Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art


If ignoring ladder and lane 1, there is the design! I tried to recreate No Face from Spirited Away.
link to benchling: https://benchling.com/s/seq-8pB9vY3uTYXRrqsIKJS6?m=slm-FP5NlW0BAaqfbSmaoUg0
DNA Design Challenge
For my homework, I decided to pick the enzyme that is the strongest candidate for facilitating luciferin synthesis. I picked my enzyme from my independent research. First I downloaded Fallon’s paper data from Fallon et al. 2018, “Firefly genomes illuminate parallel origins of bioluminescence in beetles” in eLife (DOI: 10.7554/eLife.36495. This experiment compared gene expression between the fat body (a firefly’s liver) versus the lantern (the organ that makes light) to find which genes are highly expressed in the lantern. I ran a filter in the file PPYR_OGS1.1_fatbody-vs…_test.txt texts, keeping only the statistically significant genes of TPM ≥ 50 and sleuth b ≥ 3. The TPM measures how actively a gene is being expressed in the lantern tissue, a higher TMP signaling a higher likelihood of luciferin expression. The sleuth is the statistical software Fallon used, that estimates log2 fold change. The higher the b sleuth, the more expression a gene has specifically in the lantern than fat body. I also ran qval ≤ 1e-10 to adjust for random noise.
Afterwards, I ran my candidates through Fallon’s enzyme annotation file (PPYR_OGS1.1.enzyme.ids.txt) which lists the predicted enzymes based on Fallon’s work in InterProScan. Although both filters have arbitrary thresholds with likely blind spots, it produced a good starting point.
I then ran my candidates through HMMsearch for function, and BLAST to check if the enzyme is expressed only in fireflies, or all insects (which would point to different, non-applicable functions such as exoskeleton hardening). One candidate was already discussed in Zhang (2020), but has not yet been experimentally validated. The rest are potentially novel candidates not yet found in literature.
I then did reverse BLAST to see if the top-hit organisms point back to their assigned enzyme. This was done to check if they’re true orthologs, or if BLAST was matching generic similarities. This narrowed down my search to four candidates.
I decided to go along with PPYR_02911 as my homework protein for several reasons. Firstly, it’s an oxidative enzyme (P450), which is the class you’d expect in the bottleneck. We know that firefly luciferin requires a benzoquinone precursor, therefore requiring an oxidative step facilitated by an oxidative enzyme. Secondly, it’s a tandem duplicate of one of my other candidates (PPYR_02910), being only 4kb apart. They are 87% identical, pointing to possible gene neofunctionalization. Thirdly, its BLAST hits were almost all bioluminescent species, and its Lantern TPM was the highest of the group. Lastly, the candidate proposed by Zhang (2020) likely facilitates in storage, pointing to a support function. I’m more interested in finding the missing catalytic step.
Since I already obtained the protein sequence from my own independent research, I have it on hand from Fallon’s PPYR_OGS peptide FASTA file, which contained custom protein sequences for all the predicted genes in the Common Eastern Firefly. This file was obtained from Fallon’s Github (https://github.com/photocyte/PPYR_OGS). However, for learning purposes, I pasted my PPYR_02911-PA protein sequence from Fallon’s file in NCBI, clarifying Photinus pyralis (the firefly) as my organism. My top hit was the NCBI equivalent, XP_031330391. I cross referenced XP_031330391 with PPYR_02911-PA, and confirmed it is the same gene.
XP_031330391.1 cytochrome P450 4C1-like [Photinus pyralis] MMNLVDEFAPKSALQALVPIALVTFLVWYMQYHWNRRRLYKMAALFDGPICLPFVGNGLYFVGSTSDILQNVISLVSNFK LPVRVWLGQKLFYALVDPGDLEIIMNSPHALEKDELYQYAEPIVGTGLFTAPVPKWKRHRKVIMPTFNQRILDEFVPVFA EQSEILLEQLKKQVGKGSFDIFQLVSRCTLDIICETAMGVKVEAQTTDSDYVKWANKAMEIMFTRMFNIWYHFDSIFNLT QSARDLLDVQTKMKTFTGAVVRNRREAYQRKMRERRQLPEGYVDKEAPTRKTFLQQLIELSEGGANFTDDELREEVDTFM VAGSDTTASMNSFIFIMLGMHPDVQEEVYQEVLDVLGPDRAVEAADLGRFHYMERVMRETMRIFPVGPILVRAITKDLQL ENCVIPAGSSVVMVIMQTHRSEKIWPHPLRFEPDRFLPEEVAKRHPYAWLPFSGGPRNCVGPKYAFMAMKALIATVVRRY KFKTDYKCIEDIELKADLMLKPVNGYNVSVELRE
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I clicked on XP_031330391 next to the “Sequence ID” in the XP_031330391 protein page in NCBI. From there I clicked on the DBSOURCE to get the DNA sequence, and then FASTA.
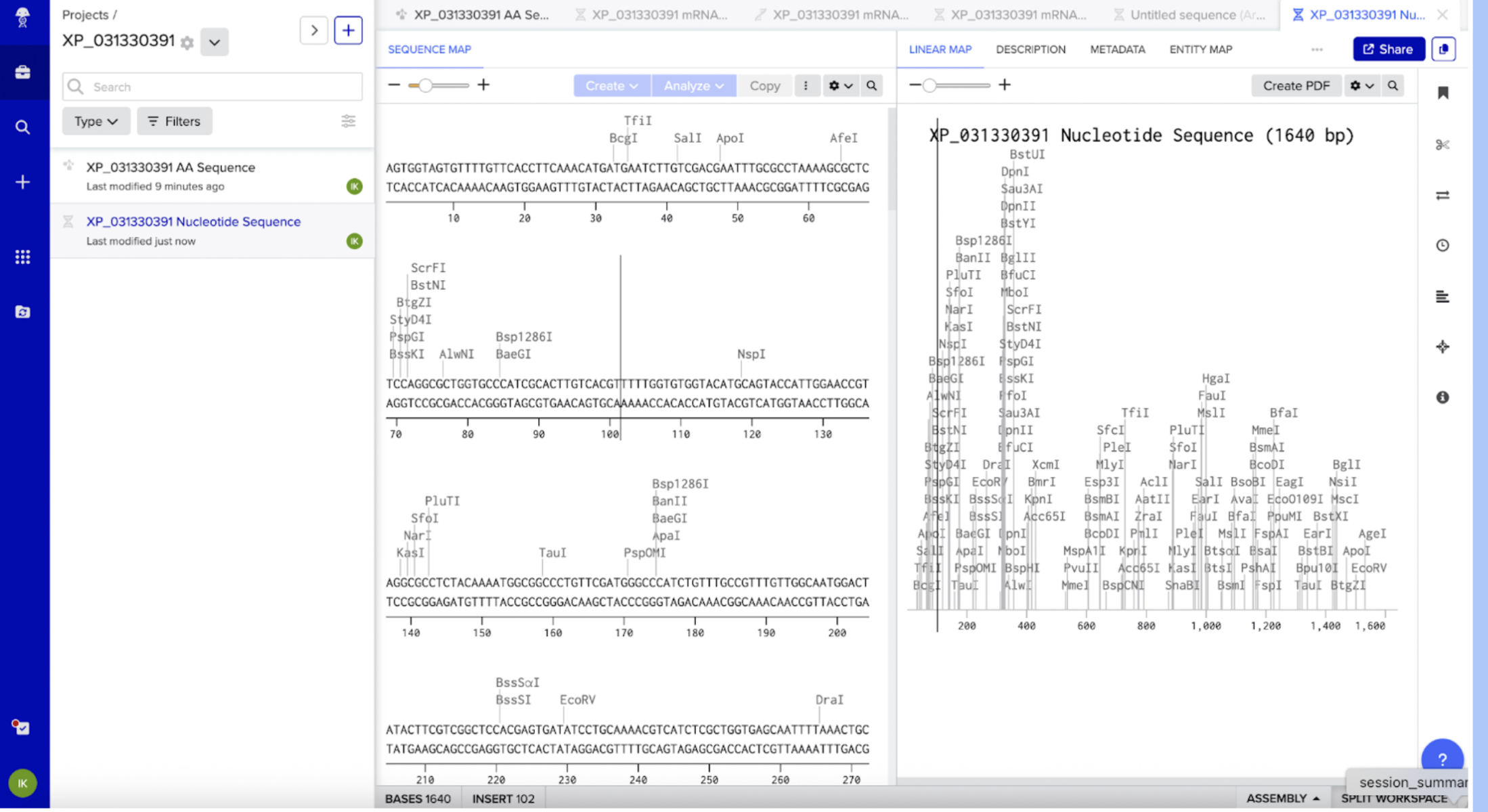
XM_031474531.1 PREDICTED: Photinus pyralis cytochrome P450 4C1-like (LOC116161249), mRNA AGTGGTAGTGTTTTGTTCACCTTCAAACATGATGAATCTTGTCGACGAATTTGCGCCTAAAAGCGCTCTC CAGGCGCTGGTGCCCATCGCACTTGTCACGTTTTTGGTGTGGTACATGCAGTACCATTGGAACCGTAGGC GCCTCTACAAAATGGCGGCCCTGTTCGATGGGCCCATCTGTTTGCCGTTTGTTGGCAATGGACTATACTT CGTCGGCTCCACGAGTGATATCCTGCAAAACGTCATCTCGCTGGTGAGCAATTTTAAACTGCCGGTGAGA GTGTGGTTGGGCCAGAAACTATTCTACGCCTTAGTTGATCCTGGAGATCTGGAAATAATCATGAATAGCC CTCACGCGCTGGAGAAGGACGAGTTGTACCAGTATGCCGAACCCATTGTGGGTACCGGATTGTTCACCGC CCCAGTGCCAAAATGGAAACGCCACCGTAAAGTGATAATGCCGACATTCAATCAGCGCATCCTCGACGAG TTTGTGCCGGTATTTGCCGAACAGTCCGAAATACTCTTGGAGCAGCTGAAGAAACAAGTTGGAAAGGGCA GCTTCGACATCTTTCAACTGGTCAGCCGTTGTACCTTGGATATTATCTGCGAGACGGCAATGGGAGTTAA GGTGGAGGCGCAGACTACAGACTCAGATTACGTCAAGTGGGCTAACAAGGCAATGGAGATTATGTTTACG AGAATGTTCAACATTTGGTACCACTTCGATTCCATATTTAACCTTACACAAAGCGCACGTGACCTTCTCG ACGTCCAGACCAAGATGAAAACGTTCACTGGAGCCGTTGTAAGAAACAGACGGGAGGCGTACCAGAGAAA GATGAGGGAACGAAGGCAACTACCGGAGGGGTACGTAGACAAAGAGGCGCCGACTCGAAAAACCTTCCTT CAACAGCTCATCGAGTTGTCCGAAGGAGGGGCCAACTTCACAGATGATGAACTGCGGGAAGAGGTCGACA CATTTATGGTGGCGGGAAGTGACACCACTGCGTCGATGAACAGTTTCATATTCATCATGCTTGGAATGCA CCCAGATGTTCAGGAAGAAGTCTACCAAGAGGTGCTAGACGTACTCGGGCCAGACAGAGCAGTAGAAGCA GCCGACCTTGGTCGTTTCCACTACATGGAGAGAGTGATGAGGGAGACCATGCGCATATTTCCCGTCGGAC CTATACTGGTTAGGGCAATCACCAAGGACCTTCAGCTAGAGAACTGCGTGATACCGGCCGGAAGTTCAGT GGTAATGGTGATAATGCAAACGCACAGAAGCGAAAAGATTTGGCCGCATCCGCTTAGGTTCGAACCCGAC CGTTTCCTACCCGAAGAGGTAGCCAAACGACATCCATACGCTTGGTTGCCATTCTCTGGTGGTCCACGTA ACTGCGTTGGTCCTAAATATGCATTTATGGCCATGAAGGCGCTCATCGCTACCGTCGTCAGGCGATACAA ATTTAAGACCGACTATAAGTGCATCGAGGATATCGAACTGAAGGCTGATTTGATGTTGAAACCGGTCAAC GGGTACAATGTTTCGGTCGAATTGAGGGAATAACAACAATTTATCAGCGCACAATTACTTTAGAACTATT CGTGTTGCAGTATAAACTTTTTTACTCGCC
3.3. Codon optimization
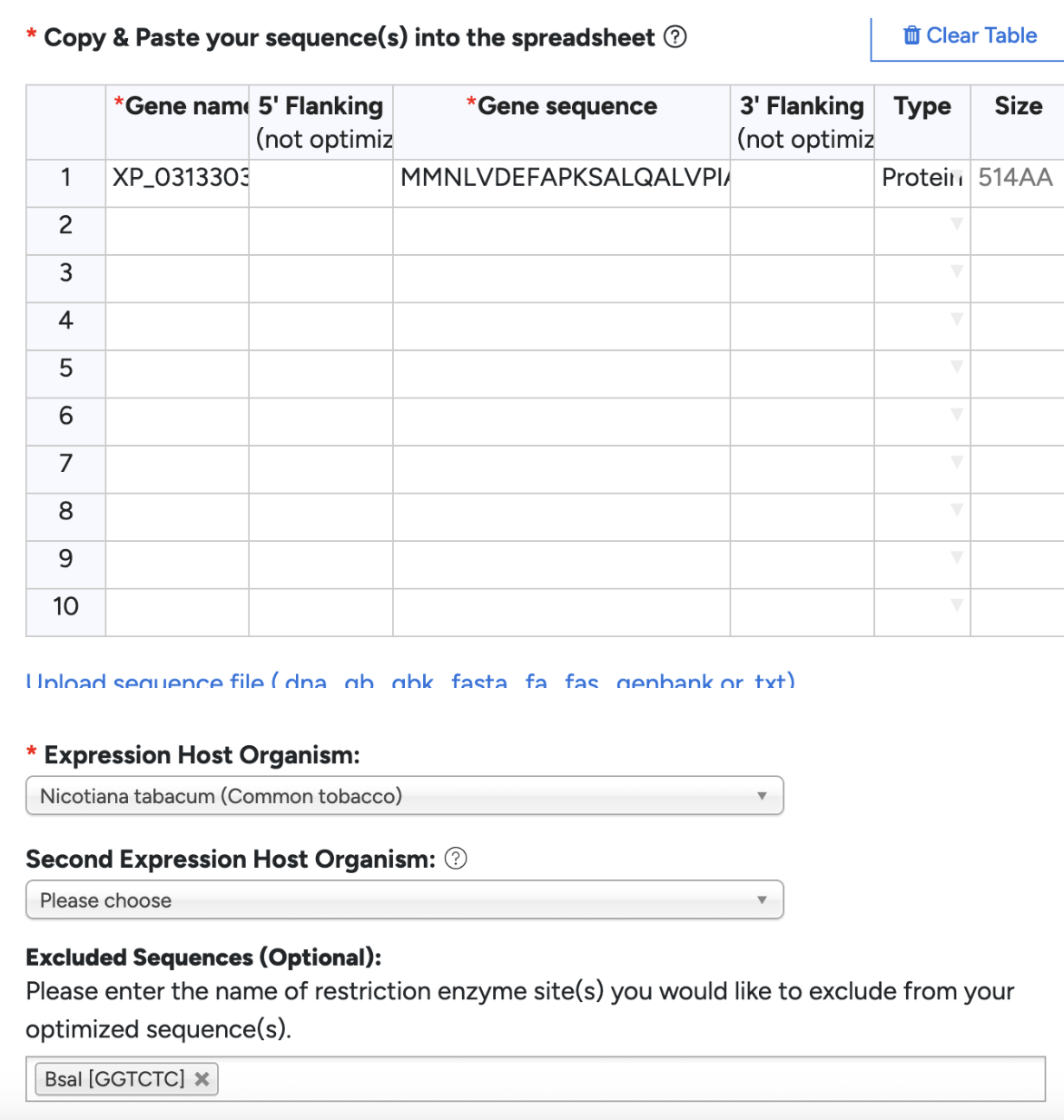
Since the same amino acid can be encoded by different codons, certain organisms prefer different codons for the same amino acid. My firefly genes have a different preference than a plant would, therefore I would need to optimize for plants to avoid truncated proteins or low production. I’m specifically optimizing for Nicotiana Rustica. I chose this species over the standard N.Bethamiana because I literally couldn’t source them anywhere in Vancouver, and there’s a slight benefit that my construct results will be more realistic for general plant species. I chose to optimize in GenSmart. Although GenSmart did not have Nicotiana Rustica, I went along with Nicotiana Tabacum as they are closely related. I excluded BsaI since I am planning to use Golden Gate Assembly.
3.4. What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein.
I will be using a cell-dependent approach since my long-term goal is to test this enzyme in plants. First I will use Golden Gate assembly to clone the gene I optimized into an expression vector. I will then heat shock this plasmid into Agrobacterium cells, and then inject the cultures into N. rustiva leaves. The plant will do the rest of the work, transcribing the DNA, and then translating it into the cytochrome p450 protein.
Part 4: Prepare a Twist DNA Synthesis Order
I inserted the optimized linear DNA sequence from GetSmart into Benchling and annotated it to XP_031330391 CDS. Since I am doing plant expression cassette, my DNA inserts will differ. I will need:
- CaMV 35S promoter
- Start Codon ATG
- XP_031330391 Coding Sequence
- Stop Codon
- NOS terminator
- No RBS needed (Plant cells don’t use RBS) and no His tag needed (I won’t need to purify).
Next I searched “CaMV 35S promoter” in AddGenes. I’m not sure how to pick the correct promoter so I had ChatGPT help me narrow it down to pP35S. Considering it says “Provides the CaMV 35S Promoter as a level 0”, it makes sense as a choice. Next I uploaded the GenBank file into Benchling, found the promoter, and inserted it into the beginning of my DNA sequence. I already had the start codon ATG in the beginning of my XP_031330391 CDS. GenSmart did not include a stop codon, so I added TAA.
I searched “NOS terminator” in AddGenes. I inferred lvl0 NOSt was the correct plasmid due to its description saying “Agrobacterium terminator for plant expression”, but double checked with ChatGPT just to be sure. Downloaded the GenBank, uploaded into Benchling, copy and pasted the terminator after XP_031330391 CDS.
Here is the link share: https://benchling.com/s/seq-aAJ0m9ksaKuRH7IAhY6i?m=slm-6QKUQA992f2toe6Y0U2a
From my understanding, I would need Gene Fragments since I am doing Golden Gate assembly, not Clonal Genes. I’ll still pick Clonal Genes for the purpose of this homework, choosing the pTwist Amp High Copy vector.
The Twist order in Benchling link: https://benchling.com/s/seq-5ic0dMqtz8CArU6mDtAJ?m=slm-ihfHt2ynzSsQTZMWejYG
5.1 DNA Read
I would use Nanopore for several reasons. Firstly, I am working out of a community lab, and this is one of the more accessible DNA sequencing technologies (that’s why it was taken to space!). Secondly, I’m still early on in my experimentation. If I need higher accuracy, I’d use Illumina+ or MGI+. Nanopore is third generation as you do not need to amplify your DNA through PCR. Therefore, my input would simply be raw DNA or RNA from my sample. To prepare my DNA for the nanopore, I’d need to extract my DNA and purify it from non-genetic material, before breaking it into smaller pieces for easier reading. Since fragmenting DNA leaves the end pieces jagged, I’d repair any blunt ends and add an adenine overhang so the adapters can grab onto the DNA. Nanopore requires a special adapter ligation onto the DNA ends to physically feed the DNA strand through the machine at a controlled pace. Lastly, I’d pipette my prepared sequence on a small flow cell plugged into the device.
The flow cell has many protein nanopores in its membrane, all of which have an electrical voltage creating a flow through each pore. As the motor feeds the DNA strands through the pores, the bases squeeze through the tiny pore, as each base blocks the current differently due to their slight differences in shapes and sizes. The machine measures these slight differences in the pore currents, creating a pattern based on the current changes and translating them into DNA letters. This simplicity allows the nanopore to be an efficient and cheaper DNA sequencing.
The output is a FASTQ file which carries the sequence and quality score (the confidence each base is correct). There is also a raw signal file of the actual electrical current data for analysis or troubleshooting.
5.2 DNA Write
As mentioned earlier, I’d like to synthesize and write my codon-optimized PPYR_02911 (XP_031330391), as I believe it is a good candidate for luciferin biosynthesis. Out of my non-novel genes, I’d like to synthesize plant-optimized BGL, Laccase ACOT1 and luciferase as they are big players in expressing autonomous bioluminescence in plants.
I’d use Twist’s phosphoramidite chemistry as it is the industry standard, and Twist developed supplementary technology to make it efficient and cheaper. I also love Twist, and want any excuse to use their website. Firstly, single-stranded DNA is built one nucleotide at a time from 3’ to 5’. Each nucleotide is a single cycle, each of which has deprotection, coupling, capping, and oxidation. Once this oligo is built, you cleave it off the solid support it was initially built on. If your gene is longer than a single oligo, Twist assembles them into a single gene with PCR. This gene then gets cloned, inserted into a plasmid vector, double-checked for verification, and shipped. As with any other writing method, there are compounding errors. Furthermore, since oligos get maxed out fairly quickly in length, stitching genes together causes the process to be more error-prone.
5.3 DNA Edit
Although glowing plants are a marvel, I would love my long term goals to be more beneficial for humanity. Plants are integral to our survival, whether it’s due to crops, materials, or our air. They are beautiful and crucial. My dream would be to take my ambition further, whether it’s to engineer nitrogen fixation in crops, help conserve plant species, or make trees more efficient at carbon sequestration. For such ambitions, you’d need the big guns: CRISPR-Cas9.
The steps of CRISPS are as follows. First you design a guide RNA, a short sequence that matches the genome region you’d like to edit. This guide RNA tells the Cas9 where to cut. This is done through Benchling or CRISPOR. You’d also prepare your donor template if using one, a sequence containing your insert with sequences matching the cut site at both ends. Therefore, the inputs are the Cas9 plasmid, sgRNA, donor template DNA and target cells. You then deliver this plasmid into wherever cell you’re utilizing. After the CAS9 makes a double stranded break, you can edit either through NHEJ or HDR. With Non-Homologous End Joining the cell glues the broken strands back together, often introducing small deletions for knocking out genes. Homology-Directed Repair uses the donor template to insert the new DNA at the cut side to knock in a gene. Lastly, verify by sequencing the targeted region.
The limitations to CRISPR: 1. Knocking in genes has a low success rate in plants. 2. The guide RNA might lead the Cas9 to the wrong sequence as sequences may look similar 3. You may have mosaicism as regenerating a whole plant with edited cells is difficult.
Work Cited
Fallon et al. 2018, “Firefly genomes illuminate parallel origins of bioluminescence in beetles” in eLife (DOI: 10.7554/eLife.36495
Ai Citations: I cannot find N.Bethamiana for my glowing plant project, world Nicotiana rustica work?
[Copy and pasted 4.2 question], what inserts would my version need since I’m optimizing for plants?
Is GAA a stop codon? [Follow-up]: what should be my stop codon?
Is nanopore first, second or third generation?
Explain how a nanopore works in detail. What are the inputs and outputs?
Explain how Twist sequences DNA in detail.
Explain how CRISPR works in detail.
Top limitations of CRISPR in Plant edits