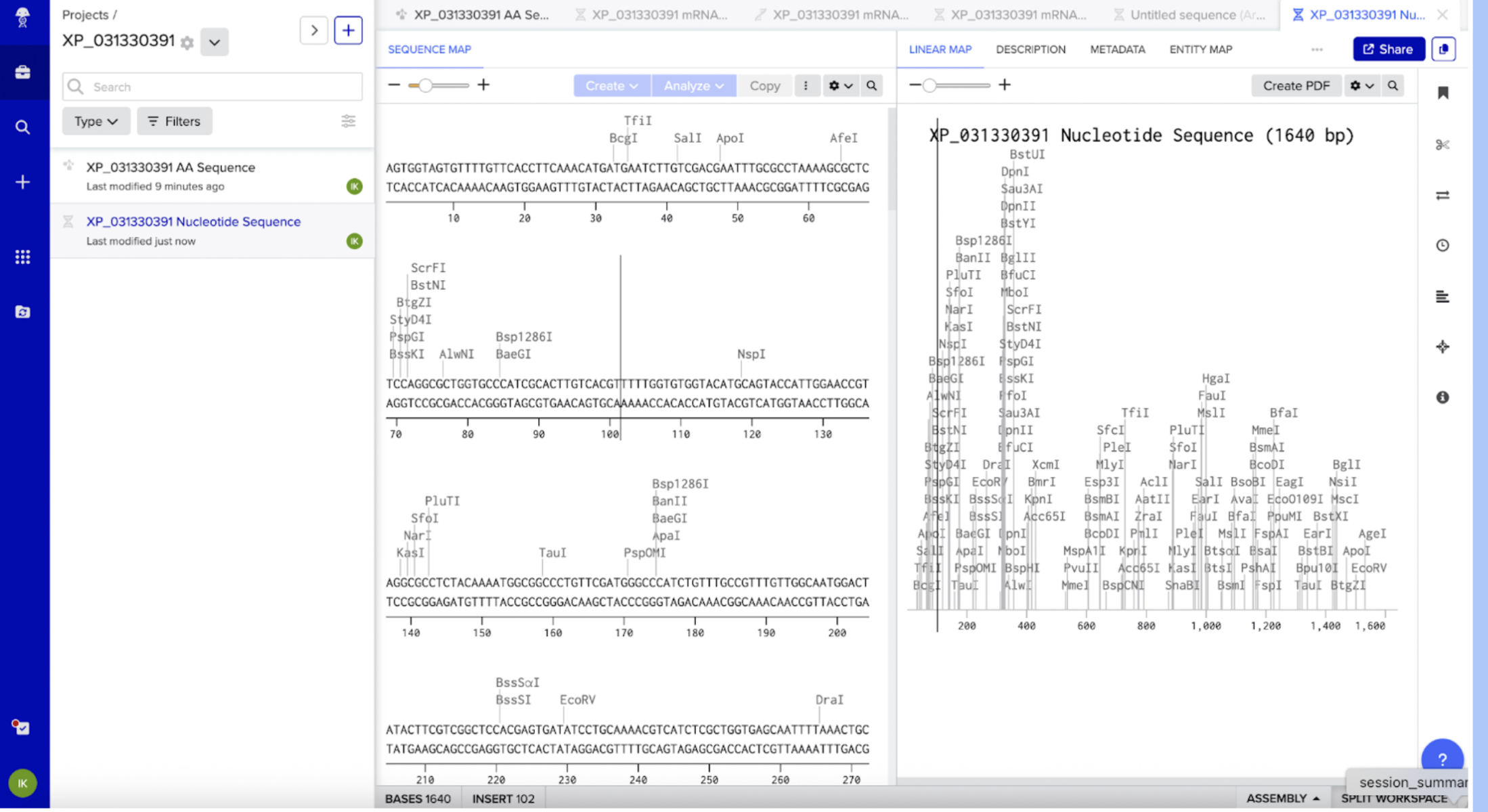
Since I already obtained the protein sequence from my own independent research, I have it on hand from Fallon’s PPYR_OGS peptide FASTA file, which contained custom protein sequences for all the predicted genes in the Common Eastern Firefly. This file was obtained from Fallon’s Github (https://github.com/photocyte/PPYR_OGS). However, for learning purposes, I pasted my PPYR_02911-PA protein sequence from Fallon’s file in NCBI, clarifying Photinus pyralis (the firefly) as my organism. My top hit was the NCBI equivalent, XP_031330391. I cross referenced XP_031330391 with PPYR_02911-PA, and confirmed it is the same gene.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I clicked on XP_031330391 next to the “Sequence ID” in the XP_031330391 protein page in NCBI. From there I clicked on the DBSOURCE to get the DNA sequence, and then FASTA.
Since the same amino acid can be encoded by different codons, certain organisms prefer different codons for the same amino acid. My firefly genes have a different preference than a plant would, therefore I would need to optimize for plants to avoid truncated proteins or low production. I’m specifically optimizing for Nicotiana Rustica. I chose this species over the standard N.Bethamiana because I literally couldn’t source them anywhere in Vancouver, and there’s a slight benefit that my construct results will be more realistic for general plant species. I chose to optimize in GenSmart. Although GenSmart did not have Nicotiana Rustica, I went along with Nicotiana Tabacum as they are closely related. I excluded BsaI since I am planning to use Golden Gate Assembly.
3.4. What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein.
I will be using a cell-dependent approach since my long-term goal is to test this enzyme in plants. First I will use Golden Gate assembly to clone the gene I optimized into an expression vector. I will then heat shock this plasmid into Agrobacterium cells, and then inject the cultures into N. rustiva leaves. The plant will do the rest of the work, transcribing the DNA, and then translating it into the cytochrome p450 protein.
Part 4: Prepare a Twist DNA Synthesis Order
I inserted the optimized linear DNA sequence from GetSmart into Benchling and annotated it to XP_031330391 CDS. Since I am doing plant expression cassette, my DNA inserts will differ. I will need:
CaMV 35S promoter
Start Codon ATG
XP_031330391 Coding Sequence
Stop Codon
NOS terminator
No RBS needed (Plant cells don’t use RBS) and no His tag needed (I won’t need to purify).
Next I searched “CaMV 35S promoter” in AddGenes. I’m not sure how to pick the correct promoter so I had ChatGPT help me narrow it down to pP35S. Considering it says “Provides the CaMV 35S Promoter as a level 0”, it makes sense as a choice. Next I uploaded the GenBank file into Benchling, found the promoter, and inserted it into the beginning of my DNA sequence. I already had the start codon ATG in the beginning of my XP_031330391 CDS. GenSmart did not include a stop codon, so I added TAA.
I searched “NOS terminator” in AddGenes. I inferred lvl0 NOSt was the correct plasmid due to its description saying “Agrobacterium terminator for plant expression”, but double checked with ChatGPT just to be sure. Downloaded the GenBank, uploaded into Benchling, copy and pasted the terminator after XP_031330391 CDS.
From my understanding, I would need Gene Fragments since I am doing Golden Gate assembly, not Clonal Genes. I’ll still pick Clonal Genes for the purpose of this homework, choosing the pTwist Amp High Copy vector.
I would use Nanopore for several reasons. Firstly, I am working out of a community lab, and this is one of the more accessible DNA sequencing technologies (that’s why it was taken to space!). Secondly, I’m still early on in my experimentation. If I need higher accuracy, I’d use Illumina+ or MGI+. Nanopore is third generation as you do not need to amplify your DNA through PCR. Therefore, my input would simply be raw DNA or RNA from my sample. To prepare my DNA for the nanopore, I’d need to extract my DNA and purify it from non-genetic material, before breaking it into smaller pieces for easier reading. Since fragmenting DNA leaves the end pieces jagged, I’d repair any blunt ends and add an adenine overhang so the adapters can grab onto the DNA. Nanopore requires a special adapter ligation onto the DNA ends to physically feed the DNA strand through the machine at a controlled pace. Lastly, I’d pipette my prepared sequence on a small flow cell plugged into the device.
The flow cell has many protein nanopores in its membrane, all of which have an electrical voltage creating a flow through each pore. As the motor feeds the DNA strands through the pores, the bases squeeze through the tiny pore, as each base blocks the current differently due to their slight differences in shapes and sizes. The machine measures these slight differences in the pore currents, creating a pattern based on the current changes and translating them into DNA letters. This simplicity allows the nanopore to be an efficient and cheaper DNA sequencing.
The output is a FASTQ file which carries the sequence and quality score (the confidence each base is correct). There is also a raw signal file of the actual electrical current data for analysis or troubleshooting.
5.2 DNA Write
As mentioned earlier, I’d like to synthesize and write my codon-optimized PPYR_02911 (XP_031330391), as I believe it is a good candidate for luciferin biosynthesis. Out of my non-novel genes, I’d like to synthesize plant-optimized BGL, Laccase ACOT1 and luciferase as they are big players in expressing autonomous bioluminescence in plants.
I’d use Twist’s phosphoramidite chemistry as it is the industry standard, and Twist developed supplementary technology to make it efficient and cheaper. I also love Twist, and want any excuse to use their website. Firstly, single-stranded DNA is built one nucleotide at a time from 3’ to 5’. Each nucleotide is a single cycle, each of which has deprotection, coupling, capping, and oxidation. Once this oligo is built, you cleave it off the solid support it was initially built on. If your gene is longer than a single oligo, Twist assembles them into a single gene with PCR. This gene then gets cloned, inserted into a plasmid vector, double-checked for verification, and shipped. As with any other writing method, there are compounding errors. Furthermore, since oligos get maxed out fairly quickly in length, stitching genes together causes the process to be more error-prone.
5.3 DNA Edit
Although glowing plants are a marvel, I would love my long term goals to be more beneficial for humanity. Plants are integral to our survival, whether it’s due to crops, materials, or our air. They are beautiful and crucial. My dream would be to take my ambition further, whether it’s to engineer nitrogen fixation in crops, help conserve plant species, or make trees more efficient at carbon sequestration. For such ambitions, you’d need the big guns: CRISPR-Cas9.
The steps of CRISPS are as follows. First you design a guide RNA, a short sequence that matches the genome region you’d like to edit. This guide RNA tells the Cas9 where to cut. This is done through Benchling or CRISPOR. You’d also prepare your donor template if using one, a sequence containing your insert with sequences matching the cut site at both ends. Therefore, the inputs are the Cas9 plasmid, sgRNA, donor template DNA and target cells. You then deliver this plasmid into wherever cell you’re utilizing. After the CAS9 makes a double stranded break, you can edit either through NHEJ or HDR. With Non-Homologous End Joining the cell glues the broken strands back together, often introducing small deletions for knocking out genes. Homology-Directed Repair uses the donor template to insert the new DNA at the cut side to knock in a gene. Lastly, verify by sequencing the targeted region.
The limitations to CRISPR: 1. Knocking in genes has a low success rate in plants. 2. The guide RNA might lead the Cas9 to the wrong sequence as sequences may look similar 3. You may have mosaicism as regenerating a whole plant with edited cells is difficult.
Work Cited
Fallon et al. 2018, “Firefly genomes illuminate parallel origins of bioluminescence in beetles” in eLife (DOI: 10.7554/eLife.36495
Ai Citations:
I cannot find N.Bethamiana for my glowing plant project, world Nicotiana rustica work?
[Copy and pasted 4.2 question], what inserts would my version need since I’m optimizing for plants?
Is GAA a stop codon? [Follow-up]: what should be my stop codon?
Is nanopore first, second or third generation?
Explain how a nanopore works in detail. What are the inputs and outputs?
Explain how Twist sequences DNA in detail.
Explain how CRISPR works in detail.
Top limitations of CRISPR in Plant edits
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
After designing in http://opentrons-art.rcdonovan.com/ (a pig with a heart in the centre), I chose to run the Collab notebook for simplicity. I used Claude Code to help generate my coordinates into proper code, before running all.
After my first simulation I noticed “WASTING BIO-INK : more aspirated than dispensed” warnings, so I edited my code. The next simulation still seemed to be wasting ink, but Claude informed me it’s an issue with floating point rounding.
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Since my project requires extensive wet-lab trial and error, I found “AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots” the most applicable paper. DNA assembly is mostly still done by hand, which makes such a meticulous process error-prone. Furthermore, it poses a high barrier to entry for those without lab or educational access. AssemblyTron is a free, open-source Python package you download for the Opentrons OT-2 that is optimized for DNA construct design protocols including: PCR setups, Golden Gate Assembly, Homology-dependent assembly, and more. The Opentron handles a wide range of tedious and time-consuming processes, allowing for better time-management and faster research progress. Lastly, as someone with a diagnosed hand tremor, I struggle constantly with contamination and resource waste (ex. repeatedly grazing tips) in the lab. This would be immensely helpful for individuals who struggle with fine-motor skills, especially for delicate procedures such as cloning methods.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
My plan requires a four-gene bioluminescence pathway through Golden Gate assembly. These are novel genes in plants, and will inevitably require trouble shooting via different combinations of promoters, luciferase variants, targeting signals, etc. Instead of assembling one construct at a time, (a real time-related bottleneck in this project), I could set up dozens of constructs in parallel. An example pseudocode could be as follows:
# Combinatorial Golden Gate Assembly for bioluminescence pathway# Testing 3 promoters × 2 targeting signals × 4 genes = many combinationspromoters=["CaMV_35S","UBQ10","NOS"]targeting=["PTS1","PTS2"]genes=["BGL","laccase","ACOT1","Ppyr_luciferase"]foreachpromoterinpromoters:foreachsignalintargeting:# OT-2 assembles the four-gene construct:pick_up_tip()aspirate(backbone_fragment)aspirate(promoter_fragment)aspirate(gene_cassette_with_signal)aspirate(BsaI+T4_ligase_master_mix)mix_and_dispense_into(thermocycler_plate)drop_tip()# Run Golden Gate thermocycler protocol# Transform into Agrobacterium# Infiltrate N. benthamiana leaves# Image bioluminescence after 3-5 days
Work Cited
Bryant, J. A., Kellinger, M., Longmire, C., Miller, R., & Wright, R. C. (2023). AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots. Synthetic Biology, 8(1), ysac032. https://doi.org/10.1093/synbio/ysac032
Ai Citations (Claude 4.6)
Generate [coordinates] into this code [your code section in Collab notebook] for my opentrons project.
Edit the code to not waste bio-ink [attached screenshot]
Is it still wasting ink? [attached screenshot]
What are the cutting edge Opentrons use cases in synthetic bio right now?
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
A dalton is a unit of mass defined as 1/12th the mass of an unbound neutral Carbon-12 atom at rest, equivalent to 1.66X10-24 grams. It is commonly used in molecular biology to calculate the mass of large molecules into grams. Meat protein is roughly 25% protein by mass, therefore 500g x 0.25 = 125g of protein since almost all amino acids are concentrated in the protein of meat.
Given the formula:
N = 125g/100 g/mol x Avogadro’s number = 7.5 x 1023 amino acid molecules
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because we all use the same amino acids as building blocks, but our DNA tells us how to use these building blocks differently. We digest the proteins into individual amino acids, after which our DNA rebuilds them into human proteins, not cow or fish proteins.
3. Why are there only 20 natural amino acids?
As mentioned in previous lectures, the true number of combinations possible through codons is vast. However, this makes life less efficient and wastefully redundant. Instead, nature has evolved to produce 20 amino acids, leading to more efficient translation, error tolerance (as multiple codons map to the same amino acids), while maintaining diversity.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, and this is an active field in synthetic biology, as long as you keep the universal backbone intact: An amino group, a carboxyl group, a central carbon, and a hydrogen on the alpha carbon. The creativity happens in a new R-group side chain. For example, while upholding the universal backbone, I could design a D-luciferin as the R-group, allowing for a possibly self-fueling luciferase, where the side chan is being used as substrate for the luciferase (no external luciferin required).
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids have the ability to form spontaneously through chemistry without the aid of enzymes, as proven by the Miller-Urey Experiment (1953), and observed from meteorites and hydrothermal vents. The theory follows that after this spontaneous accumulation of amino acids, peptides began to form, catalyze, and eventually self-replicate, eventually evolving into increasingly more efficient and controlled life.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An alpha-helix is one of the most common shapes proteins tend to fold into, where it winds into a corkscrew shape while held together by hydrogen bonds between the C=O of one amino acid and the N-H of the amino acid 4 resides ahead. The R-group sticks out from the helix (the side chains pointing away from the spiral). D-amino acids are the mirror image of the L-amino acid. Since the L-amino acids produce right handed alpha helices, D-amino acid would be the left handed version. These are less common in nature.
7. Can you discover additional helices in proteins?
Yes, in addition to alpha helixes, there can be other types of spirals/protein helices. The differentiation comes down to how far apart the hydrogen bonding partners are, as long as they remain within the physical constraints of the peptide backbone limits. However, with synthetic amino acids, you could hypothetically design helices with differing geometries than those found in nature, if they maintain the minimum stability required for the backbone.
8. Why are most molecular helices right-handed?
There are two layers to this question. The first is “why” life chose L-amino acids, to which the answer is unclear. It could have been random, inherited bias from meteorites, or a small difference in nuclear force. As life chose a preference for L-amino acids, biology adjusted to fit this specific type of arrangement, where folding is more energetically favourable due to less steric clashes between the R-group and the backbone. Similar to a nut and bolt, where the wrong bolt (R-amino acid) has trouble twisting into a nut created for L-amino acids.
9. Why do β-sheets tend to aggregate?
A beta-sheet is the other main secondary structure alongside the alpha helix (different shapes found within the same protein). Unlike the alpha helix that form into spirals, the protein chains extend into flat strands side by side, similarly held together by hydrogen bonds. They tend to aggregate because beta sheets have exposed edges of hydrogen bond donors and acceptors (N-H and C=O), unlike the alpha helixes where all the hydrogen bonds have partners. These sticky edges lead to continued chain reactions of recruitment, while the flat shape creates hydrophobic surfaces that stack on each other.
10. What is the driving force for β-sheet aggregation?
The driving force comes down to thermodynamics. Nature picks whatever is more thermodynamically favourable. Exposed edges wanting bonding partners and hydrophobic interactions lower the free energy of the system. Aggregations make the system happier and stable, as the bonds find partners, and the sheets stack on each other, hiding themselves from water. If something is more thermodynamically stable, it is inevitable.
11. Why do many amyloid diseases form β-sheets?
Amyloid diseases are diseases where proteins misfold into beta sheets and clump together into large amyloid fibrils. These fibrils damage and kill surrounding cells and tissues. They tend to fold into beta-sheets because beta sheets are the most stable misfolded state a protein can create. This is because any protein has the capability to create beta sheets (since it requires backbone atoms all protein have), the hydrogen bonding along the edges and hydrophobic stacking create a low energy structure that is very difficult to break apart, and they are self-propagating (as the edges recruit more strands, causing other proteins to misfold and join).
AI Searches:
What is a dalton?
What is the formula for calculating the number of amino acids in a given mass?
What percentage of meat is protein?
Can you make other non-natural amino acids? + what is required to make a new amino acid?
Where did amino acids come from before enzymes?
What is an alpha helix?
What are d-amino acids?
Are there other types of helices in proteins except alpha helices?
Why does nature / prefer L-amino acids?
What is a β-sheet? Why do they aggregate?
What are amyloid diseases? Why do they tend to form beta-sheets?
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
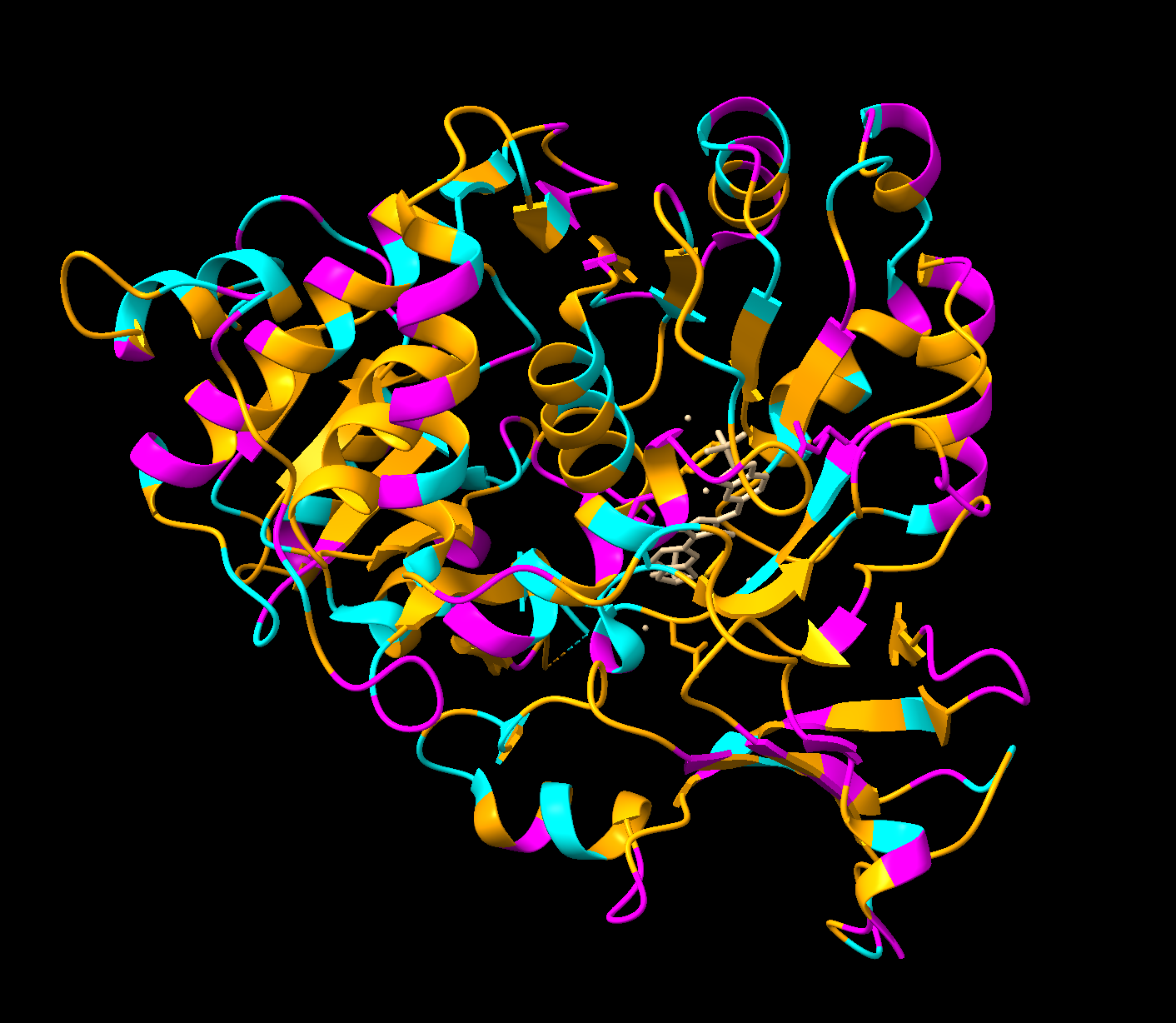
The protein I selected is firefly luciferase, as it is most applicable to my project. Its UniProt ID is P08659. The format biochemical name is Luciferin 4-monooxygenase (Luciferin = substrate it acts on, 4-monooxygenase = adds one oxygen atom at the 4th position).
How long is it? What is the most frequent amino acid?
The length is 550 amino acids, and the mass is 60,745 Da. The most common amino acid is L, which appears 52 times.
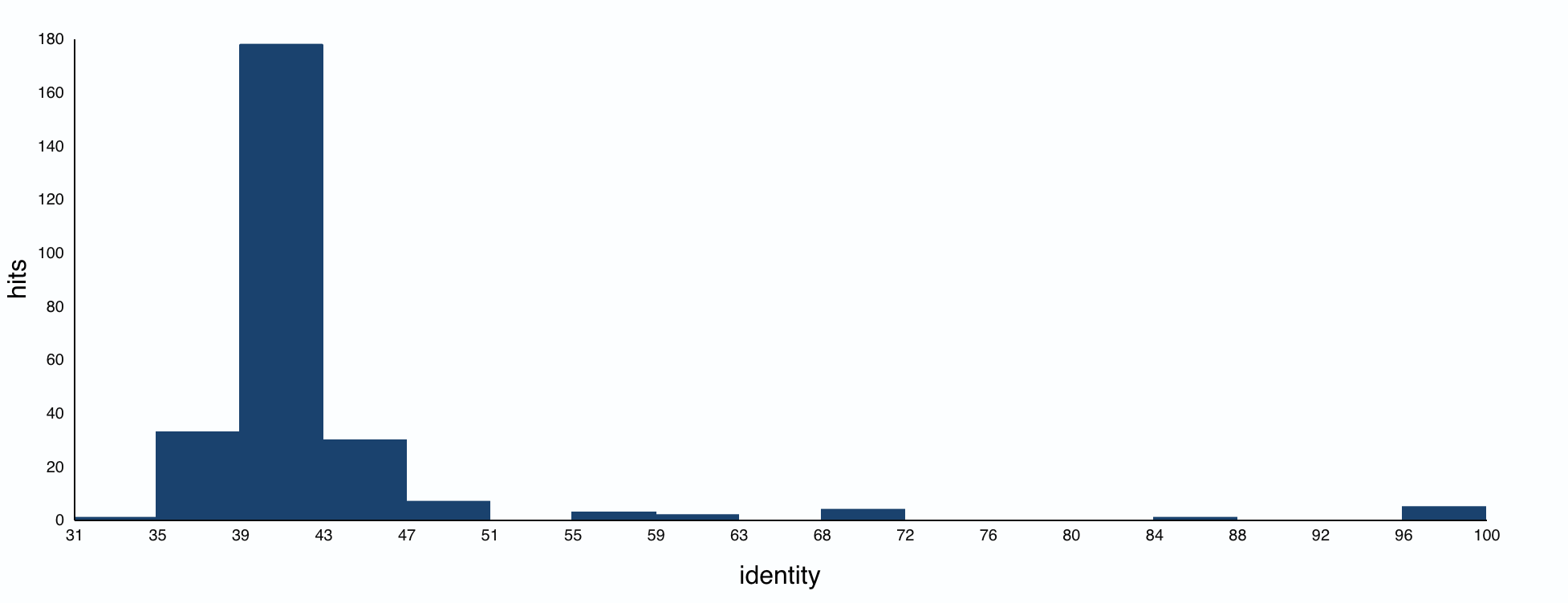
How many protein sequence homologs are there for your protein?
The BLAST search found 250 protein homologs, including luciferases from various species. Most identities were quite low, with a small cluster at 95%+.
Does your protein belong to any protein family?
The homologs are all annotated as Luciferin 4-monooxygenase. The annotations for individual homologs generally mention ATP binding and CoA-ligase activity, pointing to an AMP-binding enzyme family.
Identify the structure page of your protein in RCSB
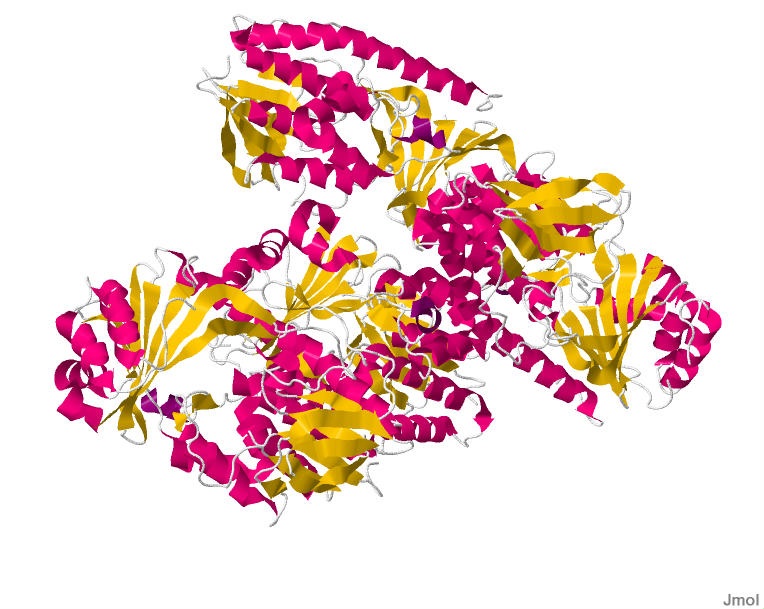
The smallest PDB under Structure is 1.70Å, 3RIX.
When was the structure solved? Is it a good quality structure?
The released date is 2011-12-07. Since the resolution is 1.70Å, it’s a good quality structure since lower resolution means more detail.
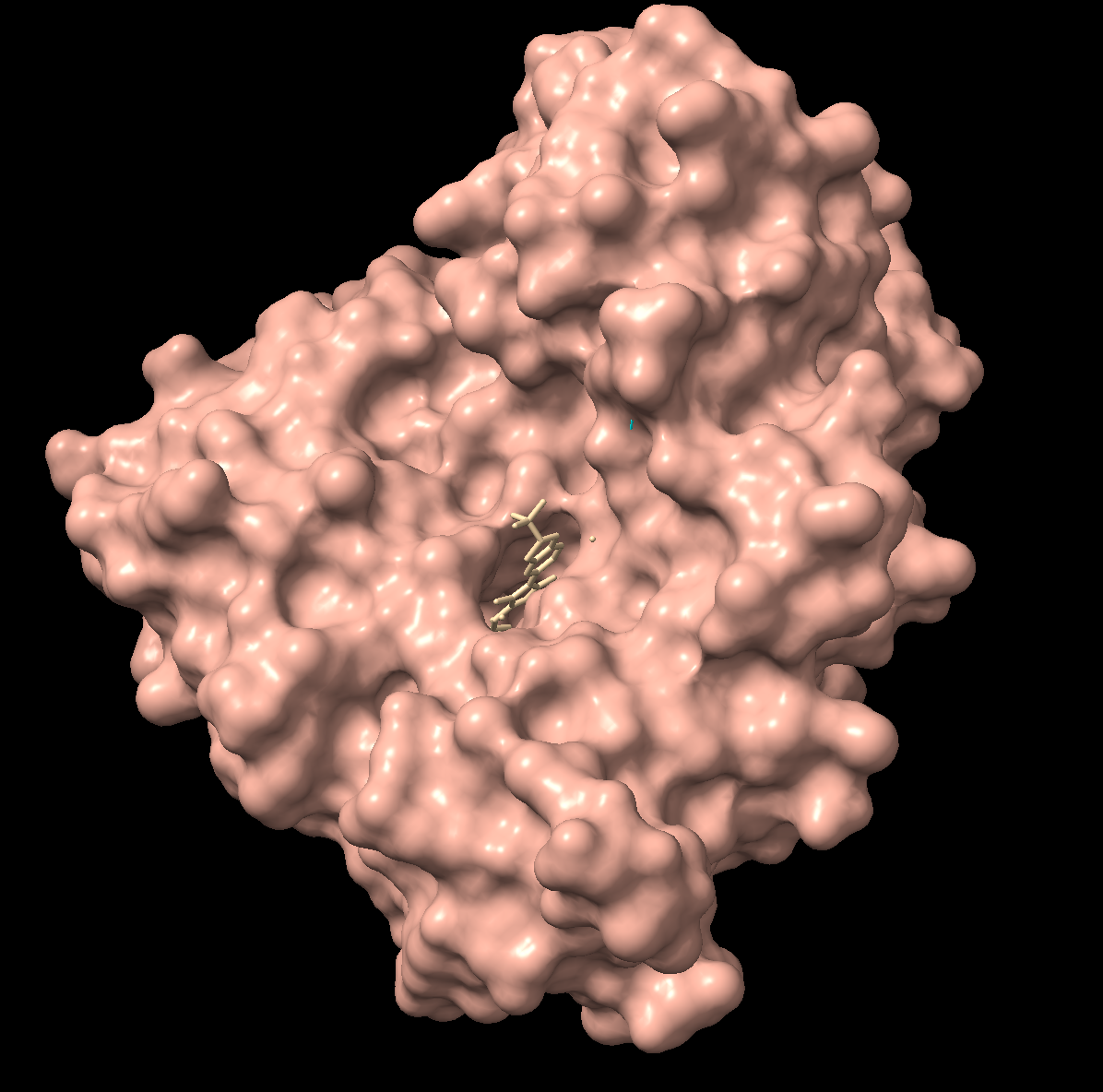
Are there any other molecules in the solved structure apart from protein?
There is one ligand in the structure, listed ID 923. It appears to be a complex organic molecule, and some sort of luciferin or substrate analog based on the ring system. Its formula is C₂₇H₂₈O₇.
Does your protein belong to any structure classification family?
Yes, the luciferase belongs to the Acetyl-CoA synthetase-like superfamily. It shares a 3D fold to other AMP-binding enzymes (enzymes that bind and use AMP), CoA ligases (attach CoA to molecules), and Acetyl-CoA synthetase (help metabolize fatty acids). This is likely because luciferase is theorized to have evolved from an ancestral CoA ligase enzyme that was repurposed for bioluminescence.
Open the structure of your protein in any 3D molecule visualization software:
Because I’ve already used ChimeraX in my research, I chose it as my molecular visualization software.
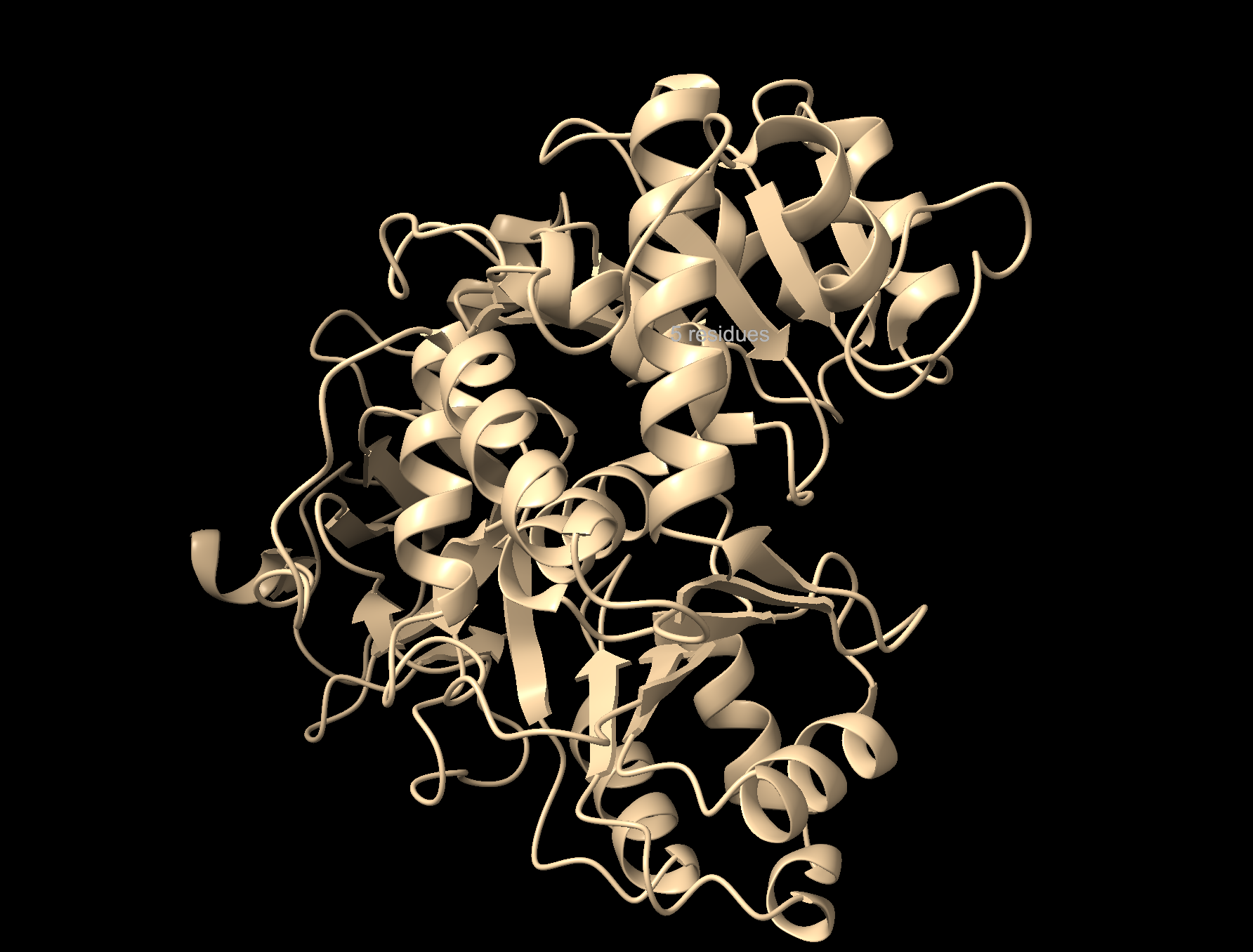
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
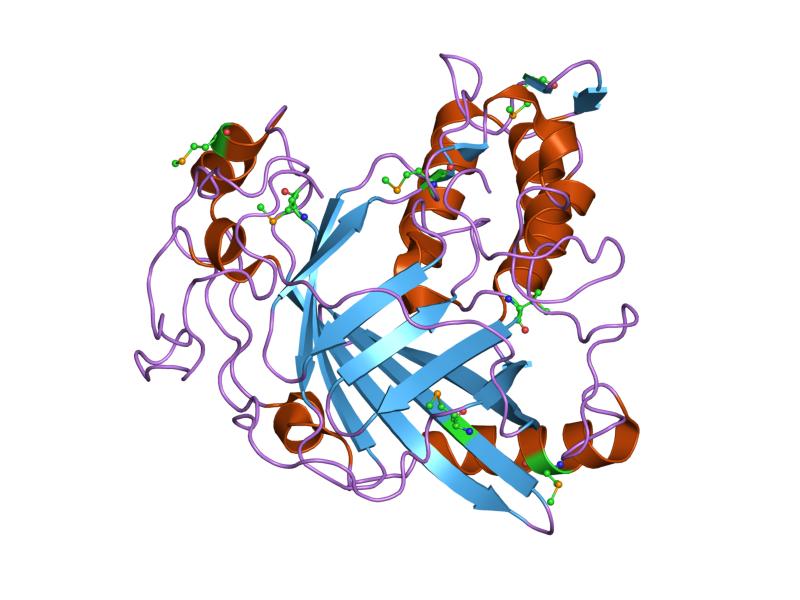
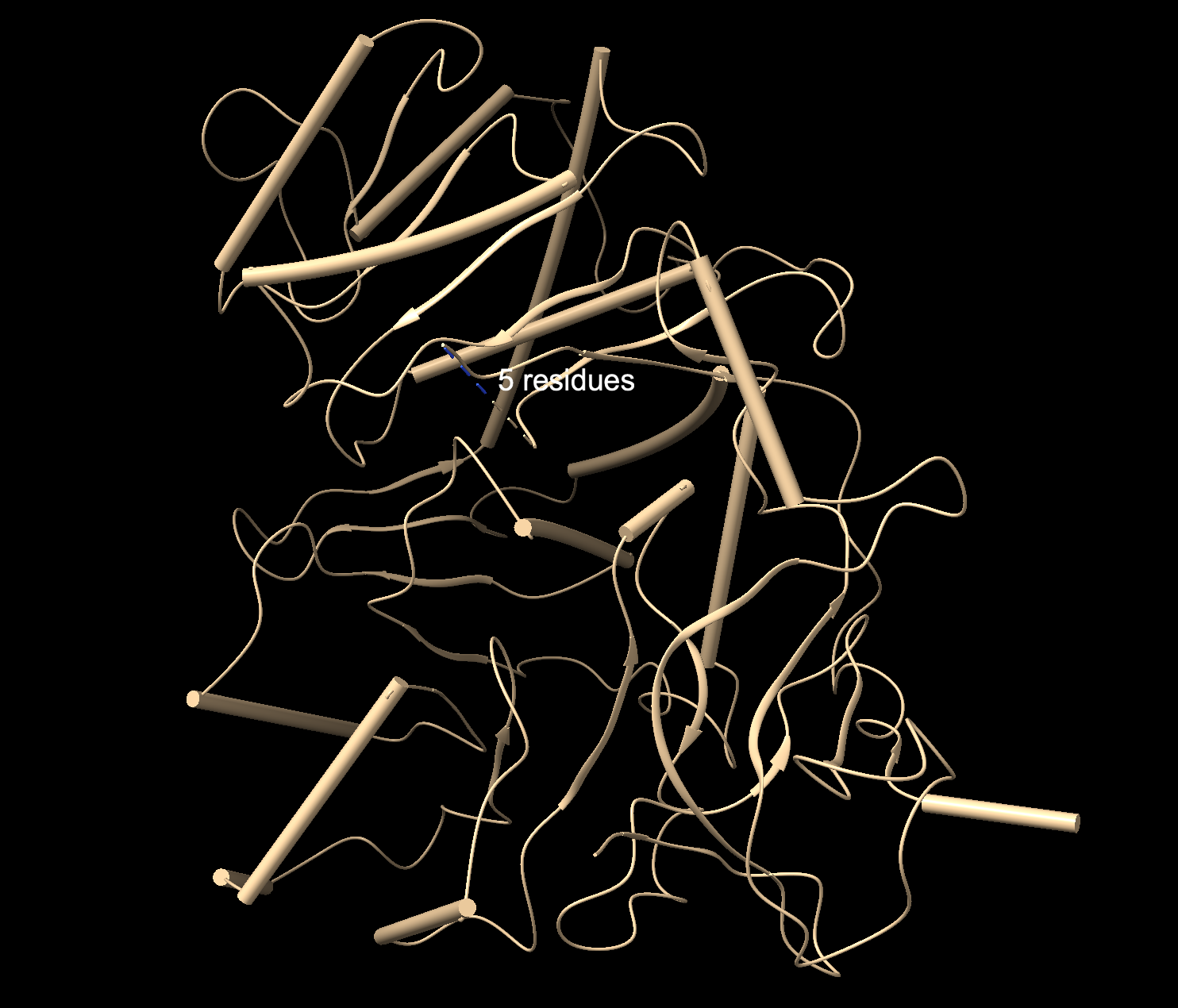
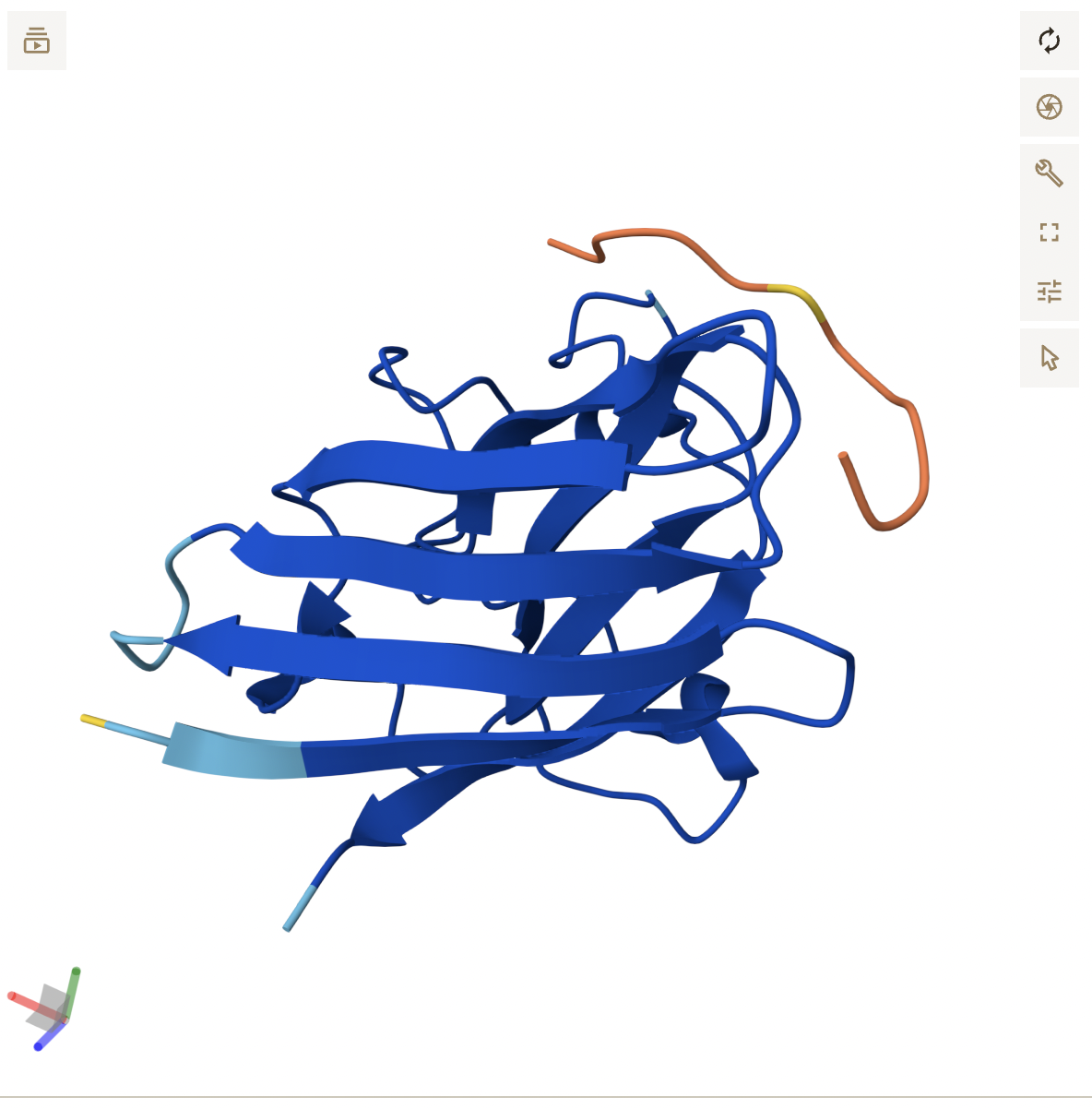
Cartoon: Overall shape and secondary structures. Luciferase has predominantly curly ribbon shapes (alpha helices) and some flat arrows (beta sheets). It’s a large protein, and quite complex. There seem to be multiple domains.
Ribbon: Backbone seen more clearly. Thick cylinders are alpha helices, flat sections are beta sheets, thin threads are loops.
Ball and stick: Shows every atom and bond in protein colored by element. As expected, the core is made out of lots of beige (as it is carbon making up the backbone and side chains). Red dots are oxygen atoms, blue dots are nitrogen atoms. Few yellow spots present, showing sulfur atoms (from the cystine and methionine residues).
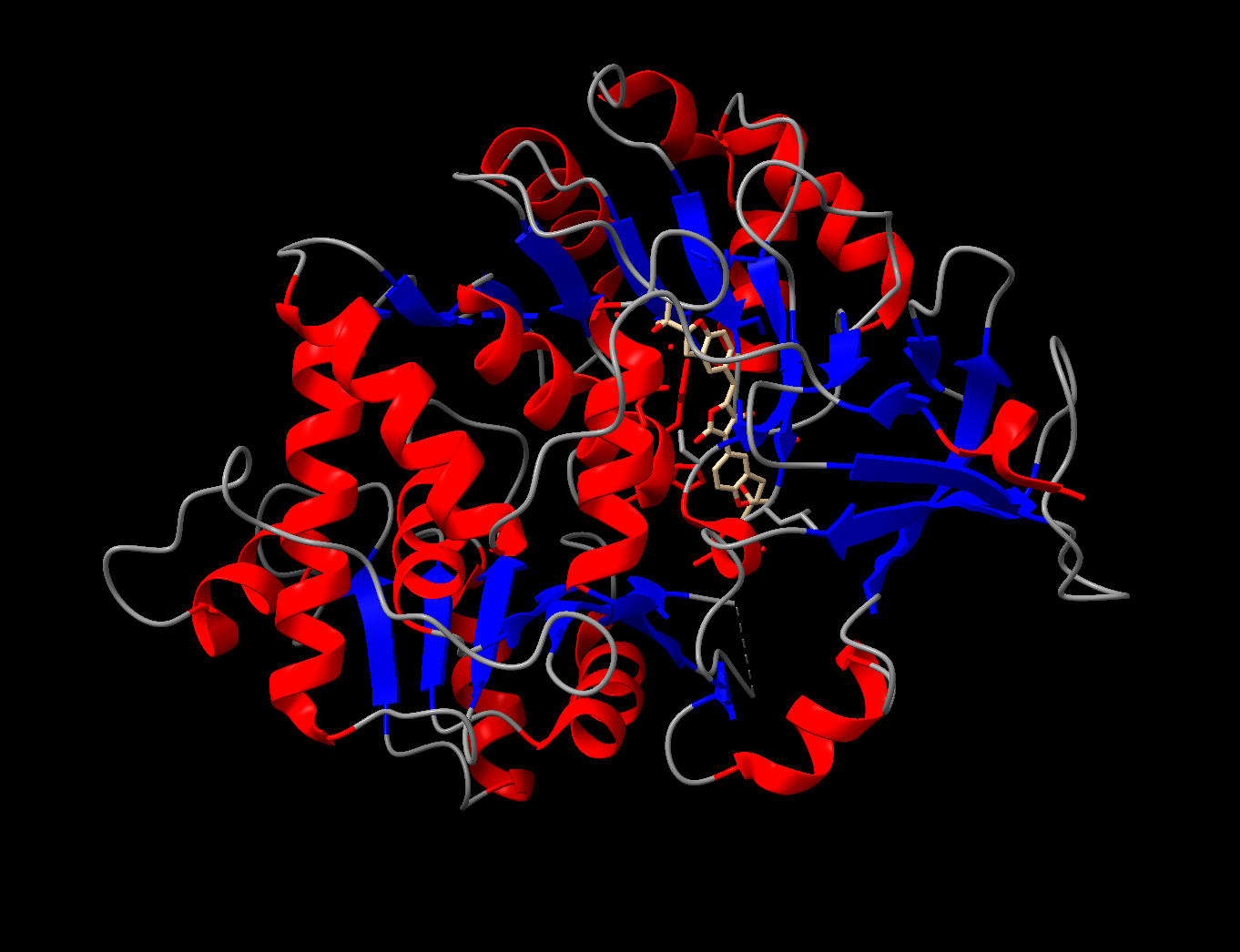
Color the protein by secondary structure. Does it have more helices or sheets?
It appears to mostly have alpha helices, although there are a good amount of beta sheets present. You can also clearly see the substrate analog (ligand 923) from earlier in the active site. That’s where the luciferin would bind.
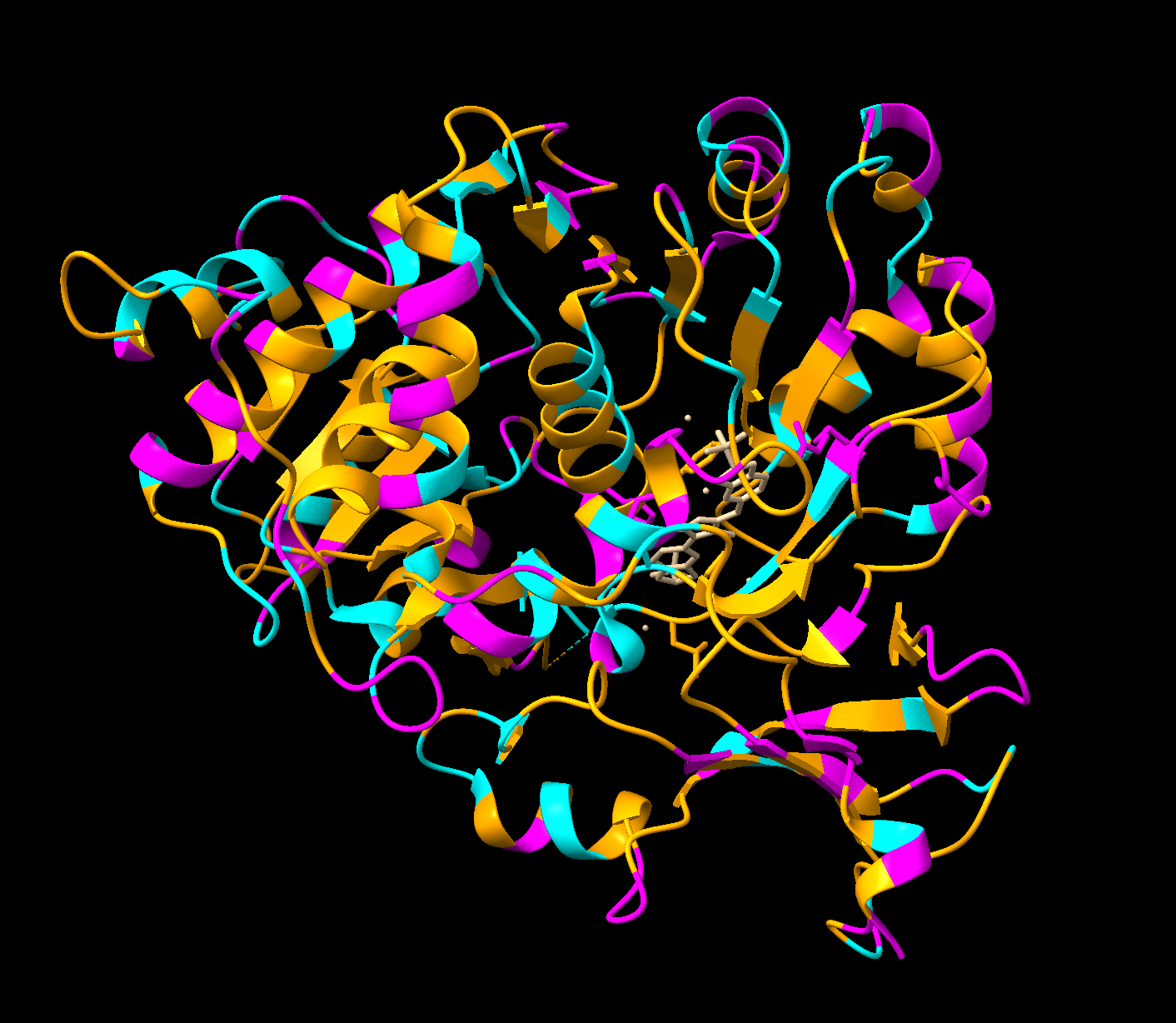
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Orange = hydrophobic residues
Cyan = polar residues
Magenta = charged residues
The hydrophobic residues are concentrated in the core of the protein, while the hydrophobic residues are concentrated in the outer loops and surface. This makes sense for soluble proteins (which luciferase is as it floats freely in water inside our inside cells), hydrophobic residues pack deeper to avoid the water while the hydrophilic face outwards.
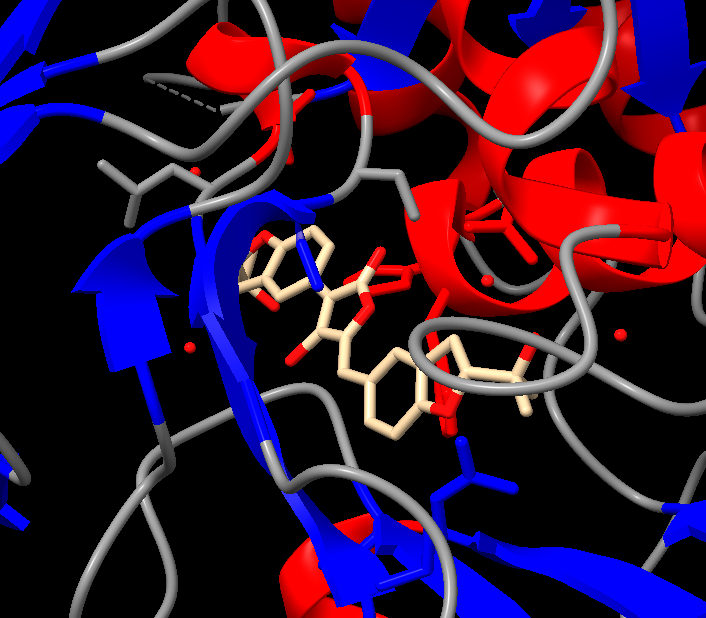
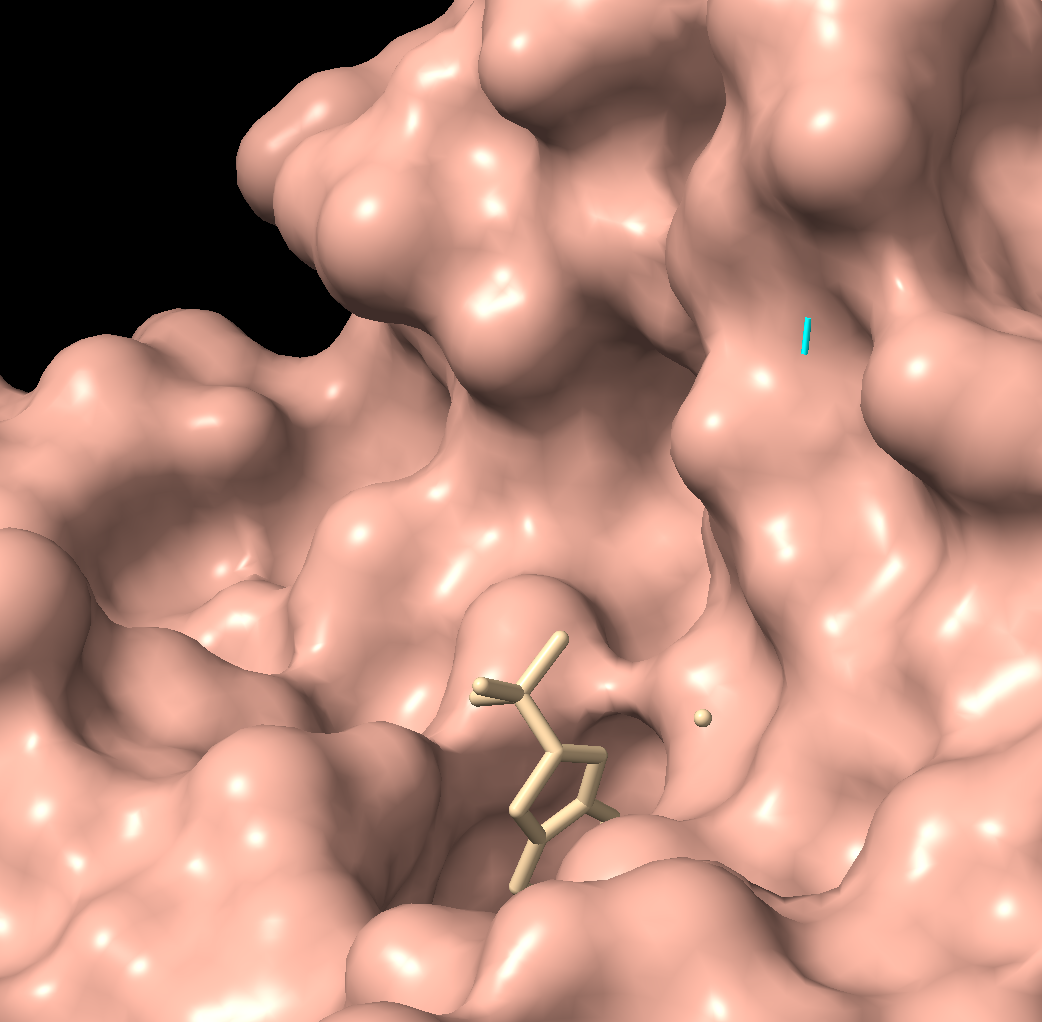
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
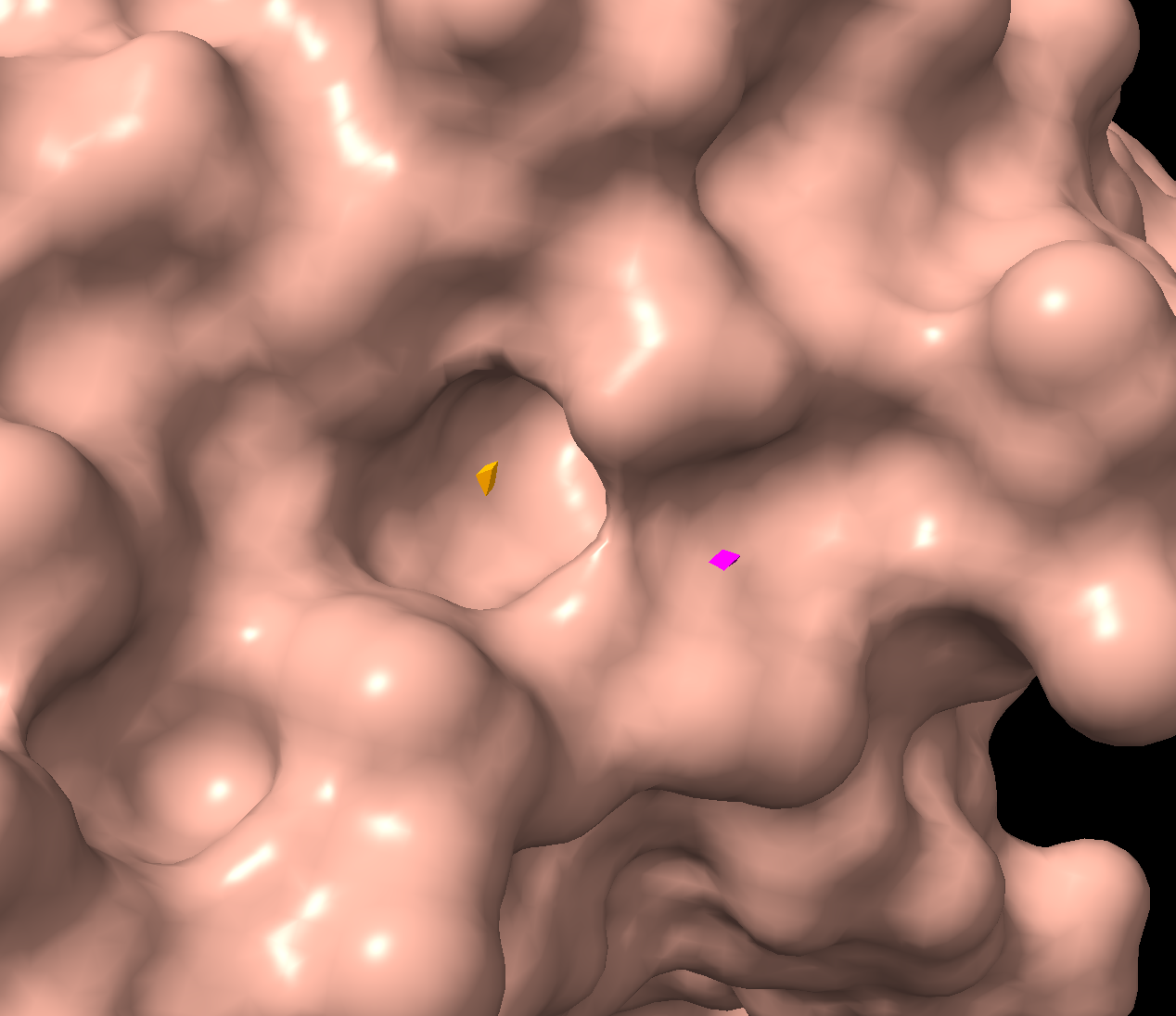
There is a clear, large binding pocket (the active site) where the D-luciferin would bind, and allow for the light-producing reaction. You can see a polar residue poking out near the active site, probably to help position the substrates through the hydrogen bonds. There are three water molecules near and inside the active site, likely involved in the reaction and guiding the substrate into the pocket. On the other side there also seems to be Valine 365 and Lysine 9 poking out of a smaller cavity entrance. Upon investigating, this region seems to be overwhelmingly hydrophobic, and does not seem to be connected to the active site. Could be a remnant of the original fatty acid binding site. Could affect how enzymes behave in plant cells where lots of fatty acids float around.
AI:
Used some help from Claude prompting ChimeraX.
Part C. Using ML-Based Protein Design Tools
Protein used: 3RIX (luciferase)
C1. Protein Language Modeling
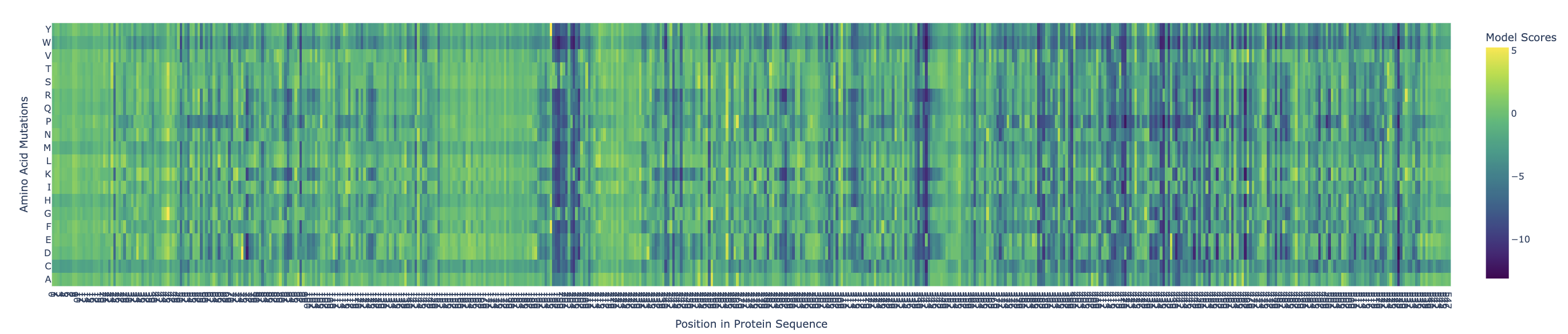
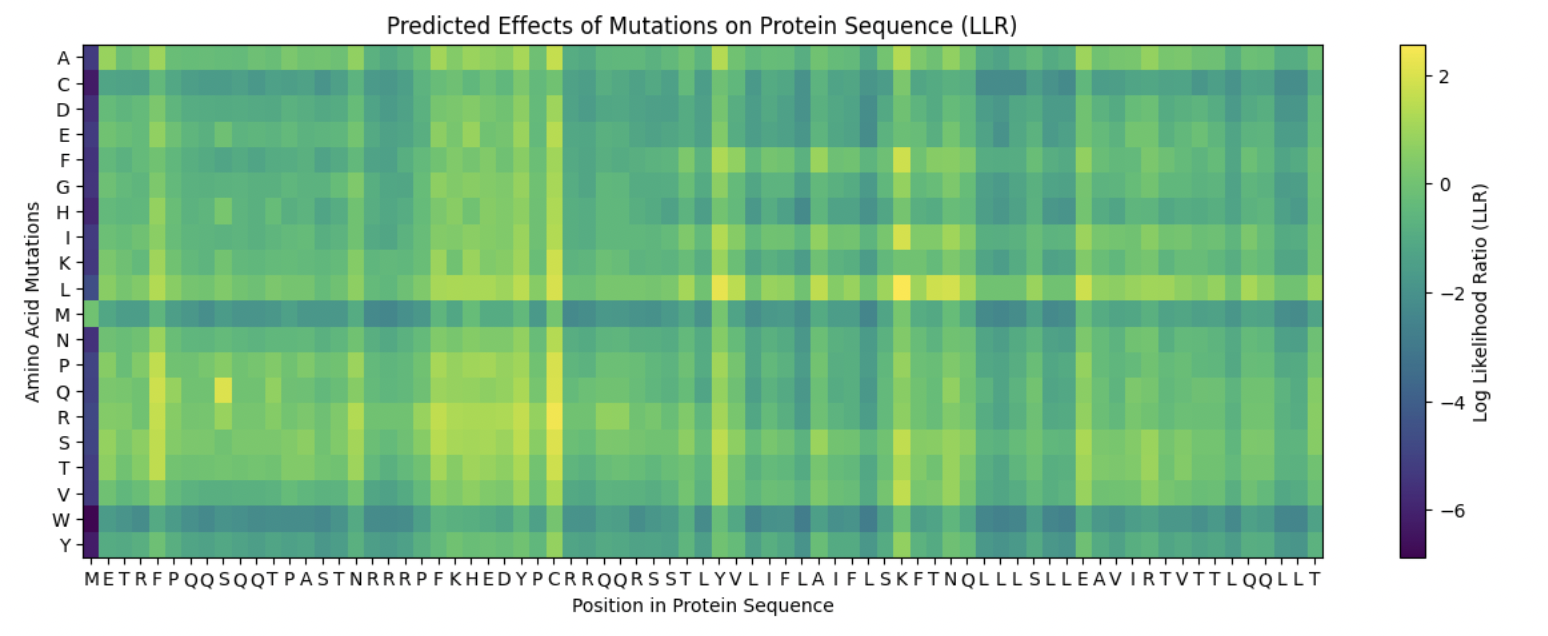
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Yellow/green (positive) = mutation is tolerated or beneficial
Teal/cyan (near zero) = neutral
Dark blue/purple (negative) = mutation is damaging
There are definitely ranges where mutations are more critical, such as:
X = 196-206
X = 338-343
X = 420
X = 435-438
X = 468
X = 527
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Examples:
x: 198, y: W, z: -11.7
Position 198 is Serine (S) a small, polar amino acid, while Tryptophan (W) is the largest amino acid. There may be no room for the Tryptophan, or is too hydrophobic, causing large disruptions in local hydrogen bonding networks.
X: 527, y: W, z:-12.0
This is near the integral C-terminal domain, which is even smaller and tightly packed. Mutating the amino acid to Tryptophan would cause large disruptions during the integral catalysis rotation phase.
X: 342
At this site nothing is tolerated except Glutamate (E) which scores neutral, even though the initial position is at Leucine (L). These are different chemically, so only E being tolerated is highly unusual for such different amino acids. This might be a limitation of the language model.
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Using Discovery of Red-Shifting Mutations in Firefly Luciferase Using High-Throughput Biochemistry, we know the experimentally validated mutations are:
N229T: red-shifts emission above 600 nm. Z = 0.06, correctly predicted these are tolerated.
T352M: red-shifts emission above 600 nm. Z = -2.4, partially wrong, experimentally enzyme still works
L286V: causes red emission
ESM2 correctly identified N229T and L286V as tolerated mutations, but scored T352M incorrectly as moderately damaging, highlighting the limitations of language models in distinguishing mutation effect.
Latent Space Analysis
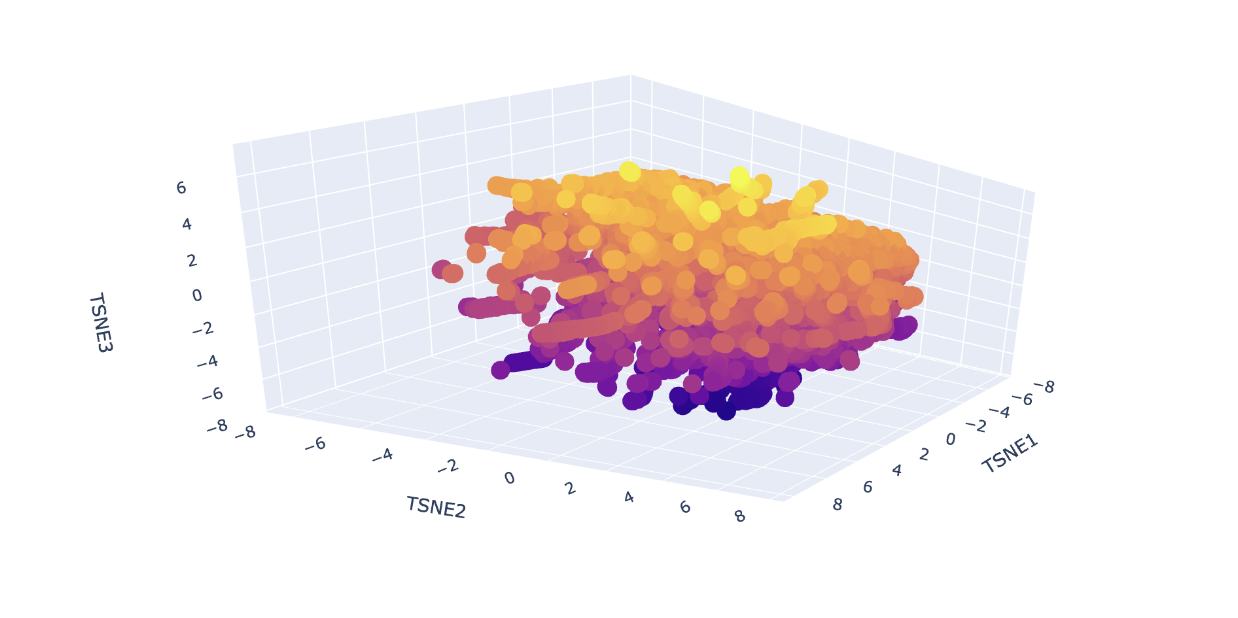
Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, I can see the clustering of proteins by similarity, indicated by colour. The colour groups seem to show the model can distinguish the different protein sequences. For example, at around TSNE1 = 4, TSNE2 = - 4 and TSNE3 = -5, we see a similar type of protein from different species (humans, rats and flies) all clustered together. This similar protein is Staphylococcus aureus Protein A which binds to mammalian antibodies, hence the clustering.
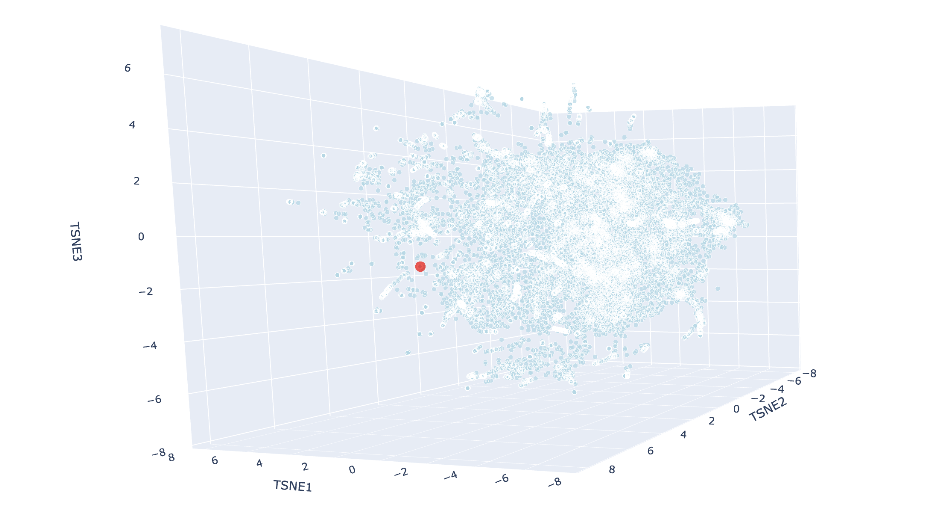
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Using Claude Code, I ran a new cell where I added my protein’s embedding to the already-computed data and rerun just the t-SNE part to save time. The red luciferase was made to appear as a big red dot. The neighbors were:
These all share the SCOP classification of c.23.5, meaning they are all in a similar fold family. Most of them are also flavodoxins, which make sense as all these (including luciferase) are nucleotide-binding enzymes, and share similar alpha and beta folds.
C2. Protein Folding
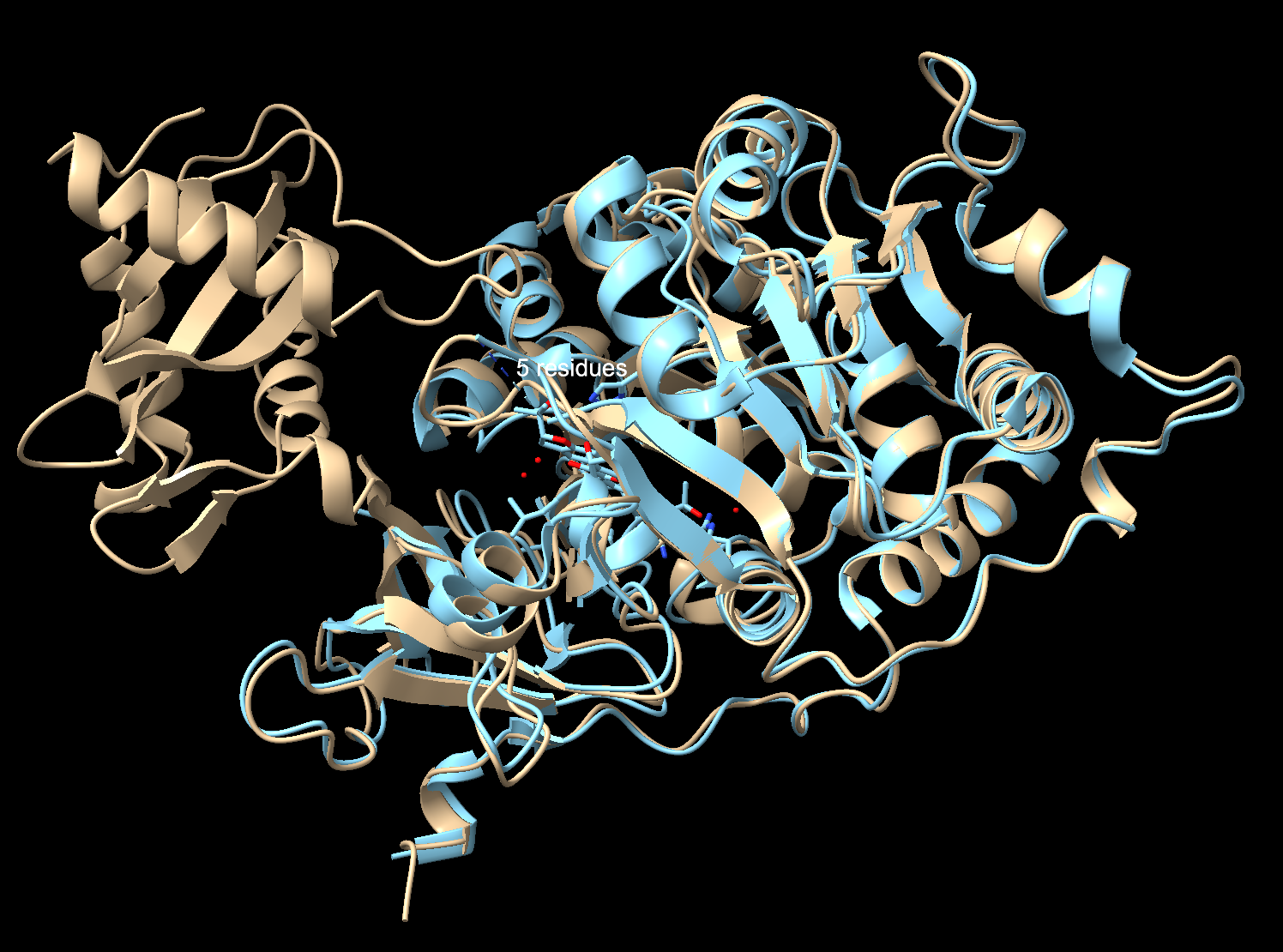
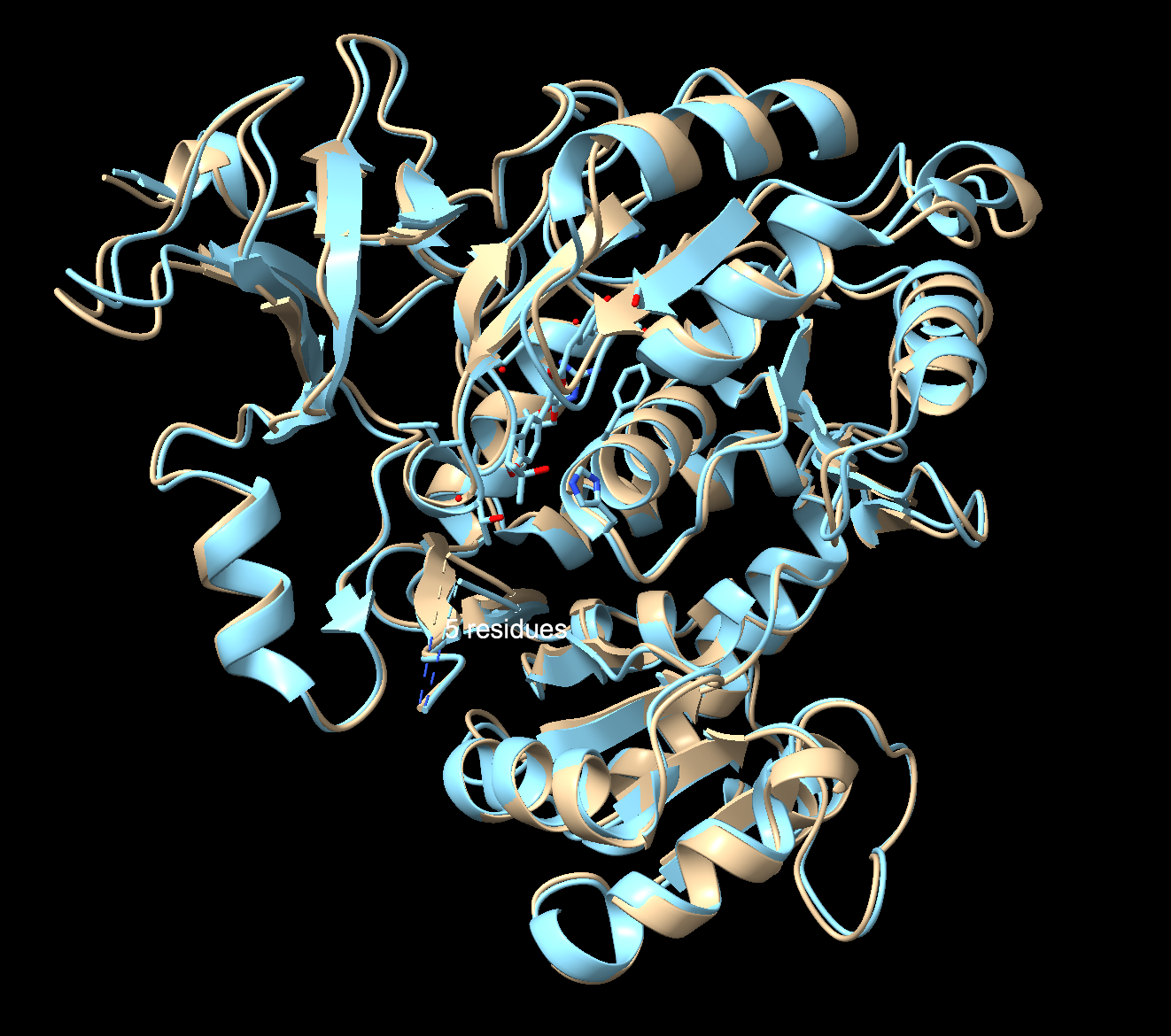
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
pTM: 0.901: Very high. ESMFold is extremely confident in the overall fold
pLDDT: 90.4: Excellent. Per-residue confidence is very high (above 90 out of 100)
To check I compared my ESMFold prediction to the real crystal structure in Chimerax. I used Claude Code to help me download the files from the notebook, and then superimpose them with my previous file in ChimeraX. Overall, they match well. The tan is the 3RIX crystal structure, the blue is the ESMFold prediction. The N-terminal matches almost exactly, where the helices and sheets overlap very well. The C-terminal has an offset between the two structures. That’s because the C-terminal domain moves catalysis, so the 3RIX only captured it in one conformation with a substrate analog bound (the protein with a molecule in the active site), while the ESMFold predicted it without the ligand in the unbound state (protein by itself with nothing bound to it). The former forces the C-terminal into a different position where it is practically closed, while the latter keeps the protein in a relaxed state.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Starting with a small mutation (Serine 198 to Tryptophane from the heatmap):
pTM: 0.905
pLDDT: 90.692
This is a very small difference from the original scores, meaning ESMFold predicts the fold is not affected by this single mutation, even though it also scored this mutation as “highly damaging” earlier. It may be that although the protein continues to fold directly, the function of the protein may be broken, and would not catalyze the reaction properly. This is a limitation of the ESMFold, where the fold may be said to occur “successfully”, but the protein is no longer viable in its function.
Removing a loop (positions 280-289):
pTM: 0.937
pLDDT: 92.6
Removing a loop made ESMFold more confident than the single mutation above, suggesting the region I deleted was a floppy loop that was actually lowering the confidence score. Its removal made the structure more stable.
Trying a larger change, removing a big chunk from 200-300:
pTM: 0.768
pLDDT: 86.6
The score has dropped significantly from the larger mutation, showing the structure cannot handle large deletions.
The luciferase protein structure seems to be quite resilient (at least in folding ability) to small mutations, but suffers immensely from larger mutations. Interestingly, removing certain floppy regions actually makes the folding process more stable.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
This section had a lot of bugs, so I used ClaudeCode to help ensure my code was running smoothly.
Seq_recovery = 0.5164, design sequence 51.6% identical to the original.
Score = 0.7081, ProteinMPNN confident the sequence would fold correctly. This is higher than the original score of 1.4079.
Then I ran code:
**Input this sequence into ESMFold and compare the predicted structure to your original.**
original = "DAKNIKKGPAPFYPLEDGTAGEQLHKAMKRYALVPGTIAFTDAHIEVNITYAEYFEMSVRLAEAMKRYGLNTNHRIVVCSENSLQFFMPVLGALFIGVAVAPANDIYNERELLNSMNISQPTVVFVSKKGLQKILNVQKKLPIIQKIIIMDSKTDYQGFQSMYTFVTSHLPPGFNEYDFVPESFDRDKTIALIMNSXXXXXLPKGVALPHRTACVRFSHARDPIFGNQIIPDTAILSVVPFHHGFGMFTTLGYLICGFRVVLMYRFEEELFLRSLQDYKIQSALLVPTLFSFFAKSTLIDKYDLSNLHEIASGGAPLSKEVGEAVAKRFHLPGIRQGYGLTETTSAILITPEGDDKPGAVGKVVPFFEAKVVDLDTGKTLGVNQRGELCVRGPMIMSGYVNNPEATNALIDKDGWLHSGDIAYWDEDEHFFIV"
designed = "DPANIRRGPAPAEPLPPGTAGQLLFDALKKFAKEPGKIAFIDAETGEKMTYKEFYETAVKMAAALKNYGLDKNDVIAVISKNSLEYLIPVLAALMIGIAVAPVDPNYNVEELYHVLSLAKPKVVFTSKENLEKVLKVKEKLPIIKEIIVLDSEEEYKGYPSIKTFMESHLPEGFNPWEFKPEEFDPEETVAFLLEDXXXXXKPRLVELPHLALRHNLVVARDPVFGYPVEPNTVILNSLPLHKNVGLFTTLGAIYNGFTIVLLSEYDEDLFLKTIQDYKVNIVYLSPEQAELLAKSTKISKYDISSLKEIVVGGAPLDKEVAEKVAKKFGLPGIRTGYGKTELSHAFLITPRGEEVPGSVGRVVPGHEARVVDPTTGAVLGVNEVGELEVRGPMLMRGYRDDPAATAARVDADGWYHTGDLAYFDENGHWFIV"
match = sum(1 for a, b in zip(original, designed) if a == b)
total = len(original)
print(f"Identical positions: {match}/{total} ({100*match/total:.1f}%)")
# Show position-by-position comparison
print("\nPosition-by-position (. = match, X = different):")
comparison = ''.join(['.' if a == b else 'X' for a, b in zip(original, designed)])
print(comparison)
# Count matches in different regions
n = len(original)
third = n // 3
region1 = sum(1 for a, b in zip(original[:third], designed[:third]) if a == b)
region2 = sum(1 for a, b in zip(original[third:2*third], designed[third:2*third]) if a == b)
region3 = sum(1 for a, b in zip(original[2*third:], designed[2*third:]) if a == b)
print(f"N-terminal third (1-{third}): {region1}/{third} ({100*region1/third:.1f}%) match")
print(f"Middle third ({third+1}-{2*third}): {region2}/{third} ({100*region2/third:.1f}%) match")
print(f"C-terminal third ({2*third+1}-{n}): {region3}/{n-2*third} ({100*region3/(n-2*third):.1f}%) match")
# What types of residues are conserved?
hydrophobic = set('AILMFWVP')
charged = set('DEKR')
polar = set('STNQYC')
conserved_hydro = sum(1 for a, b in zip(original, designed) if a == b and a in hydrophobic)
conserved_charged = sum(1 for a, b in zip(original, designed) if a == b and a in charged)
conserved_polar = sum(1 for a, b in zip(original, designed) if a == b and a in polar)
print(f"\nConserved hydrophobic: {conserved_hydro}")
print(f"Conserved charged: {conserved_charged}")
print(f"Conserved polar: {conserved_polar}")
The terminal most conserved is the C-terminal, while the least conserved region is the middle region. Furthermore, the hydrophobic residues seem to be the most conserved, as they are packed the closest to the core, and therefore, have more specific shapes.
Input this sequence into ESMFold and compare the predicted structure to your original.
pTM: 0.932
pLDDT: 92.4
Both numbers scored higher than the original full length sequence, implying a higher folding confidence with the predicted structure. Comparing the original to the predicted structure in ChimeraX revealed that despite being half identical, it folds essentially into the same 3D structure. The fold is robust, and multiple sequences can encode into a similar protein shape. Great news for redesigning luciferase!
Ai:
Why is changing Serine to Tryptophan in Position 198 of 3RIX a detrimental mutation? + What about at position 527?
Why is changing Leucine to Glutamate at position 342 a detrimental mutation?
Find a paper with sequences for which we have experimental scans that are about luciferase.
Used ClaudeCode for Latent Space section to add protein’s embedding to the already-computed data and rerun just the t-SNE part.
How are the (neighbor proteins) similar to luciferase?
Why doesn’t the C-terminal match between the crystal structure and ESMFold?
Used ClaudeCode in the C3 section as the section had lots of coding errors.
Part D. Group Brainstorm on Bacteriophage Engineering
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Notestranslation begins with Methionine in eukaryotes, therefore the position of translation is technically 5 (A -> V). Protein language models train on amino acid sequences. Masked language modelling trains sequences by randomly masking some positions, and training the model to fill in the masked positions based on context. PepMLM does the same thing but to peptide binder design, where it masks all the peptides in the proteins you give, iteratively fills in those masked positions from most to least confident. Low perplexity = more confident, high perplexity = model uncertain.
Sequence
Pseudo Perplexity
WHYYPVAVELKK
14.221520879021000
WHSPPAAARLWE
19.212231606782200
WLYYAVGAALWX
12.951986018963500
WHSPVVAVEHKX
10.69098885919460
FLYRWLPSRRGG
-
Part 2: Evaluate Binders with AlphaFold3
Number
Binder
Pseudo Perplexity
ipTM
Peptide 1
WHYYPVAVELKK
14.221520879021000
0.22
Peptide 2
WHSPPAAARLWE
19.212231606782200
0.27
Peptide 3
WRSGAVGAAHKK
8.089792
0.58
Peptide 4
WRYYAVAARLGK
10.665547
0.29
Peptide 5
FLYRWLPSRRGG
-
0.37
Peptide 1 has a low confidence score, and consistent with its low ipTM, appears to be floating off to the side, not even engaging with SOD1. It does not localize near the N-terminus, nor engage with β-barrel region or dimer interface.
Peptide 2 similarly has a poor ipTM score. It is not localized near the N-terminus, instead, seemingly loosely associated with the c-terminal region.
Peptide 3 reflects its higher score, appearing as a continuous chain that is in close proximity to the β-barrel region, around the middle of the protein and potentially near the dimer interface. It does not seem to be localized near the N-terminus. While the backbone appears to be hovering right near SOD1, it is close enough where side chain interactions likely exist, but are not displayed in the current, skeleton view.
Peptide 4 formed a helix, but still seems to be floating above SOD1, not making any clear contact. It floats to the side of the N-terminus, with no clear engagement, reflecting its poor score.
Peptide 5 appears to be a continuous strand that slightly wraps around the C-terminus region, suggesting it does interact with the surface. It seems surface bound, and complementary to the SOD1 shape.
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
It seems PepMLM generally creates weak peptide sequences where strong, usable sequences are rarer to come across. Generally, high perplexity sequences did correlate with poor ipTM scores. My ipTM scores were generally low, but I did have one outlier (peptide 3) that outperformed the known binder FLYRWLPSRRGG by 0.21 points. However, this tool is still extremely valuable, just with additional time and testing.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Summary: A few interesting results pop-up. Firstly, peptides with higher ipTM do not show stronger binding affinity. In fact, the peptide with the strongest binding affinity (6.502) had a very weak ipTM score (0.29), while the peptide with the highest ipTM (0.58) had the weakest binding affinity (4.991). This may be due to the inheritance difference in the purpose of these tools: AlphaFold is asking if you can physically fit together the peptide and target, while PeptiVerse is asking how strongly the peptide BIOCHEMICALLY interacts with the target.There was no variation in the hemolysis and solubility columns across the peptides. The peptide that best balances these properties is peptide 3 (WRSGAVGAAHKK). All the peptides were in the “weak binding” category. Structure is more meaningful, of which peptide 3 displayed the highest confidence, while balancing good hemolysis and solubility.
Choose one peptide you would advance and justify your decision briefly.
All peptides had good solubility, hemolysis and weight. The decision comes down to net charge and binding affinity. Peptide 4 has the strongest binding affinity of 6.502 and a reasonable, positive charge. However, its ipTM is one of the lowest. Therefore a happy medium is peptide 3, highest ipTM, positive charge, even with a weaker binding affinity (but all 5 had “weak binding” so the decision is less relevant based on binding affinity).
Part 4: Generate Optimized Peptides with moPPIt
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Region used:
β-barrel region near WRSGAVGAAHKK as that is what it closest to: 95-105
0.966 ← Hemolysis
6.356 ← Affinity
0.850 ← Solubility
0.814 ← Motif score
PepMLM’s prompting is a lot more loose compared to moPPIt. The former needed to just generate something plausible, while the latter had to optimize for multiple factors in a specific range. Although moPPIt did produce better affinity and solubility, it did seem to sacrifice hemolysis in the process. Before putting this into clinical studies, I would validate this in a wet lab (including tests such as SPR, ITC and red blood cell lysis assay to test for the predictions), specifically putting emphasis on testing the high hemolysis score, as it may be a dealbreaker before clinical trials. I would also consider running more computational tests on the hemolysis, and perhaps pivoting to a different peptide (such as WRSGAVGAAHKK) as to not waste time and resources. If the wet lab results are promising, the next steps would be ALS motor neuron cell models before human trials.
Ai:
Is the A4V mutation for the SOD1 sequence done at the 5th out 5th position? The fourth position in my sequence is not A.
Why is there an extra Methionine? Is this in all organisms? Is it always cleaved off?
Explain PepMLM in detail.
For peptides that will bond to SOD1, what molecular weight is ideal? What net charge is ideal? (Why?)
If I have a peptide I think may help the A4V mutation in SOD1, what are my next validating steps before sending it to human trials?
Part C: Final Project: L-Protein Mutants
Note: I was unable to find a group in time for this assignment. I will continue searching for a group going forward, but completed this assignment on my own to avoid falling behind.
As I was most familiar with Option 1, I chose it for my analysis. I started with picking out mutations from the L-Protein Mutants file. I made sure to pick mutations where both the protein and lysis levels are 1, so they are experimentally confirmed to work and the protein is made properly. My picks are as follows:
Soluble region: P13L (position 13)
Base pair change: C→T at position 38
Lysis = 1, Protein Levels = 1
Soluble region: R18G (position 18)
Base pair change: A→G at position 52
Lysis = 1, Protein Levels = 1
Anywhere: R30Q (position 30)
Base pair change: G→A at position 89
Lysis = 1, Protein Levels = 1
I also initially picked mutations at 44 and 46, but swapped them due to the cluster omega results.
ClusterOmega: Checked every mutation against the output. 44 and 46 were conserved, so I swapped them out for mutations at 45 and 47, and cross referenced them with the L-Protein Mutants file:
A45P (pos 45) TM Region Lysis=1
F47Y (pos 47) TM Regio Lysis=1
A few things came up after using Protein Language Model Notebook:
I swapped out the A45P to the A45L as ESM2 liked the L better, giving a higher score and placing it in the top 30 ( ESM2 score 1.539).
A45P (pos 45) TM Region Lysis=1 -> SWAPPED TO A45L
The rest of the mutations were not the top ESM2 contenders. I tried to swap them out for better contenders that scored in the top 30, but the L-Protein Mutants either had no experimental data on them, or gave bad scores for the mutations.
In general, the heat map scores were favorable. A45L had the lightest colour, while the rest had medium, darker greens, but not near dark blue.
Summary:
I selected my five mutations for enhanced lysis function based on the cross-reference of three resources: 1) experimental lysis data from the L-Protein mutants file 2) CluterOmega evolutionary conservation, ESM2 LLR scores from the language model. Since I chose option 1, I prioritized experimental data first (where Lysis = 1 and Protein Levels = 1) before adjusting my chosen mutations based on their ESM2 scores. I do want to note that the top ESM2 candidates were not in the dataset. My final mutations are as follows:
P13L (position 13, soluble region): Proline → Leucine. Lysis = 1, Protein Levels = 1. Was not conserved in ClusterOmega. Neutral heatmap score.
R30Q (position 30, anywhere): Arginine → Glutamine. Lysis = 1, Protein Levels = 1. Not conserved, acceptable heatmap.
A45L (position 45, transmembrane region): Alanine → Leucine. Lysis = 1, Protein Levels = 1. Chosen to replace A45P due to better ESM2.
F47Y (position 47, transmembrane region): Phenylalanine → Tyrosine. Lysis = 1, Protein Levels = 1. Chosen replacement due to better ClusterOmega. Not conserved, acceptable ESM2 scores.
A45L is the most promising mutation as it has confirmed experimental lysis, is non-conserved, and has a good ESM2 score (placing in top 30). The other mutations have good experimental data and conservation, but less promising ESM2 scores. This highlights an interesting disconnect between ESM2 rankings and experimental lysis outcomes.
Ai:
used ai when running cluster omega to make sure I was counting the position right, and so whether every position was conserved was calculated correctly.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Components included are Phusion DNA Polymerase, deoxynucleotides (dNTPs), and HF Reaction Buffer containing MgCl₂. Physion DNA polymerase is a Pyrococcus-like enzyme. The polymerase domain reads the strand and synthesizes it by adding dNTPs one at a time (through phosphodeister bond formation). This requires a primer to already be present in the template. The enhancing domain fused onto the polymerase keeps the enzyme attached to the DNA template, speeding up the process. The exonuclease domain reads this newly synthesized stand and corrects any mistakes. Deoxynucleotides (dNTPs) are the building blocks of DNA at optimized concentrations. The HF reaction buffer helps control the pH for optimal activity, while the MgCl₂ positions the dNTPs correctly and stabilizes the transition state during bond formation.
What are some factors that determine primer annealing temperature during PCR?
Primer length: Optimal length is between 18-22bp to allow for adequate specificity while allowing for binders to bind easily. However, overhangs can be 20-22 bp.
GC content: should be 40-60% of total primer sequence, directly influencing Temp more since G-C pairs are more thermodynamically stable than A-T
GC clamp:1-3 G/C bases during the last 5 bases at the 3’ end improve binding but more than 3 should be avoided as that can cause non-specific annealing.
Secondary structures: hairpin structures and primer dimers reduce how much primer is available to anneal to the template, recommended to keep Gibbs free energy above -10kcal/mol.
Note the ideal temperature specified is 52-58 Celcius, where about 65 Celcus can cause secondary annealing issues.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR uses a polymerized enzyme to synthesize new copies of DNA region, where the region is defined stand to end using primers, and the machine cycles through denaturation, annealing and extension to amplify that specific region into many copies. This requires multiple temperature cycles, specific reagents (including polymerase, primers, dNTPs, buffer solution), and 1-2 hours. Restriction enzyme digests cut existing DNA at specific recognition sites, where different enzymes recognize difference sequences and cleave at that region. There is no new DNA created, it’s simply cut. This requires one temperature, regents of restriction enzymes and buffer, and larger amounts of starting plasmids and DNA. PCR creates blunt ends or overhangs depending on your polymerase, and is better for amplifying specific regions from a larger piece of DNA. It’s also useful in cases where you need to add overhangs for Gibson Assembly, you have little amounts of starting DNA, or your sequence doesn’t have convenient restriction sites. Restriction digests usually produce sticky overhand ends, and are better for when you want to cut a plasmid backbone at specific sites, you want sticky ends, or you have a large fragment that is too impractical for PCR. It’s also preferred in verifying band sizes on a gel, due to PCR being simpler to do.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To be appropriate for Gibson cloning, the fragments must have correct overhangs at the end of each fragment. For PCR, you must ensure each fragment has a unique, 20-22 overhang that is identical to the adjacent fragment, and has enough GC pairs. For the digests, the enzymes must cut at the locations that expose the sequence you want to overlap with PCR (which can be checked in the Benchling cut sites). It’s very important the orientation of the overlaps is correct in the 5->3 direction. To verify the cut was at the intended site and not anywhere else in the backbone, gel or sequencing may be used.
How does the plasmid DNA enter the E. coli cells during transformation?
DNA can enter through electroporation (though not discussed in the lab manual, heat shock may also be used). Cells are placed in a cuvette with the plasmid DNA, followed by a brief, high voltage electrical pulse that destabilizes the cell membrane, creating pores through which DNA can enter (driven by the electrical potential). The membrane then reseals, trapping the DNA inside. After the electric pulse, cells are incubated in media to recover for about an hour.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden gate assembly uses Type IIS restriction enzymes to generate custom overhangs of 4 bp in DNA fragments that can then be ligated together in a custom order. This allows for assembling multiple fragments simultaneously. The Type IIS enzymes cut downstream of their recognized site, allowing for these customizable overhangs, and removing the recognition site itself from the DNA. DNA is then ligated together with the complementary overhangs in a one tube reaction. This allows for very precise cutting due to the unique 4bp overhangs.
Model this assembly method with Benchling or Asimov Kernel!
All my notes and simulations are under the repository called: Iman_Karibzhanova_HM6 in the HTGAA Victoria Node account. Below is a general summary.
My simulation of the demo:
Demo’s simulation:
My first construct:
The simulation is as expected. pTetR is fully active with nothing suppressing it, so RNA shoots up. Protein concentrations plateaus at around 2.5. RBS, TetR and LacI are at maximum output. This is how genes act when there is nothing controlling its output.
My second construct:
The results are interesting. The plateau happens at a lower concentration (around 1.1) due to the repressor capping expression from suppression of its own promoter. The RNA dips down quickly in the beginning of the simulation, due to the LacI suppressor kicking in from production. This shows autoregulation could prevent toxic accumulation in my project.
My third construct:
The simulation makes sense based off of construct 1 and 2. Lac1 goes up to around 2.5, similarly to the first experiment, which makes sense since pTetR is not repressed. LambdaCI spikes when LacI is not yet present to repress pLacI, and then swiftly falls as LacI shuts the pLacI promoter down, eventually silencing LambdaCI. Gene one remains on, while the second gene is shut off. This is an important dynamic to keep in mind when constructing multi-gene pathways.
List of parts given in Asimov Repository: Which are the simplest to use in my constructs to test designs?
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs (Intracellular Artificial Neural Networks) have several advantages over traditional genetic circuits. Booleans inherently work in a binary fashion: there is either a statement ment that makes it true/false, and therefore, either on/off. IANNs have the ability to process continuous signals, allowing for inputs to be weighted against each other before an output. This makes the system more sensitive to differences, not just whether something is true or false. This is great for cells as molecular concentrations exist on a spectrum. Another reason is its reconfigurability: you can reprogram the IANN circuit without needling to completely rebuild it. IANNs also make multi-input easier to configure, as an arbitrary number of inputs can be collapsed into one step, instead of building onto each other in booleans. Lastly, boolean circuits are static, with inherently fixed behaviour after it is built. IANNs are defined by weights, allowing for greater flexibility if you were to modify the weight thresholds.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNs are very applicable to my final project of autonomously, bioluminescent plants. My plan has five constructs, where my enzymes will likely be fixed due to my promoter choices, when optimal ratios of expression depend on the plant, not my assembly. Hypothetically, an IANN that senses multiple metabolic signals, and therefore adjusts expression levels of each enzyme pathway would be more efficient. Inputs could be luciferin precursors, ATP concentrations, CoA levels, reactive oxygen species, sensed by engineered biosensor modules, that can convert concentrations into molecular signals for the IANN to read. For an example of an output, if low luciferin is detected, its precursors would be upregulated, allowing for a graded output that adjusts expression levels. This would work better than boolean logic, as I have several metabolic signals all interacting with various variables. Optimal response to one metabolic process depends on the others. The limitations of this idea include training (no training in living plant cells has been mentioned or achieved), sensor availability for my specific metabolic pathways, signal crosstalk and noise (as many other pathways are being performed in a plant cell at all times), and scalability (the paper doesn’t mention five inputs). Another limitation is metabolic burden, (on top of an already pre-existing problem of bioluminescence metabolically loading the plant cells).
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
X1 encodes for Csy4 endoribonuclease. Csy4 cleaves X2’s mRNA acting as inhibitory weight.
X2 encodes for endoribonuclease 2. It has a recognition site for Csy4. The mRNA that survives Csy4 gets translated into endoribonuclease 2 giving the output for layer 1.
X3 encodes for fluorescent protein. It has a recognition site for endoribonuclease 2. The endoribonuclease 2 from before is the inhibitory weight. Whatever mRNA survives the endoribonuclease is translated into fluorescent protein output Y.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are becoming increasingly more popular in various sectors. A prominent example includes the fashion industry, where mycelium leather is becoming more popular with luxury designers, including Hermes and Stella McCartney. While production includes less toxic chemicals and water needed, it’s still difficult to scale outside of luxury/avant-garde products, and is a bit finicky in production. Another industry adapting fungal materials includes furniture/delivery. An example includes IKEA committing to using mycelium styrofoam for packaging. As regular styrofoam degrades slowly and is toxic to the environment, this offers a more eco-friendly solution. However, it has similar limitations, and slacking economics are difficult, and the mycelium is sensitive to water absorption damage during shipment. Lastly, mycelium is an interesting construction material, offering a non toxic solution to fiberglass. However, it remains limited in its structural usage due to its sensitivity to damage from water, and weak compressive strength.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Interestingly, LightBio is a company that already produces glowing plants through the use of fungal pathways. There’s an obvious application to my project where it has been proven as plausible. However, its efficiency in converting energy to photons remains weak. Beyond bioluminescence, fungi still offer a wide variety of engineering purposes such as living biofertilizers, biocontrol agents (instead of more toxic insecticides), and biosensors for metal contamination or plant disease. Fungi has many advantages over bacteria in plant engineering. Firstly, it has the ability to penetrate root cells for metabolite exchange, something bacteria cannot do. The pathway to deliver engineered metabolites into plants already exists specifically in fungi. Furthermore, fungi share many similarities in protein folding, translations, etc. allowing for better protein processing than bacteria. These similarities translate to organelles, as fungi and plants share similar structures that bacteria lack. Lastly, fungi is already adapted to fluctuating soil conditions, making it a more stable and faithful medium.
Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 HW: Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis provides multiple advantages over traditional methods due to its lack of membrane. You don’t have to cross the membrane, and the cell’s homeostasis controls don’t resist any experimentations. Furthermore, since everything is now a controllable variable, you have better control over composition (such as concentrations) of amino acids, substrates, etc. You also maintain better folding control and template flexibility (since you don’t have to clone into plasmids). Cases where this is beneficial include space travel (since cell-free synthesis allows for freeze-drying, which is more shelf-stable in storage), and anywhere else resources may be limited to run a fermenter (war zones, rural clinics, etc.) Another case is the expression of “unhostable” proteins, which would in other cases, kill its living cell. The cell-free environment means you bypass the actual cell host.
Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free system is essentially a cell with no membrane, and no homeostasis-related regulation. Components include:
DNA Template (you still need parts the system can read such as promoters, RBS, CDS, terminator)
RNA polymerase (for synthesizing mRNA in the cell-free system)
Ribosomes (for translation, added back individually and generally most expensive component of cell-free systems)
tRNAs (for reading the codons and delivering the amino acids)
Amino acids (all canonical ones supplied so proteins are synthesized)
NTPs (required monomers for transcription)
Energy regeneration system (translation burns a lot of ATP, some sort of system required to keep system running)
Salts and cofactors that are critical for ribosomes and polymerase activity
Cell extract (lysed E.Coli containing most of the listed above that end up in the cell-free system)
Buffer (“background” holding everything together)
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Translation is very energy “expensive”, requiring multiple high-energy phosphate bonds for one peptide bond. Without some sort of energy regeneration, the cell-free system would function very slowly and inefficiently (as ATP → ADP + Pi, eventually most of the system is ADP instead of what the cell needs). A way you could keep ATP flowing is a high-energy phosphate donor + its kinase, where a phosphate is transferred back onto an ADP to make ATP again. This phosphate can be donated from other molecules, (called a high-energy phosphate donor). Common donors include phosphoenolpyruvate, creatine phosphate, and glucose.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cells are generally the fastest and cheapest (generally in synthetic biology), allowing for greater efficiency and affordability. Furthermore, because prokaryotic cells are generally simpler, it’s easier to prototype and use. However, that simplicity means no post-translational modifications, and certain proteins cannot form properly due to lack of disulfide bonds. Also, if expressing a eukaryotic gene, it lacks basic infrastructure needed.
Protein example: Luc2 (used in my final project), a cytosolic enzyme from Photinus pyralis that folds well in E. coli. If you add D-luciferin and ATP to it, you get bioluminescence proportional to its translation rate. This would be ideal to prototype in a tube before agroinfiltration (rehydrate a BioBits-style pellet with a linear PCR product of 35S+Ω → luc2+SKL → NOS, add luciferin, light appears)
Eukaryotic cell systems are generally more expensive, and often output lower yield with slower reactions, but do offer more variations and resources. For plants, a popular choice is wheat germ extract (WGE), as it handles eukaryotic protein folding well due to its eukaryotic translation factors and plant-derived folding machinery.
Protein example: The expression of a novel candidate (such as the one I’m using in my second translational unit), in a wheat germ extract. Since I am expressing cytochrome P450s, I know they are difficult to express in prokaryotic cells. Therefore, it is better to express in a eukaryotic system, despite the higher costs.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Since CFPS have no membrane, you’d design a system environment specifically suited for that specific type of membrane protein. Challenges include the obvious lack of membrane (therefore, nowhere for the protein to insert), translation issues (since many membrane proteins translate as they are being inserted into the membrane), orientation of the membranes (since membrane proteins have a defined position in the membrane), and having the correct lipids and confactors. I think the most practical solution that should theoretically solve all these problems is a fake membrane added in the system. These “fake”, lipid-rich membranes would not surround anything, but float freely allowing for a location for the membrane protein to insert into. They would be added into the reaction while the membrane proteins are being translated.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
One: Not enough energy (ATP)/Energy is running out too early
If translation is burning ATP faster than it’s being created, even with an energy regenerative system, reactions would plateau or slow down. To troubleshoot you’d adjust concentrations of PEP, and/or add sodium oxalate to suppress the phosphatases that are destroying the PEPs. You can also upgrade to a continuous-exchange CFPS (CECF), where you have two compartments separated by a dialysis membrane, so that there is continuous exchange between the “fuel” energy supply and waste supply.
Two: Low magnesium ion concentrations
If the concentration of Mg²⁺ is too low or high, ribosomes and polymerase don’t function correctly. Troubleshooting involves running a Mg²⁺ titration to to find the optimal concentration.
Three: Bad lysate batch
A problem with CFPS is that freeze-dried batches are not consistent. Extracts can vary even with the same protocol, and ribosome activity is sensitive to freeze/thaw cycles. Troubleshooting would involve never re-freezing, strict aliquoting, or a new batch.
AI
How could you keep energy flowing for continuous ATP supply in a cell-free system?
Prokaryotic vs eukaryotic cell free expression systems?
How can you add more energy to cell-free systems if your energy regenerative system is still sluggish?
What factors are cell-free protein synthesis systems sensitive to?
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows: What would your synthetic cell do? What is the input and what is the output?
Detect luciferin precursors and produce visible light (photo emission) in response (for example, in a plant if it detects all the metabolic precursors needed, it lights up). This is useful because we don’t know all the precursors for firefly bioluminescence. This is a helpful debugging tool in case my novel candidates are correct, but don’t glow due to folding errors, expression errors, etc. The input would be p-coumaric acid (at least for the example of my cytochrome P450s candidate), and output would be visible light at 560nm.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes but with losses from the lack of encapsulation. The chemistry wouldn’t suffer much, but the application (deploying the genes into living plant tissue) could be problematic as you need encapsulation to deploy. Also, encapsulation protects the luc2 genes from nucleases, which may reduce light output.
Could this function be realized by genetically modified natural cell?
You could express luc2 in E.coli or directly in the plant, but synthetic clinical cells may be more efficient at converting higher light output, since you wouldn’t be losing ATP for cell housekeeping and resources due to other pathways. In theory, this would give a clearer, defined system where the only chemistry happening is luciferin-related.
Describe the desired outcome of your synthetic cell operation.
When the minimal cell encounters a p-coumaric acid or other precursor, light output rises depending on the concentration detected. This would validate the enzyme I am testing, to see if it actually generates luciferin precursors before committing to an agroinfiltration pipeline.
Design all components that would need to be part of your synthetic cell. What would be the membrane made of?
Phosphatidylcholine (POPC) to mimic the cell membrane and cholesterol to stiffen the bilayer enough to hold a pore stably without leakage.
What would you encapsulate inside? Enzymes, small molecules.
I would encapsulate a bacterial cell-free system (E.Coli lysate), plasmid encoding luc2, plasmid encoding α-hemolysin, plasmid encoding my novel candidate (PPYR_02911), ATP starting pool, PEP for energy regeneration, and Mg²⁺.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
It would come from bacterial organisms (E.Coli) lysate since the luc2 (firefly luciferase) I’m using from iGEM folds well into E.Coli and does not need eukaryotic post-translational modifications. I do not need a mammalian system since I’m not using any mammalian parts.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
P-coumaric acid (what I’m testing for) diffuses into the membrane through α-hemolysin pores (which are also narrow enough to retain luc2 and lysate machinery). Light would exit passively since photons are not affected by lipid bilayers. Therefore, no export channel is needed for output (since it’s photons), allowing for a simpler design.
Experimental details. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: POPC, cholesterol (at a 70:30 molar ratio)
Genes:
Luc2 (BBa_K389004, firefly luciferase)
Hla (α-hemolysin, membrane pore)
PPYR_02911 (candidate CYP4C cytochrome)
How will you measure the function of your system?
The function of my system will be measured by a luminometer (measured over time after adding higher concentrations of p-coumaric - to a suspension synthetic minimal cell. Another measurement can be captured with a camera to image the cells as bright spots.
Ai
I am designing a useful synthetic minimal cell for my firefly plant project that detects luciferin precursors and produces visible light in response (to test my novel candidates. Lets use PPYR_02911). What should the membrane be made of? Should we use bacterial since we’re using luc2?
Why a 70:30 ratio for POPC and cholesterol?
Homework question from Peter Nguyen
Write a one-sentence summary pitch sentence describing your concept.
Freeze-dried cell-free coating embedded in clothing fabric that produces bioluminescence when activated by sweat, transforming clubwear, entertainment, and performance garments into living light at clubs/concerts/nightlife.
How will the idea work, in more detail? Write 3-4 sentences or more.
The fabric is coated (you can shade them onto the fabric with each use) with the freeze-dried cell-free reaction pellets containing the luciferase expression system discussed earlier. These are all lyophilized into a stable powder bound into the textile amtrak with water-soluble polymers. When the wearer sweats (or is misted with water), the pellets are rehydrated, triggering the luciferase-driven light emission. The pattern follows the pattern of moisture, where the back may glow lighter, following the dancer’s movements. The garment’s lifecycle is a single night out, growing brightest when the person wearing the garment is most active, and dimming with the night winding down.
What societal challenge or market need will this address?
Clubwear is already saturated with glowing garments, but with expensive LED-embedded garments that are difficult to wash, are heavy, require batteries, and fail often. A bioluminescent textile is light, soft, batteryless, washable, and biodegradable, being kinder for the environment. This taps into broader movements against single-use plastics/batteries, and biofabricated materials, where the garment is part of the larger story of how it’s made (“my garment is alive and reacts to me”). It could also be used with runners for safety wear (being visible in the dark), and other sport events where you need to track athletes.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
This idea has many limitations, but all can be addressed:
Activation (If the wearer does not sweat, water can be applied externally with sprays or other means. Water is provided everywhere in nightlife due to safety.)
Shelf life (The garment could be on the shelf for many months or years before first activation. This is well within the shelf-stability of lyophilized cell-free systems)
One-time use (Design the garments with refillable pockets for freeze-fried pellets cartridges where the wearer refills them between uses.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting.
Long-duration spaceflights of space colonies require the crew to grow their own food. However, plants often show stress under microgravity, with impaired growth, weakened cell walls, and other unpredictable responses. Although we have successfully grown certain vegetables in space, our monitoring of plant stress is still heavily reliant on freezing samples, and sending them back to earth. I propose a field-deployable sensor that reports plant stress directly on orbit, allowing for early intervention, and protecting food security on long missions.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches.
I propose to study the Arabidopsis jasmonic acid responsive transcripts (Jaz1), which is an early mRNA marker of plant dress and wounds. This would be detected via biosensor.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses.
Jasmonic acid signaling is one of the fastest plant stress responses, being activated within minutes of abiotic stress or wounds. JAZ1 transcripts spike sharply during this response making it an almost real-time readout of plant stress. Detecting JAZ1 through a tiny leaf punch (adjusted for the leaf punch response) would tell astronauts right away if the plant is healthy, or if the stress response is currently active. This could also explore if microgravity inherently affects this pathway in plants.
Clearly state your hypothesis or research goal and explain the reasoning behind it.
Hypothesis: Arabidopsis plants that are grown in microgravity exhibit chronically elevated JAZ1 transcripts, reflecting the chronic low-grade stress plants inhibit in response to space flight. This can be detected using freeze-fried BioBits switch biosensors.
Reasoning: Previous studies of spaceflight grown plants have implied chronic stress and cell-wall remodeling. However, these samples are freeze-dried and return to earth, where samples may be getting affected by storage and re-entry. A direct measurement during orbit of JAZ1 in fresh tissue can inform us how much is spaceflight stress, and how much is sampling-related. The number of JAZ1 is proportional to transcript abundance. Therefore, it can also allow astronauts real-time, same-day data on the stress of their crops throughout the course of their mission.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc.
The JAZ1 targeting switch plasmid would be designed on earth, and then freeze fried into BioBits pellets. During the orbit, astronauts collect small leaf punches from the Arabidopsis, which scientists on earth collect in parallel with ground-control plants. The total RNA is then extracted using a simple column kit, and then rehydrated with BioBits reactions. Fluorescence is measured over a few hours and imaged with an iPad. Controls include: 1) BioBits + water 2) BioBits + synthetic JAZ1 RNA 3) BioBits + mismatched trigger RNA 4) wounded ground control leaves
Ai
I am designing a proposal that incorporates BioBits cell-free protein expression system. My idea is a biosensor that detects stress signals in plants in space. What is a reliable gene I can target with my biosensor?
How does JAZ1 work?
Homework Part B: Individual Final Project
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1 (DONE)
Submit this Final Project selection form if you have not already.(DONE)
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.