Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Components included are Phusion DNA Polymerase, deoxynucleotides (dNTPs), and HF Reaction Buffer containing MgCl₂. Physion DNA polymerase is a Pyrococcus-like enzyme. The polymerase domain reads the strand and synthesizes it by adding dNTPs one at a time (through phosphodeister bond formation). This requires a primer to already be present in the template. The enhancing domain fused onto the polymerase keeps the enzyme attached to the DNA template, speeding up the process. The exonuclease domain reads this newly synthesized stand and corrects any mistakes. Deoxynucleotides (dNTPs) are the building blocks of DNA at optimized concentrations. The HF reaction buffer helps control the pH for optimal activity, while the MgCl₂ positions the dNTPs correctly and stabilizes the transition state during bond formation.
What are some factors that determine primer annealing temperature during PCR?
Primer length: Optimal length is between 18-22bp to allow for adequate specificity while allowing for binders to bind easily. However, overhangs can be 20-22 bp.
GC content: should be 40-60% of total primer sequence, directly influencing Temp more since G-C pairs are more thermodynamically stable than A-T
GC clamp:1-3 G/C bases during the last 5 bases at the 3’ end improve binding but more than 3 should be avoided as that can cause non-specific annealing.
Secondary structures: hairpin structures and primer dimers reduce how much primer is available to anneal to the template, recommended to keep Gibbs free energy above -10kcal/mol.
Note the ideal temperature specified is 52-58 Celcius, where about 65 Celcus can cause secondary annealing issues.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR uses a polymerized enzyme to synthesize new copies of DNA region, where the region is defined stand to end using primers, and the machine cycles through denaturation, annealing and extension to amplify that specific region into many copies. This requires multiple temperature cycles, specific reagents (including polymerase, primers, dNTPs, buffer solution), and 1-2 hours. Restriction enzyme digests cut existing DNA at specific recognition sites, where different enzymes recognize difference sequences and cleave at that region. There is no new DNA created, it’s simply cut. This requires one temperature, regents of restriction enzymes and buffer, and larger amounts of starting plasmids and DNA. PCR creates blunt ends or overhangs depending on your polymerase, and is better for amplifying specific regions from a larger piece of DNA. It’s also useful in cases where you need to add overhangs for Gibson Assembly, you have little amounts of starting DNA, or your sequence doesn’t have convenient restriction sites. Restriction digests usually produce sticky overhand ends, and are better for when you want to cut a plasmid backbone at specific sites, you want sticky ends, or you have a large fragment that is too impractical for PCR. It’s also preferred in verifying band sizes on a gel, due to PCR being simpler to do.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To be appropriate for Gibson cloning, the fragments must have correct overhangs at the end of each fragment. For PCR, you must ensure each fragment has a unique, 20-22 overhang that is identical to the adjacent fragment, and has enough GC pairs. For the digests, the enzymes must cut at the locations that expose the sequence you want to overlap with PCR (which can be checked in the Benchling cut sites). It’s very important the orientation of the overlaps is correct in the 5->3 direction. To verify the cut was at the intended site and not anywhere else in the backbone, gel or sequencing may be used.
How does the plasmid DNA enter the E. coli cells during transformation?
DNA can enter through electroporation (though not discussed in the lab manual, heat shock may also be used). Cells are placed in a cuvette with the plasmid DNA, followed by a brief, high voltage electrical pulse that destabilizes the cell membrane, creating pores through which DNA can enter (driven by the electrical potential). The membrane then reseals, trapping the DNA inside. After the electric pulse, cells are incubated in media to recover for about an hour.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden gate assembly uses Type IIS restriction enzymes to generate custom overhangs of 4 bp in DNA fragments that can then be ligated together in a custom order. This allows for assembling multiple fragments simultaneously. The Type IIS enzymes cut downstream of their recognized site, allowing for these customizable overhangs, and removing the recognition site itself from the DNA. DNA is then ligated together with the complementary overhangs in a one tube reaction. This allows for very precise cutting due to the unique 4bp overhangs.
Model this assembly method with Benchling or Asimov Kernel!
Link to Benchling assembly: https://benchling.com/s/seq-3zLNvguw0VFkZfiLAYqq?m=slm-D5TDZGbXt7lUpfKGzo5N
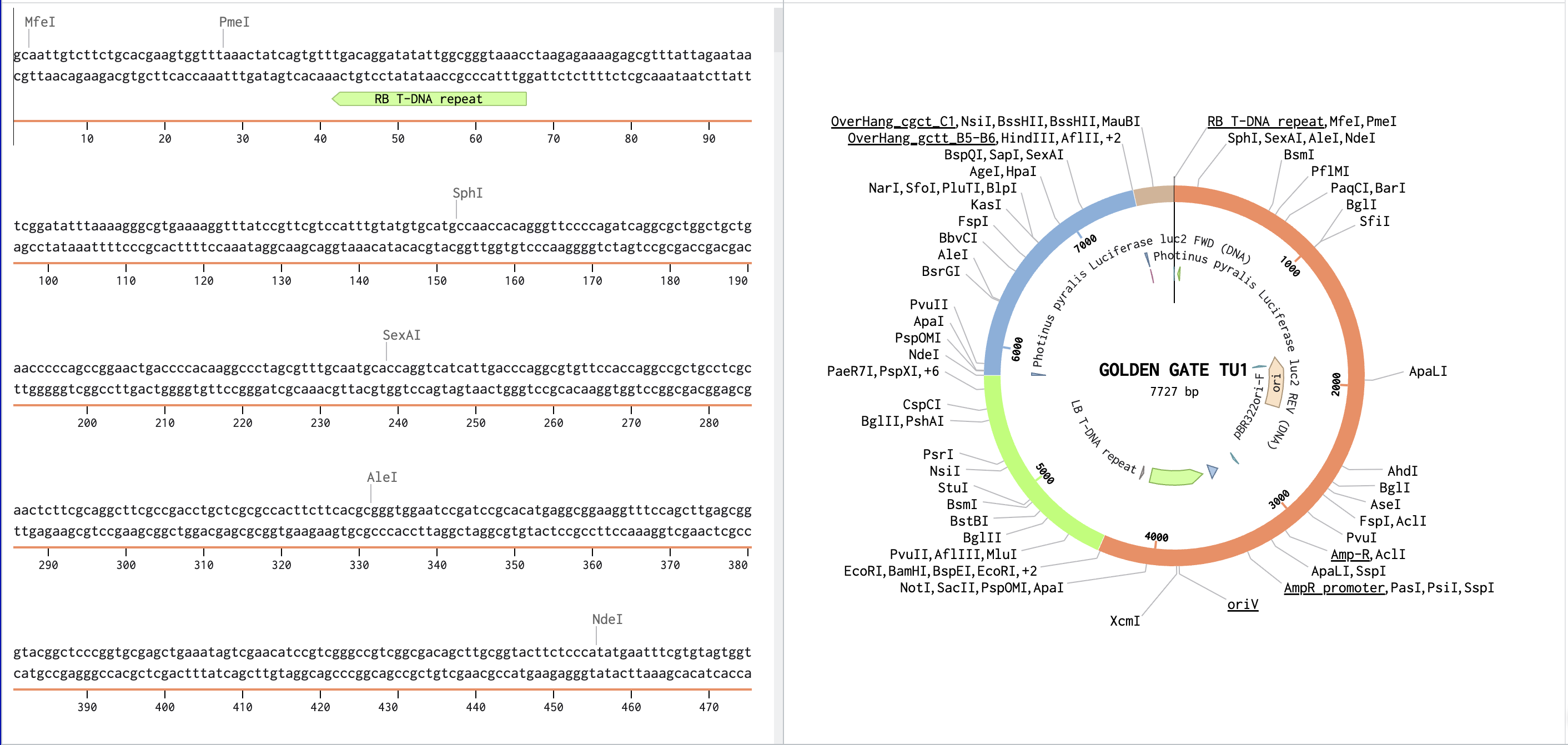
Assignment: Asimov Kernel
All my notes and simulations are under the repository called: Iman_Karibzhanova_HM6 in the HTGAA Victoria Node account. Below is a general summary.
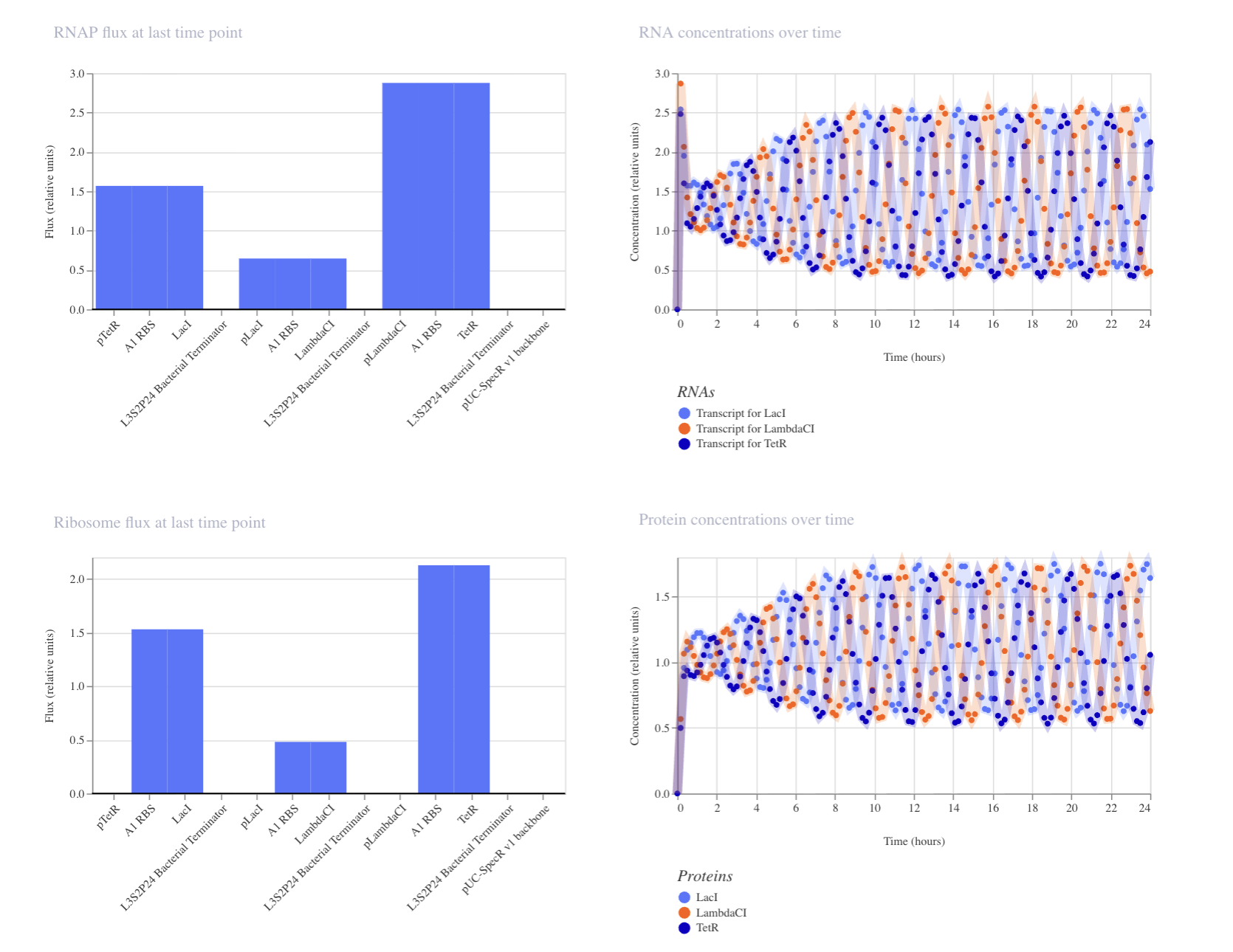
My simulation of the demo:
Demo’s simulation:
My first construct:
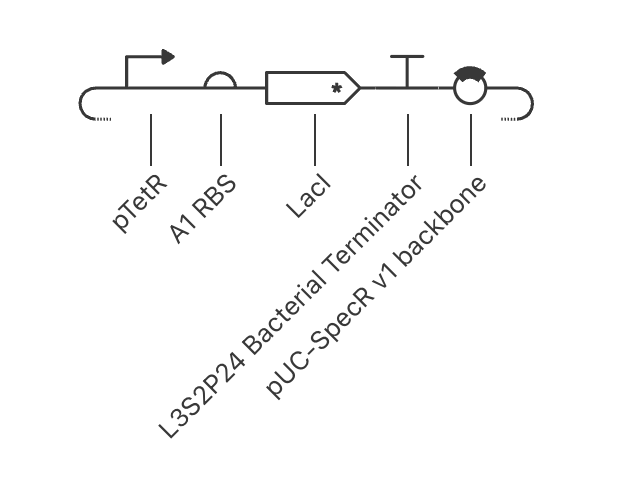
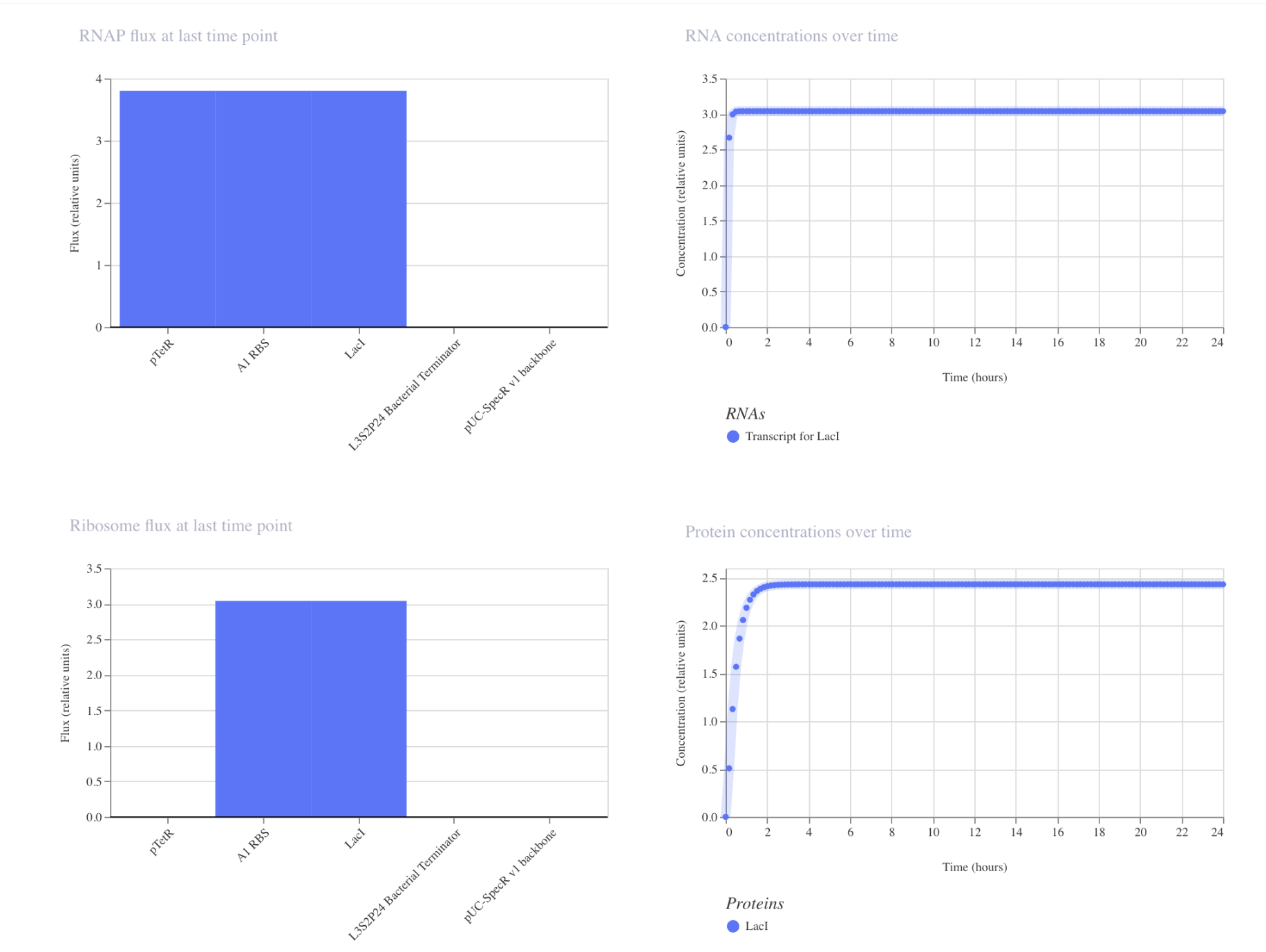
The simulation is as expected. pTetR is fully active with nothing suppressing it, so RNA shoots up. Protein concentrations plateaus at around 2.5. RBS, TetR and LacI are at maximum output. This is how genes act when there is nothing controlling its output.
My second construct:
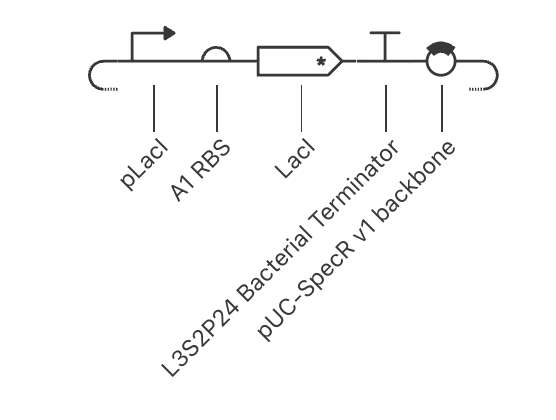
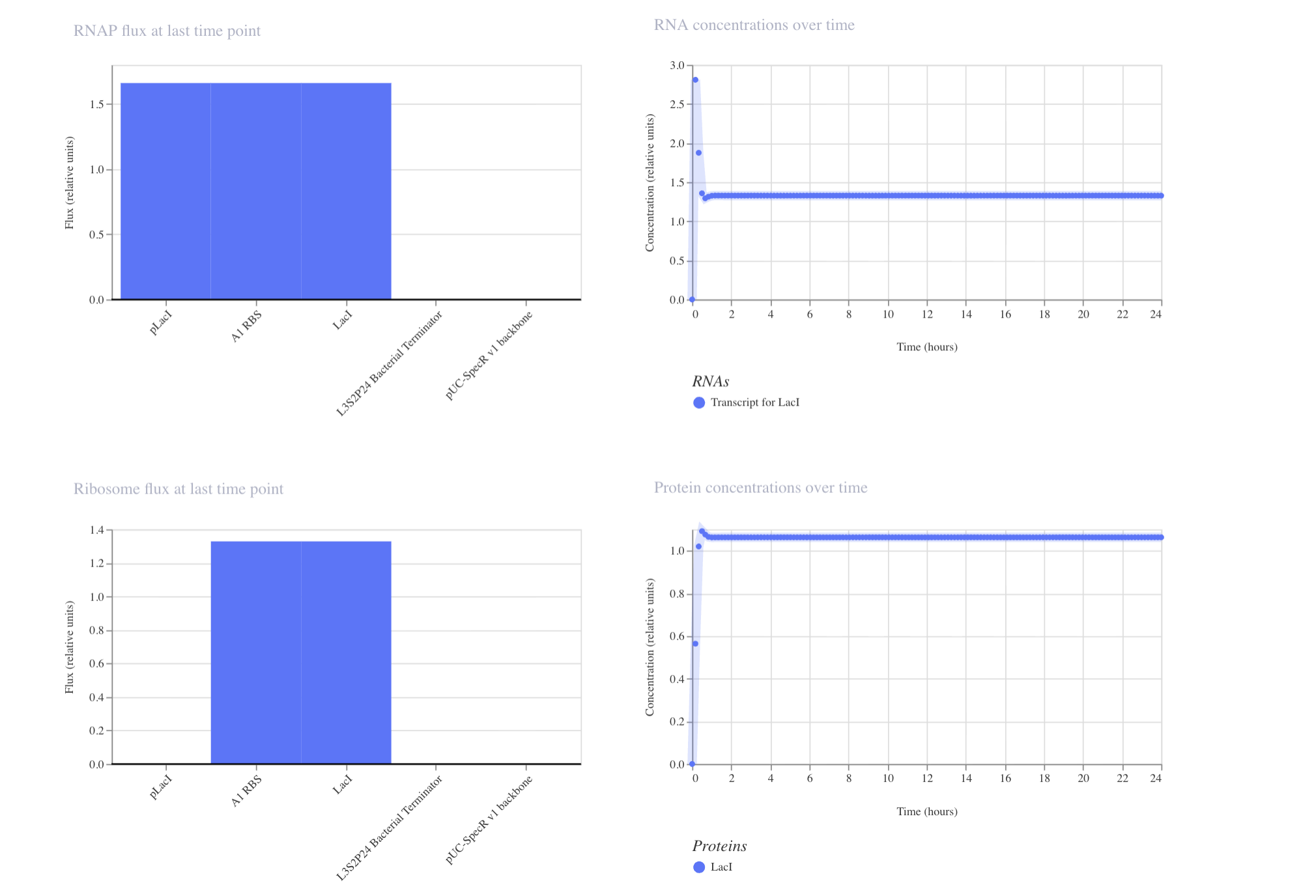
The results are interesting. The plateau happens at a lower concentration (around 1.1) due to the repressor capping expression from suppression of its own promoter. The RNA dips down quickly in the beginning of the simulation, due to the LacI suppressor kicking in from production. This shows autoregulation could prevent toxic accumulation in my project.
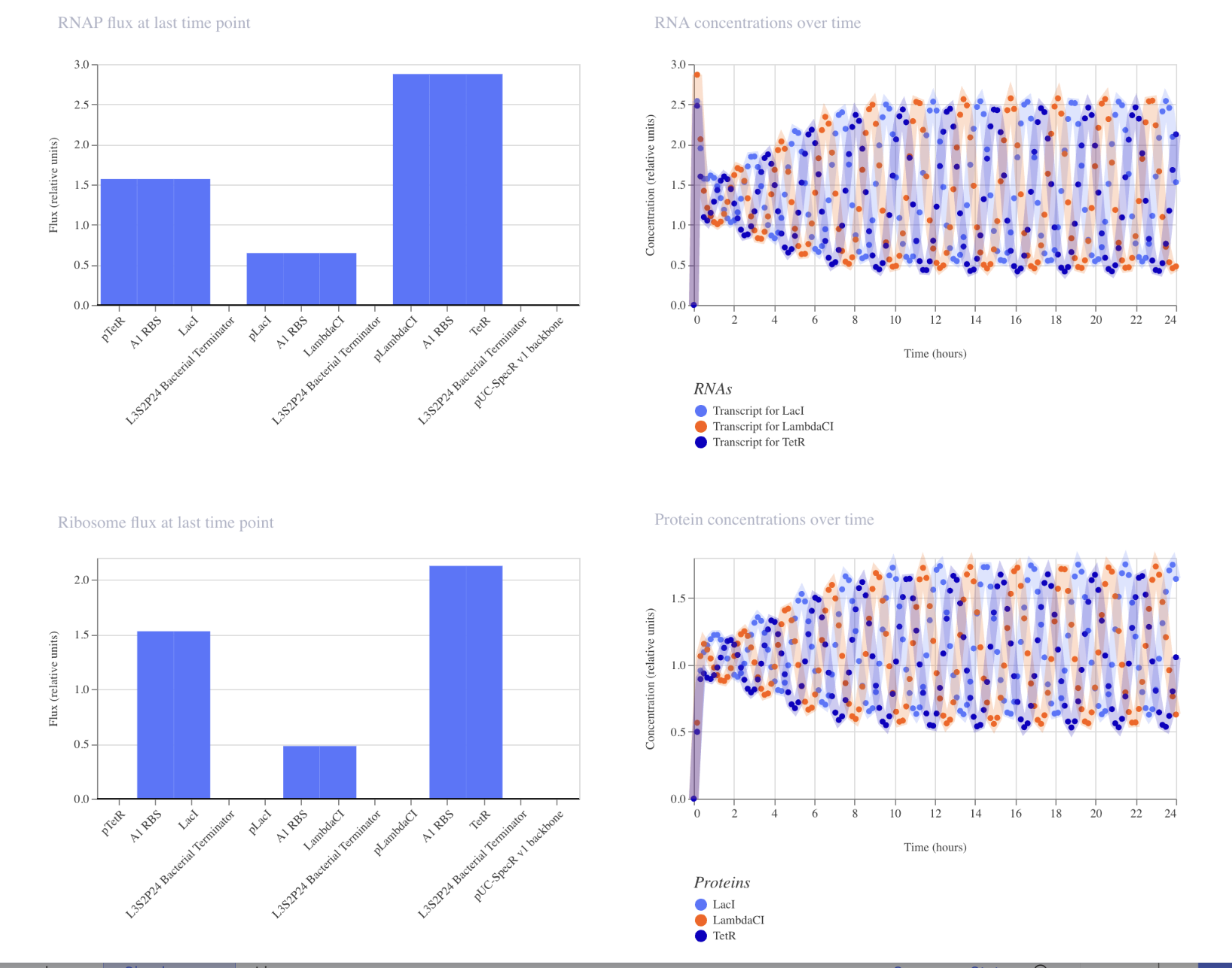
My third construct:
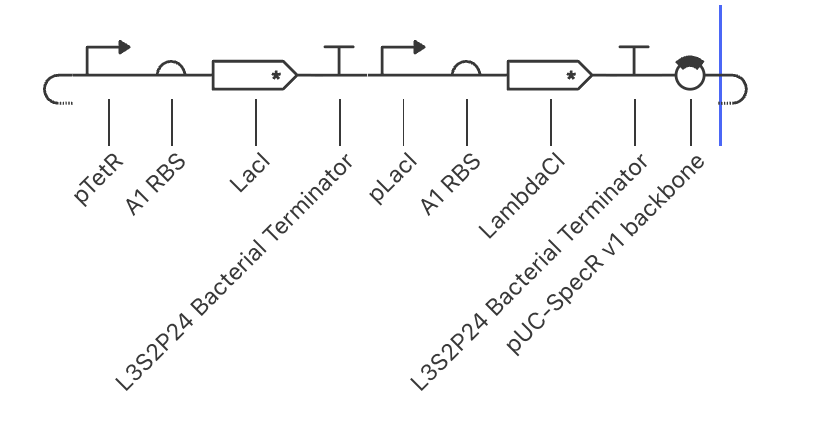
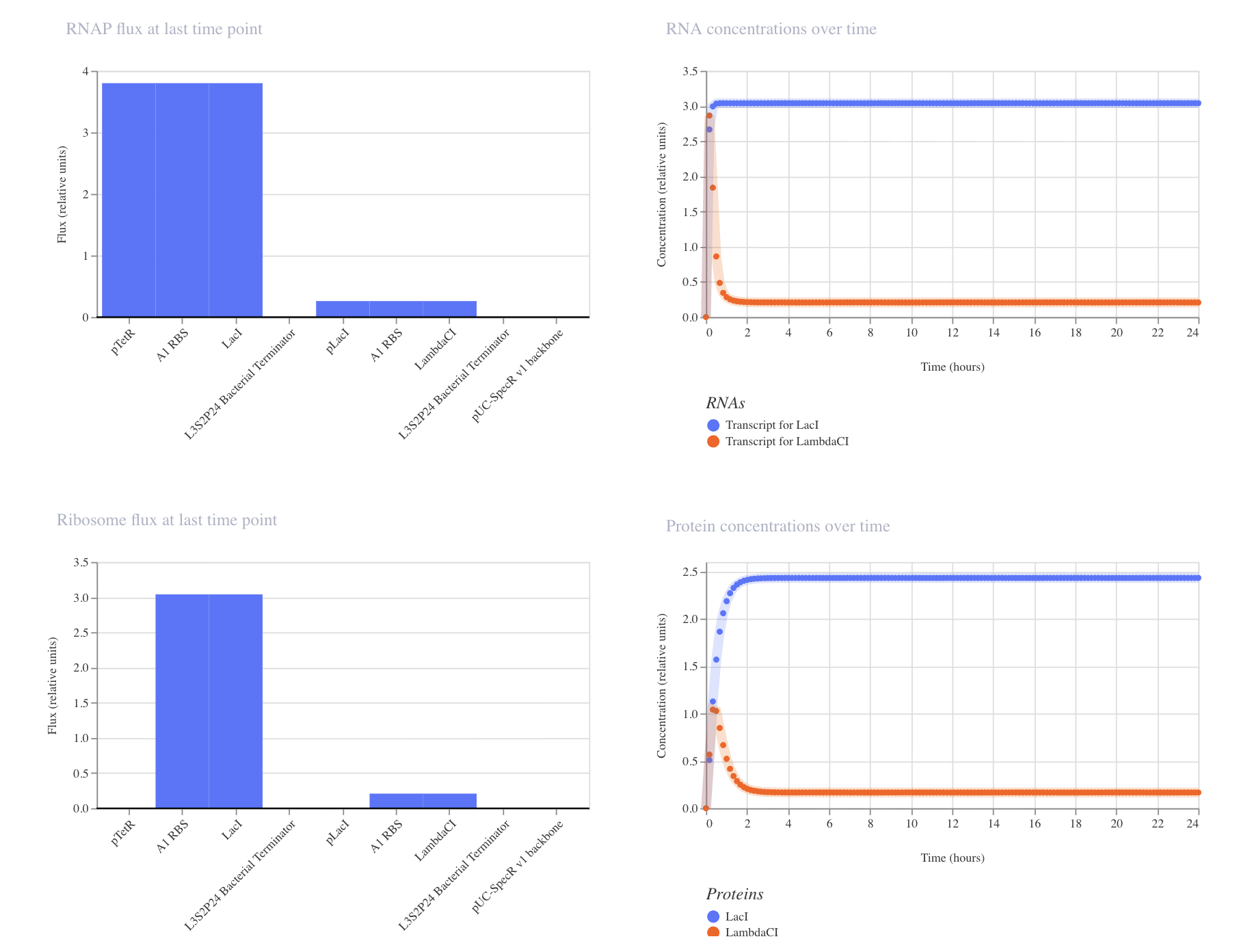
The simulation makes sense based off of construct 1 and 2. Lac1 goes up to around 2.5, similarly to the first experiment, which makes sense since pTetR is not repressed. LambdaCI spikes when LacI is not yet present to repress pLacI, and then swiftly falls as LacI shuts the pLacI promoter down, eventually silencing LambdaCI. Gene one remains on, while the second gene is shut off. This is an important dynamic to keep in mind when constructing multi-gene pathways.
Work Cited
https://docs.google.com/document/d/1OuFjkFD7O29v9Zzekam66o0gKQ8iqJSPhEKEIhU1y_s/edit?tab=t.0
AI:
List of parts given in Asimov Repository: Which are the simplest to use in my constructs to test designs?