Week 4 HW: Protein Design Part I
Important
PART A: Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
On average raw meat contains 20% protein by mass (but this varies depending on the type of meat)
500g of meat x 20% = 100g of protein
The average molecular weight of an amino acid is approx 100 Da (daltons the mass of a single molecule)
Da has the same numerical value as g/mol
Therefore 100 Da = 100 g/mol
Therefore, 1 mole of amino acids weighs 100g
1 mole = 6.022 × 10²³ particles (Avogadro constant: the ratio between an amount of substance and the number of particles that it contains)
If 100g = 1 mole
consuming 100g of protein = consuming 6.022 × 10²³ amino acid molecules.
Therefore if you consumed 500g of meat, you took 6.022 × 10²³ amino acid molecules.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not become the animals they eat because the our bodies only absorb the molecular building blocks from food, not the genetic information. Proteins from meat are broken down into amino acids and DNA is broken down into nucleotides before being absorbed in digestion. These molecules are then reassembled to make human proteins, tissues, and organs according to the human genome. The DNA in the food does not alter human cells, so humans remain human.
Why are there only 20 natural amino acids?
According to Bywater et all (2018), the standard 20 amino acids were selected during prebiotic chemical evolution based on the principle of “parsimony whereby the simplest possible structures that have value in terms of function are retained.” Between the 20 amino acids Bywater et al show that significant “function space” is covered, properties such as different polarities, hydrophobicity and reactivity are present in the 20. Other possible amino acids were rejected due to being too complex, redundant or difficult to form naturally. Therefore, the 20 selected form the simplest set of molecules that together would provide sufficient chemical diversity for all protein structures and functions.
As all organisms share a common ancestor, the protein synthesis machinery (e.g ribosomes, tRNAs and enzymes) evolved using the same set of 20 amino acids. This selection likely became evolutionarily “locked in”, or a “frozen accident” as proposed by Francis Crick in 1968, since changing the set would require re-engineering the entire translation system, something strongly discouraged by natural selection (Ribas de Pouplana et al, 2017).
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids formed before life on earth through abiotic (non-biological) chemical reactions occuring on the early earth or in space. Simple molecules carbon dioxide, methane, ammonia, and water reacted with energy from lightning, ultraviolet radiation or volcanic activity to produce amino acids. In 1953 the Miller- Urey experiments demonstrated this through the production of biomolecules from simple gaseous starting materials that simulated the primordial Earth (Parker et al, 2014).
Amino acids may also have been formed in space in asteroids and comets (planetesimals). Planetesimals contained water ice and organic compounds such as methanol, carbon monoxide, and ammonia etc. When radioactive elements heated them, liquid water was produced (termed aqueous alterations) enabling reactions such as Strecker synthesis and Formose-like reactions that form amino acids within asteroids which were delivered later to earth through collision (Cowing, 2023)
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
a left-handed a-helix
Biological proteins use L-amino acids (left handed enantiomers) and form right handed a-helices. Synthetic D-amino acids (right handed enantiomers) form left handed a-helices. This preference for one handedness over the other is called homochirality (Ozturk and Sasselov, 2025).
Can you discover additional helices in proteins?
Yes, a-helices are the most common but scientists have discovered that proteins can fold into many different helical structures such as the π- helix, 3₁₀-helix, Polyproline helix or Collagen triple helix usually identified by analysing the 3D geometry of the protein backbone, (Cao et al, 2015).
Why are most molecular helices right-handed?
Most molecular helices are right-handed due to evolutionary selection and structural stability. Research from Scripps Research (Yu, Deng, & Blackmond, 2024) found that in early Earth chemistry, “kinetic resolution” acted as a filter. Chemical reactions in pre-biotic conditions favoured the production of L-amino acids by depleting their right-handed counterparts. These findings suggest that the initial dominance of left-handed enantiomers resulted in the pure, single-handed building blocks necessary for life. Because natural proteins are built from these L-amino acids they naturally twist into right-handed coils to minimise steric hindrance (clashing) between the side chains and the backbone. Moreover, right-handed structures are preferred because their side chains point away from the backbone, making them more stable than the alternative (Banerjee et al., 1996). Finally, Cole and Bystroff (2009) identify a kinetic trapping mechanism where the folding helix exerts a “mechanical torque” on the protein. This torque reinforces right-handed connections while physically pulling apart left-handed ones, ensuring the right-handed form is more dominant.
Why do β-sheets tend to aggregate?
Beta-sheets tend to aggregate because they have sticky edges with unsatisfied hydrogen bonds looking for a compatible protein strand. This allows them to link onto other Beta-strands or sheets indefinitely to form long, stable structures like amyloid fibrils (Niu et al 2024). In a folded protein, beta-sheets are usually protected, however if a protein misfolds or denatures, the backbone amide amide and carbonyl groups are exposed. These groups seek stability by forming intermolecular hydrogen bonds with other available beta-strands.
What is the driving force for β-sheet aggregation?
The primary driving force is the backbone’s need to satisfy hydrogen bonds. The edge of a beta-sheet need to bond with any compatible protein strand to complete its structural requirements.
Another force is Hydrophobicity. Beta-sheets often have one side that is hydrophobic. To avoid contact with water, these hydrophobic faces aggregate into beta-sheets with a dry internal core. Moreover, in many aggregates, the side chains of two opposing sheets interlock tightly in whats termed a steric zipper. This excludes water from the center, creating a dry and stable core (Matthes et al, 2012).
Why do many amyloid diseases form β-sheets?
Many amyloid diseases involve proteins that misfold and form β-sheets because these structures are thermodynamically stable and can stack together to resist degradation. In Beta-sheets, an extensive network of intermolecular hydrogen bonds and tightly packed side chains (steric zippers) provde stability. This creates a rigid, sheet-like structure, stacked further into fibrils, which are insoluble and resistant to degradation causing the toxic aggregates seen in diseases such as Alzheimers (Makin et al, 2005)
Can you use amyloid β-sheets as materials?
These same properties e.g rigidity, stability, resistance to degradation and self-assembly make amyloid β-sheets useful as materials. Scientists are exploring their purpose as programmable scaffolds for tissue engineering, templates for conductive nanowires in electronics and robust membranes for filtering heavy metals from wastewater etc.
References
Bywater RP. Why twenty amino acid residue types suffice(d) to support all living systems. PLoS One. 2018 Oct 15;13(10):e0204883. doi: 10.1371/journal.pone.0204883. PMID: 30321190; PMCID: PMC6188899.
Ribas de Pouplana L, Torres AG, Rafels-Ybern À. What Froze the Genetic Code? Life (Basel). 2017 Apr 5;7(2):14. doi: 10.3390/life7020014. PMID: 28379164; PMCID: PMC5492136.
Cowing, Keith. How were amino acids formed before the origin of life on earth: https://astrobiology.com/2023/04/how-were-amino-acids-formed-before-the-origin-of-life-on-earth.html. 2023.
Parker ET, Cleaves JH, Burton AS, Glavin DP, Dworkin JP, Zhou M, Bada JL, Fernández FM. Conducting miller-urey experiments. J Vis Exp. 2014 Jan 21;(83):e51039. doi: 10.3791/51039. PMID: 24473135; PMCID: PMC4089479.
Ozturk SF, Sasselov DD. Life’s homochirality: Across a prebiotic network. Proc Natl Acad Sci U S A. 2025 Aug 26;122(34):e2505126122. doi: 10.1073/pnas.2505126122. Epub 2025 Aug 19. PMID: 40828029; PMCID: PMC12403148.
Cao C, Xu S, Wang L. An Algorithm for Protein Helix Assignment Using Helix Geometry. PLoS One. 2015 Jul 1;10(7):e0129674. doi: 10.1371/journal.pone.0129674. PMID: 26132394; PMCID: PMC4488512.
Cole BJ, Bystroff C. Alpha helical crossovers favor right-handed supersecondary structures by kinetic trapping: the phone cord effect in protein folding. Protein Sci. 2009 Aug;18(8):1602-8. doi: 10.1002/pro.182. PMID: 19569186; PMCID: PMC2776948.
Banerjee, A., Datta, S.A., Pramanik, A., Shamala, N., & Balaram, P. (1996). Heterogeneity and stability of helical conformations in peptides: crystallographic and NMR studies of a model heptapeptide. Journal of the American Chemical Society, 118, 9477-9483.
Yu, J., Darú, A., Deng, M., & Blackmond, D. G. (2024). Prebiotic access to enantioenriched amino acids via peptide-mediated transamination reactions. Proceedings of the National Academy of Sciences, 121(7), e2315447121. https://doi.org/10.1073/pnas.2315447121
Z. Niu, X. Gui, S. Feng, B. Reif, Chem. Eur. J. 2024, 30, e202400277. https://doi.org/10.1002/chem.202400277
Petersson, E., Williams, M. A., & Shea, J.-E. (2012). Driving forces and structural determinants of steric zipper peptide oligomer formation elucidated by atomistic simulations. Journal of Molecular Biology, 421(2–3), 390–416. https://doi.org/10.1016/j.jmb.2012.02.007
O.S. Makin, E. Atkins, P. Sikorski, J. Johansson, & L.C. Serpell, Molecular basis for amyloid fibril formation and stability, Proc. Natl. Acad. Sci. U.S.A. 102 (2) 315-320, https://doi.org/10.1073/pnas.0406847102 (2005).
PART B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
For this exercise I have chosen the same protein I explored in Week 2 which is a silk worm protein, Bombyx mori Fibroin heavy chain (FibH). It is the primary component of Bombyx mori silk consisting of 75% of its weight. Fibroin has amazing mechanical properties used by insects to construct various marvellous structures including a cocoon, nest, and egg case (Zhang et al. 2024). Therefore, it would be interesting to experiment with for the production of biomaterials and bio-inks to create transparent, complex or bioactive structures. Moreover, Fibroin is non-toxic and biodegradable offering an interesting material alternative to traditional sculptural materials such as glass, resin or plastic. For this task it will also be a great pick as it has a clear relationship between its unique repetitive amino acid sequence and beta sheet secondary structure, responsible for the high tensile strength, elasticity, and toughness of silk fibers.

Scarlett Yang Serecin Protein Bioplastic
- Identify the amino acid sequence of your protein.
Its incredibly long. You can also find it on Uniprot
Important
MRVKTFVILCCALQYVAYTNANINDFDEDYFGSDVTVQSSNTTDEIIRDASGAVIEEQITTKKMQRKNKNHGILGKNEKMIKTFVITTDSDGNESIVEEDVLMKTLSDGTVAQSYVAADAGAYSQSGPYVSNSGYSTHQGYTSDFSTSAAVGAGAGAGAAAGSGAGAGAGYGAASGAGAGAGAGAGAGYGTGAGAGAGAGYGAGAGAGAGAGYGAGAGAGAGAGYGAGAGAGAGAGYGAGAGAGAGAGYGAGAGAGAGAGYGAASGAGAGAGYGQGVGSGAASGAGAGAGAGSAAGSGAGAGAGTGAGAGYGAGAGAGAGAGYGAASGTGAGYGAGAGAGYGGASGAGAGAGAGAGAGAGAGYGTGAGYGAGAGAGAGAGAGAGYGAGAGAGYGAGYGVGAGAGYGAGYGAGAGSGAASGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGTGAGSGAGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGAGYGAGAGAGYGAGAGVGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGVGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVANGGYSRSDGYEYAWSSDFGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGVGYGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGVGSGAGAGSGAGAGVGYGAGAGVGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGVGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGAGYGAGYGAGAGAGYGAGAGSGAASGAGSGAGAGSGAGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGVGYGAGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVAHGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGVGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGVGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVANGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVAHGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAAYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGYGAGAGAGYGAGYGAGAGAGYGAGAGTGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVAHGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGAGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGTGSGAGAGSGAGAGYGAGVGAGYGAGAGSGAAFGAGAGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGVGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVANGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGYGVGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGVGAGYGAGAGSGAASGAGAGSGAGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGVGAGYGAGAGVGYGAGAGAGYGAGAGSGAASGAGAGAGSGAGAGTGAGAGSGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVANGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGSGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGVGAGYGVGYGAGAGAGYGVGYGAGAGAGYGAGAGSGTGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGYGVGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGVGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVANGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGYGAGYGAGVGAGYGAGAGVGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGSGAGAGSGAGAGAGSGAGAGYGAGYGIGVGAGYGAGAGVGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGVGAGYGAGAGVGYGAGAGAGYGAGAGSGAASGAGAGAGAGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVNGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGYGAGYGAGVGAGYGAGAGVGYGAGAGAGYGAGAGSGAASGAGAGSGSGAGSGAGAGSGAGAGSGAGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGAGSGAASGAGAGAGAGAGTGSSGFGPYVANGGYSGYEYAWSSESDFGTGSGAGAGSGAGAGSGAGAGYGAGVGAGYGAGYGAGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGAGAGAGYGAGAGVGYGAGAGAGYGAGAGSGAGSGAGAGSGSGAGAGSGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGYGIGVGAGYGAGAGVGYGAGAGAGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGYGAGAGVGYGAGAGSGAASGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGYGAGYGAGVGAGYGAGAGYGAGYGVGAGAGYGAGAGSGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGAGSGAGSGAGAGYGAGAGAGYGAGAGAGYGAGAGSGAASGAGAGAGAGSGAGAGSGAGAGSGAGSGAGAGSGAGAGYGAGAGSGAASGAGAGSGAGAGAGAGAGAGSGAGAGSGAGAGYGAGAGSGAASGAGAGAGAGTGSSGFGPYVANGGYSRREGYEYAWSSKSDFETGSGAASGAGAGAGSGAGAGSGAGAGSGAGAGSGAGAGGSVSYGAGRGYGQGAGSAASSVSSASSRSYDYSRRNVRKNCGIPRRQLVVKFRALPCVNC
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is: 5263 amino acids. The most common amino acid is: G, Glycine which appears 2415 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
The Fibroin protein sequence is really long! and consists largely of highly repetitive, low-complexity patterns that were causing BLAST to time out across all my devices. Instead, I decided to narrow the homologs search to the N-terminal domain (residues 1–151).
MRVKTFVILCCALQYVAYTNANINDFDEDYFGSDVTVQSSNTTDEIIRDASGAVIEEQITTKKMQRKNKNHGILGKNEKMIKTFVITTDSDGNESIVEEDVLMKTLSDGTVAQSYVAADAGAYSQSGPYVSNSGYSTHQGYTSDFSTSAAVGAGAG
This domain is the non-repetitive ‘molecular switch’ responsible for pH-dependent silk assembly before the silk is secreted by the insect.
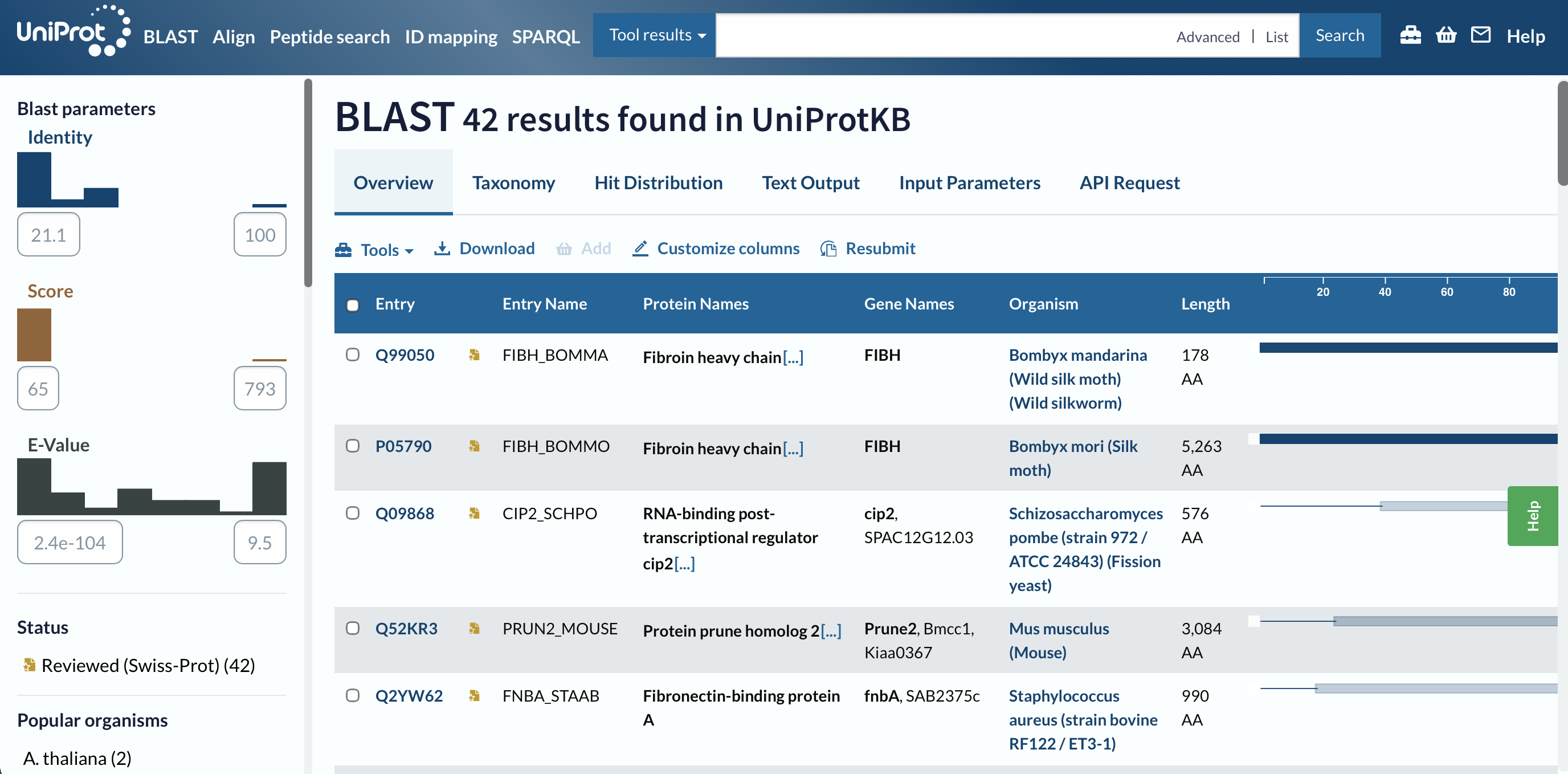
I got 42 results.
The E-values range from 2.4e-104 (basically 0, significant matches ) to 9.5 (really insignificant)
The BLAST search returned 42 results with identities ranging from 21.1% to 100%. The top hit Fibroin heavy chain from Bombyx mandarina (Wild silk moth)showed a maximum score of 793 and an E-value of 2.4×10-104 confirming a highly significant evolutionary relationship with other silk-producing insect species.
Does your protein belong to any protein family?
UniProt and the PANTHER database classify it specifically under the Fibroin Heavy Chain Fib-H Like Protein 1 family. It is defined by its signature Fib-H N terminal domain.
This family is central to the production of silk fibers in insects and spiders and the N-terminal domain mediates the pH responsive assembly of silk proteins as they are secreted.
Identify the structure page of your protein in RCSB
Entry: 3UA0 N-Terminal Domain of Bombyx mori Fibroin Mediates the Assembly of Silk in Response to pH Decrease.
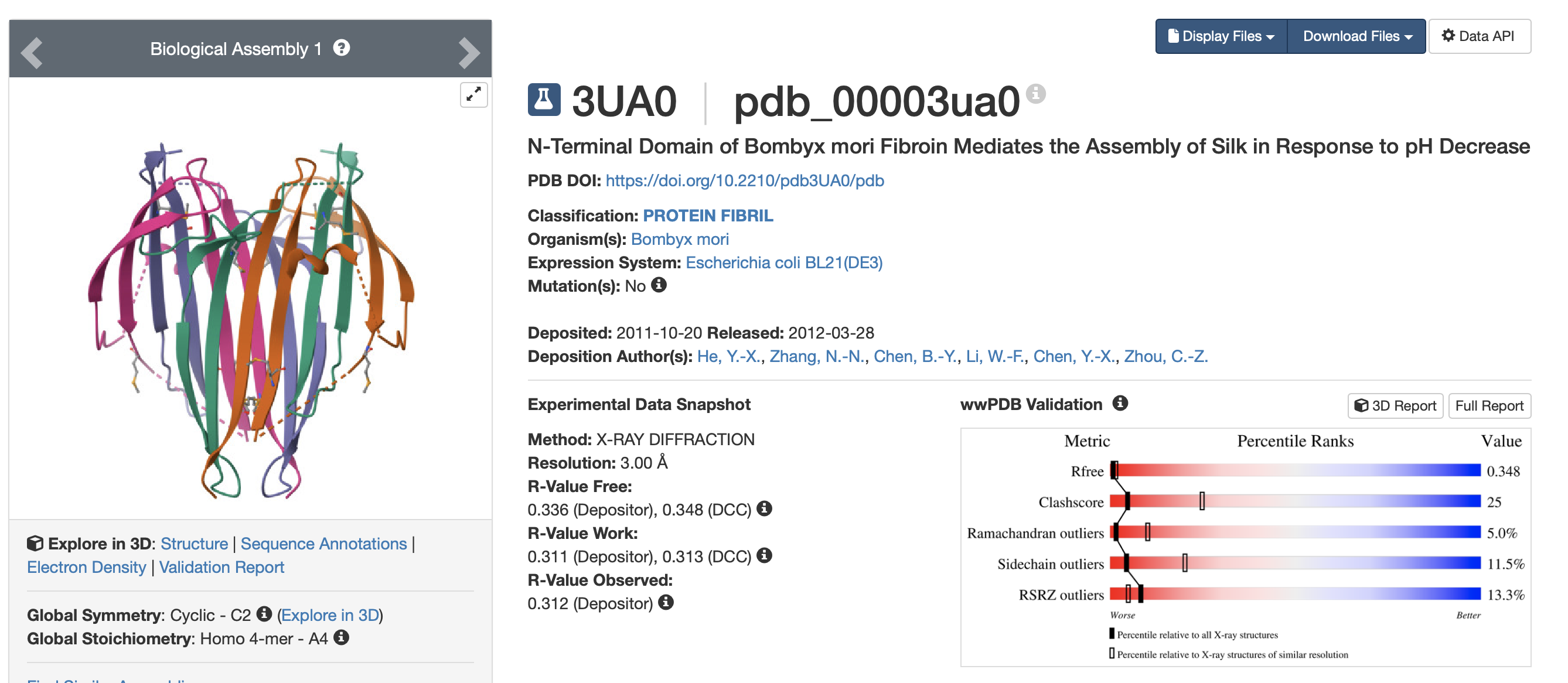
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure was solves on the 20/10/2011 and released publicly on the 28/03/2012. The resolution is moderate to low at 3.00Å using X-ray diffraction.
Are there any other molecules in the solved structure apart from protein?
MSE (Selenomethionine) a naturally occuring amino acid in some plant materials such as cereal grains, soybeans and enriched yeast.
Does your protein belong to any structure classification family?
No SCOP 2 Classification was available for this protein.
Open the structure of your protein in any 3D molecule visualisation software:
First I used the code “fetch 3UA0” to load the 3UA0 | pdb_00003ua0 from RCSB.
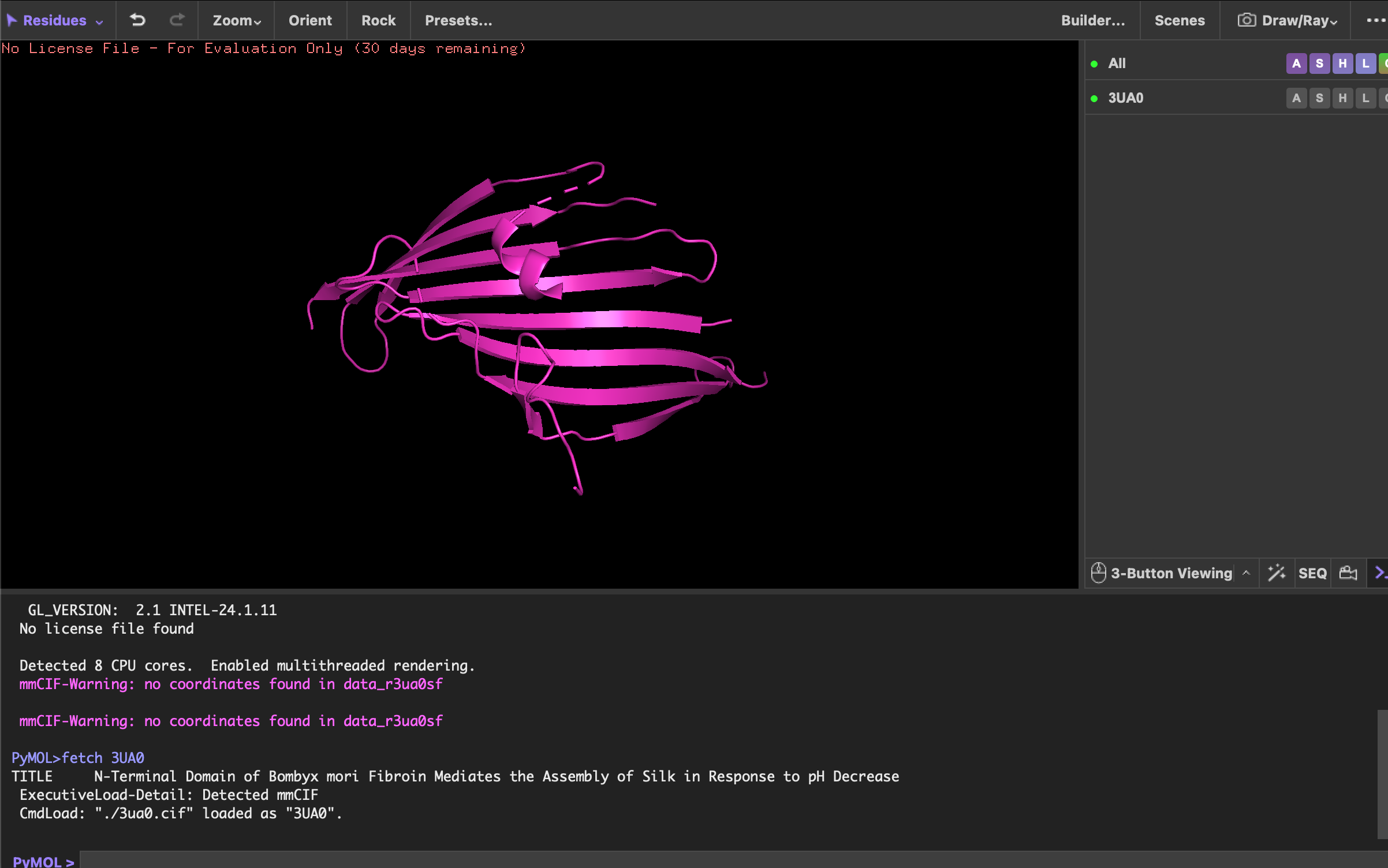
Visualise the protein as “cartoon”, “ribbon” and “ball and stick”.
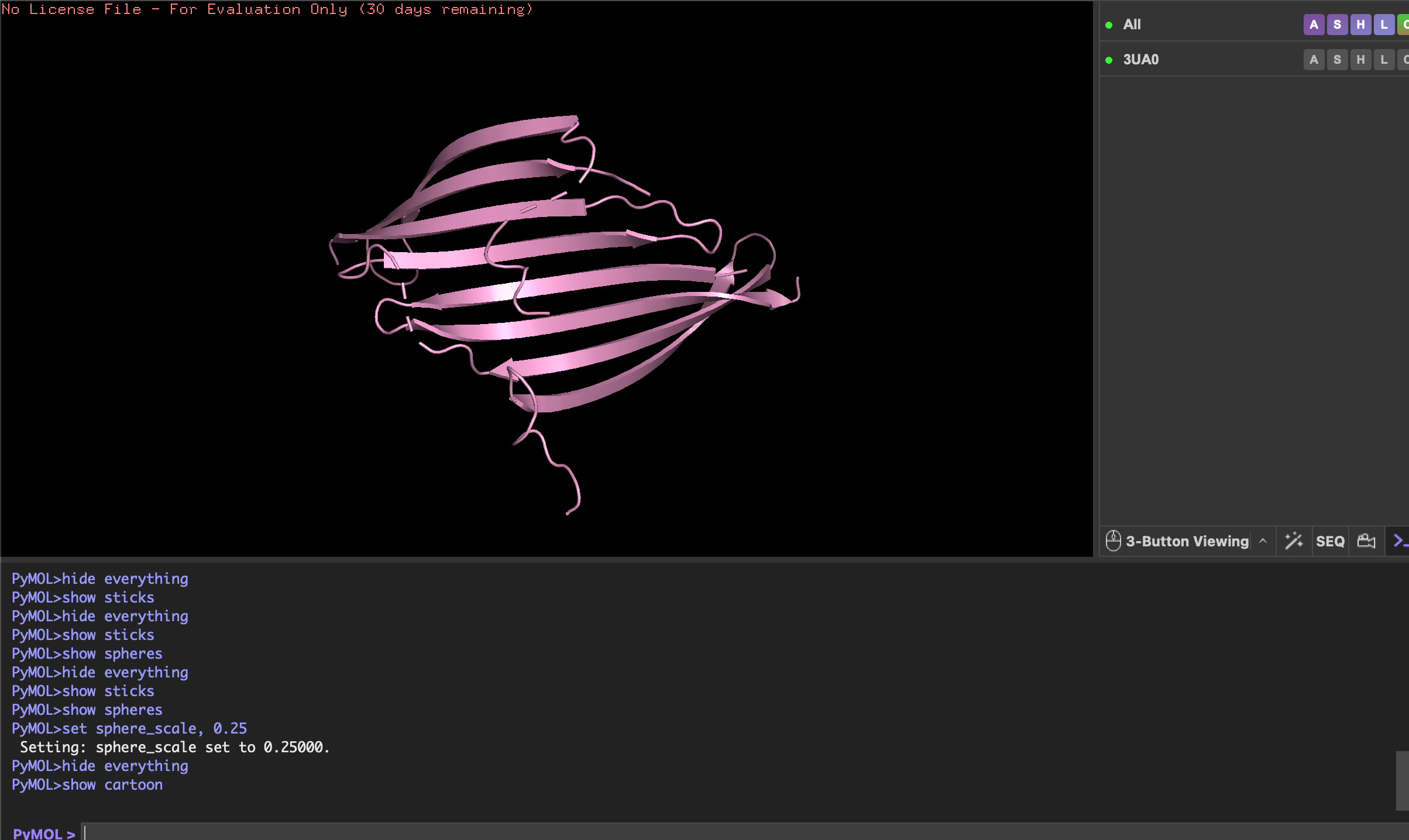
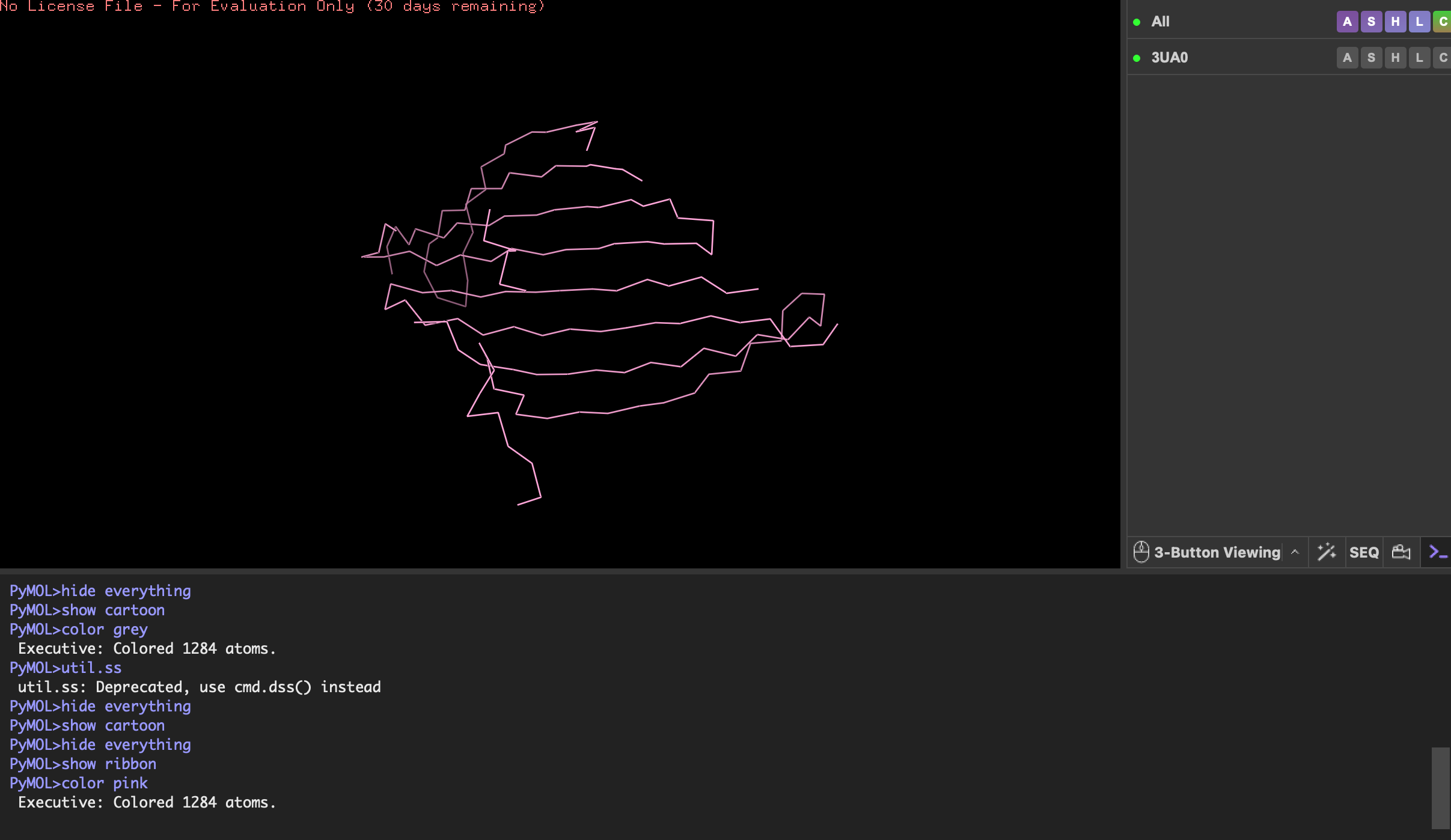
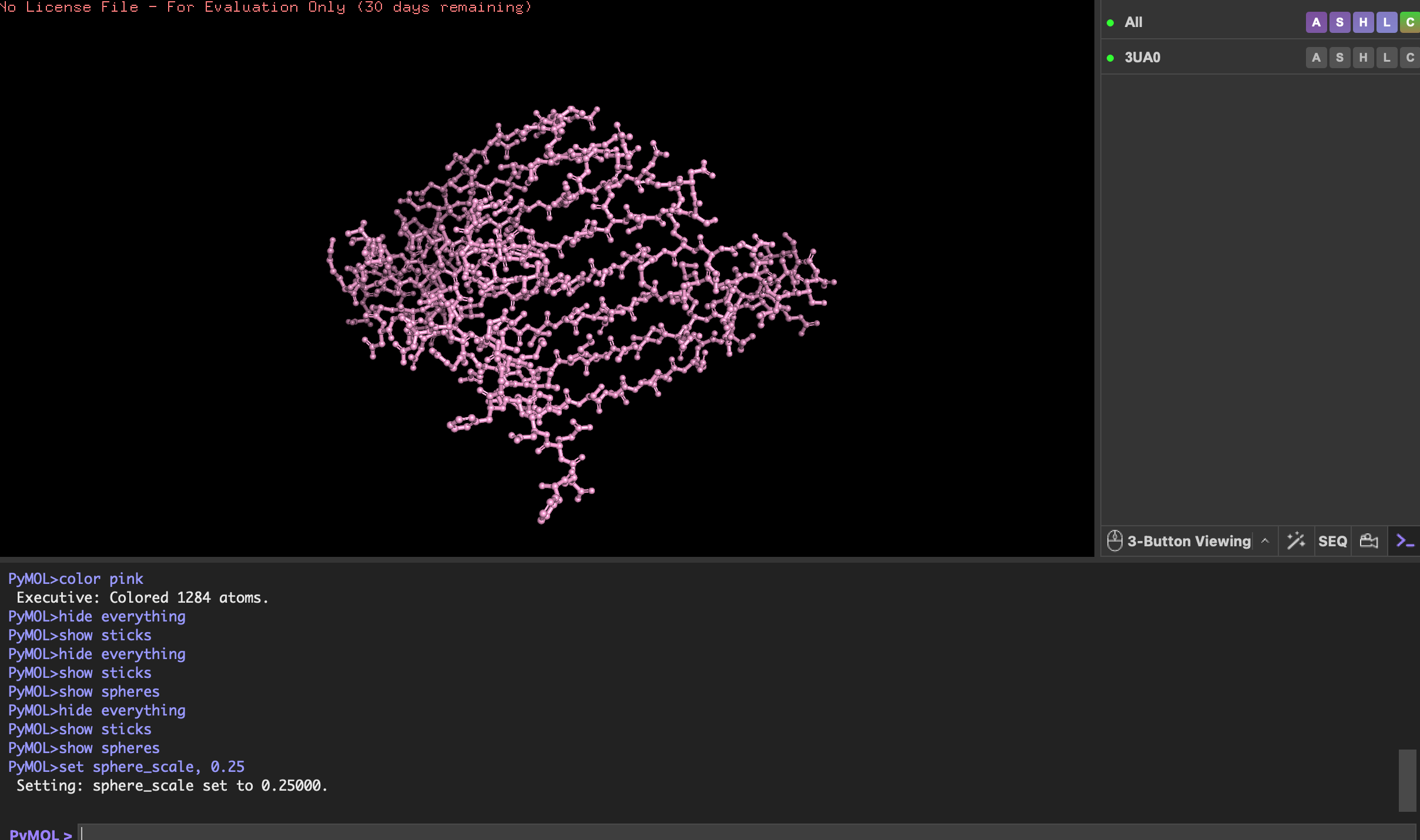
Color the protein by secondary structure. Does it have more helices or sheets?
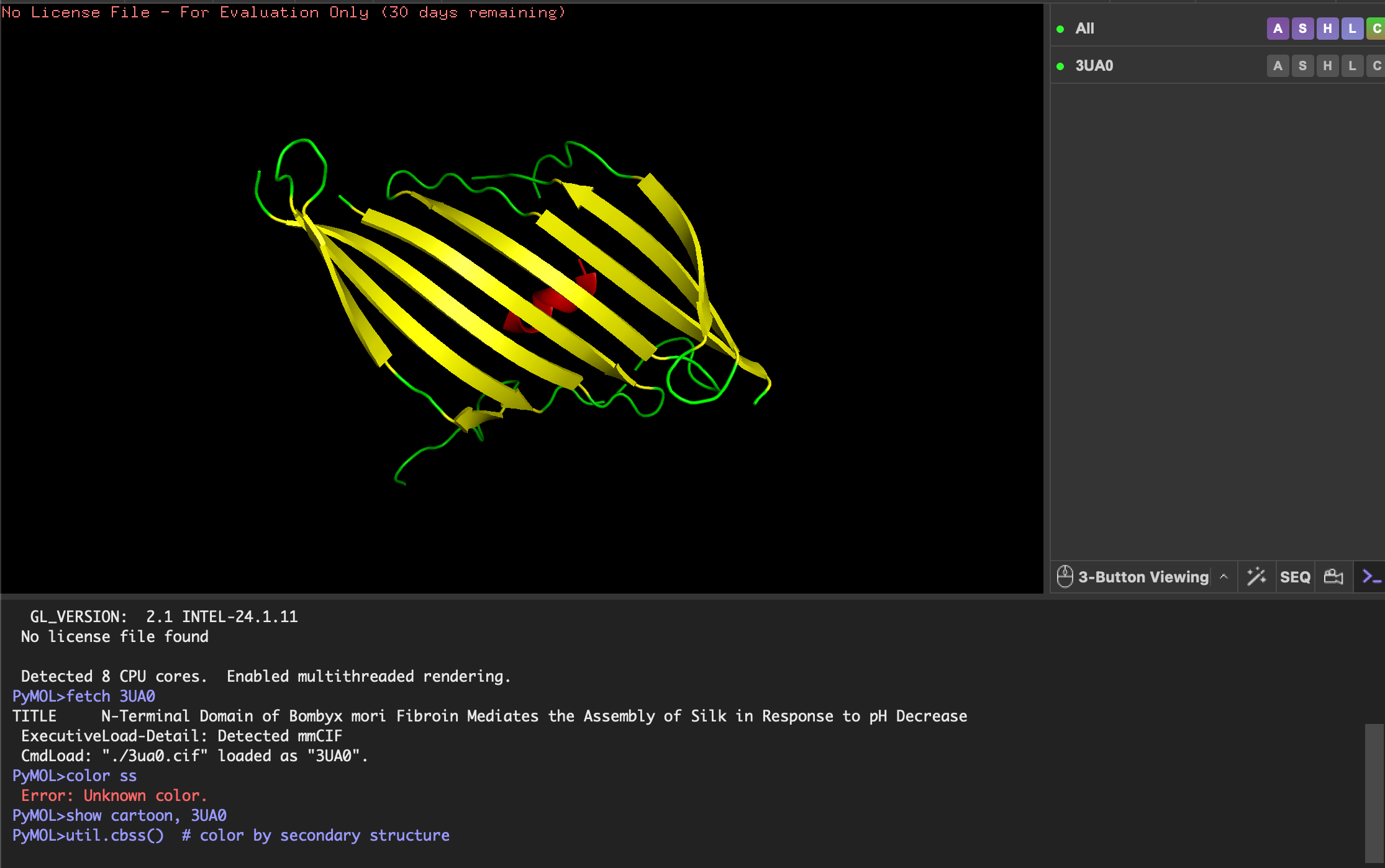
- Helices: red
- Beta-sheets: yellow
- Loops/coils: green
As expected the Beta-sheets are the most abundant!
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
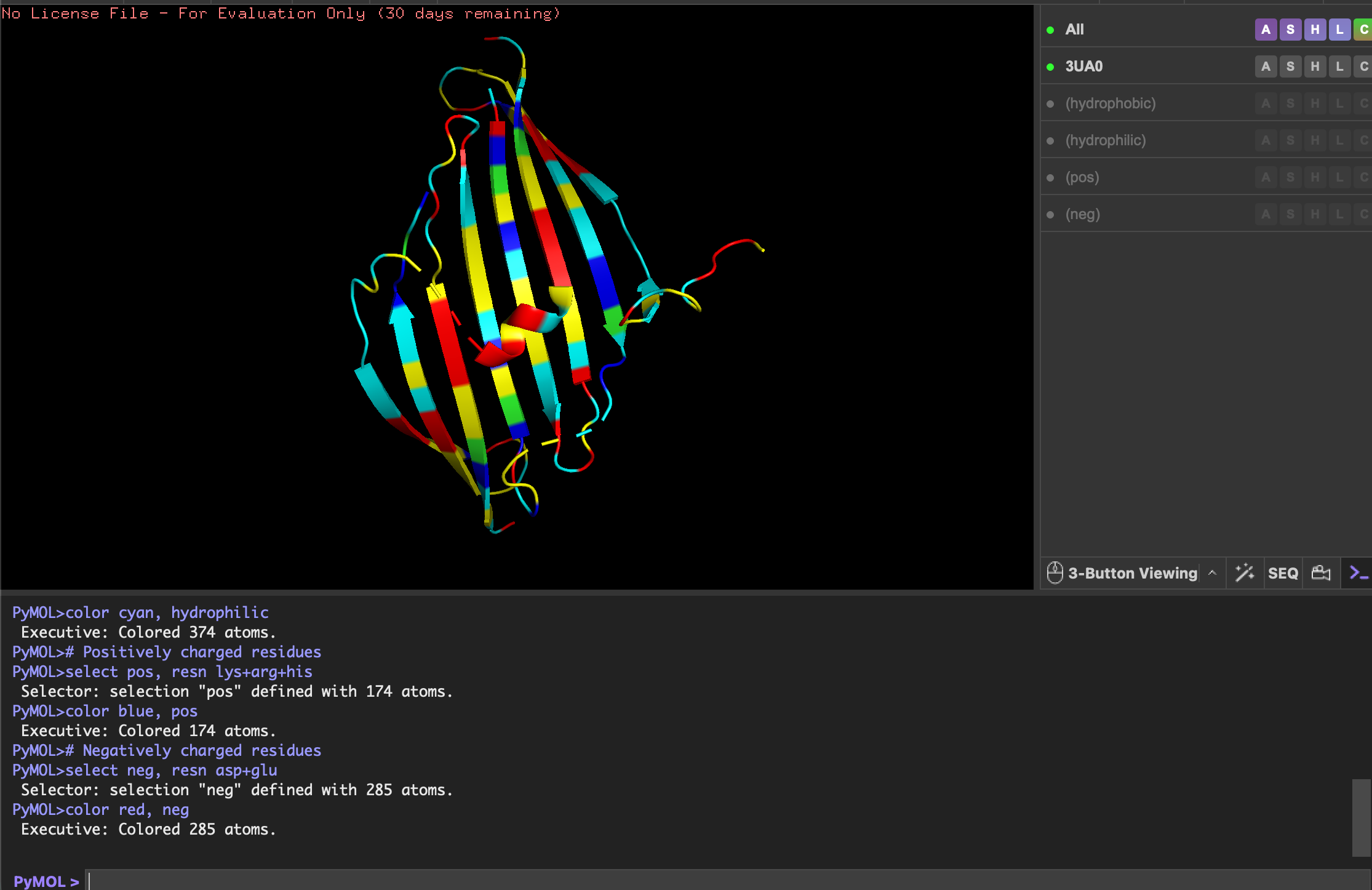
- Yellow: hydrophobic
- Cyan: hydrophilic
- Blue: positively charged
- Red: negatively charged
Although quite evenly distributed, the hydrophilic residues (cyan) are more exposed on the ends of sheets and loops, interacting with water to keep protein soluble. The hydrophobic residues (yellow) are more buried in the center of the sheets and helix, stabilising the fold.
Visualise the surface of the protein. Does it have any “holes” (aka binding pockets)?
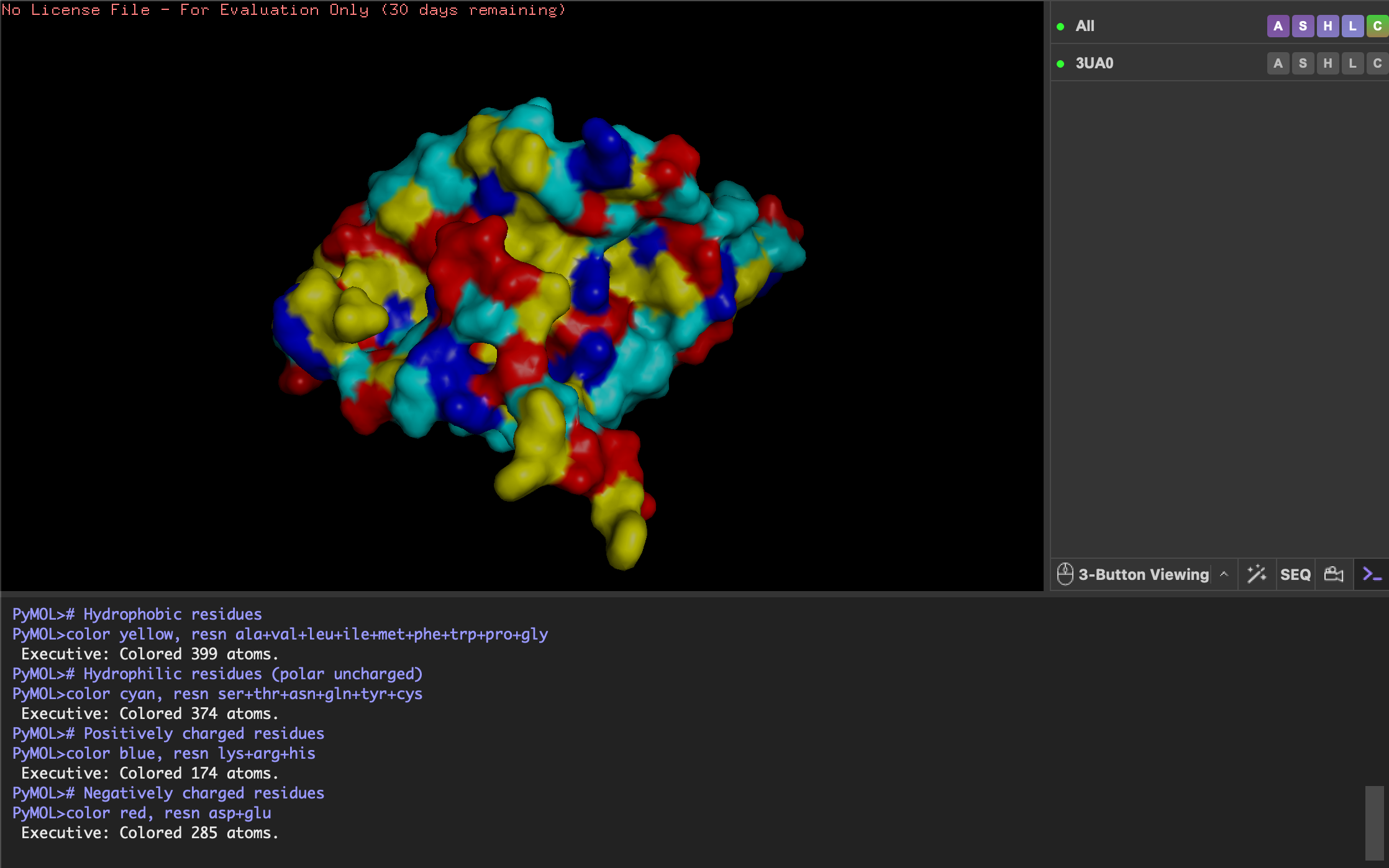
The protein surface does not show any obvious holes, but there are several deep grooves and clefts. These grooves are likely substrate-binding or interaction sites where ligands or substrates can fit. These grooves (binding pockets) are lined with hydrophobic (yellow) and positively charged (blue) residues so are potentially involved in interacting with hydrophobic and negatively charged molecules. This would make sense for Fibroin known for its hydrophobic core lending to its stability.
PART C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Choose your favorite protein from the PDB.
Lysozyme: An enzyme that breaks down bacterial cell walls.
1LYZ, pbd 00001lyz
SEQUENCE:
KVFGRCELAAAMKRHGLDNYRGYSLGNWVCAAKFESNFNTQATNRNTDGSTDYGILQINSRWWCNDGRTPGSRNLCNIPCSALLSSDITASVNCAKKIVSDGNGMNAWVAWRNRCKGTDVQAWIRGCRL
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
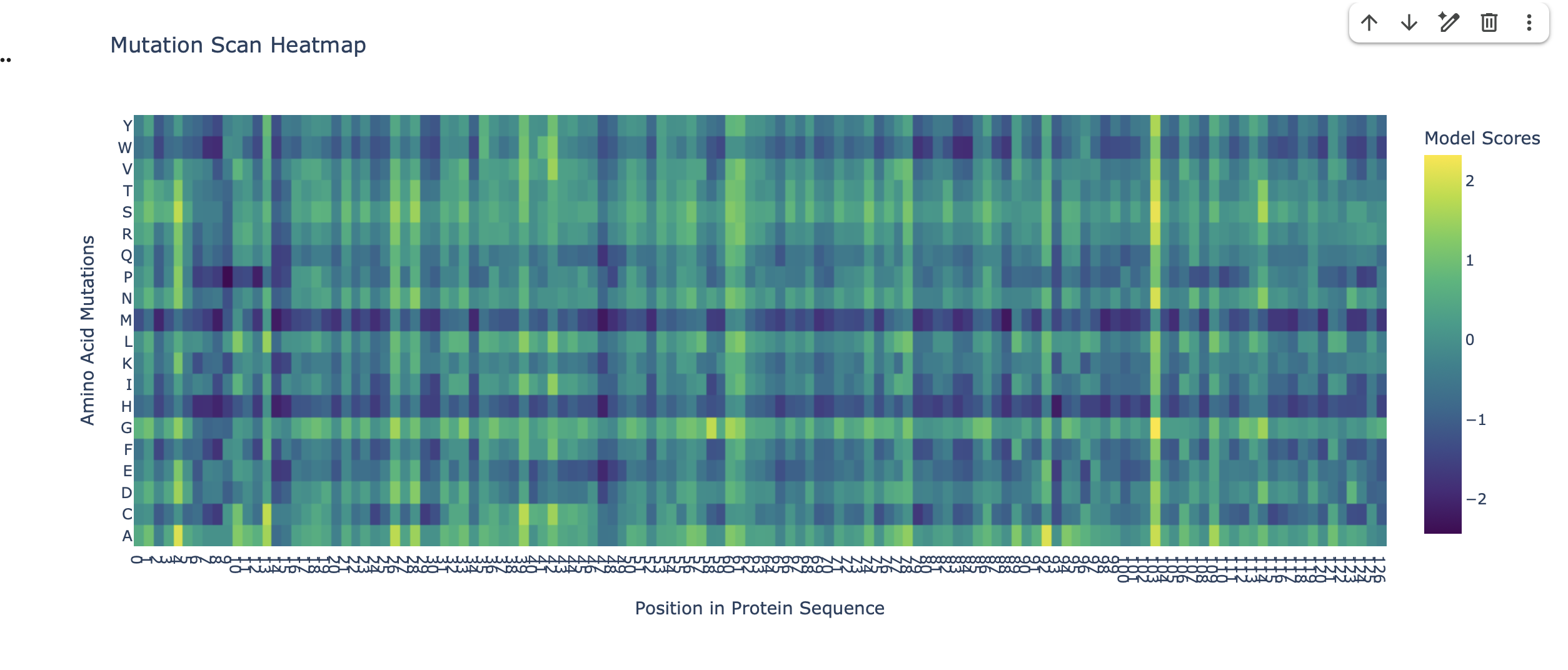
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The heatmap as a visual guide to how ’tolerant’ the protein is to changes in its amino acids at different positions. Each colored square tells us about the mutational tolerance at that specific spot for a specific amino acid change.
Bright Colors: Indicate high mutational tolerance. This means the protein can easily accept (or even prefer) that particular amino acid change without negatively affecting its function or stability.
Dark Colors (like deep blue/purple): Indicate low mutational tolerance. This means the protein is very sensitive to that amino acid change, and it’s likely to cause problems, making the protein less functional or unstable.
Stand outs:
Consistent Low Mutational Tolerance for Methionine (dark blue horizontal line on M row). This signifies that, for many different residues in the protein, mutating to a Methionine is predicted to be highly unfavorable(low LLR). This suggests a generally low mutational tolerance for introducing Methionine into the protein structure.
Consistent High Mutational Tolerance at residue position 104 (which corresponds to x axis 102 on the heatmap). The wild type amino acid at this position in the sequence is Glycine (G). The entire vertical column 102 is yellow and green. This is a strong indicator of high mutational tolerance at residue position 104, the Glycine can accommodate a wide variety of amino acid mutations as they are predicted by the model to be either favorable (high LLRs) or at least neutral (LLR around 0).
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Using the Collab to embed proteins in reduced dimensionality, I first downloaded a dataset of protein sequences in FASTA format. Then, I processed these sequences through the ESM2 language model to generate high-dimensional embeddings for each protein. Finally, I used t-SNE, a dimensionality reduction technique to transform these high-dimensional embeddings into a 3D representation, making them suitable for visualszation and analysis in a lower-dimensional space.
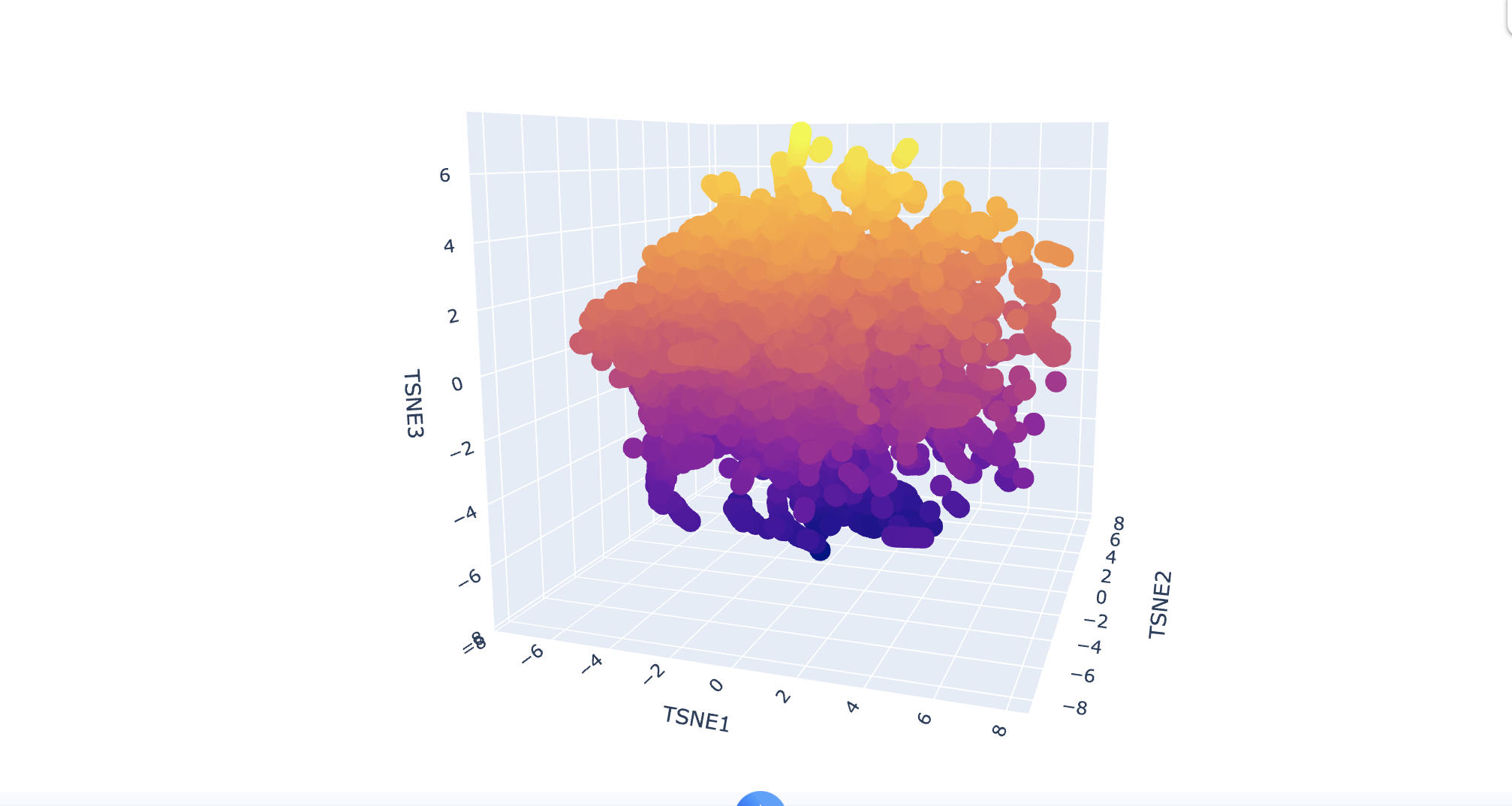
Analyze the different formed neighborhoods: do they approximate similar proteins?
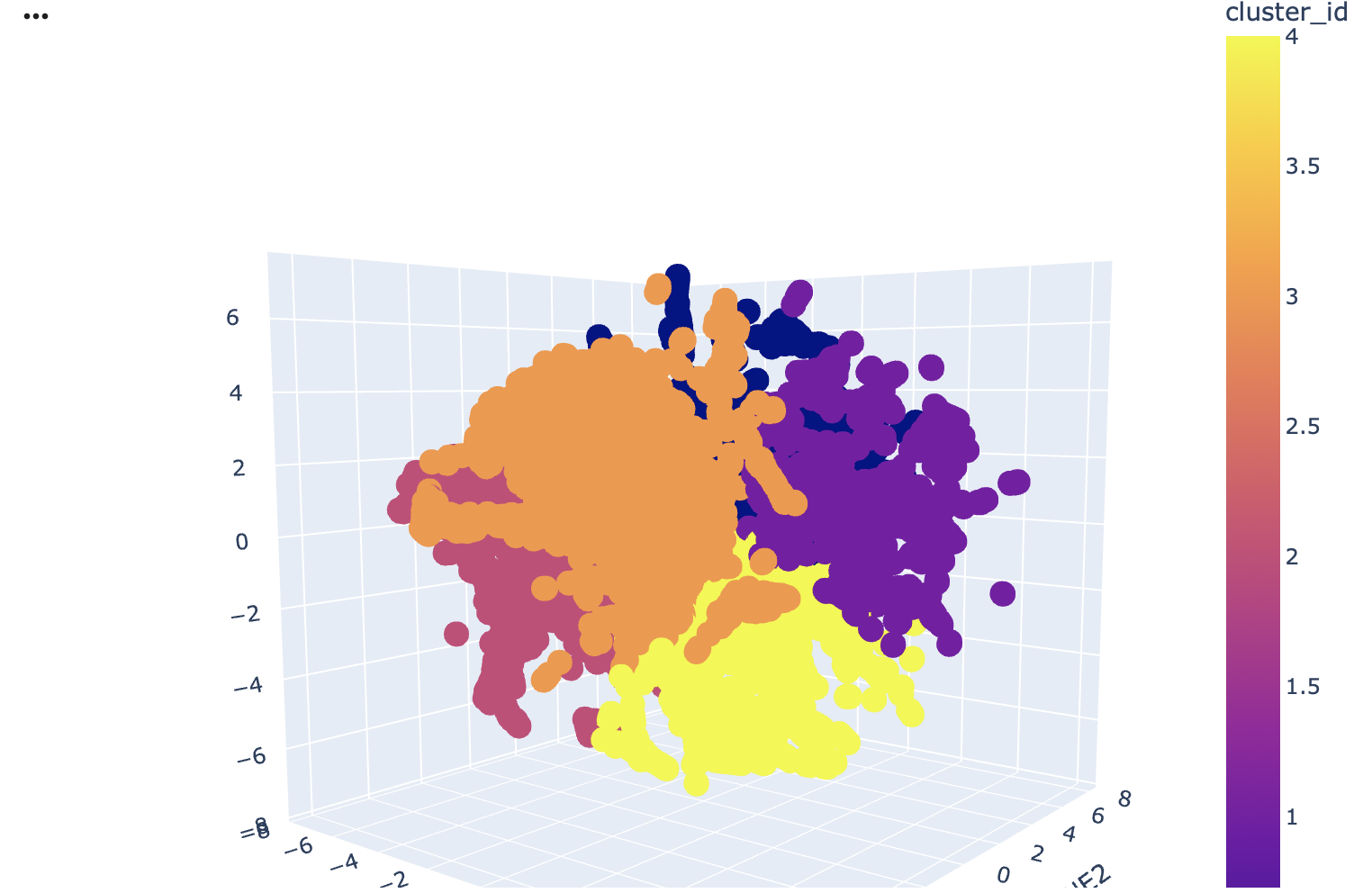
Yes, the latent space neighbourhoods as represented by the t-SNE clusters do approximate similar proteins. I used the gemini add on in Collab to do an analysis of keyword frequencies in the protein annotations within each neighbouring cluster reveals that proteins are grouped primarily by their organismal origin and shared structural or functional characteristics. You can see the clusters used in the image above.
For instance Cluster 2 (pink) is predominantly composed of human proteins, as indicated by the high frequency of keywords like human, homo, and sapiens. Additionally, the term domain appears with significant frequency within this cluster. A protein domain refers to a distinct functional within a larger protein, this suggests that the proteins in Cluster 2 have complex, multi-domain architectures.

Place your protein in the resulting map and explain its position and similarity to its neighbours.
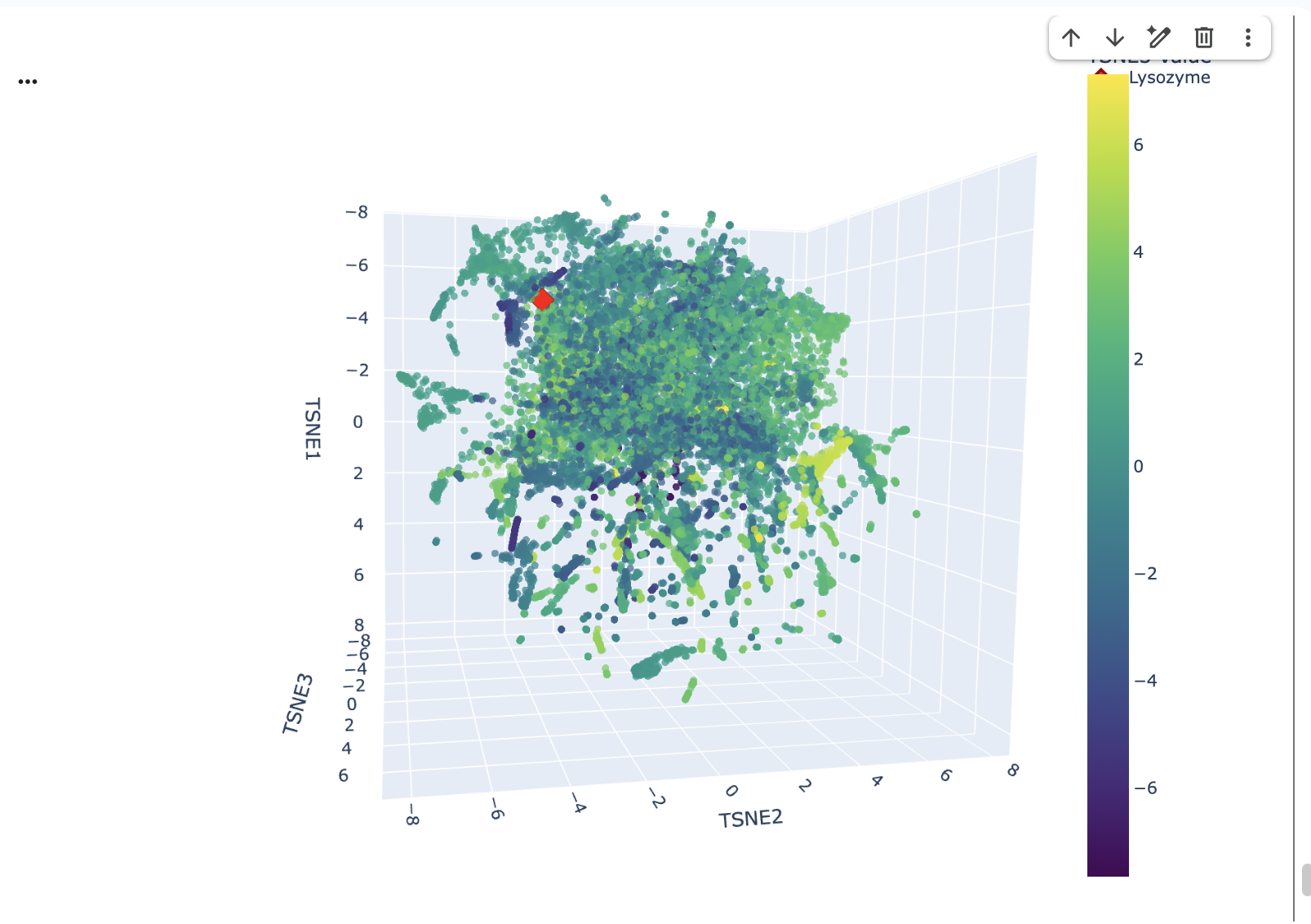
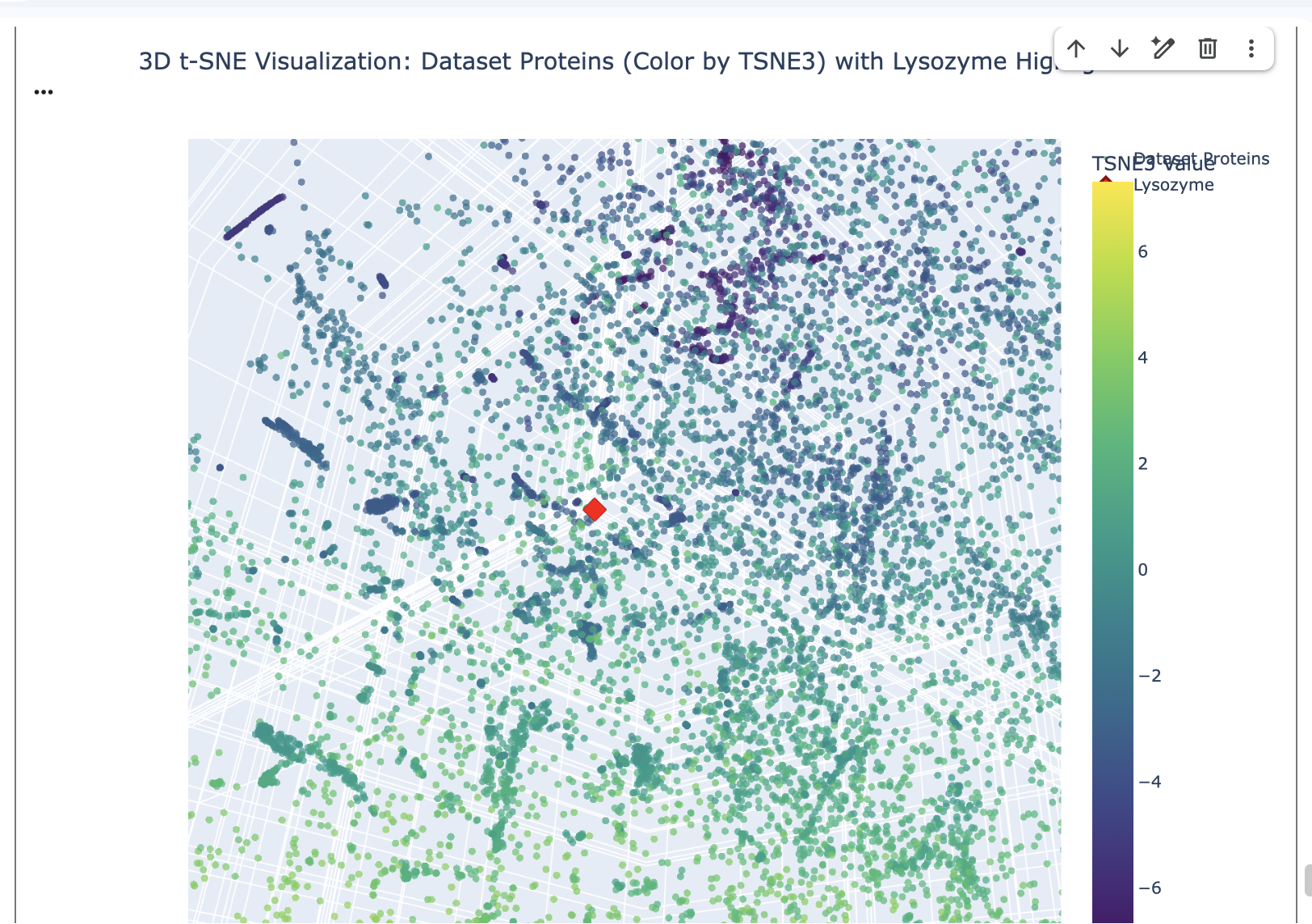
The position of Lysozyme on the t-SNE map is in close proximity to other Lysozyme proteins and grouped with proteins with similar functions related to antimicrobial activity or general proteolytic degradation.
Lysozyme’s single closest neighbour (cosine distance of 0.5614) is another Lysozyme (d2vb1a_ d.2.1.2 (A:) Lysozyme {Chicken (Gallus gallus)}). This demonstrates that the ESM-2 embeddings are highly effective at identifying direct homologs placing them very close together in the latent space.
- 2nd Nearest (Distance: 1.0996): d1kp6a_ d.58.25.1 (A:) Killer toxin KP6 alpha-subunit {Smut fungus (Ustilago maydis)}
- 3rd Nearest (Distance: 1.1177): d4jp6a_ b.52.1.2 (A:) automated matches {Papaya (Carica papaya)}
- 5th Nearest (Distance: 1.1413): d1deua_ d.3.1.1 (A:) (Pro)cathepsin X {Human (Homo sapiens)}
Other nearby proteins include the Killer toxin KP6 alpha-subunit, a protein from Carica papaya, and the pro-domain of Cathepsin X. Although these proteins originate from different organisms and perform different biological roles, they share several structural characteristics with lysozyme. In particular, they are relatively small, compact proteins that are often secreted and stabilised by disulphide bonds. As a result, they can display similar secondary-structure arrangements, cysteine patterns and surface properties.
Functionally, there are also some broad parallels. Lysozyme acts as an antimicrobial enzyme that degrades bacterial cell walls, while killer toxins are secreted proteins that inhibit competing microorganisms. Similarly, Cathepsin X is a lysosomal cysteine protease involved in protein degradation. While their specific biological roles differ, all three proteins participate in processes involving extracellular defense or proteolytic activity.
C2. Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
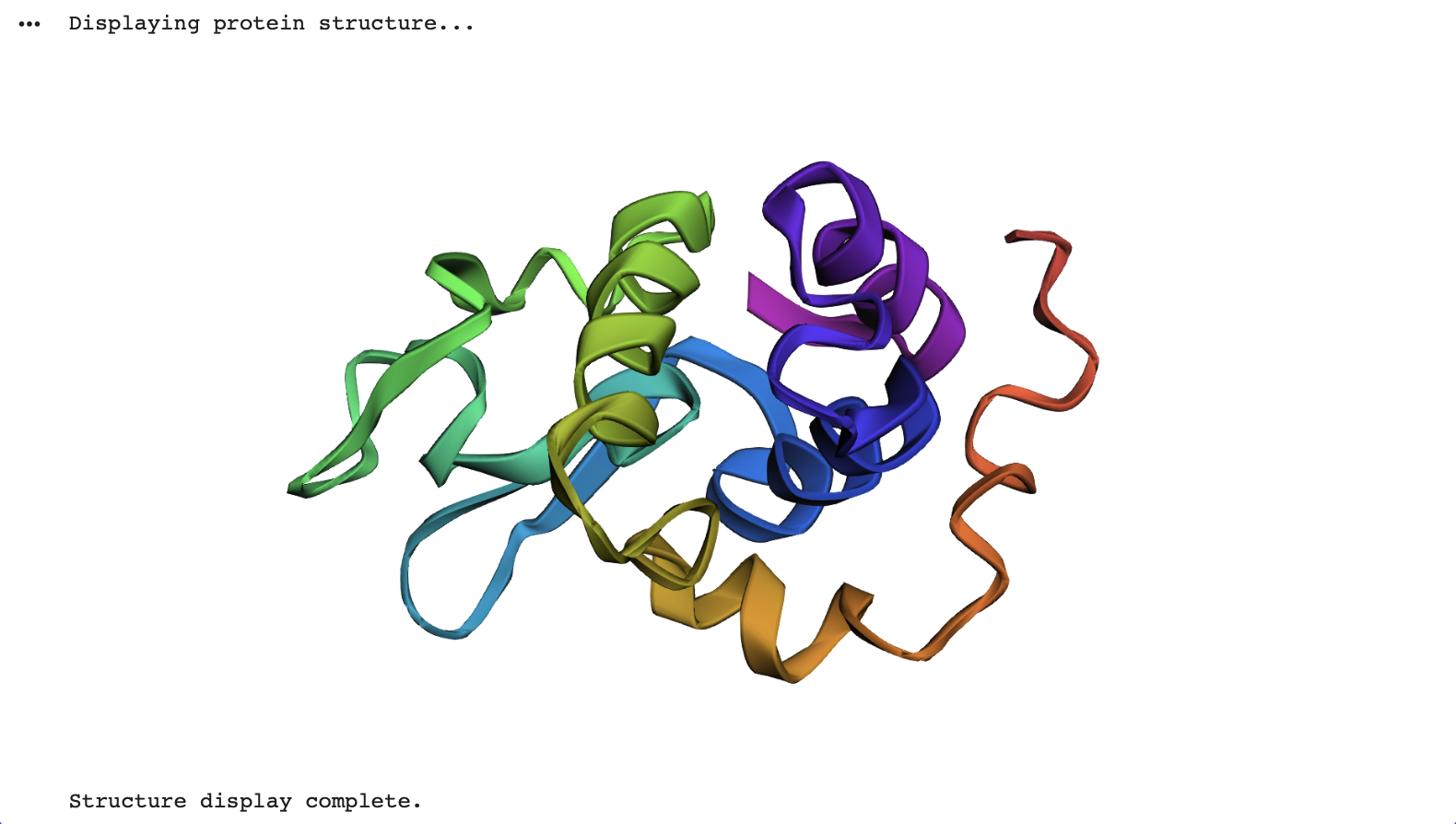

Yes, the predicted coordinates closely match the original structure. The Root Mean Square Deviation (RMSD) calculated in the collab between the C-alpha atomic co-ordinates extracted from the original pdb co-ordinates and the ESM Fold predicted Lysozyme structures is 0.553 Angstroms (low = more accurate), indicating a very high degree of structural similarity.
In the below visualisation, both the original and predicted structures contained 129 C-alpha atoms, ensuring a consistent basis for comparison. The original (blue) and predicted (green) structures were successfully superimposed and visualized using py3Dmol, allowing for a clear visual confirmation of their alignment.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I generated three point mutated versions of Lysozyme and predicted their structures using ESMFold.
- Lysozyme_K1A_mutant (single mutation: Lysine at position 1 to Alanine)
- Lysozyme_F3L_G4A_mutant (double mutation: Phenylalanine at position 3 to Leucine, Glycine at position 4 to Alanine)
- Lysozyme_V2I_W28Y_mutant (double mutation: Valine at position 2 to Isoleucine, Tryptophan at position 28 to Tyrosine)
Structural comparisons of these mutated proteins against the original 1LYZ using Gemini showed consistently low RMSD values:
Lysozyme_K1A_mutant: RMSD = 0.555 ÅLysozyme_F3L_G4A_mutant: RMSD = 0.596 ÅLysozyme_V2I_W28Y_mutant: RMSD = 0.608 Å
These low RMSD values (all below ~0.6 Å)(Root Mean Square Deviation (RMSD), which quantifies the structural difference along with the visualisation of the overlat indicate a high degree of structural similarity between the original Lysozyme, the initial ESMFold prediction and the mutated structures.
The ptm and plddt scores for the mutated proteins were also high (e.g., ptm ~0.906-0.909, plddt ~94.7-95.2), indicating high confidence in ESMFold’s predictions for these variants.
This suggests that these specific single or double point mutations do not drastically alter the overall global fold of the protein, which is expected for minor changes in a well-folded, stable protein like Lysozyme.

Based on the analysis of these point mutations, the Lysozyme protein structure appears highly resilient to these specific 1-2 point mutations.
Large Segment Mutation
I replaced a 15-residue segment of the original Lysozyme sequence (from position 50 to 64) with a new segment, “AAAAAGGGGGPPPPP”, resulting in a segment-mutated Lysozyme sequence of the same length (129 residues).
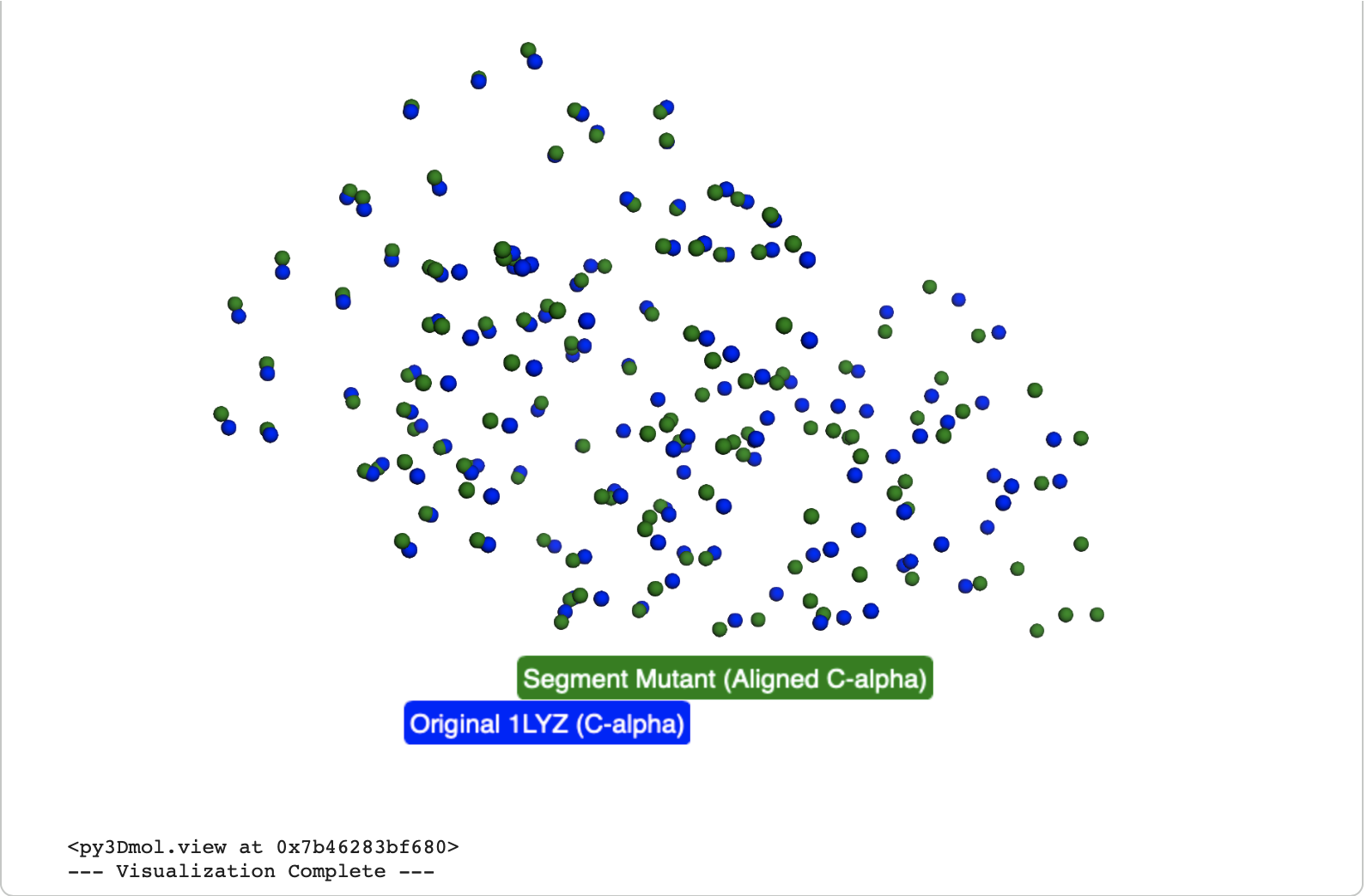
The Root Mean Square Deviation (RMSD) calculated between the original Lysozyme and the Lysozyme_segment_mutant C-alpha atoms after structural alignment is 3.163 Å.
The visual shows significant difference between the original (blue ) and the transformed segment mutant (green ) structures. While some parts of the protein backbone might still align, the region corresponding to the large segment mutation shows clear structural differences, leading to a much higher RMSD compared to the point mutants.
The ptm and plddt scores for the segment mutant were noticeably lower (ptm ~0.768, plddt ~75.559) compared to the original ESMFold prediction (ptm: 0.907, plddt: 95.138) and the point mutants which indicates a lower confidence in the predicted structure and a less well-folded.
Based on these results, the Lysozyme protein structure is not resilient to large segment mutations of this magnitude.
Inverse-Folding a protein: Using the backbone of Lysozyme to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. 2. Input this sequence into ESMFold and compare the predicted structure to your original.
The heatmap generated visualises the probability distribution for each amino acid at every position along the 5MBA lysozyme backbone. For each position, ProteinMPNN assigns a high probability to one specific amino acid, which forms the basis of its generated sequence.
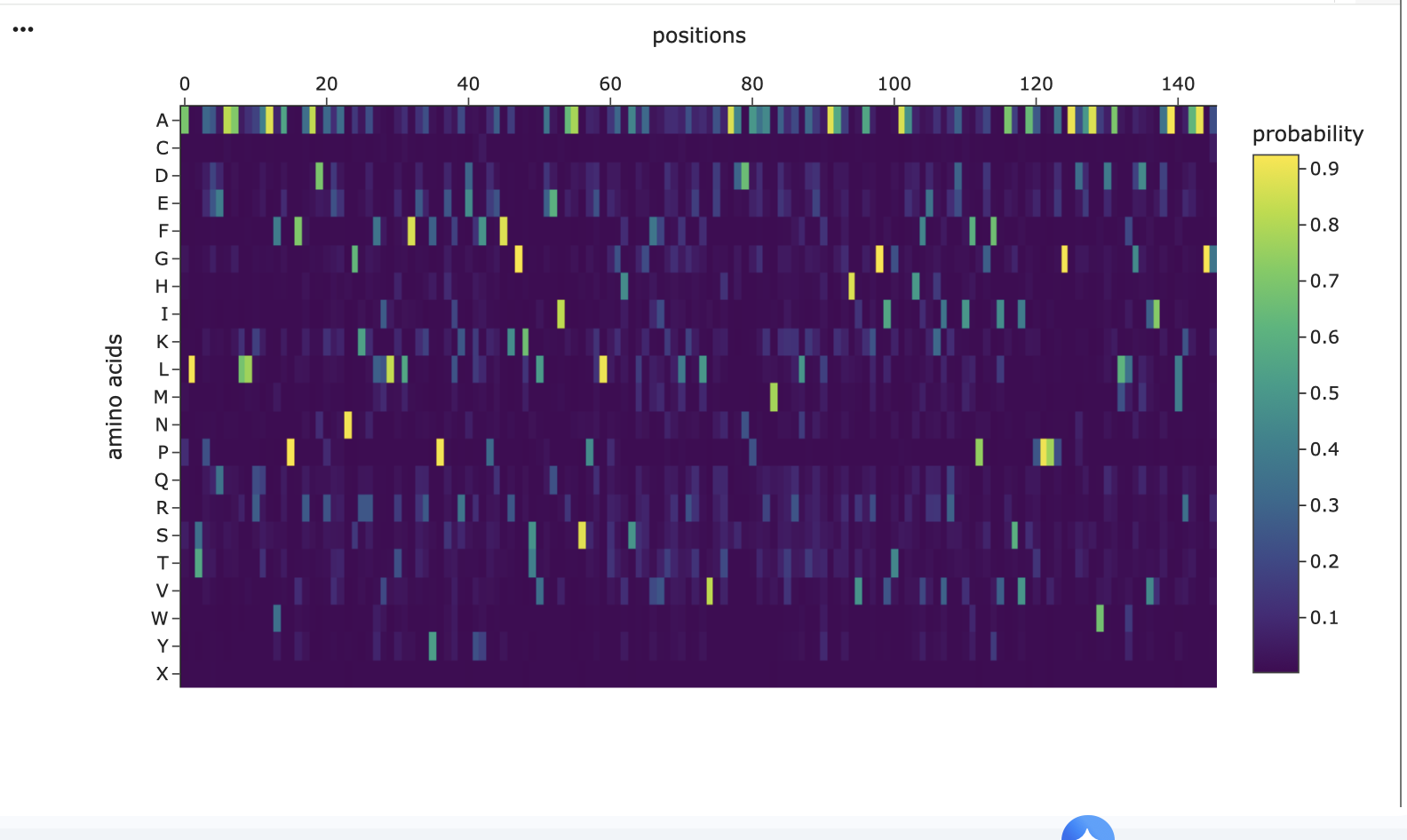
Original Lysozyme Sequence (Length129):
KVFGRCELAAAMKRHGLDNYRGYSLGNWVCAAKFESNFNTQATNRNTDGSTDYGILQINSRWWCNDGRTPGSRNLCNIPCSALLSSDITASVNCAKKIVSDGNGMNAWVAWRNRCKGTDVQAWIRGCRLProteinMPNN Generated Sequence (Length 146):
ALTPEEAALLRAAAAPVFADREANGRAFVLRLFEAYPELAELFPEFKGKTLAEIAASPALGAIAGAIMDGLATLVEHADDPARMATLLAALAAAHRARGITAAHFERIRALFPGFIASVAPPPPGADAAWDRLLGLVIDAMRAAGG
Differences
Out of the 129 positions in the original Lysozyme sequence, 124 positions show differences when compared to the ProteinMPNN-generated sequence. This means the model proposed a significantly different amino acid at almost every position. This creates a very low sequence recovery rate of 3.88%, indicating that only a small fraction of the residues in the generated sequence match the original Lysozyme sequence at corresponding positions.
This means ProteinMPNN found a very different sequence that it believes is stable for the backbone of Lysozyme which is to be expected given I didn’t give the model any sequence constraints.
I then took this newly generated sequence and fed it into ESMFold to predict its structure
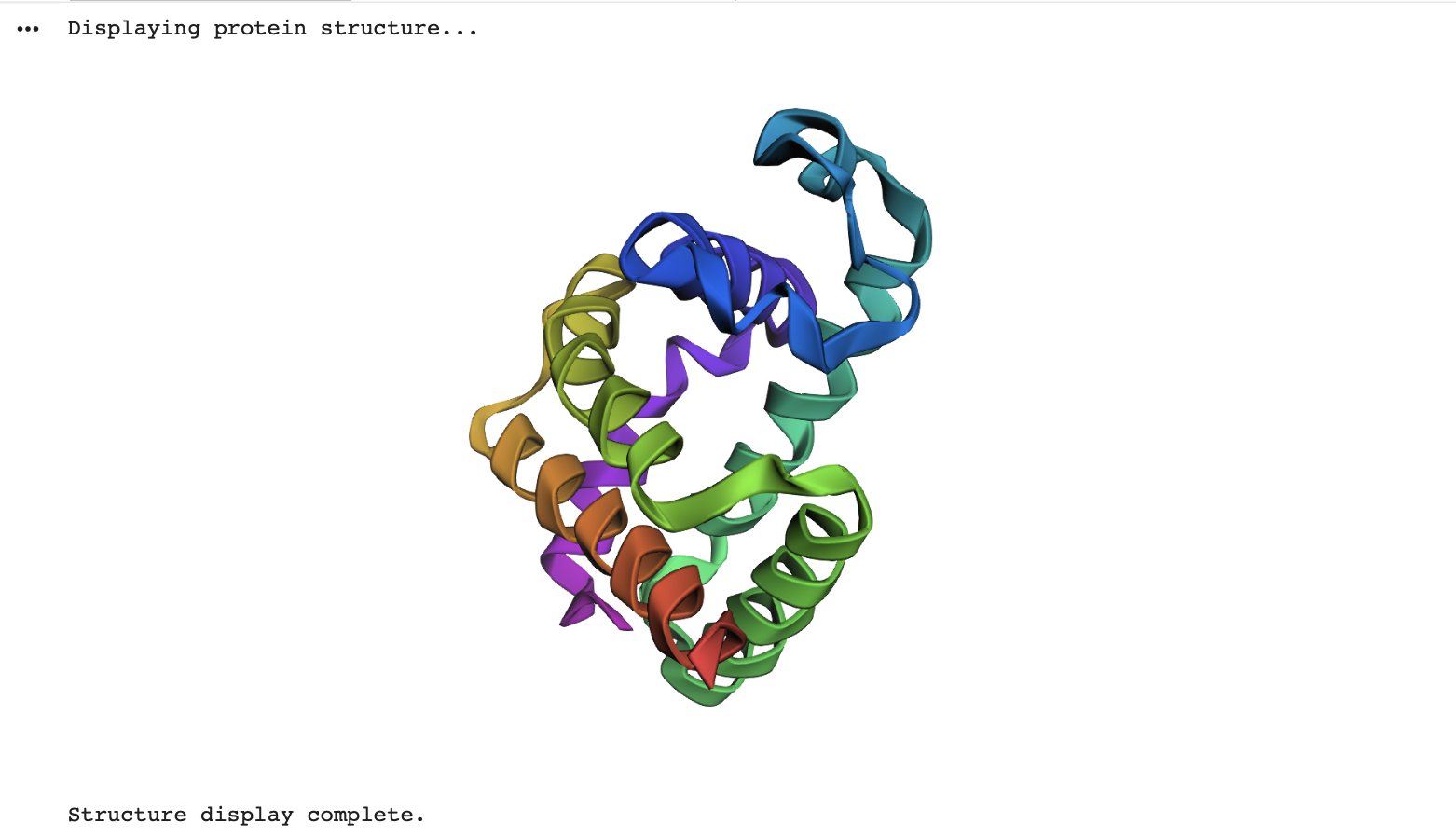
Structural Comparison to Original
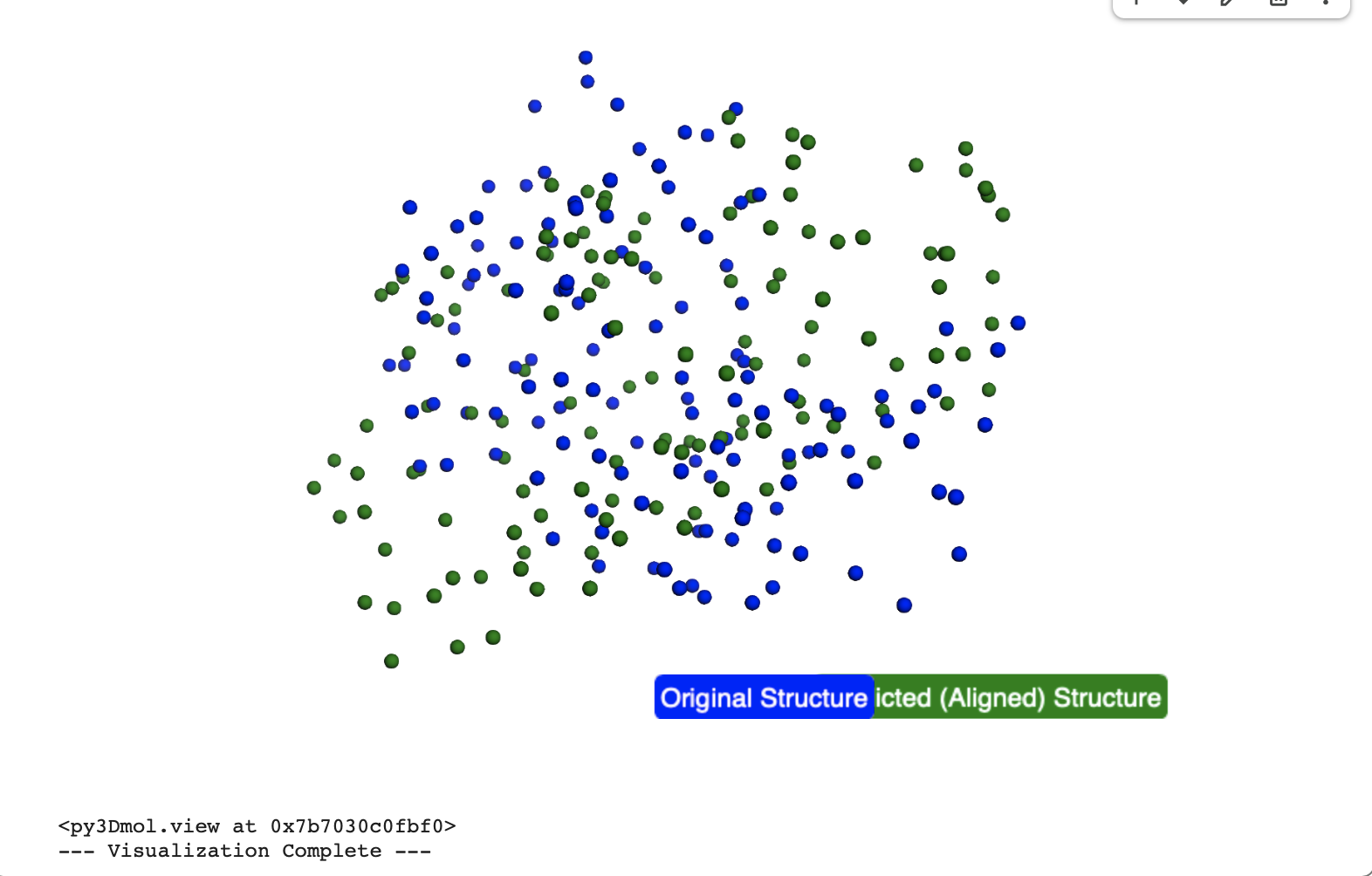
Root Mean Square Deviation (RMSD): After performing structural alignment of the C-alpha atoms between the original Lysozyme structure and the ESMFold predicted structure of the ProteinMPNN-generated sequence, an RMSD of 14.459 Å was calculated which is very high.
Visual Overlay: The 3D visualisation displayed the original Lysozyme structure (blue) superimposed with the ESMFold-predicted structure of the new sequence (green). Visually, these two structures were indeed very different.
References
Zhang T, Ma S, Zhang Z, Guo Y, Yang D, Lu W. Overview and Evolution of Insect Fibroin Heavy Chain (FibH). Int J Mol Sci. 2024 Jun 29;25(13):7179. doi: 10.3390/ijms25137179. PMID: 39000286; PMCID: PMC11241164.
PART D: Bacteriophage Engineering
Schematic:
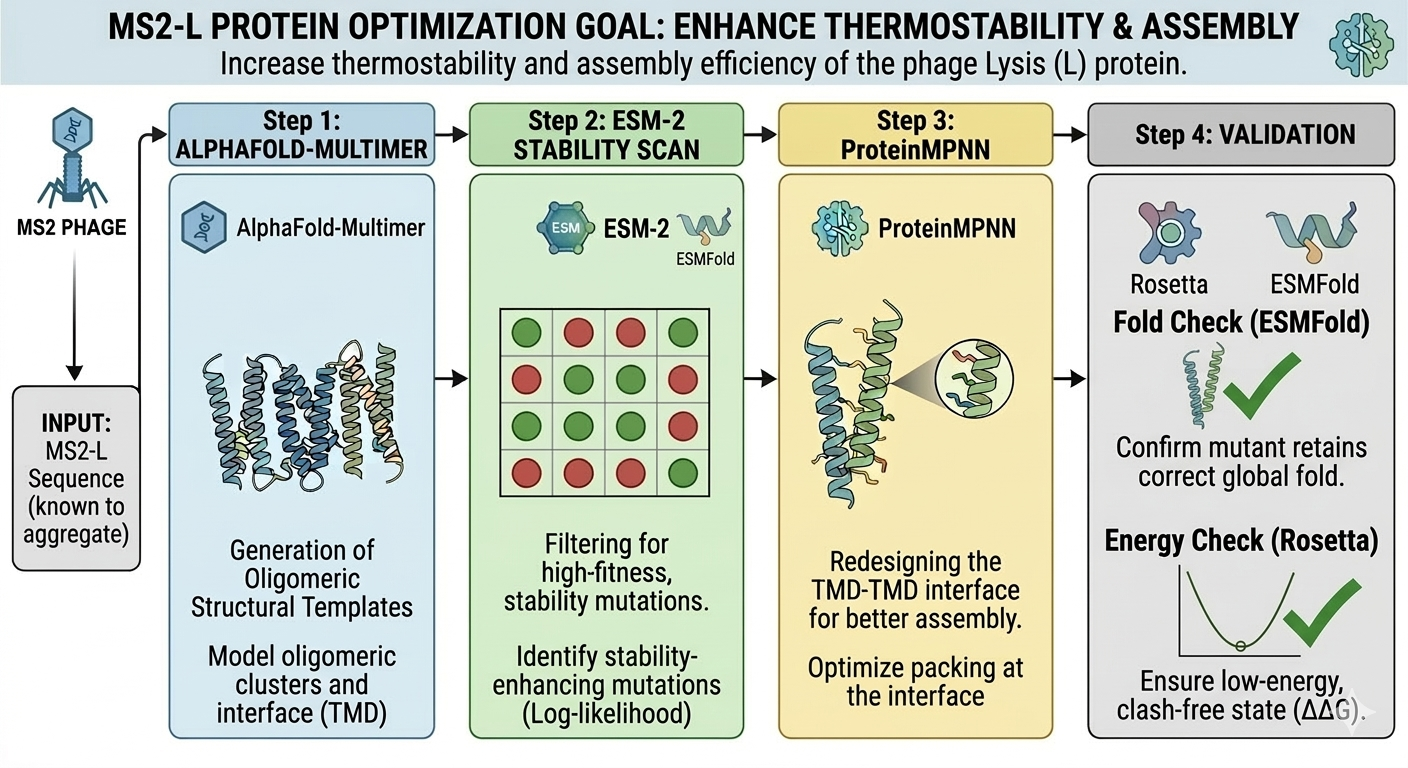
Schematic made with Gemini