Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
SOD1 A4V mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Notestranslation begins with Methionine in eukaryotes, therefore the position of translation is technically 5 (A -> V). Protein language models train on amino acid sequences. Masked language modelling trains sequences by randomly masking some positions, and training the model to fill in the masked positions based on context. PepMLM does the same thing but to peptide binder design, where it masks all the peptides in the proteins you give, iteratively fills in those masked positions from most to least confident. Low perplexity = more confident, high perplexity = model uncertain.
| Sequence | Pseudo Perplexity |
|---|---|
| WHYYPVAVELKK | 14.221520879021000 |
| WHSPPAAARLWE | 19.212231606782200 |
| WLYYAVGAALWX | 12.951986018963500 |
| WHSPVVAVEHKX | 10.69098885919460 |
| FLYRWLPSRRGG | - |
Part 2: Evaluate Binders with AlphaFold3
| Number | Binder | Pseudo Perplexity | ipTM |
|---|---|---|---|
| Peptide 1 | WHYYPVAVELKK | 14.221520879021000 | 0.22 |
| Peptide 2 | WHSPPAAARLWE | 19.212231606782200 | 0.27 |
| Peptide 3 | WRSGAVGAAHKK | 8.089792 | 0.58 |
| Peptide 4 | WRYYAVAARLGK | 10.665547 | 0.29 |
| Peptide 5 | FLYRWLPSRRGG | - | 0.37 |
- Peptide 1 has a low confidence score, and consistent with its low ipTM, appears to be floating off to the side, not even engaging with SOD1. It does not localize near the N-terminus, nor engage with β-barrel region or dimer interface.
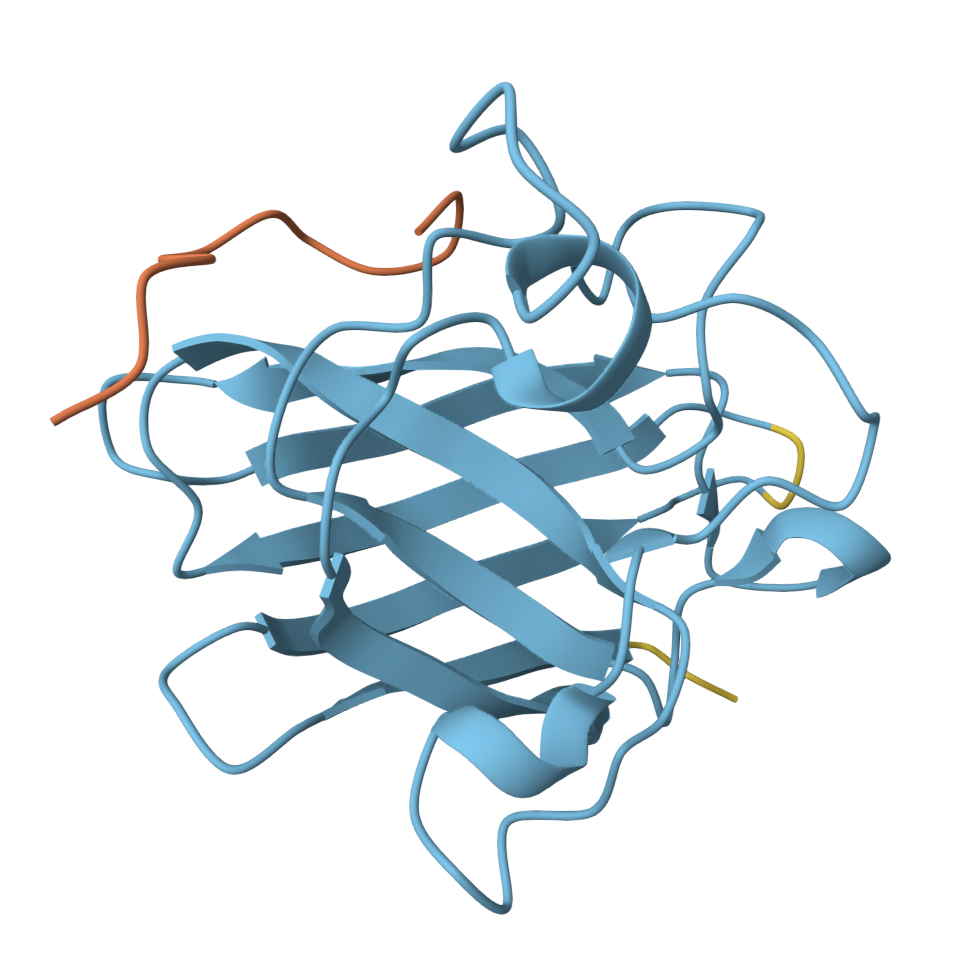
- Peptide 2 similarly has a poor ipTM score. It is not localized near the N-terminus, instead, seemingly loosely associated with the c-terminal region.
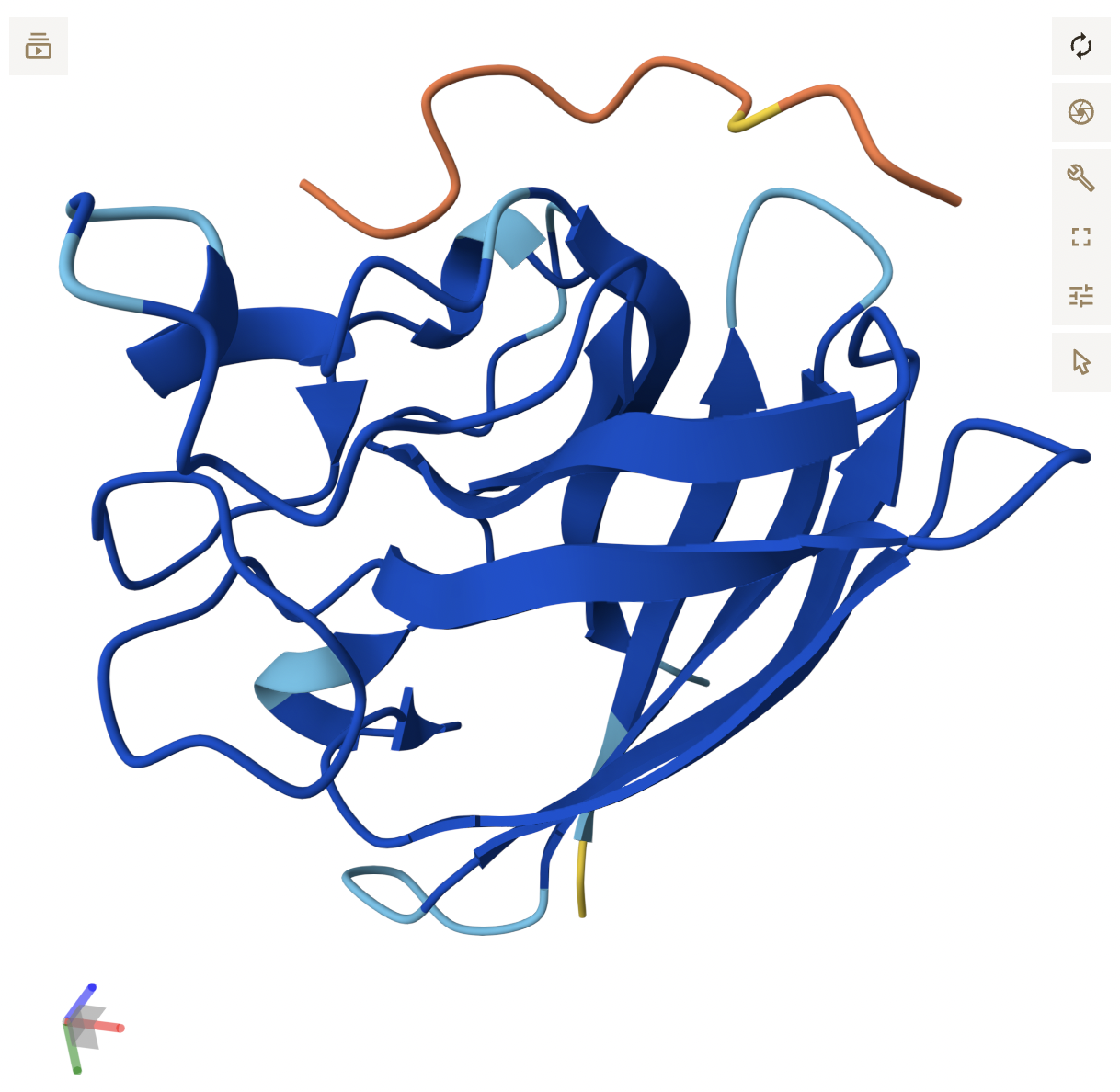
- Peptide 3 reflects its higher score, appearing as a continuous chain that is in close proximity to the β-barrel region, around the middle of the protein and potentially near the dimer interface. It does not seem to be localized near the N-terminus. While the backbone appears to be hovering right near SOD1, it is close enough where side chain interactions likely exist, but are not displayed in the current, skeleton view.
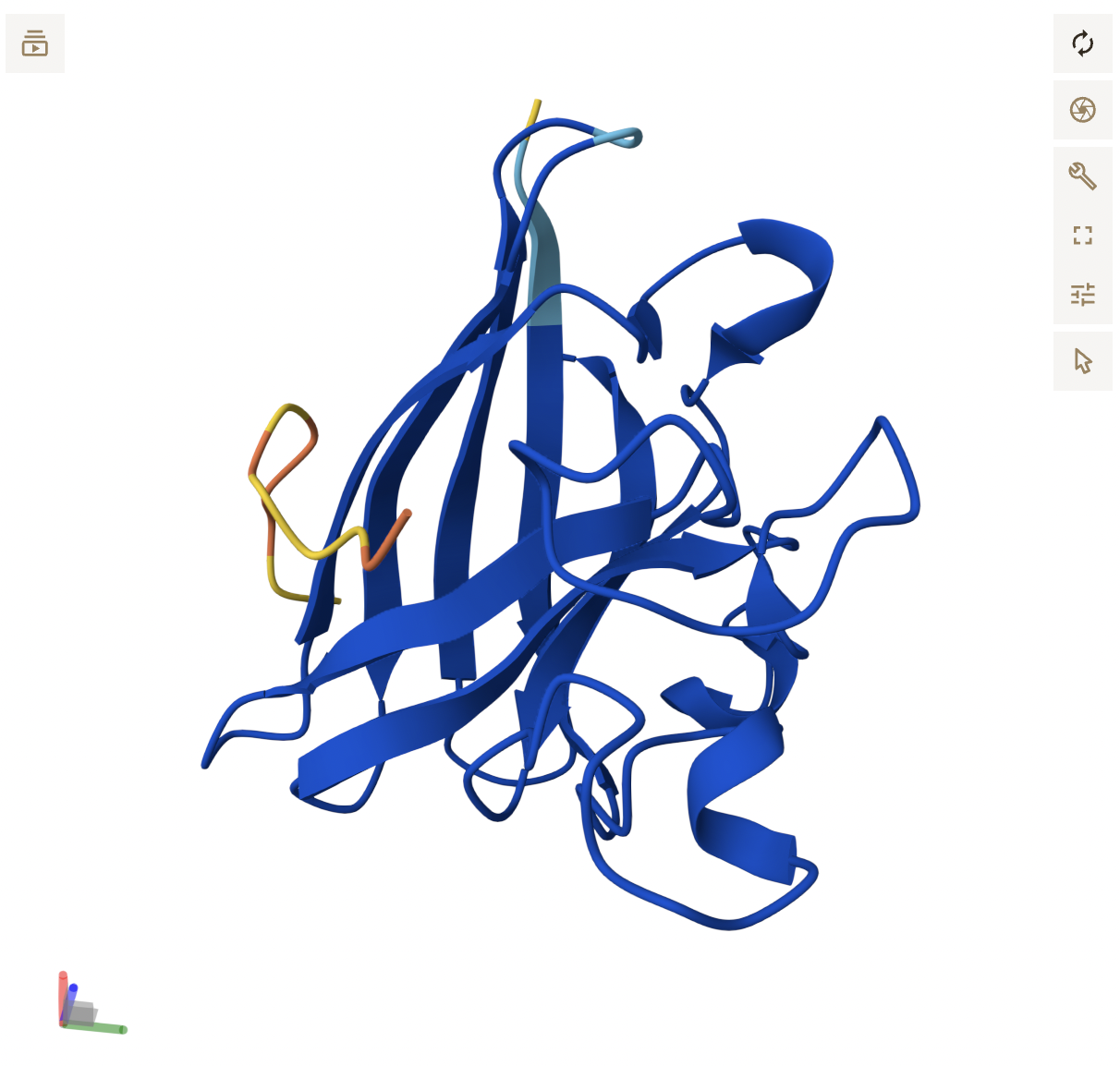
- Peptide 4 formed a helix, but still seems to be floating above SOD1, not making any clear contact. It floats to the side of the N-terminus, with no clear engagement, reflecting its poor score.
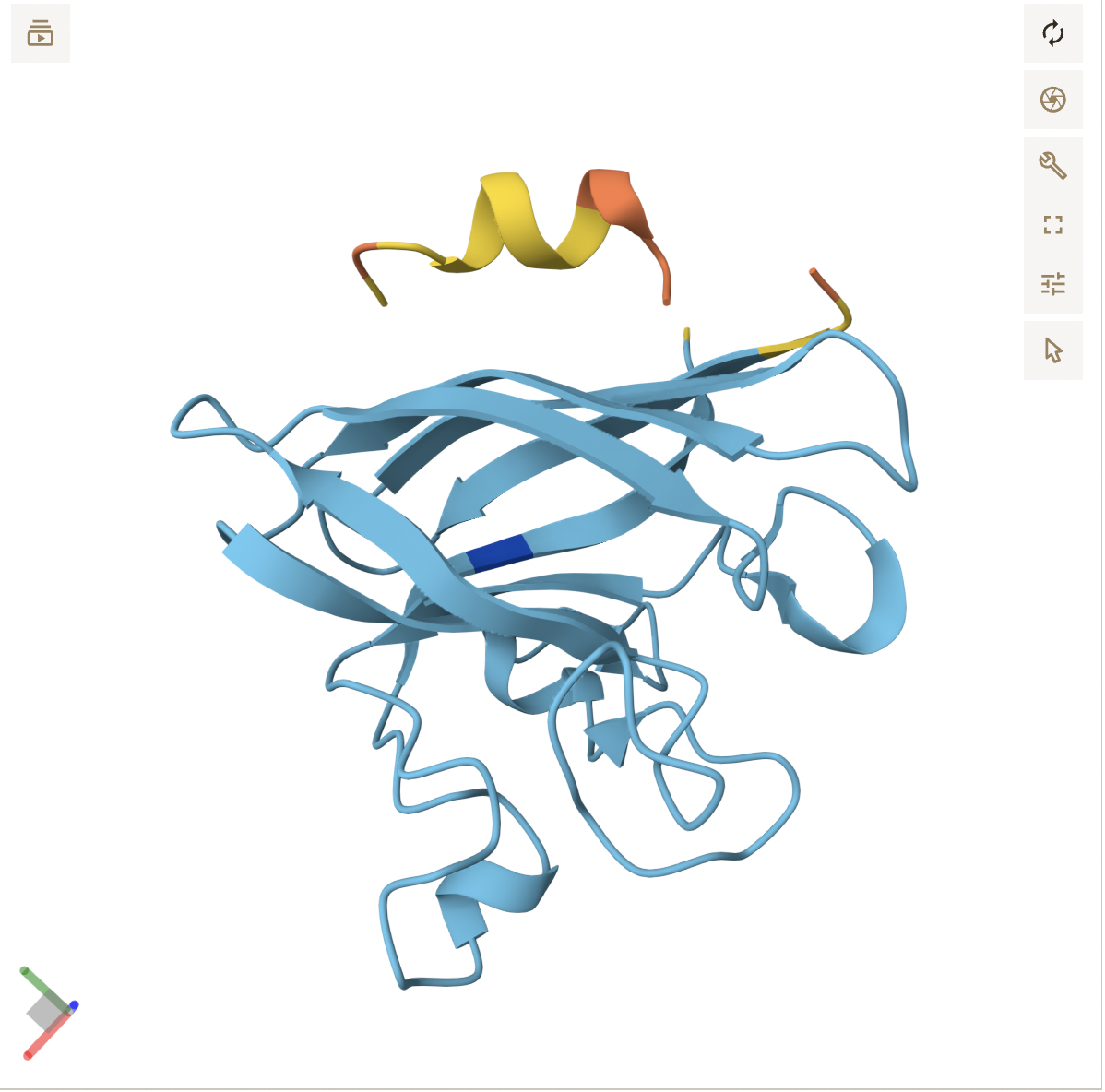
- Peptide 5 appears to be a continuous strand that slightly wraps around the C-terminus region, suggesting it does interact with the surface. It seems surface bound, and complementary to the SOD1 shape.
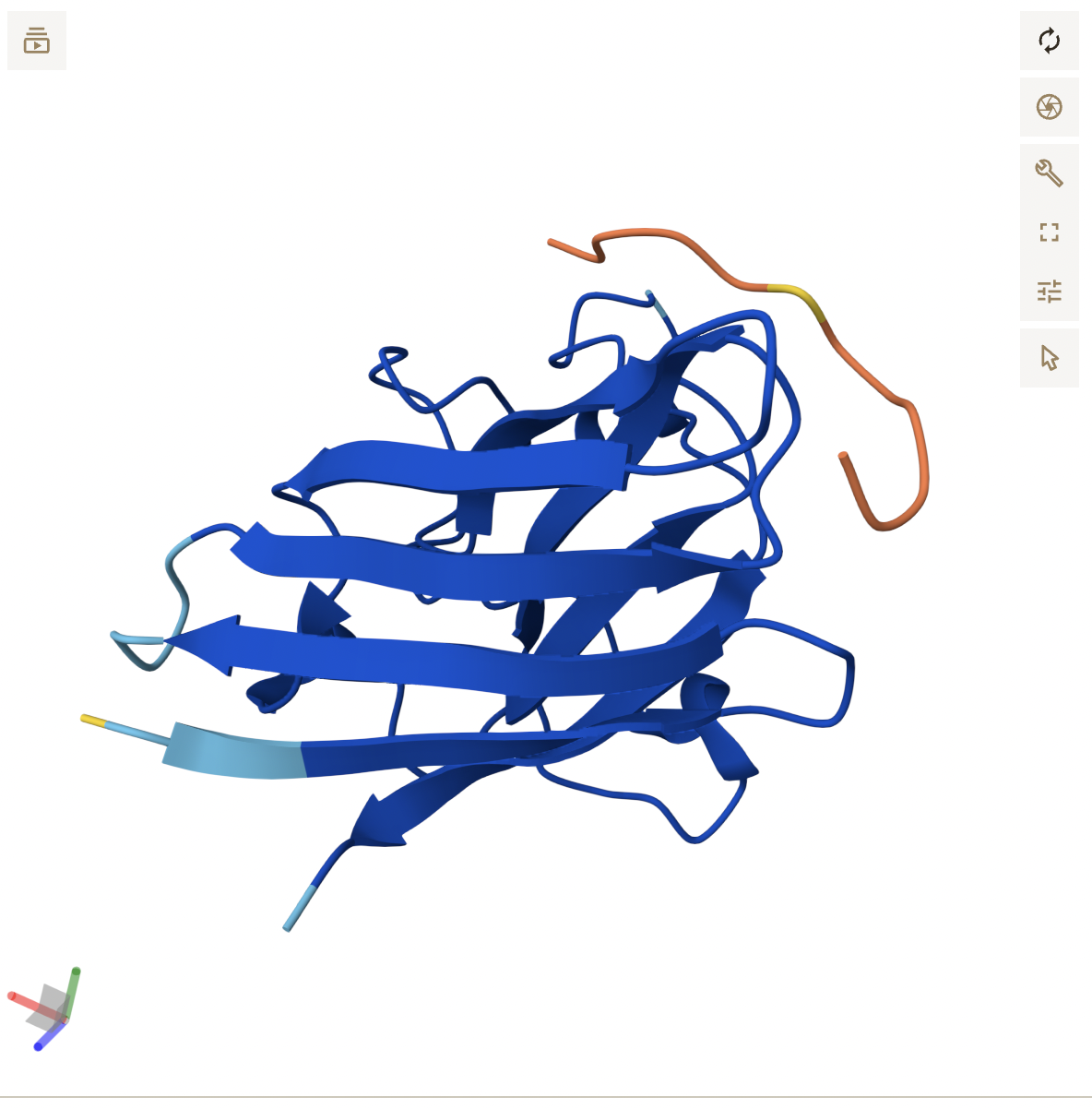
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
It seems PepMLM generally creates weak peptide sequences where strong, usable sequences are rarer to come across. Generally, high perplexity sequences did correlate with poor ipTM scores. My ipTM scores were generally low, but I did have one outlier (peptide 3) that outperformed the known binder FLYRWLPSRRGG by 0.21 points. However, this tool is still extremely valuable, just with additional time and testing.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide 1
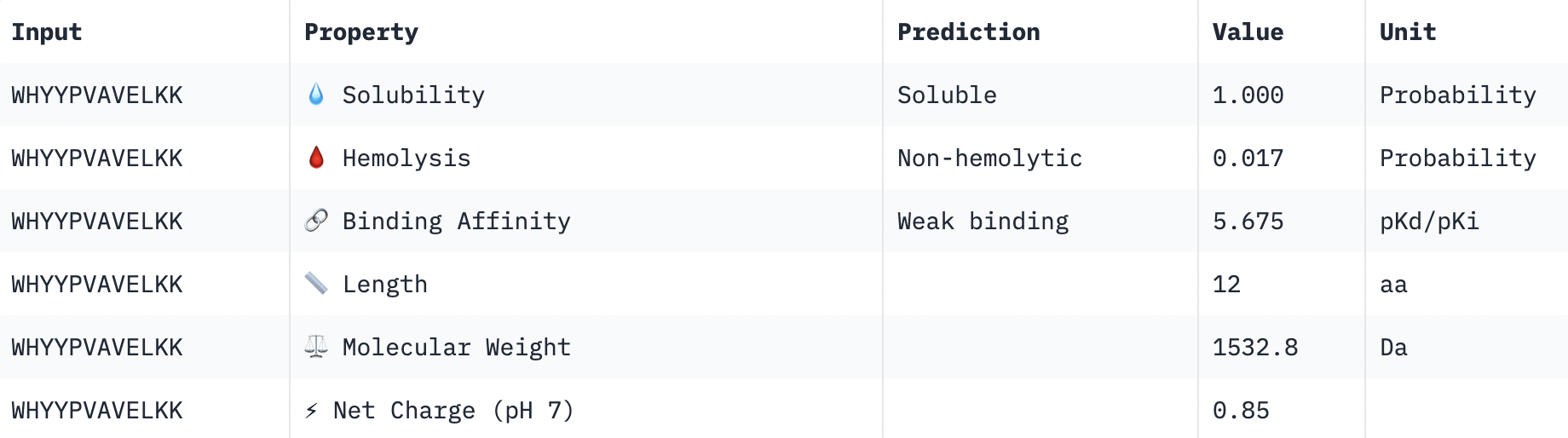
Perfect solubility, safe hemolysis, weak binding, reasonable weight, positive net charge.
Peptide 2
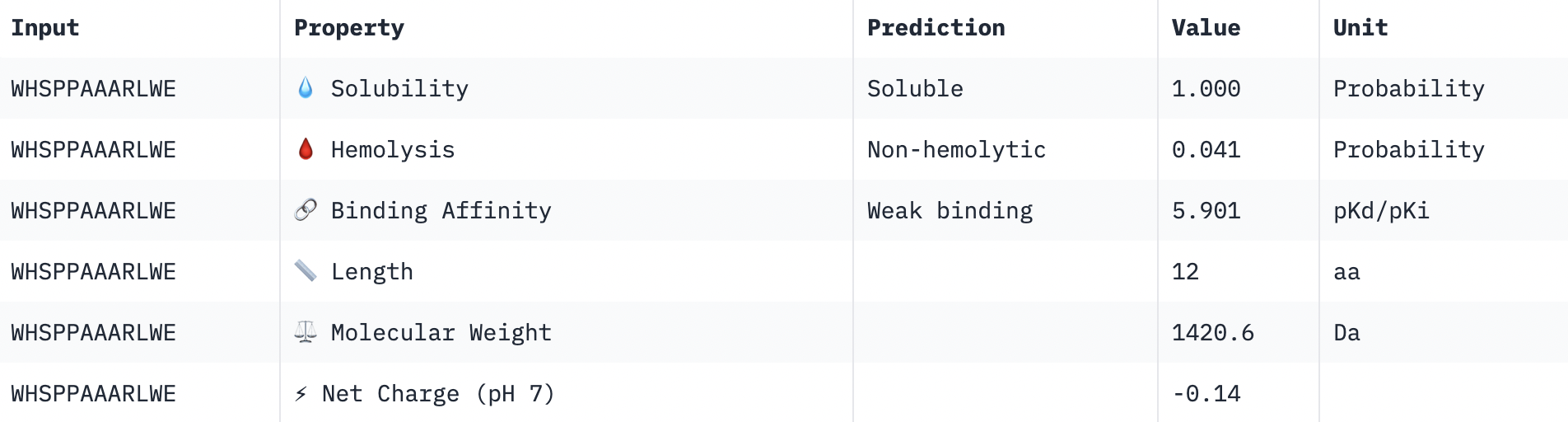
Perfect solubility, safe hemolysis, weak binding, reasonable weight, almost neutral net charge.
Peptide 3
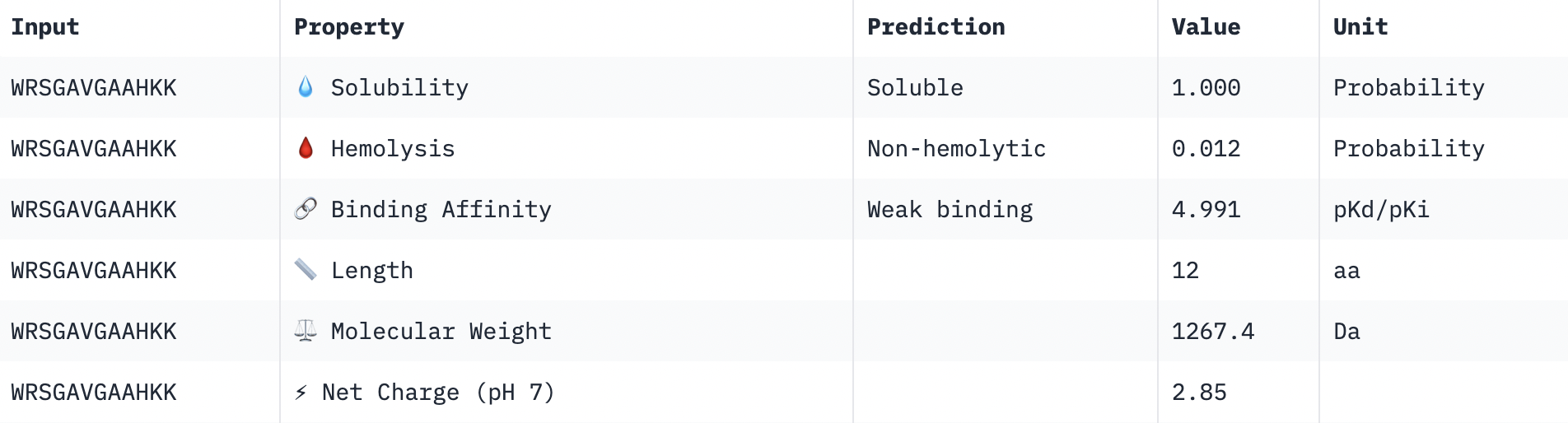
Perfect solubility, safe hemolysis, weak binding, reasonable weight, positive net charge.
Peptide 4
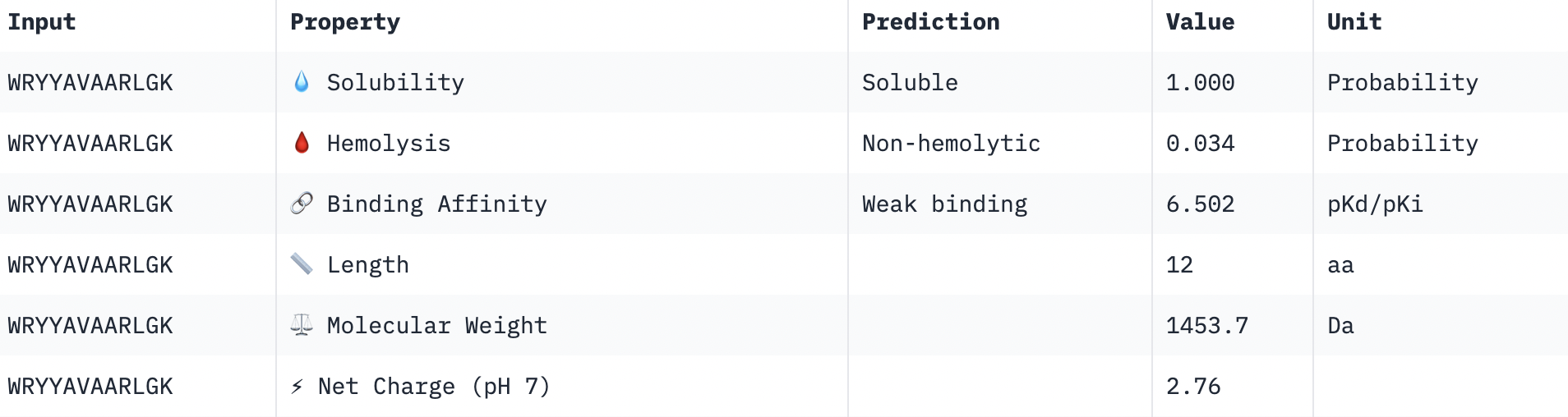
Perfect solubility, safe hemolysis, weak binding, reasonable weight, positive net charge.
Peptide 5

Perfect solubility, safe hemolysis, weak binding, reasonable weight, positive net charge.
Summary: A few interesting results pop-up. Firstly, peptides with higher ipTM do not show stronger binding affinity. In fact, the peptide with the strongest binding affinity (6.502) had a very weak ipTM score (0.29), while the peptide with the highest ipTM (0.58) had the weakest binding affinity (4.991). This may be due to the inheritance difference in the purpose of these tools: AlphaFold is asking if you can physically fit together the peptide and target, while PeptiVerse is asking how strongly the peptide BIOCHEMICALLY interacts with the target.There was no variation in the hemolysis and solubility columns across the peptides. The peptide that best balances these properties is peptide 3 (WRSGAVGAAHKK). All the peptides were in the “weak binding” category. Structure is more meaningful, of which peptide 3 displayed the highest confidence, while balancing good hemolysis and solubility.
Choose one peptide you would advance and justify your decision briefly.
All peptides had good solubility, hemolysis and weight. The decision comes down to net charge and binding affinity. Peptide 4 has the strongest binding affinity of 6.502 and a reasonable, positive charge. However, its ipTM is one of the lowest. Therefore a happy medium is peptide 3, highest ipTM, positive charge, even with a weaker binding affinity (but all 5 had “weak binding” so the decision is less relevant based on binding affinity).
Part 4: Generate Optimized Peptides with moPPIt
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Region used: β-barrel region near WRSGAVGAAHKK as that is what it closest to: 95-105

0.966 ← Hemolysis
6.356 ← Affinity
0.850 ← Solubility
0.814 ← Motif score
PepMLM’s prompting is a lot more loose compared to moPPIt. The former needed to just generate something plausible, while the latter had to optimize for multiple factors in a specific range. Although moPPIt did produce better affinity and solubility, it did seem to sacrifice hemolysis in the process. Before putting this into clinical studies, I would validate this in a wet lab (including tests such as SPR, ITC and red blood cell lysis assay to test for the predictions), specifically putting emphasis on testing the high hemolysis score, as it may be a dealbreaker before clinical trials. I would also consider running more computational tests on the hemolysis, and perhaps pivoting to a different peptide (such as WRSGAVGAAHKK) as to not waste time and resources. If the wet lab results are promising, the next steps would be ALS motor neuron cell models before human trials.
Ai:
Is the A4V mutation for the SOD1 sequence done at the 5th out 5th position? The fourth position in my sequence is not A.
Why is there an extra Methionine? Is this in all organisms? Is it always cleaved off?
Explain PepMLM in detail.
For peptides that will bond to SOD1, what molecular weight is ideal? What net charge is ideal? (Why?)
If I have a peptide I think may help the A4V mutation in SOD1, what are my next validating steps before sending it to human trials?
Part C: Final Project: L-Protein Mutants
Note: I was unable to find a group in time for this assignment. I will continue searching for a group going forward, but completed this assignment on my own to avoid falling behind.
As I was most familiar with Option 1, I chose it for my analysis. I started with picking out mutations from the L-Protein Mutants file. I made sure to pick mutations where both the protein and lysis levels are 1, so they are experimentally confirmed to work and the protein is made properly. My picks are as follows:
Soluble region: P13L (position 13)
Base pair change: C→T at position 38
Lysis = 1, Protein Levels = 1
Soluble region: R18G (position 18)
Base pair change: A→G at position 52
Lysis = 1, Protein Levels = 1
Anywhere: R30Q (position 30)
Base pair change: G→A at position 89
Lysis = 1, Protein Levels = 1
I also initially picked mutations at 44 and 46, but swapped them due to the cluster omega results.
ClusterOmega: Checked every mutation against the output. 44 and 46 were conserved, so I swapped them out for mutations at 45 and 47, and cross referenced them with the L-Protein Mutants file:
A45P (pos 45) TM Region Lysis=1
F47Y (pos 47) TM Regio Lysis=1
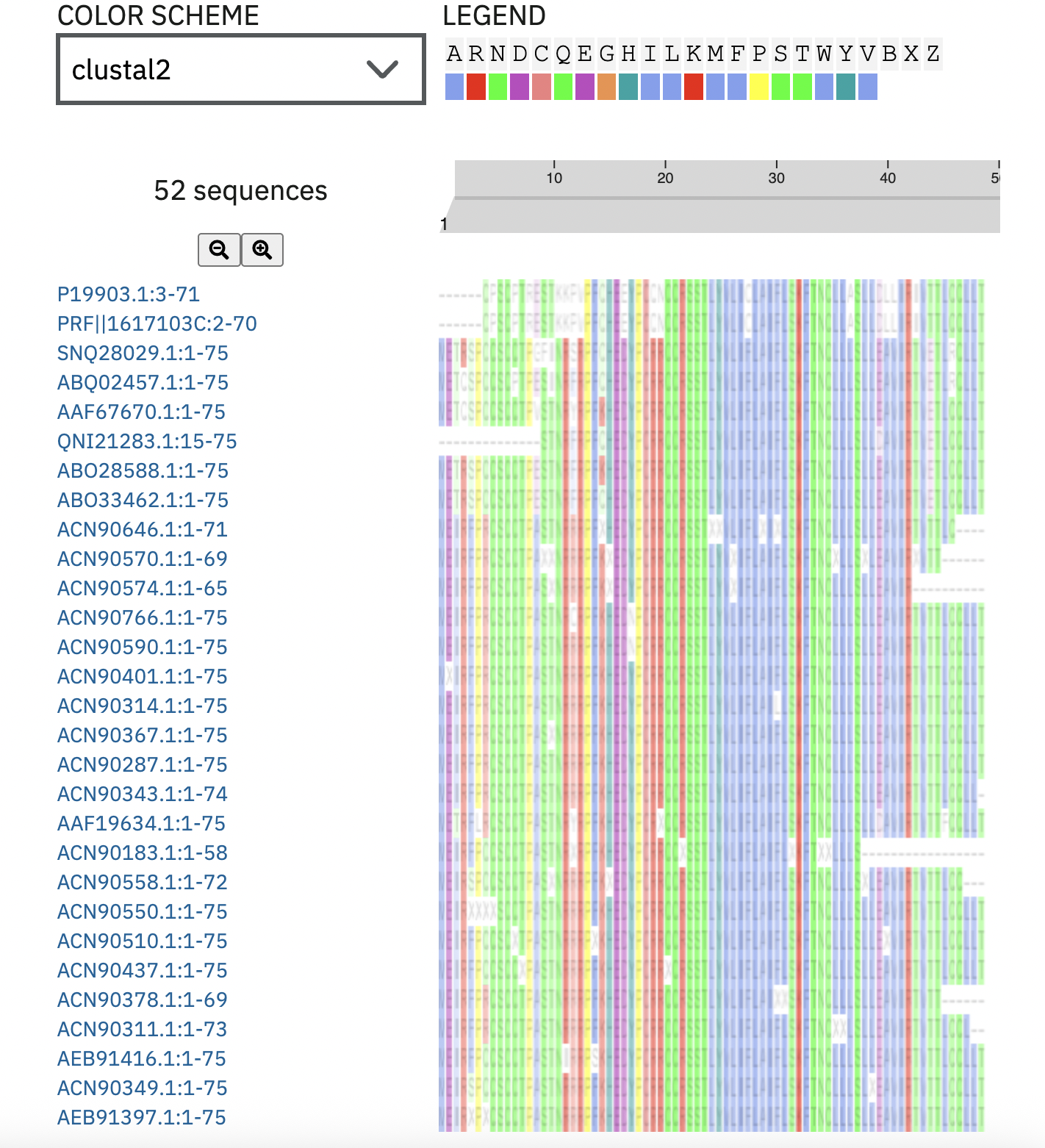
A few things came up after using Protein Language Model Notebook:
- I swapped out the A45P to the A45L as ESM2 liked the L better, giving a higher score and placing it in the top 30 ( ESM2 score 1.539).
A45P (pos 45) TM Region Lysis=1 -> SWAPPED TO A45L
- The rest of the mutations were not the top ESM2 contenders. I tried to swap them out for better contenders that scored in the top 30, but the L-Protein Mutants either had no experimental data on them, or gave bad scores for the mutations.
- In general, the heat map scores were favorable. A45L had the lightest colour, while the rest had medium, darker greens, but not near dark blue.
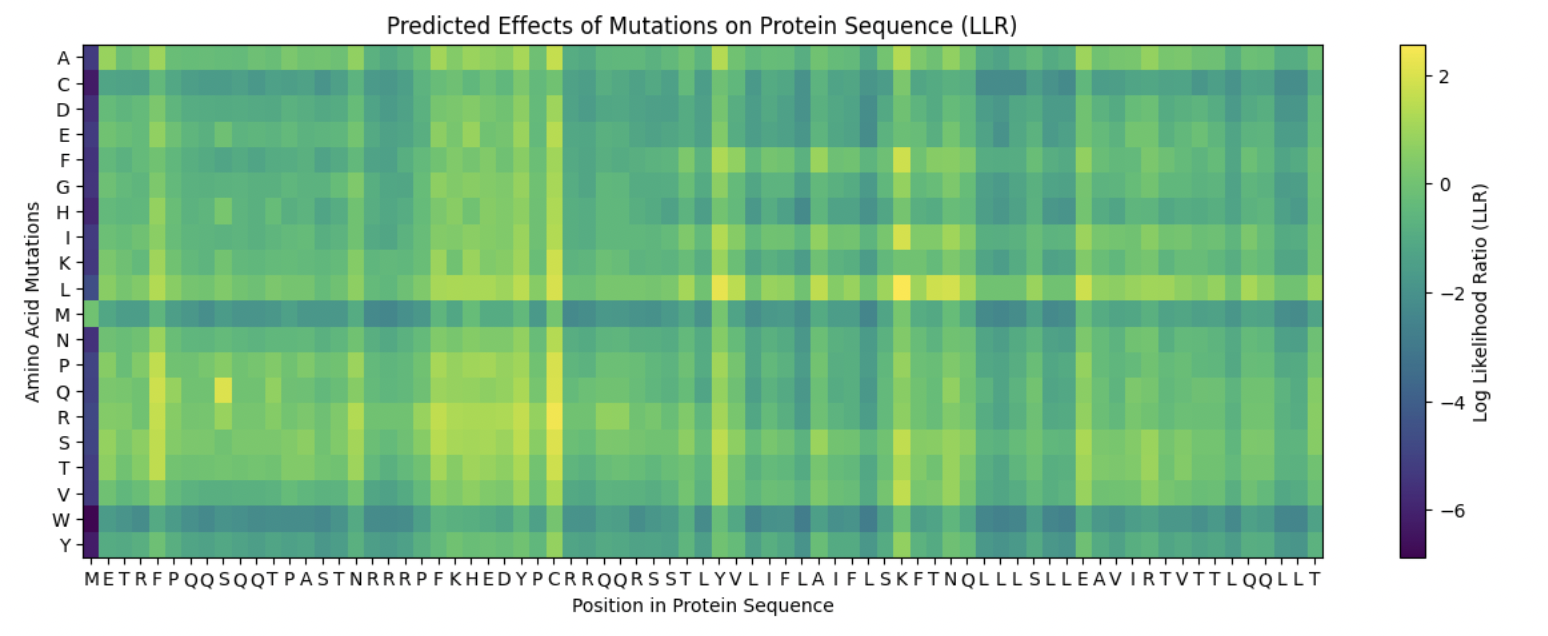
Summary:
I selected my five mutations for enhanced lysis function based on the cross-reference of three resources: 1) experimental lysis data from the L-Protein mutants file 2) CluterOmega evolutionary conservation, ESM2 LLR scores from the language model. Since I chose option 1, I prioritized experimental data first (where Lysis = 1 and Protein Levels = 1) before adjusting my chosen mutations based on their ESM2 scores. I do want to note that the top ESM2 candidates were not in the dataset. My final mutations are as follows:
P13L (position 13, soluble region): Proline → Leucine. Lysis = 1, Protein Levels = 1. Was not conserved in ClusterOmega. Neutral heatmap score.
R18G (position 18, soluble region): Arginine → Glycine. Lysis = 1, Protein Levels = 1. No conservation. Acceptable heat map score.
R30Q (position 30, anywhere): Arginine → Glutamine. Lysis = 1, Protein Levels = 1. Not conserved, acceptable heatmap.
A45L (position 45, transmembrane region): Alanine → Leucine. Lysis = 1, Protein Levels = 1. Chosen to replace A45P due to better ESM2.
F47Y (position 47, transmembrane region): Phenylalanine → Tyrosine. Lysis = 1, Protein Levels = 1. Chosen replacement due to better ClusterOmega. Not conserved, acceptable ESM2 scores.
A45L is the most promising mutation as it has confirmed experimental lysis, is non-conserved, and has a good ESM2 score (placing in top 30). The other mutations have good experimental data and conservation, but less promising ESM2 scores. This highlights an interesting disconnect between ESM2 rankings and experimental lysis outcomes.
Ai:
used ai when running cluster omega to make sure I was counting the position right, and so whether every position was conserved was calculated correctly.