Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) A dalton is a unit of mass defined as 1/12th the mass of an unbound neutral Carbon-12 atom at rest, equivalent to 1.66X10-24 grams. It is commonly used in molecular biology to calculate the mass of large molecules into grams. Meat protein is roughly 25% protein by mass, therefore 500g x 0.25 = 125g of protein since almost all amino acids are concentrated in the protein of meat.
Given the formula:
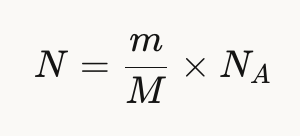
N = 125g/100 g/mol x Avogadro’s number = 7.5 x 1023 amino acid molecules
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because we all use the same amino acids as building blocks, but our DNA tells us how to use these building blocks differently. We digest the proteins into individual amino acids, after which our DNA rebuilds them into human proteins, not cow or fish proteins.
3. Why are there only 20 natural amino acids?
As mentioned in previous lectures, the true number of combinations possible through codons is vast. However, this makes life less efficient and wastefully redundant. Instead, nature has evolved to produce 20 amino acids, leading to more efficient translation, error tolerance (as multiple codons map to the same amino acids), while maintaining diversity.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, and this is an active field in synthetic biology, as long as you keep the universal backbone intact: An amino group, a carboxyl group, a central carbon, and a hydrogen on the alpha carbon. The creativity happens in a new R-group side chain. For example, while upholding the universal backbone, I could design a D-luciferin as the R-group, allowing for a possibly self-fueling luciferase, where the side chan is being used as substrate for the luciferase (no external luciferin required).
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids have the ability to form spontaneously through chemistry without the aid of enzymes, as proven by the Miller-Urey Experiment (1953), and observed from meteorites and hydrothermal vents. The theory follows that after this spontaneous accumulation of amino acids, peptides began to form, catalyze, and eventually self-replicate, eventually evolving into increasingly more efficient and controlled life.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
An alpha-helix is one of the most common shapes proteins tend to fold into, where it winds into a corkscrew shape while held together by hydrogen bonds between the C=O of one amino acid and the N-H of the amino acid 4 resides ahead. The R-group sticks out from the helix (the side chains pointing away from the spiral). D-amino acids are the mirror image of the L-amino acid. Since the L-amino acids produce right handed alpha helices, D-amino acid would be the left handed version. These are less common in nature.
7. Can you discover additional helices in proteins?
Yes, in addition to alpha helixes, there can be other types of spirals/protein helices. The differentiation comes down to how far apart the hydrogen bonding partners are, as long as they remain within the physical constraints of the peptide backbone limits. However, with synthetic amino acids, you could hypothetically design helices with differing geometries than those found in nature, if they maintain the minimum stability required for the backbone.
8. Why are most molecular helices right-handed?
There are two layers to this question. The first is “why” life chose L-amino acids, to which the answer is unclear. It could have been random, inherited bias from meteorites, or a small difference in nuclear force. As life chose a preference for L-amino acids, biology adjusted to fit this specific type of arrangement, where folding is more energetically favourable due to less steric clashes between the R-group and the backbone. Similar to a nut and bolt, where the wrong bolt (R-amino acid) has trouble twisting into a nut created for L-amino acids.
9. Why do β-sheets tend to aggregate?
A beta-sheet is the other main secondary structure alongside the alpha helix (different shapes found within the same protein). Unlike the alpha helix that form into spirals, the protein chains extend into flat strands side by side, similarly held together by hydrogen bonds. They tend to aggregate because beta sheets have exposed edges of hydrogen bond donors and acceptors (N-H and C=O), unlike the alpha helixes where all the hydrogen bonds have partners. These sticky edges lead to continued chain reactions of recruitment, while the flat shape creates hydrophobic surfaces that stack on each other.
10. What is the driving force for β-sheet aggregation?
The driving force comes down to thermodynamics. Nature picks whatever is more thermodynamically favourable. Exposed edges wanting bonding partners and hydrophobic interactions lower the free energy of the system. Aggregations make the system happier and stable, as the bonds find partners, and the sheets stack on each other, hiding themselves from water. If something is more thermodynamically stable, it is inevitable.
11. Why do many amyloid diseases form β-sheets?
Amyloid diseases are diseases where proteins misfold into beta sheets and clump together into large amyloid fibrils. These fibrils damage and kill surrounding cells and tissues. They tend to fold into beta-sheets because beta sheets are the most stable misfolded state a protein can create. This is because any protein has the capability to create beta sheets (since it requires backbone atoms all protein have), the hydrogen bonding along the edges and hydrophobic stacking create a low energy structure that is very difficult to break apart, and they are self-propagating (as the edges recruit more strands, causing other proteins to misfold and join).
AI Searches:
What is a dalton?
What is the formula for calculating the number of amino acids in a given mass?
What percentage of meat is protein?
Can you make other non-natural amino acids? + what is required to make a new amino acid?
Where did amino acids come from before enzymes?
What is an alpha helix?
What are d-amino acids?
Are there other types of helices in proteins except alpha helices?
Why does nature / prefer L-amino acids?
What is a β-sheet? Why do they aggregate?
What are amyloid diseases? Why do they tend to form beta-sheets?
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
The protein I selected is firefly luciferase, as it is most applicable to my project. Its UniProt ID is P08659. The format biochemical name is Luciferin 4-monooxygenase (Luciferin = substrate it acts on, 4-monooxygenase = adds one oxygen atom at the 4th position).
How long is it? What is the most frequent amino acid?
The length is 550 amino acids, and the mass is 60,745 Da. The most common amino acid is L, which appears 52 times.
How many protein sequence homologs are there for your protein?
The BLAST search found 250 protein homologs, including luciferases from various species. Most identities were quite low, with a small cluster at 95%+.
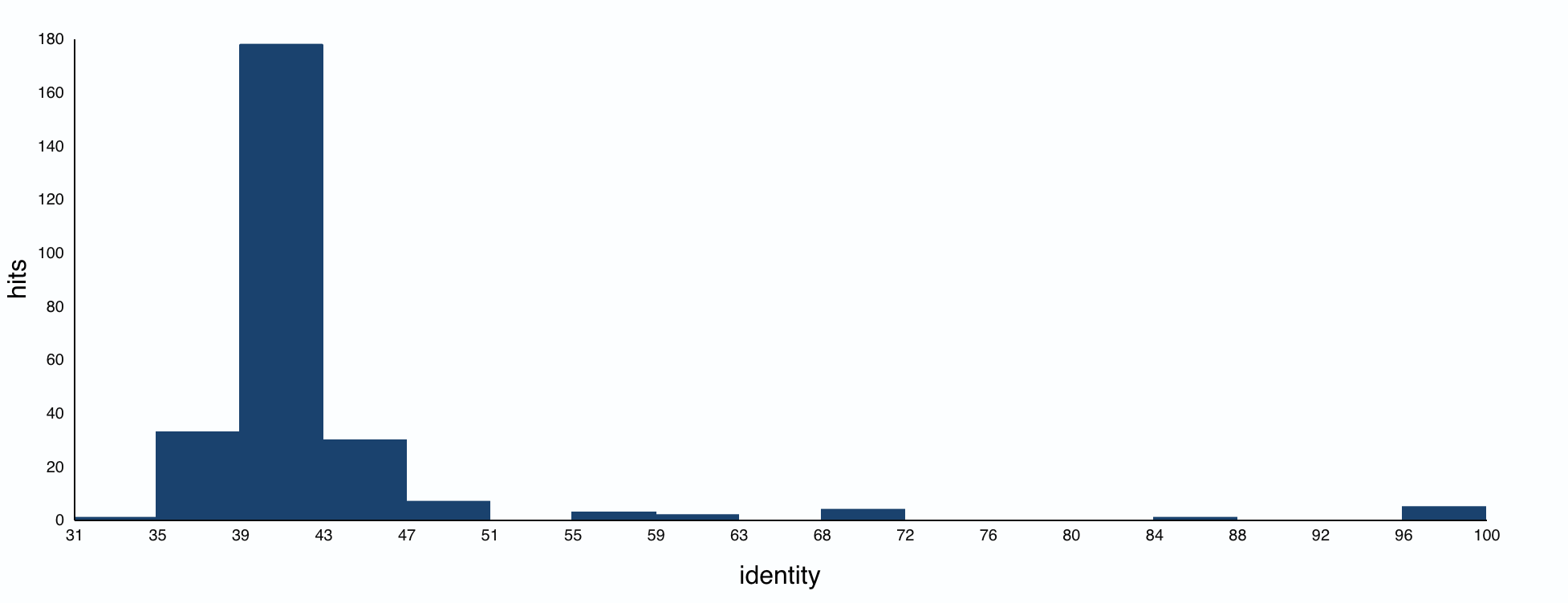
Does your protein belong to any protein family?
The homologs are all annotated as Luciferin 4-monooxygenase. The annotations for individual homologs generally mention ATP binding and CoA-ligase activity, pointing to an AMP-binding enzyme family.
Identify the structure page of your protein in RCSB
The smallest PDB under Structure is 1.70Å, 3RIX.
When was the structure solved? Is it a good quality structure?
The released date is 2011-12-07. Since the resolution is 1.70Å, it’s a good quality structure since lower resolution means more detail.
Are there any other molecules in the solved structure apart from protein?
There is one ligand in the structure, listed ID 923. It appears to be a complex organic molecule, and some sort of luciferin or substrate analog based on the ring system. Its formula is C₂₇H₂₈O₇.
Does your protein belong to any structure classification family?
Yes, the luciferase belongs to the Acetyl-CoA synthetase-like superfamily. It shares a 3D fold to other AMP-binding enzymes (enzymes that bind and use AMP), CoA ligases (attach CoA to molecules), and Acetyl-CoA synthetase (help metabolize fatty acids). This is likely because luciferase is theorized to have evolved from an ancestral CoA ligase enzyme that was repurposed for bioluminescence.

Open the structure of your protein in any 3D molecule visualization software:
Because I’ve already used ChimeraX in my research, I chose it as my molecular visualization software.
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon: Overall shape and secondary structures. Luciferase has predominantly curly ribbon shapes (alpha helices) and some flat arrows (beta sheets). It’s a large protein, and quite complex. There seem to be multiple domains.
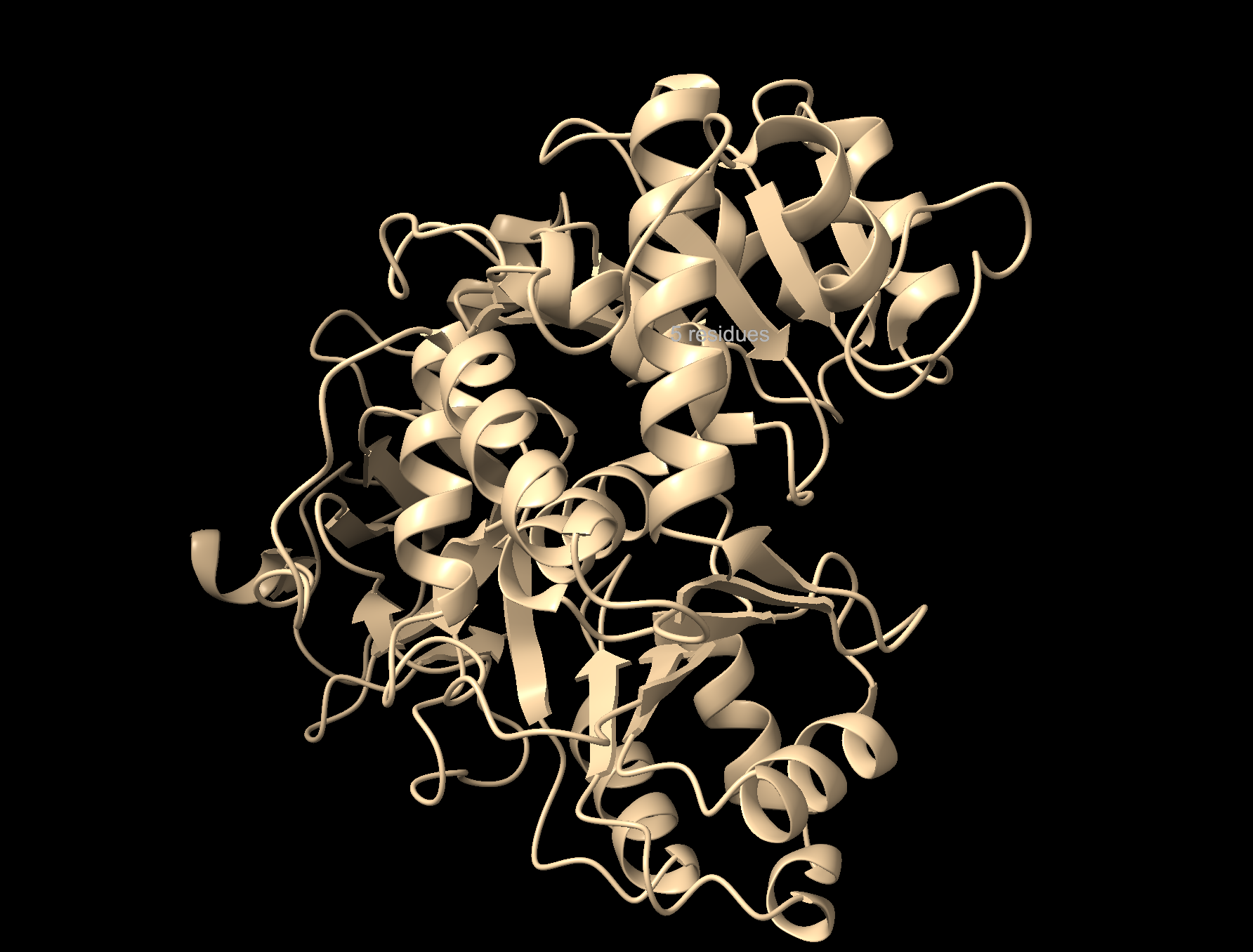
Ribbon: Backbone seen more clearly. Thick cylinders are alpha helices, flat sections are beta sheets, thin threads are loops.
Ball and stick: Shows every atom and bond in protein colored by element. As expected, the core is made out of lots of beige (as it is carbon making up the backbone and side chains). Red dots are oxygen atoms, blue dots are nitrogen atoms. Few yellow spots present, showing sulfur atoms (from the cystine and methionine residues).

Color the protein by secondary structure. Does it have more helices or sheets?
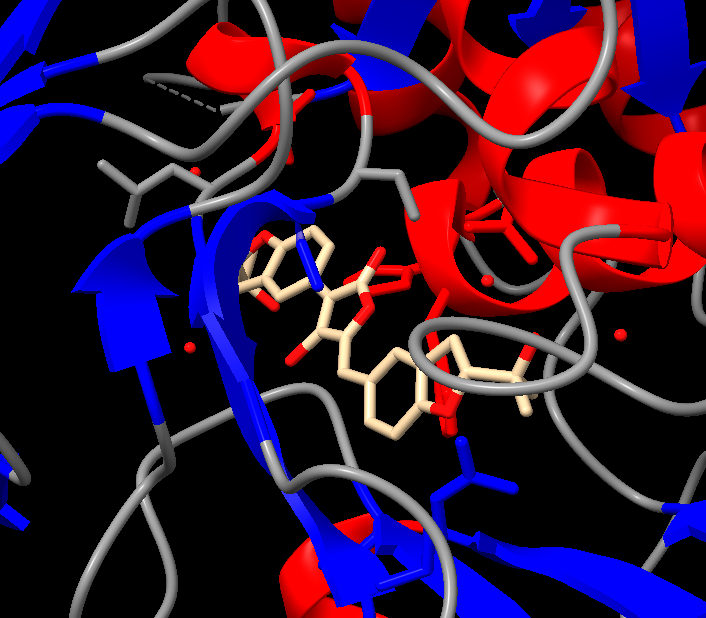
It appears to mostly have alpha helices, although there are a good amount of beta sheets present. You can also clearly see the substrate analog (ligand 923) from earlier in the active site. That’s where the luciferin would bind.
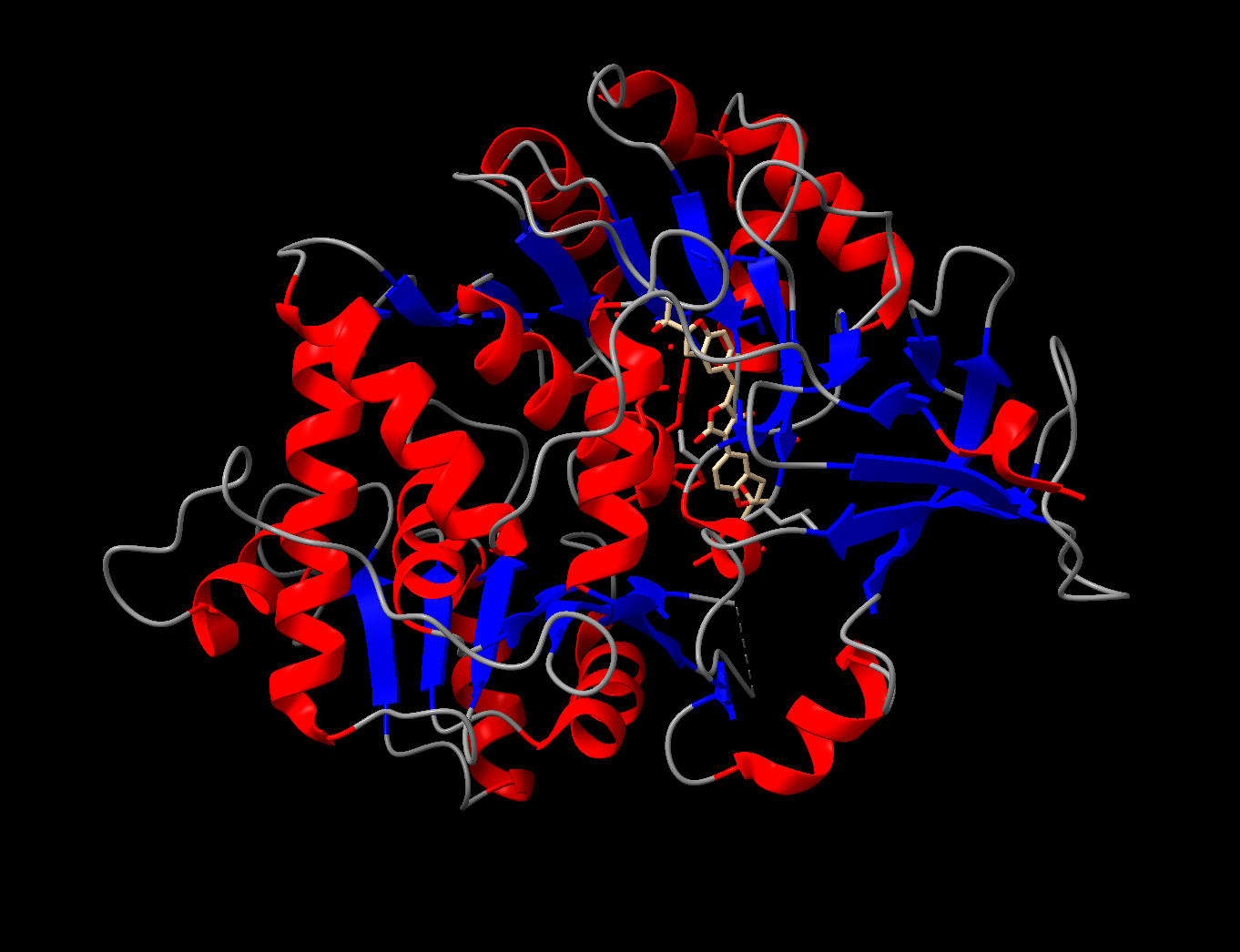
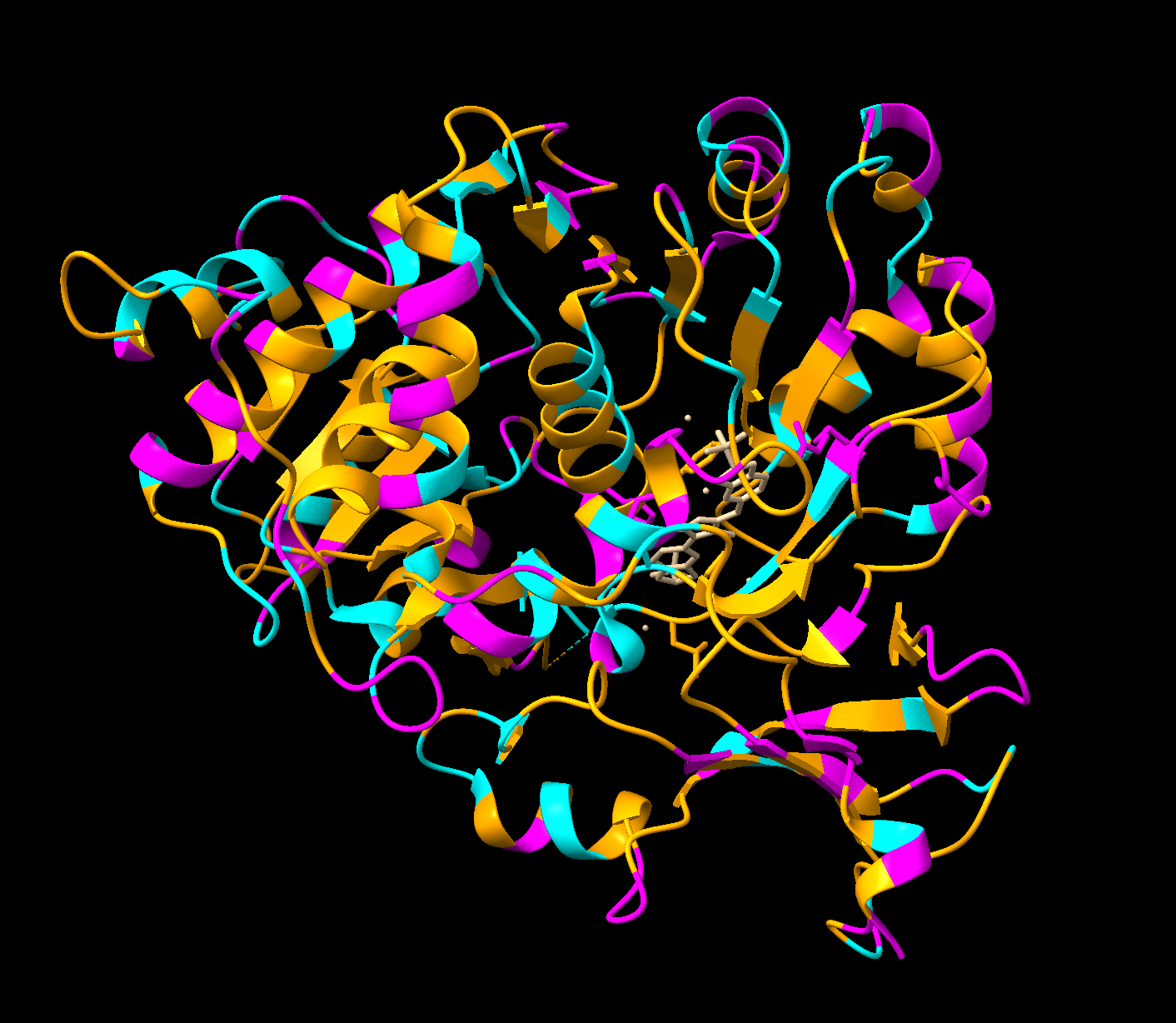
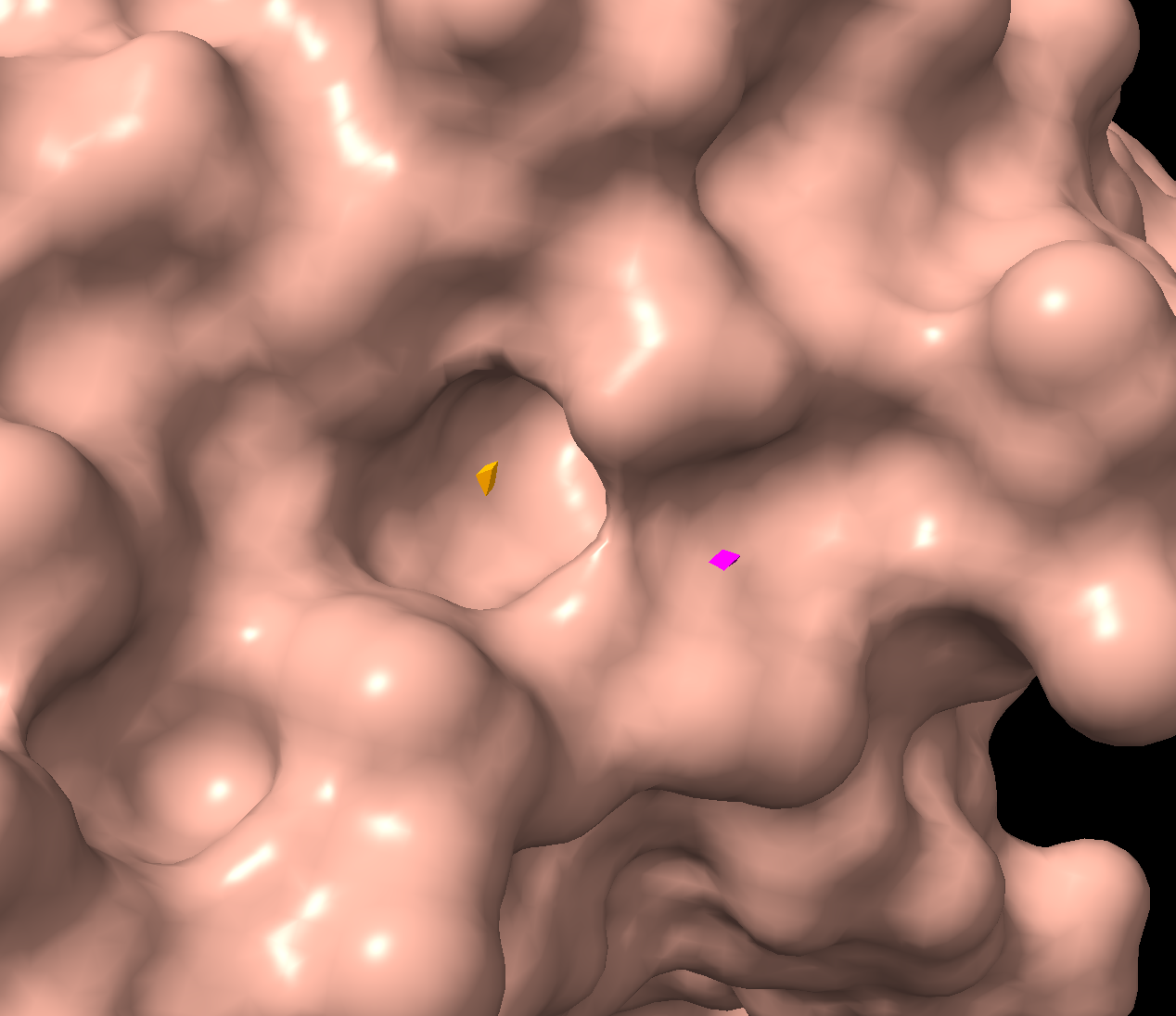
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Orange = hydrophobic residues
Cyan = polar residues
Magenta = charged residues
The hydrophobic residues are concentrated in the core of the protein, while the hydrophobic residues are concentrated in the outer loops and surface. This makes sense for soluble proteins (which luciferase is as it floats freely in water inside our inside cells), hydrophobic residues pack deeper to avoid the water while the hydrophilic face outwards.
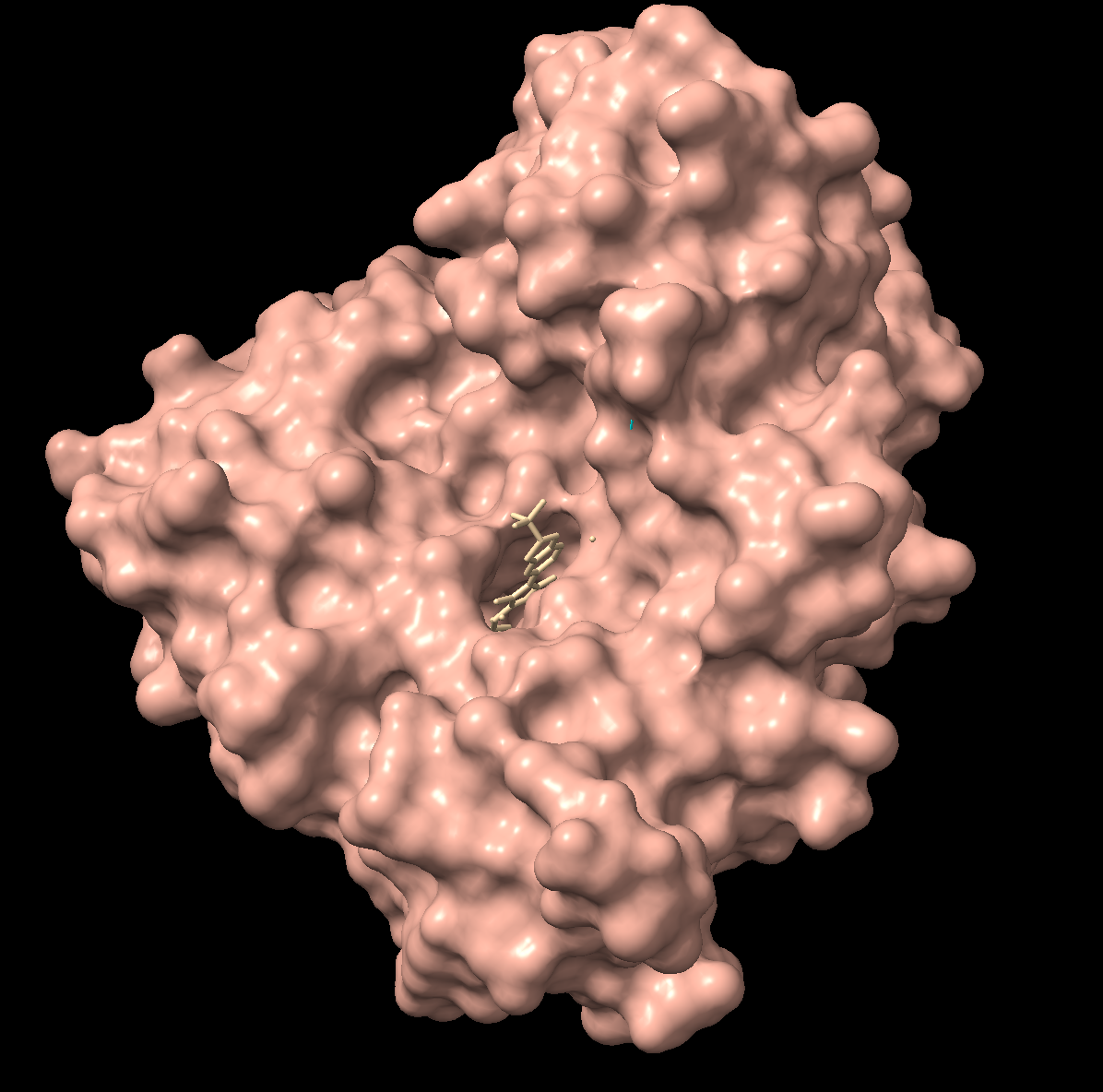
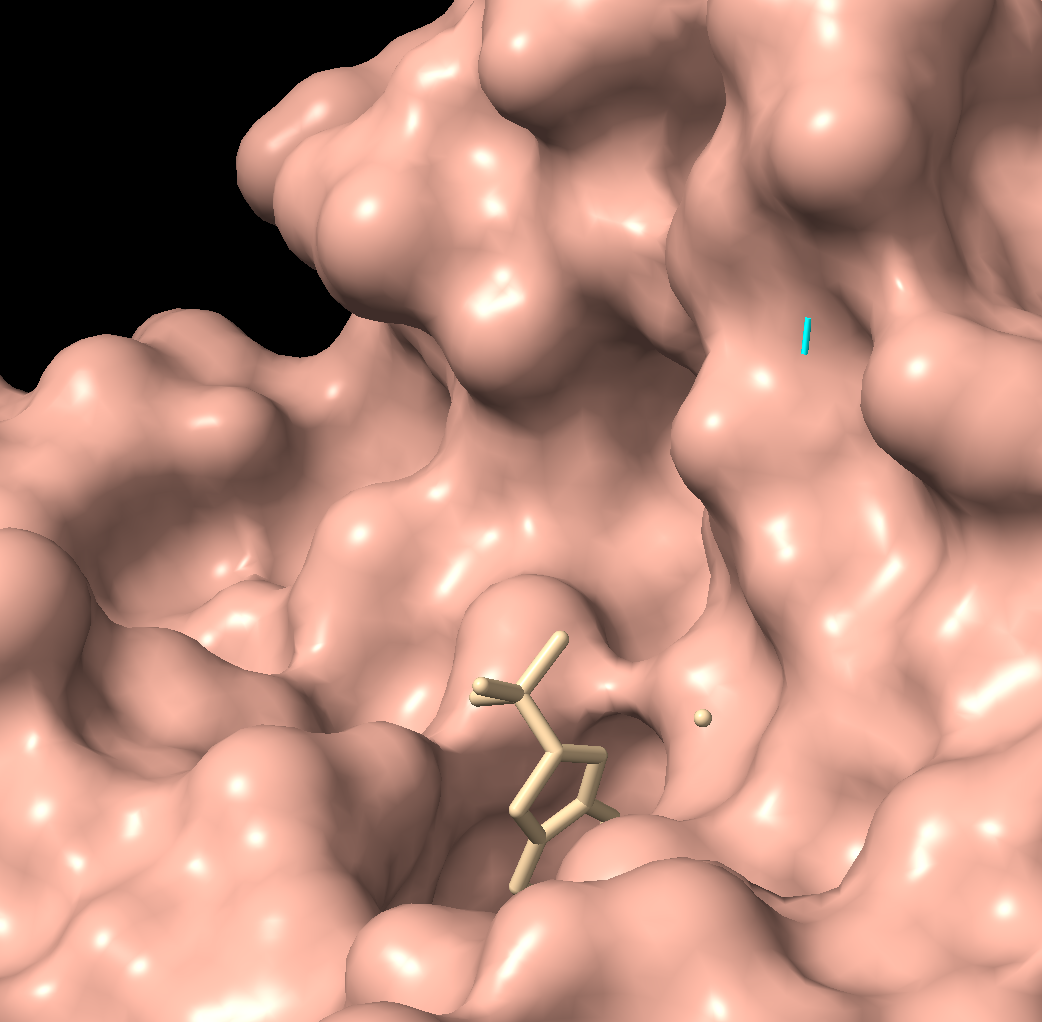
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
There is a clear, large binding pocket (the active site) where the D-luciferin would bind, and allow for the light-producing reaction. You can see a polar residue poking out near the active site, probably to help position the substrates through the hydrogen bonds. There are three water molecules near and inside the active site, likely involved in the reaction and guiding the substrate into the pocket. On the other side there also seems to be Valine 365 and Lysine 9 poking out of a smaller cavity entrance. Upon investigating, this region seems to be overwhelmingly hydrophobic, and does not seem to be connected to the active site. Could be a remnant of the original fatty acid binding site. Could affect how enzymes behave in plant cells where lots of fatty acids float around.
AI:
Used some help from Claude prompting ChimeraX.
Part C. Using ML-Based Protein Design Tools
Protein used: 3RIX (luciferase)
C1. Protein Language Modeling
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Yellow/green (positive) = mutation is tolerated or beneficial
Teal/cyan (near zero) = neutral
Dark blue/purple (negative) = mutation is damaging
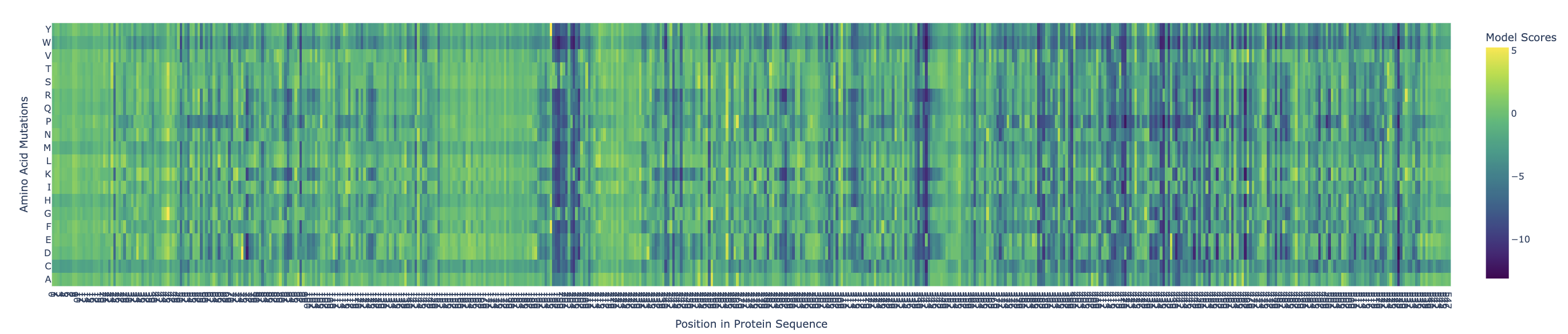
There are definitely ranges where mutations are more critical, such as:
X = 196-206
X = 338-343
X = 420
X = 435-438
X = 468
X = 527
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Examples:
x: 198, y: W, z: -11.7
Position 198 is Serine (S) a small, polar amino acid, while Tryptophan (W) is the largest amino acid. There may be no room for the Tryptophan, or is too hydrophobic, causing large disruptions in local hydrogen bonding networks.
X: 527, y: W, z:-12.0
This is near the integral C-terminal domain, which is even smaller and tightly packed. Mutating the amino acid to Tryptophan would cause large disruptions during the integral catalysis rotation phase.
X: 342
At this site nothing is tolerated except Glutamate (E) which scores neutral, even though the initial position is at Leucine (L). These are different chemically, so only E being tolerated is highly unusual for such different amino acids. This might be a limitation of the language model.
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
https://pmc.ncbi.nlm.nih.gov/articles/PMC10956436/
Using Discovery of Red-Shifting Mutations in Firefly Luciferase Using High-Throughput Biochemistry, we know the experimentally validated mutations are:
N229T: red-shifts emission above 600 nm. Z = 0.06, correctly predicted these are tolerated.
T352M: red-shifts emission above 600 nm. Z = -2.4, partially wrong, experimentally enzyme still works
L286V: causes red emission
ESM2 correctly identified N229T and L286V as tolerated mutations, but scored T352M incorrectly as moderately damaging, highlighting the limitations of language models in distinguishing mutation effect.
Latent Space Analysis
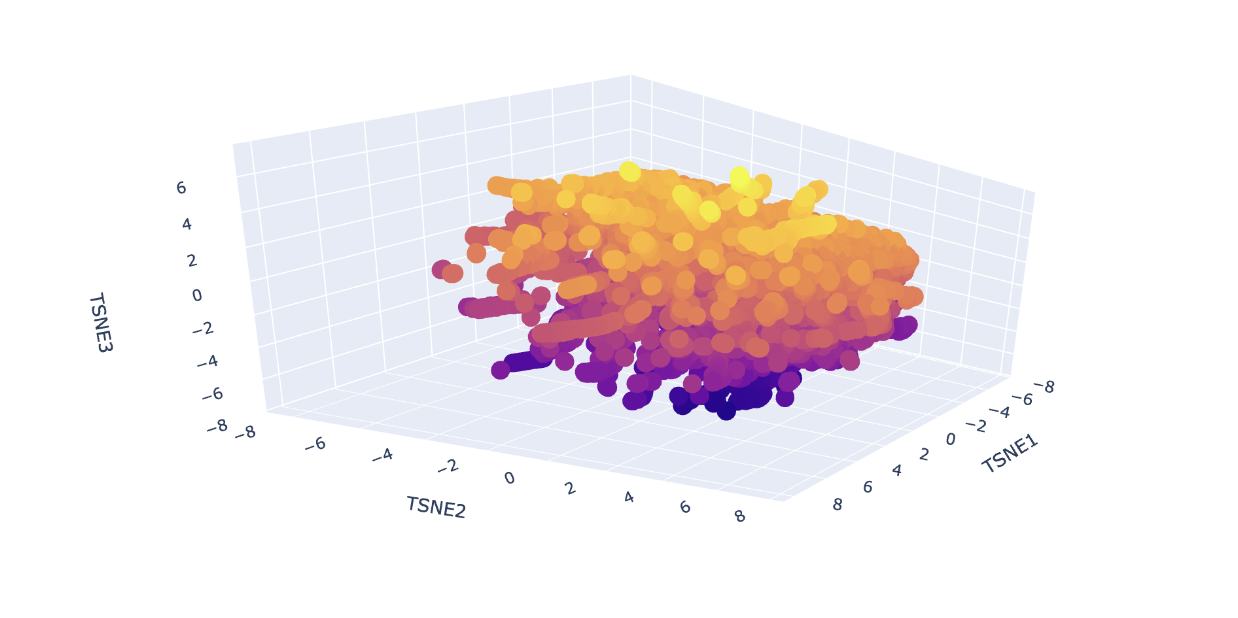
Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, I can see the clustering of proteins by similarity, indicated by colour. The colour groups seem to show the model can distinguish the different protein sequences. For example, at around TSNE1 = 4, TSNE2 = - 4 and TSNE3 = -5, we see a similar type of protein from different species (humans, rats and flies) all clustered together. This similar protein is Staphylococcus aureus Protein A which binds to mammalian antibodies, hence the clustering.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
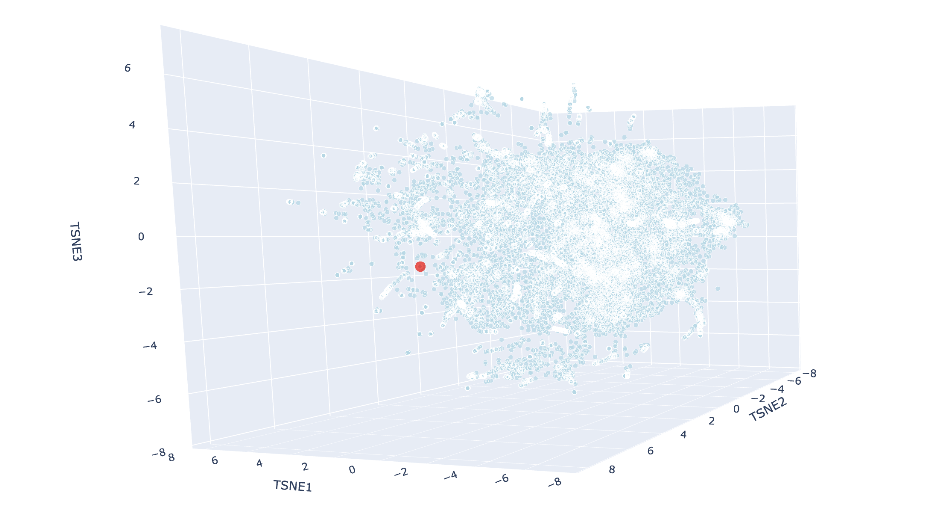
Using Claude Code, I ran a new cell where I added my protein’s embedding to the already-computed data and rerun just the t-SNE part to save time. The red luciferase was made to appear as a big red dot. The neighbors were:
Flavodoxin FldA: Aquifex aeolicus (SCOP: c.23.5.8)
Flavodoxin: Desulfovibrio vulgaris (SCOP: c.23.5.1)
Hypothetical protein YwqN: Bacillus subtilis (SCOP: c.23.5.6)
ROO-like flavoprotein TM0755: hermotoga maritima (SCOP: c.23.5.1)
Automated matches: Fusobacterium nucleatum (SCOP: c.23.5.0)
These all share the SCOP classification of c.23.5, meaning they are all in a similar fold family. Most of them are also flavodoxins, which make sense as all these (including luciferase) are nucleotide-binding enzymes, and share similar alpha and beta folds.
C2. Protein Folding
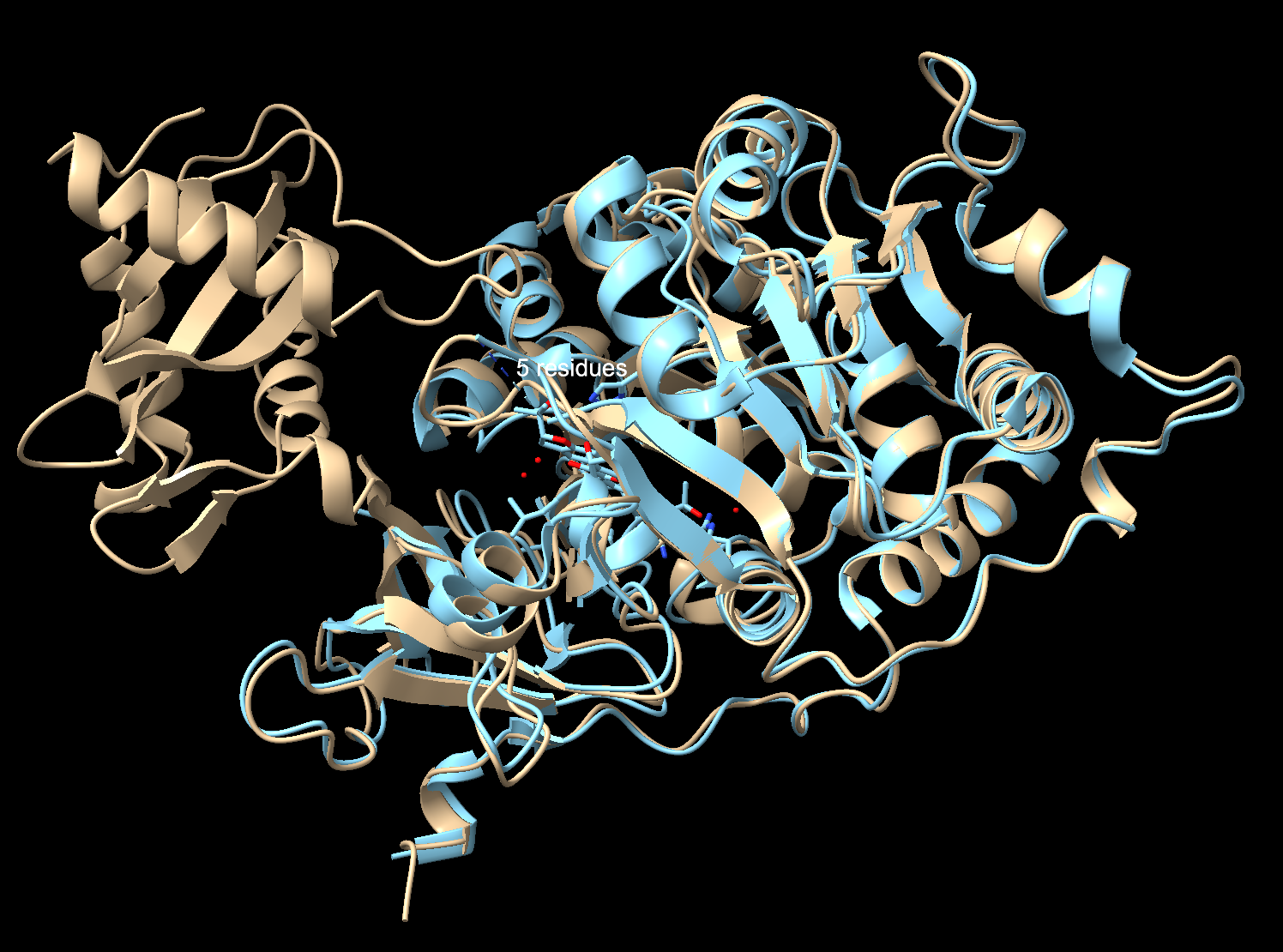
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
pTM: 0.901: Very high. ESMFold is extremely confident in the overall fold
pLDDT: 90.4: Excellent. Per-residue confidence is very high (above 90 out of 100)
To check I compared my ESMFold prediction to the real crystal structure in Chimerax. I used Claude Code to help me download the files from the notebook, and then superimpose them with my previous file in ChimeraX. Overall, they match well. The tan is the 3RIX crystal structure, the blue is the ESMFold prediction. The N-terminal matches almost exactly, where the helices and sheets overlap very well. The C-terminal has an offset between the two structures. That’s because the C-terminal domain moves catalysis, so the 3RIX only captured it in one conformation with a substrate analog bound (the protein with a molecule in the active site), while the ESMFold predicted it without the ligand in the unbound state (protein by itself with nothing bound to it). The former forces the C-terminal into a different position where it is practically closed, while the latter keeps the protein in a relaxed state.
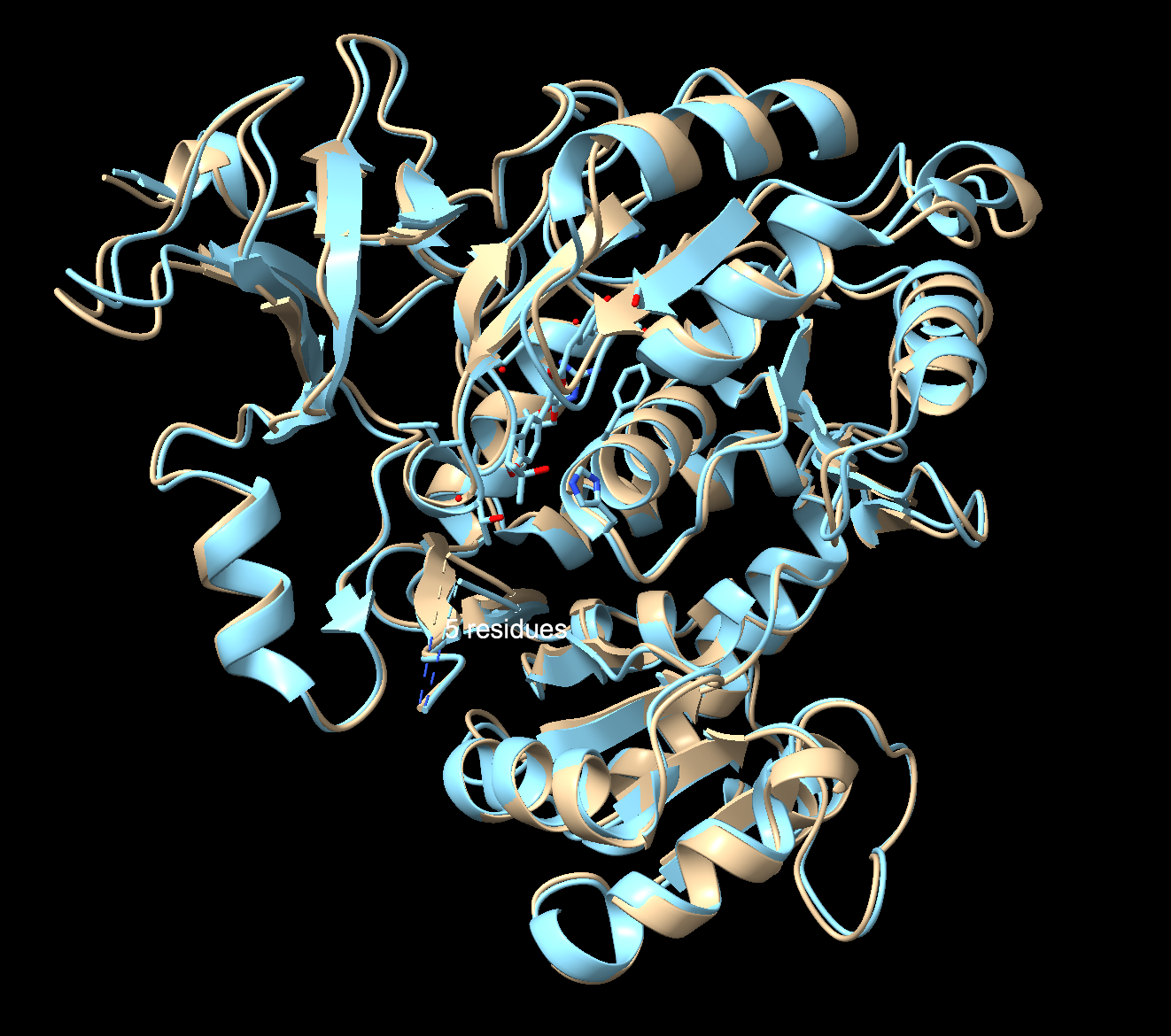
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Starting with a small mutation (Serine 198 to Tryptophane from the heatmap):
pTM: 0.905
pLDDT: 90.692
This is a very small difference from the original scores, meaning ESMFold predicts the fold is not affected by this single mutation, even though it also scored this mutation as “highly damaging” earlier. It may be that although the protein continues to fold directly, the function of the protein may be broken, and would not catalyze the reaction properly. This is a limitation of the ESMFold, where the fold may be said to occur “successfully”, but the protein is no longer viable in its function.
Removing a loop (positions 280-289):
pTM: 0.937
pLDDT: 92.6
Removing a loop made ESMFold more confident than the single mutation above, suggesting the region I deleted was a floppy loop that was actually lowering the confidence score. Its removal made the structure more stable.
Trying a larger change, removing a big chunk from 200-300:
pTM: 0.768
pLDDT: 86.6
The score has dropped significantly from the larger mutation, showing the structure cannot handle large deletions.
The luciferase protein structure seems to be quite resilient (at least in folding ability) to small mutations, but suffers immensely from larger mutations. Interestingly, removing certain floppy regions actually makes the folding process more stable.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
This section had a lot of bugs, so I used ClaudeCode to help ensure my code was running smoothly.
Original sequence:
3RIX, score=1.4079, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020
DAKNIKKGPAPFYPLEDGTAGEQLHKAMKRYALVPGTIAFTDAHIEVNITYAEYFEMSVRLAEAMKRYGLNTNHRIVVCSENSLQFFMPVLGALFIGVAVAPANDIYNERELLNSMNISQPTVVFVSKKGLQKILNVQKKLPIIQKIIIMDSKTDYQGFQSMYTFVTSHLPPGFNEYDFVPESFDRDKTIALIMNSXXXXXLPKGVALPHRTACVRFSHARDPIFGNQIIPDTAILSVVPFHHGFGMFTTLGYLICGFRVVLMYRFEEELFLRSLQDYKIQSALLVPTLFSFFAKSTLIDKYDLSNLHEIASGGAPLSKEVGEAVAKRFHLPGIRQGYGLTETTSAILITPEGDDKPGAVGKVVPFFEAKVVDLDTGKTLGVNQRGELCVRGPMIMSGYVNNPEATNALIDKDGWLHSGDIAYWDEDEHFFIV
ProteinMPNN’s proposed sequence:
T=0.1, sample=0, score=0.7081, seq_recovery=0.5164
DPANIRRGPAPAEPLPPGTAGQLLFDALKKFAKEPGKIAFIDAETGEKMTYKEFYETAVKMAAALKNYGLDKNDVIAVISKNSLEYLIPVLAALMIGIAVAPVDPNYNVEELYHVLSLAKPKVVFTSKENLEKVLKVKEKLPIIKEIIVLDSEEEYKGYPSIKTFMESHLPEGFNPWEFKPEEFDPEETVAFLLEDXXXXXKPRLVELPHLALRHNLVVARDPVFGYPVEPNTVILNSLPLHKNVGLFTTLGAIYNGFTIVLLSEYDEDLFLKTIQDYKVNIVYLSPEQAELLAKSTKISKYDISSLKEIVVGGAPLDKEVAEKVAKKFGLPGIRTGYGKTELSHAFLITPRGEEVPGSVGRVVPGHEARVVDPTTGAVLGVNEVGELEVRGPMLMRGYRDDPAATAARVDADGWYHTGDLAYFDENGHWFIV
Seq_recovery = 0.5164, design sequence 51.6% identical to the original. Score = 0.7081, ProteinMPNN confident the sequence would fold correctly. This is higher than the original score of 1.4079.
Then I ran code:
Output:
Identical positions: 226/433 (52.2%)
Position-by-position (. = match, X = different):
.XX..XX….XX..XX….XX.XX.X.XX.XX..X…X..XXXXXX..X.XX.XX.XX.X.X.X…XX.XX.X.X.X…XXXX…X..X..X….XXXX..XX..XXXXXXXX.X…X..XX.X.X.X.XX…..XX..XX..XXX.X.XX.XX..XX….X…XXX.X..X..XXX.X.XXXXX…..X.XX.X…XXXXXXXXX….X..XXXX.X.X..XXX.X.XXX.X…..XXXX..XX..XXXXX.X…XXX….XXXXX.X.XXXXXX….X.X…X.X.X..XX…..X…X.X…X.X…..X…X..XXX.X….X.XXX..X..X…XX..X…XX..XX….XX…X…..X.X..XXX.X..X.XX.X…X.X..X..X..XX.X…
N-terminal third (1-144): 72/144 (50.0%) match
Middle third (145-288): 65/144 (45.1%) match
C-terminal third (289-433): 89/145 (61.4%) match
Conserved hydrophobic: 116
Conserved charged: 37
Conserved polar: 36
The terminal most conserved is the C-terminal, while the least conserved region is the middle region. Furthermore, the hydrophobic residues seem to be the most conserved, as they are packed the closest to the core, and therefore, have more specific shapes.
Input this sequence into ESMFold and compare the predicted structure to your original.
pTM: 0.932
pLDDT: 92.4
Both numbers scored higher than the original full length sequence, implying a higher folding confidence with the predicted structure. Comparing the original to the predicted structure in ChimeraX revealed that despite being half identical, it folds essentially into the same 3D structure. The fold is robust, and multiple sequences can encode into a similar protein shape. Great news for redesigning luciferase!
Ai:
Why is changing Serine to Tryptophan in Position 198 of 3RIX a detrimental mutation? + What about at position 527?
Why is changing Leucine to Glutamate at position 342 a detrimental mutation?
Find a paper with sequences for which we have experimental scans that are about luciferase.
Used ClaudeCode for Latent Space section to add protein’s embedding to the already-computed data and rerun just the t-SNE part.
How are the (neighbor proteins) similar to luciferase?
Why doesn’t the C-terminal match between the crystal structure and ESMFold?
Used ClaudeCode in the C3 section as the section had lots of coding errors.
Part D. Group Brainstorm on Bacteriophage Engineering
Write a 1-page proposal (bullet points or short paragraphs) describing:
- Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
- Why do you think those tools might help solve your chosen sub-problem?
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
- Include a schematic of your pipeline.