Week 5 HW: Protein Design Part II
PART 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
SOD1 Sequence from Uniprot is:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutations in SOD1 causes familial Amyotrophic Lateral Sclerosis (ALS). Among these mutations, one that causes a very aggressive form of the disease is the A4V mutation (where residue 4 is changed from Alanine → Valine). Introducing A4V mutation makes the sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
From this A4V mutated sequence, I generate four peptides of length 12 amino acids using the PpPMLM Colab
MKLLLLELQLKIMKKLLLELRKILSSKILLEAQLKKSSTLLEQLLLLK
I added the known SOD1-binding peptide FLYRWLPSRRGG to the list for comparison with perplexity scores that indicate PepMLM’s confidence in the binder.
- Binder 1: MKLLLLELQLKI | Perplexity Score: 15.09267260097674
- Binder 2: MKKLLLELRKIL | Perplexity Score: 9.666623275735994
- Binder 3: SSKILLEAQLKK | Perplexity Score: 11.727098975582301
- Binder 4: SSTLLEQLLLLK | Perplexity Score: 10.224047521763415
- Control Binder: FLYRWLPSRRGG | Perplexity Score: 13.767869
2 of the generated binders have a lower perplexity (higher confidence) than the control and the highest confidence binder is no. 2 but they are all pretty moderate to low!
Part 2: Evaluate Binders with AlphaFold3
For each peptide, I submitted the mutant SOD1 sequence followed by the peptide sequence into Alphafold to model the protein-peptide complex.
Binder 1: MKLLLLELQLKI
Alpha Fold Predicted Structure:
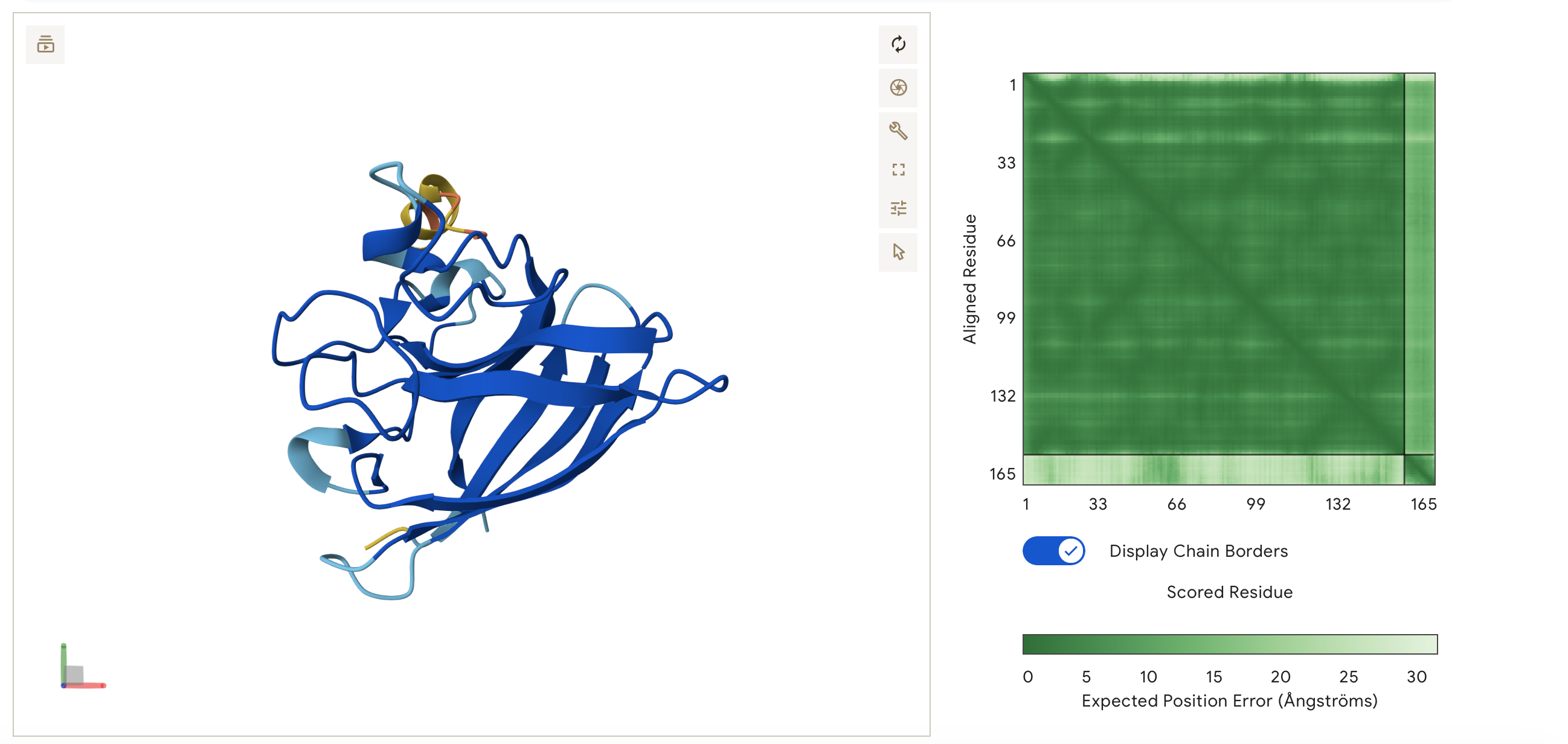
ipTM = 0.35, pTM = 0.83
Low Confidence: A ipTM score of 0.35 suggests that AlphaFold is struggling to find a stable, locking interaction between the peptide and the SOD1 protein. AlphaFold is not confident that the peptide is actually binding to it in a meaningful way as approx 0.5 score or higher would suggest a real interaction.
High pTM: However, the high pTM of 0.83 means AlphaFold is very confident that the SOD1 protein itself is folded correctly.
This is reflected in the visualisation. The peptide MKLLLLELQLKI does not appear to bind. It is predicted by Alphafold to float in the solvent space near the protein. It is not localised near the N-terminus and remains disassociated from the B-barrel and dimer interface.
Binder 2: MKKLLLELRKIL
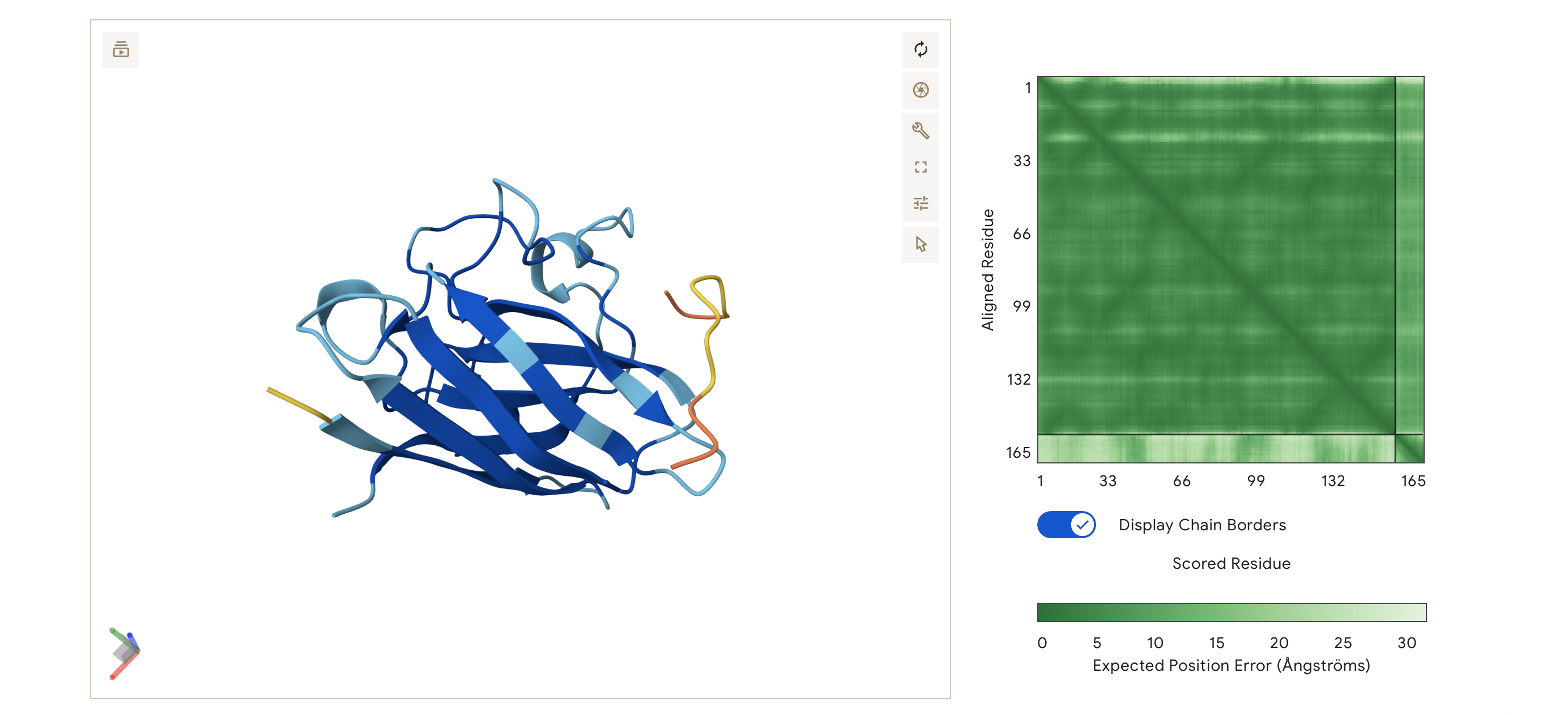
ipTM = 0.46, pTM = 0.78
Low confidence: Binder 2’s ipTM score is slightly higher at 0.46 but still a non-binder and the peptide is disassociated for the main SOD1 protein body.
It does not localise to any specific region it is distant from both terminuses and is unbound from the protein surface.
Binder 3: SSKILLEAQLKK
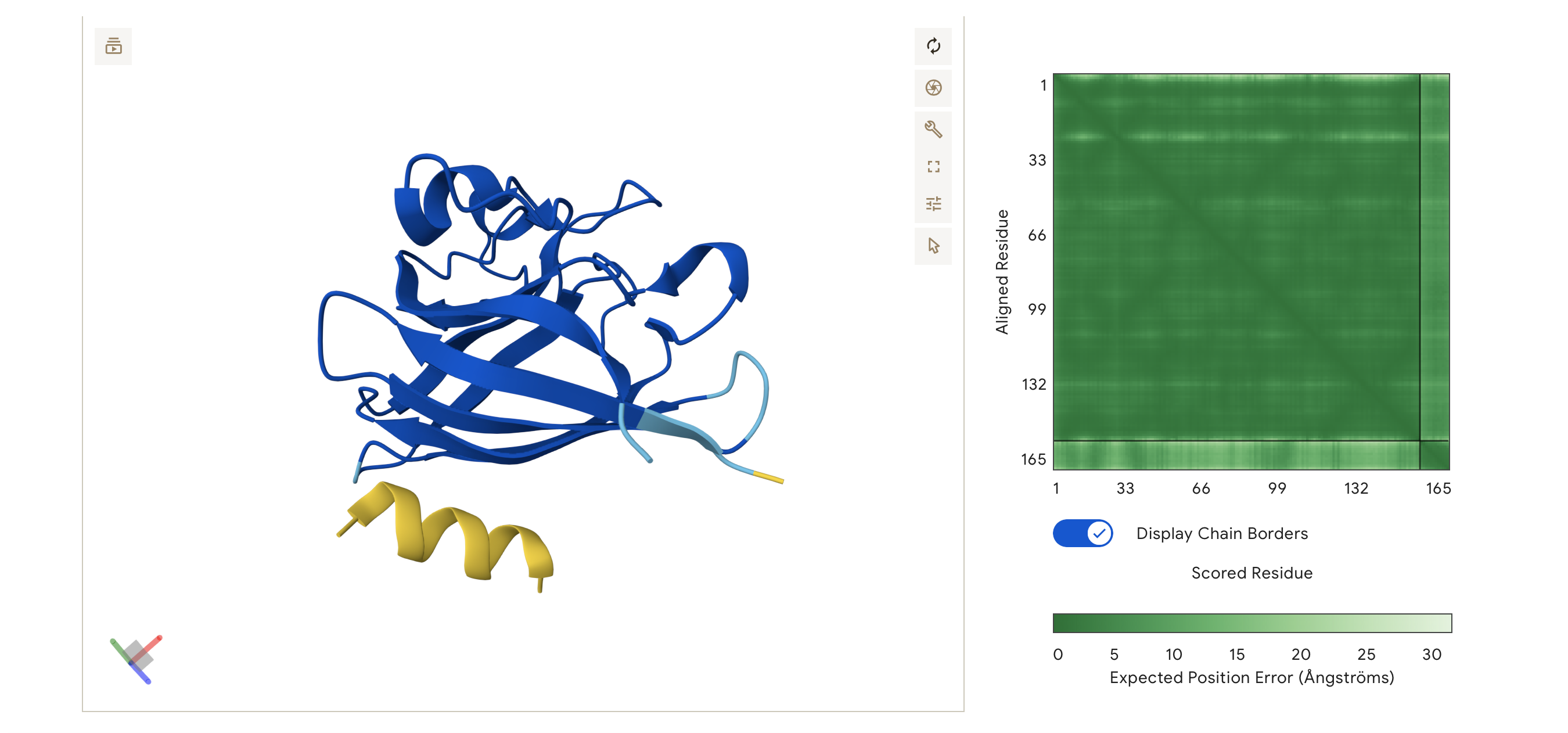
ipTM = 0.64, pTM = 0.9
Moderate Confidence Even though the 3D viewer shows the peptide and the protein structure as separate, the ipTM score is 0.64 which is significantly higher than my previous binders scores, showing that AlphaFold has found a statistically likely docking spot but the peptide might be loosely held.
The pTM is also very high, meaning the SOD1 structure is extremely stable in this simulation.
The peptide appears to localise near the N-terminus of the SOD1 protein and the A4V mutation. It hovers parallel to the first few flexible loops of the N-terminus and the initial strands of the b-barrel. The peptide is surface-bound, following the contour of the protein surface but maintaining a slight gap in the predicted structure.
Binder 4: SSTLLEQLLLLK
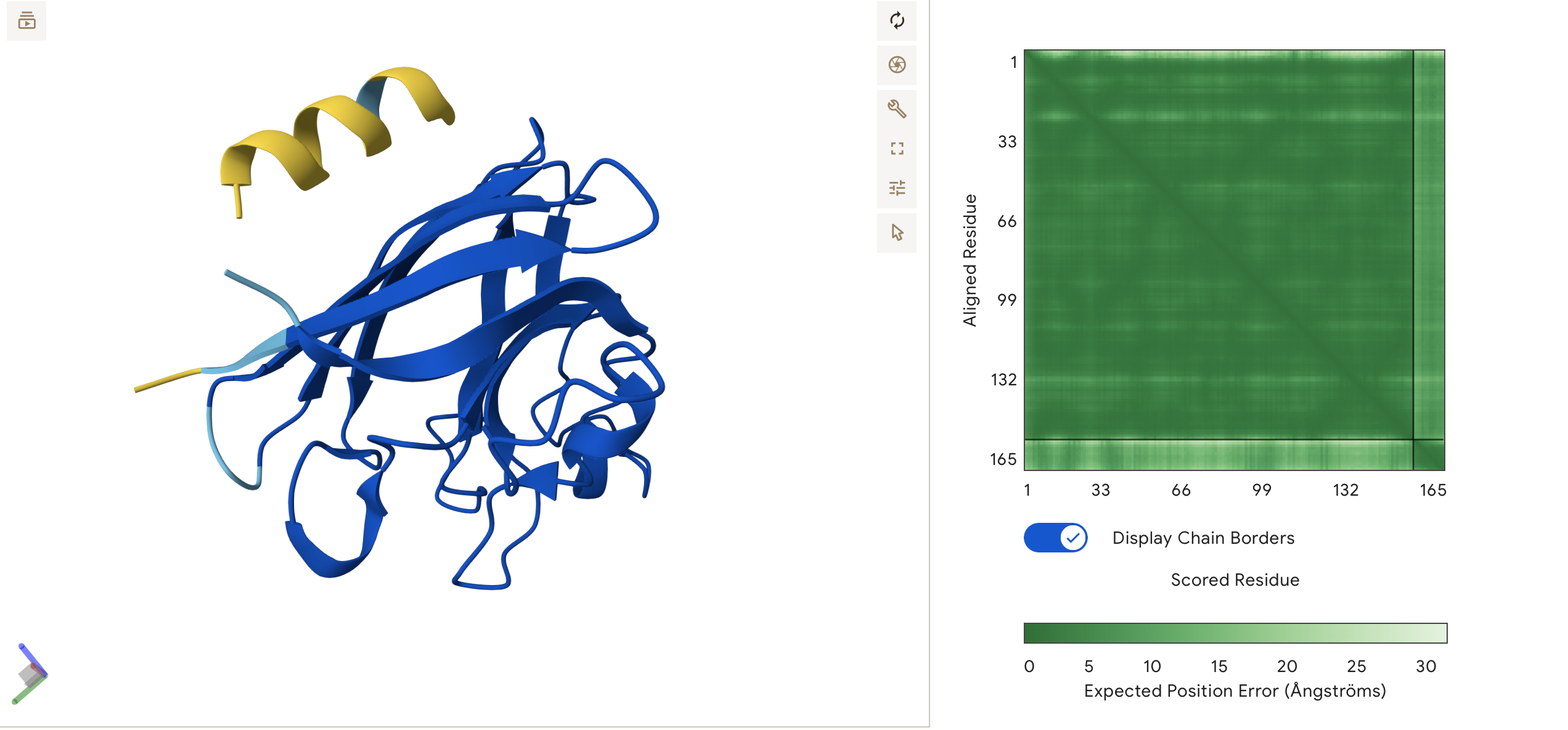
ipTM = 0.6, pTM = 0.9
Very similar results to Binder 3. Again a moderate confidence suggesting more docking potential between the binder and the protein.
This peptide localizes near to the N-terminus and A4V mutation. The peptide forms a helix that sits parallel to the first few beta-sheets of the barrel, remaining surface-bound and floating.
The consistency between Binders 3 and 4 suggests a clear preference for the N-terminal region when using these Serine/Leucine-rich sequences.
Control Binder: FLYRWLPSRRGG
ipTM = 0.35, pTM = 0.83
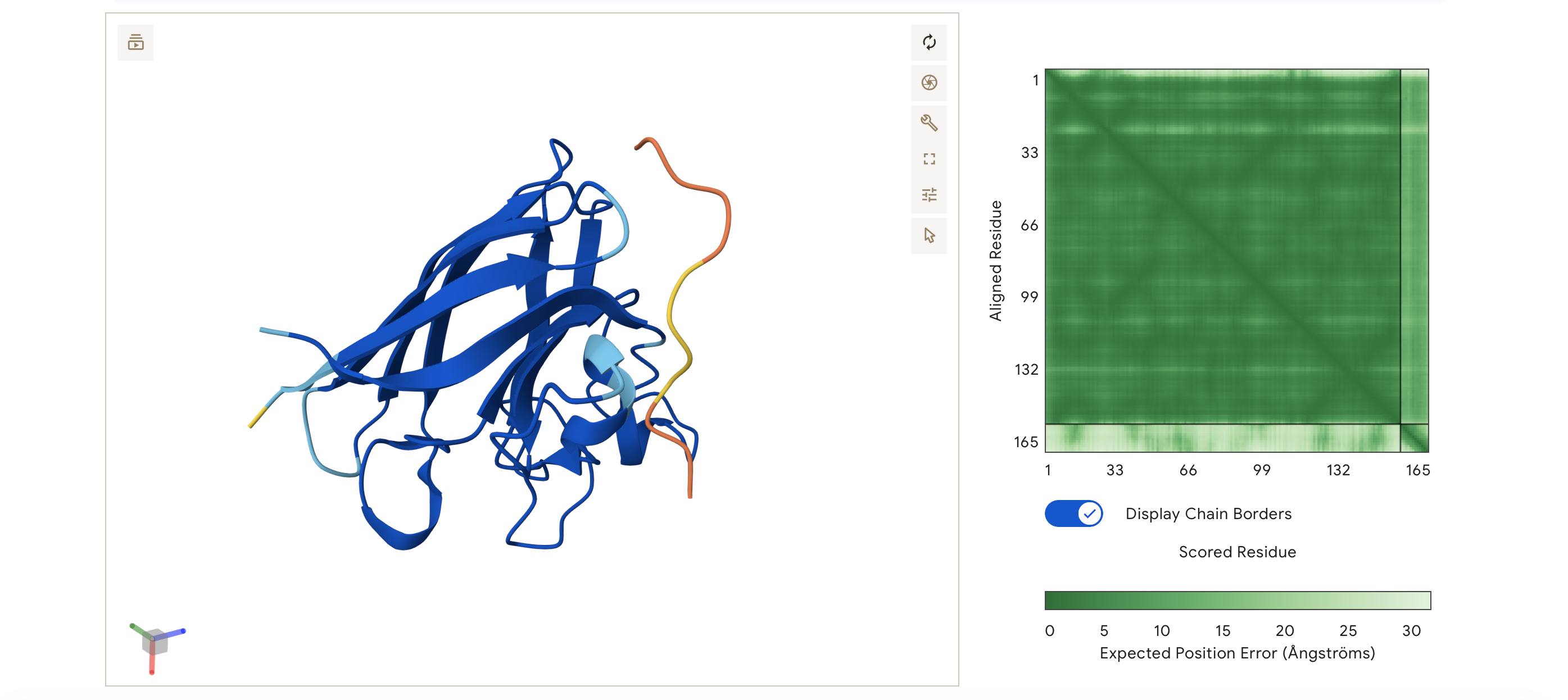
ipTM = 0.35, pTM = 0.83
The peptide does not localise near to the N-terminus. Instead, it hovers on the opposite side of the protein, away from the A4V mutation site. Like Binders 1 and 2, it appears dissociated/unbound in this specific simulation, not engaging the β-barrel.
Despite being a known binder, the control has the lowest score of the ones I have tested all my generated peptides matched or exceeded the control binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
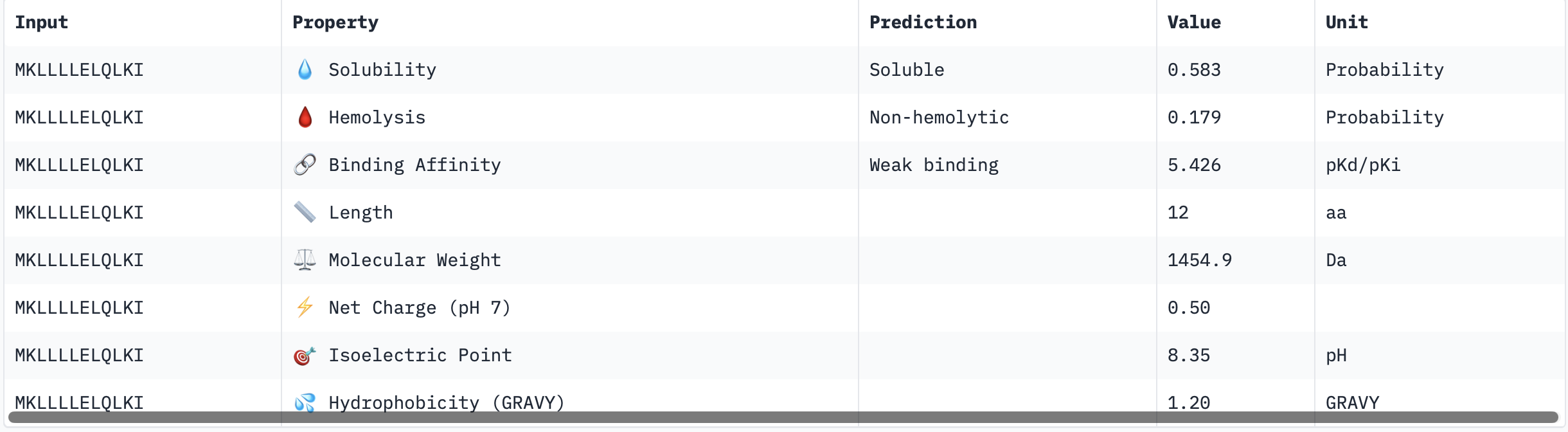
Binder 1: MKLLLLELQLKI
Binder 2: MKKLLLELRKIL
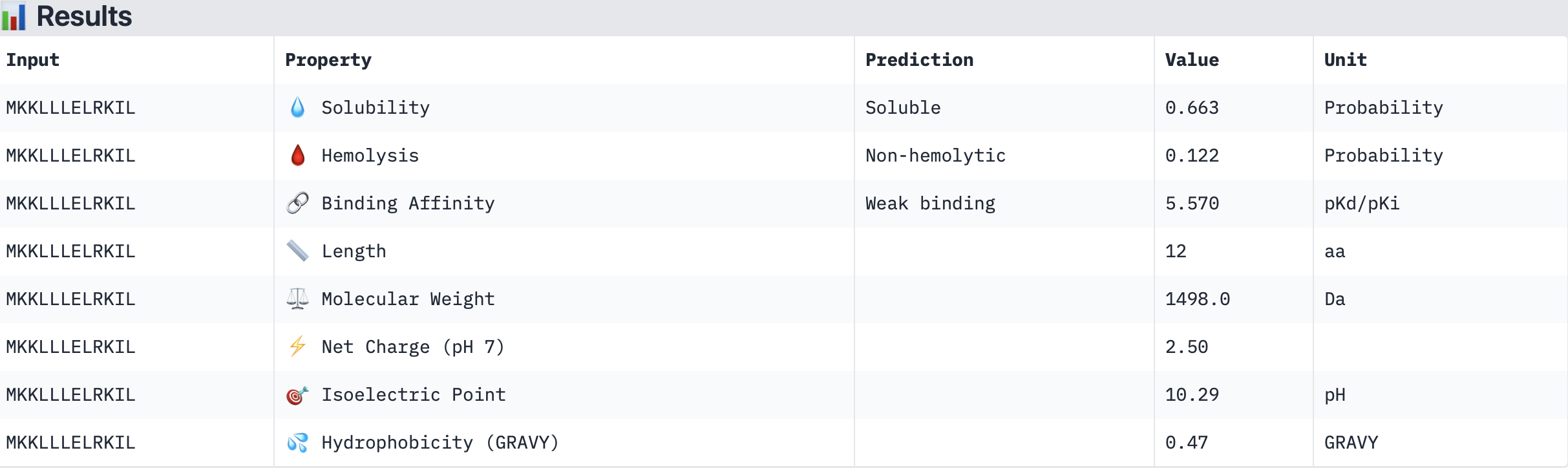
Binder 3: SSKILLEAQLKK
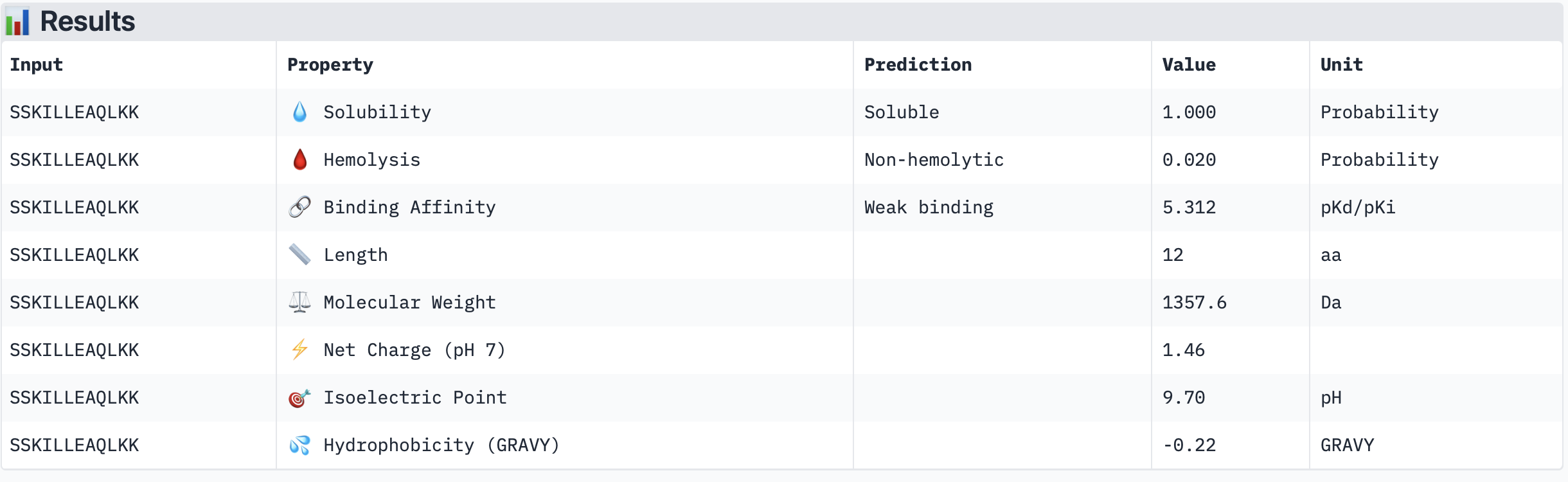
Binder 4: SSTLLEQLLLLK
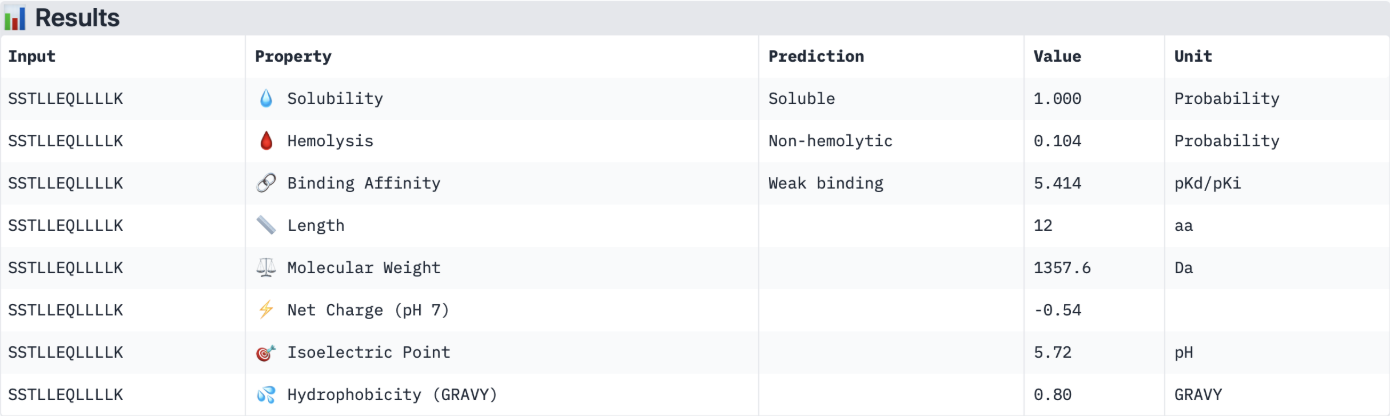
The PeptiVerse results for predicted affinity show a divergence with the structural confidence scores from AlphaFold. Binder 3 (SSKILLEAQLKK) has the highest structural confidence (ipTM = 0.64) of my generated binders and a clear localisation near to the N-terminus. However, PeptiVerse suggests it has the lowest predicted binding affinity (5.312 pKd/pKi). Conversely, Binder 2 (MKKLLLELRKIL) had the highest predicted affinity (5.570 pKd/pKi) but the lower structural confidence (ipTM = 0.46) and a unbound structure in AlphaFold.
All four peptides are predicted to be soluble and non-hemolytic, with Binders 3 and 4 reaching perfect solubility scores (1.000 probability). While Binder 2 offers the highest binding affinity, its lower ipTM suggests it may not target the A4V mutation site specifically. In contrast, Binder 3 offers a superior structural fit near the N terminus and possesses a favourable cationic charge (+1.46) and the lowest hemolysis risk (0.020), making it the most stable and safe candidate in an aqueous physiological environment.
Therefore, I would advance Binder 3 (SSKILLEAQLKK) for further development. While its predicted chemical affinity is slightly lower than Binder 2, its high structural confidence (ipTM 0.64) and specific localization to the A4V mutation site (N-terminus) make it a more promising lead for targeted ALS therapy. Furthermore, its perfect solubility and negligible hemolytic probability suggest an excellent safety profile, providing a stable scaffold that can be chemically optimized to enhance its binding strength in future iterations.
Part 4: Generate Optimized Peptides with moPPIt
While PepMLM generates binders based on the general language of the protein sequence, I used moPPIt to engineer a peptide that specifically recognises the A4V mutation site. By setting the target motif to residues 1-6 (mutation in position 4), I focused the design on the motif around the N-terminal where the toxic mis-folding begins in diseases such as ALS.
I selected 3 samples and chose the objectives:
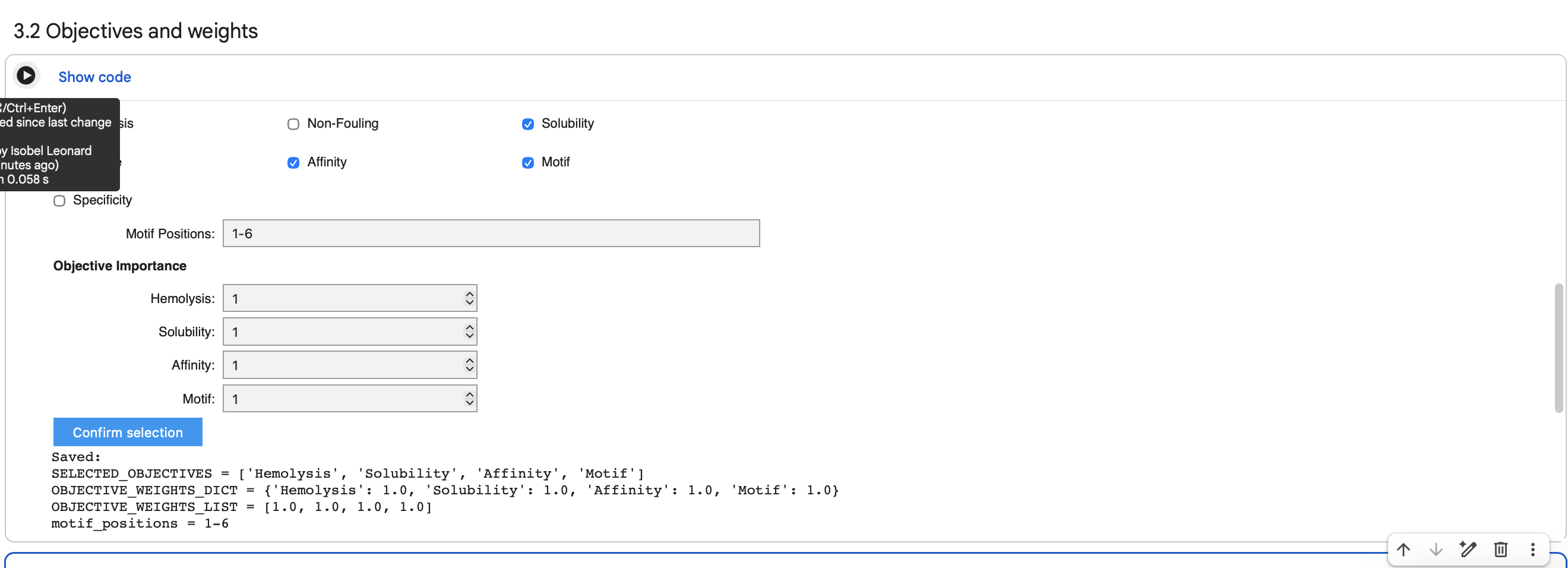
Affinity: Prioritized sequences with high predicted binding strength.
Motif: Forced the interaction to occur at residues 1-6 (the N-terminus).
Solubility: Ensured the peptide remains stable in aqueous environments.
Hemolysis: Filtered out sequences with potential toxicity to red blood cells.
Binder Generation:
| Binder | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| KWTFKFEKQKQK | 0.9835032857954502 | 0.75 | 5.448083400726318 | 0.8681516647338867 |
| KKKISVTAKNGY | 0.9790930487215519 | 0.75 | 6.005250930786133 | 0.5721415281295776 |
| LQKCIELKLTTP | 0.9543265551328659 | 0.5833333134651184 | 5.929729461669922 | 0.8600120544433594 |
Briefly describe how these moPPit peptides differ from your PepMLM peptides.
Compared with the PepMLM peptides the moPPIt peptides look quite different. The earlier PepMLM candidates were heavily enriched in Leucine (L) (e.g., MKLLLLELQLKIand MKKLLLELRKIL), which were likely chosen to mimic common natural hydrophobic cores. In contrast, moPPIt binder 1 (KWTFKFEKQKQK) and moPPIt binder 2 (KKKISVTAKNGY) are heavily enriched with Lysine and Arginine. This shifts towards a high positive charge density, possibly as a result of choosing to target optimise for solubility and hemolysis.
This shift makes sense because moPPIt was run with an explicit multi objective targeting objective (Affinity, Solubility, Hemolysis, and Motif) rather than using sequence conditioned generation based on behaviour most like general surface binders, where as moPPIt was used here to focus peptide design toward the N-terminal A4V mutation region of SOD1.
How would you evaluate these peptides before advancing them to clinical studies?
Predict their structures with AlphaFold-Multimer to verify the high motif scores (0.86). Check if moPPIt binders achieve an ipTM score higher than 0.64 (my previous best from PepMLM) and translate to a physical dock at the residues 1–6 N-terminal site.
Check the new binders with PeptiVerse, check the Net Charge, Molecular weight and Isoelectric Point (pI) of the new binders as well as re-validating the solubility, affinity and hemolysis scores from moPPit.
If the computational results hold, use Surface Plasmon Resonance (SPR) to measure the actual Equilibrium Dissociation Constant. Compare the binding affinity of the new binders to wild-type SOD1 vs the A4V Mutant.
Perform a standardised red blood cell assay to confirm the moPPIt prediction of non-hemolytic. Additionally, perform a Serum Stability Assay by incubating the peptides in human serum to determine their half-life and susceptibility to protease degradation.
Test the peptides in SOD1 aggregation assays (cell-free or cell-based) to see if binding actually prevents the formation of toxic protein aggregates.
The most successful candidates would advance to an ALS mouse model.
PART C: Final Project: L-Protein Mutants
The objective is to improve the stability and autofolding of the lysis protein.
Option 1: Mutagenesis
Lysis Protein Sequence (UniProtKB ID: https://www.uniprot.org/uniprotkb/P03609/entry) (75 residues)
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (last 35 residues).
Transmembrane protein affects the lysis activity.
The soluble domain is the domain responsible for interaction with DnaJ.
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYV(Soluble: 1–40)LIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT(Transmembrane: 41–75)
| Domain | Residues | Sequence Length | Goal |
|---|---|---|---|
| Soluble Domain | 1 – 40 | 40 residues | Overcome DnaJ dependency (Folding) |
| Transmembrane | 41 – 75 | 35 residues | Increase Lysis Speed (Pore formation) |
DnaJ sequence (UniProtKB ID: https://www.uniprot.org/uniprotkb/P03609/entry)
MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Generating mutated sequences:
First I input the L-protein sequence into the ESM collab and ran a zero-shot mutational scan. This will result in a log-likelihood ratio (LLR) score for substituting each amino acid at each position in the L-protein sequence.
The LLR score can either be positive or negative for the protein.
Positive scores (>0): The protein language model has confidence in the mutation vs the existing amino acid. It suggests the change is likely stabilising or fits well within the protein
Negative score (<0): The protein language model has low confidence in the mutation. It suggests the change is likely destabilising or could break the protein’s structure.
This is the heat map generated. The colour relates to the predicted LLR score meaning the results range from brighter yellow as most favourable to the darker purple as least favourable.
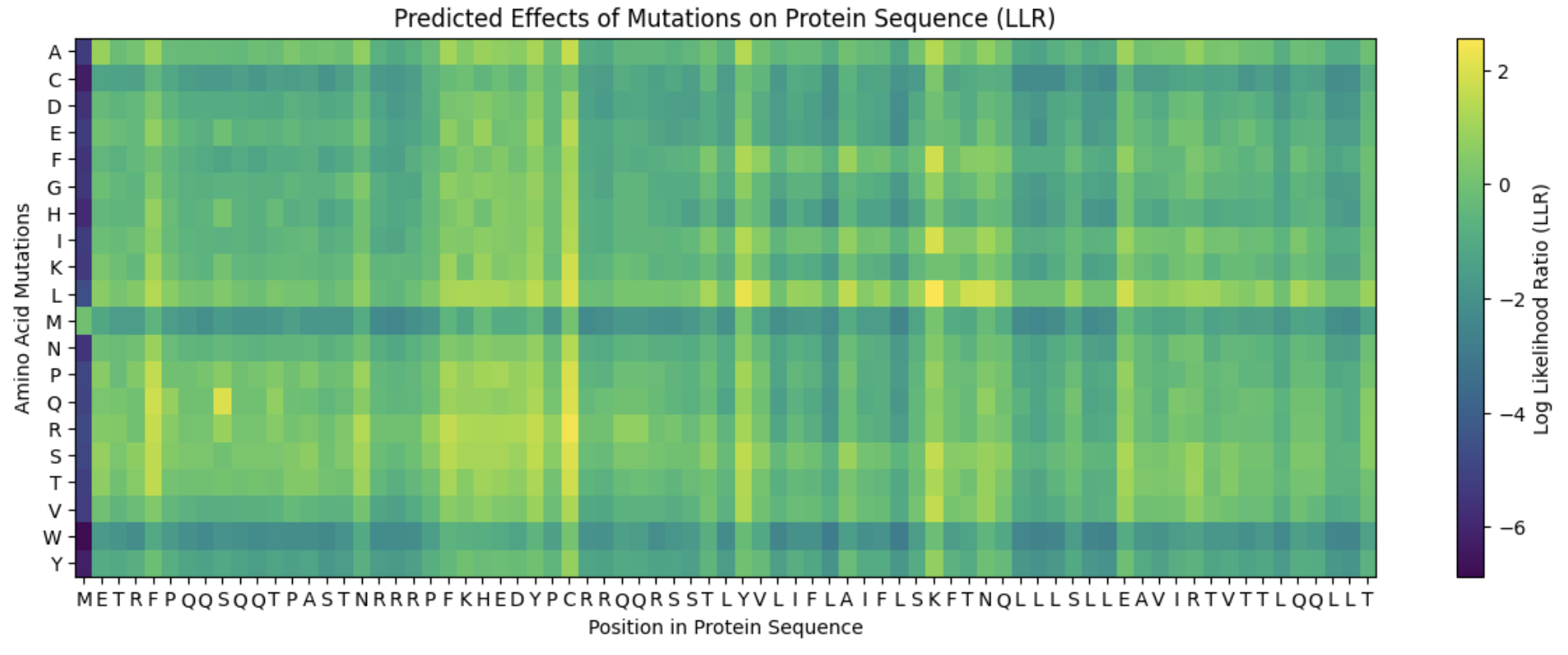
Top 20 L-protein Mutation Scores
| Position | Wild Type AA | Mutation AA | LLR Score | Domain |
|---|---|---|---|---|
| 50 | K | L | 2.561 | TM |
| 29 | C | R | 2.395 | Soluble |
| 39 | Y | L | 2.242 | Soluble |
| 29 | C | S | 2.043 | Soluble |
| 9 | S | Q | 2.014 | Soluble |
| 29 | C | Q | 1.997 | Soluble |
| 29 | C | P | 1.971 | Soluble |
| 29 | C | L | 1.961 | Soluble |
| 50 | K | I | 1.929 | TM |
| 53 | N | L | 1.865 | TM |
| 61 | E | L | 1.818 | TM |
| 52 | T | L | 1.814 | TM |
| 50 | K | F | 1.802 | TM |
| 29 | C | T | 1.797 | Soluble |
| 29 | C | K | 1.796 | Soluble |
| 5 | F | Q | 1.795 | Soluble |
| 5 | F | R | 1.660 | Soluble |
| 29 | C | A | 1.649 | Soluble |
| 27 | Y | R | 1.628 | Soluble |
| 22 | F | R | 1.602 | Soluble |
| 5 | F | P | 1.597 | Soluble |
| 50 | K | V | 1.595 | TM |
| 50 | K | S | 1.575 | TM |
| 5 | F | T | 1.559 | Soluble |
| 5 | F | S | 1.556 | Soluble |
| 45 | A | L | 1.539 | TM |
| 39 | Y | S | 1.517 | Soluble |
| 27 | Y | S | 1.497 | Soluble |
| 40 | V | L | 1.478 | Soluble (End) |
| 27 | Y | L | 1.475 | Soluble |
| 22 | F | S | 1.423 | Soluble |
| 29 | C | E | 1.383 | Soluble |
| 39 | Y | A | 1.365 | Soluble |
| 29 | C | N | 1.363 | Soluble |
| 50 | K | A | 1.358 | TM |
| 29 | C | I | 1.344 | Soluble |
| 5 | F | L | 1.333 | Soluble |
| 17 | N | R | 1.324 | Soluble |
| 39 | Y | I | 1.320 | Soluble |
| 39 | Y | T | 1.303 | Soluble |
| 26 | D | R | 1.269 | Soluble |
| 29 | C | H | 1.246 | Soluble |
| 39 | Y | F | 1.246 | Soluble |
| 39 | Y | V | 1.244 | Soluble |
| 23 | K | R | 1.237 | Soluble |
| 25 | E | R | 1.229 | Soluble |
| 24 | H | R | 1.228 | Soluble |
| 50 | K | T | 1.222 | TM |
| 27 | Y | Q | 1.219 | Soluble |
| 27 | Y | T | 1.216 | Soluble |
The highest LLR of 2.561 was scored at position 50 where the wild type K (Lysine) is changed to L (Leucine).
Positions with the many high scoring mutations were positions 50, 29 and 39suggesting they are “hotspots” for positive redesign as the Wild-Type AA is predicted as quite unoptimised for that specific spot.
Experimental Dataset
Next, I checked if the ESM scores (theoretical fitness) correlated with this L-Protein Mutants Experimental Dataset which included results of different mutations and their effects on lysis.
Using Gemini I checked whether the data correlated by comparing the LLR score from ESM against the functional outcomes (Lysis and Protein expression) in the experimental dataset.
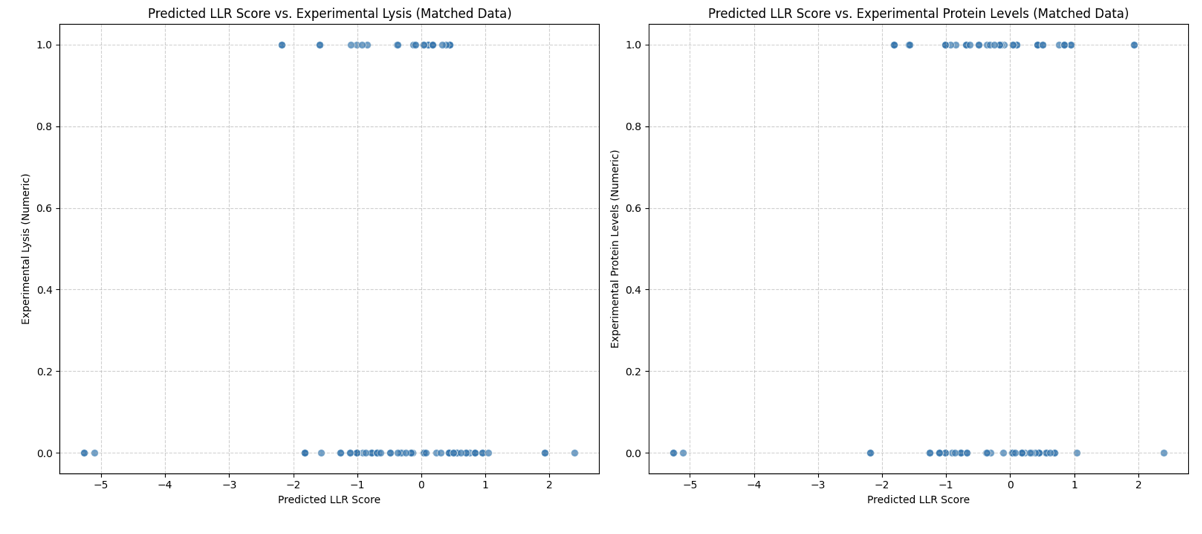
Gemini found weak statistical correlation. The Pearson correlation coefficients was 0.0922 for the Lysis Activity with the LLR and 0.0602 for Protein Levels with the LLR.
I was concerned this outcome was due to the binary vales of the experimental dataset vs the range of scores in the LLR. So I binarised the LLR scores into positive and negative but even then the correlations remained weak.
Significantly, the highest performing predicted mutations had N.D results or no experimental result which meant they lacked direct experimental validation in the provided dataset making the comparison limited.
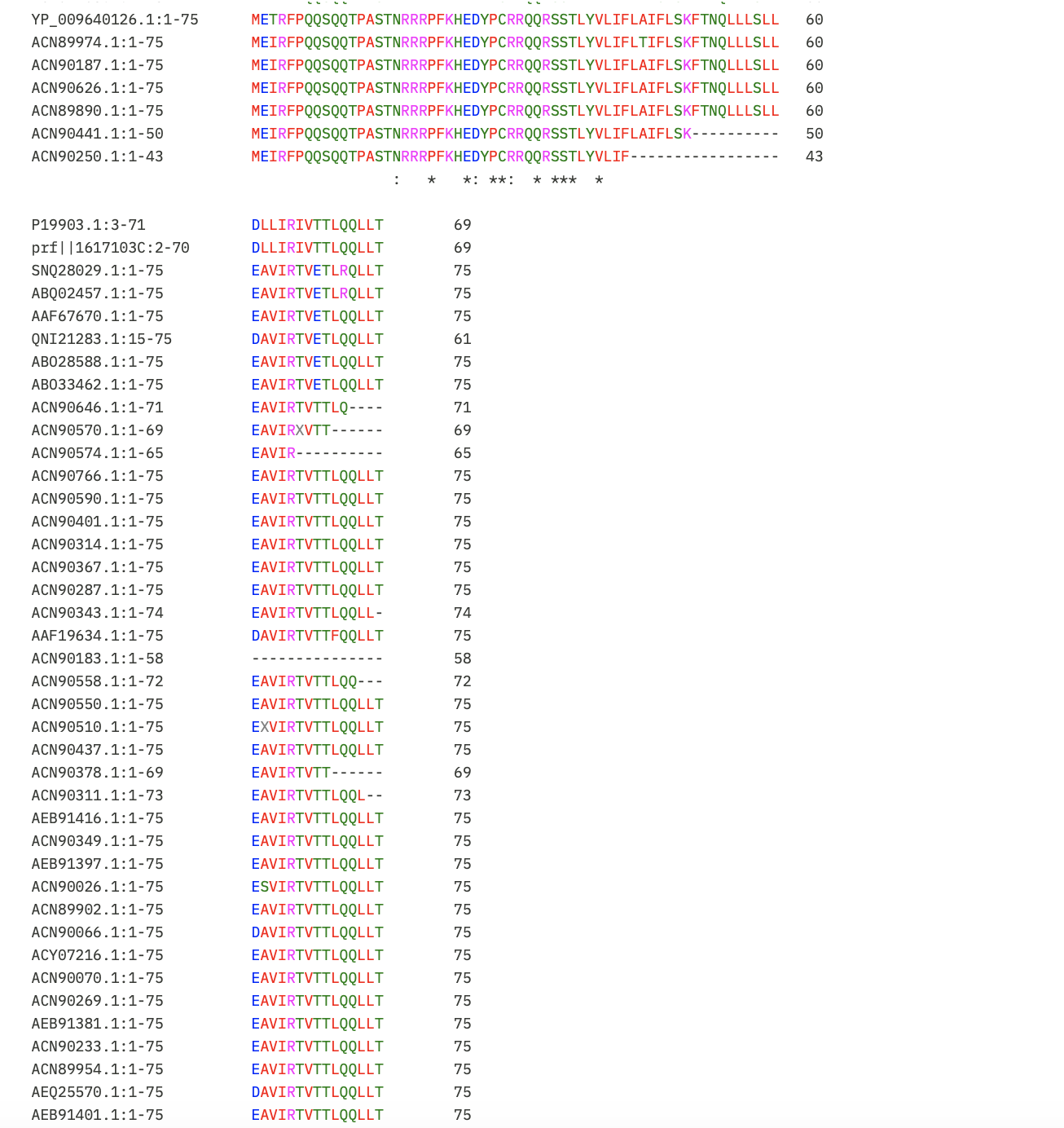
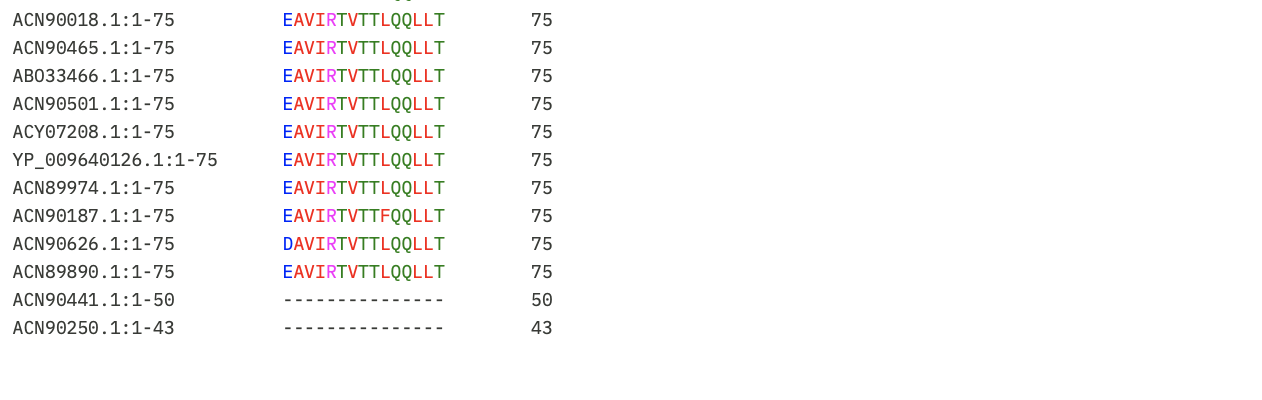
pBlast and Clustal Omega: Multiple Sequence Alignment
Next, I put the pBLAST results for Lysis Protein into Clustal Omega to identify the conserved regions so the mutations recommended don’t impact protein function.
I learnt using Gemini to interpret the results that:
- Asterisk (*): Perfectly conserved across all 52 sequences. Do not mutate these.
- Colon (:): Strong conservation of chemical properties.
- Period (.): Weak conservation.
- Blank space: Highly variable. These are the safest regions to mutate.
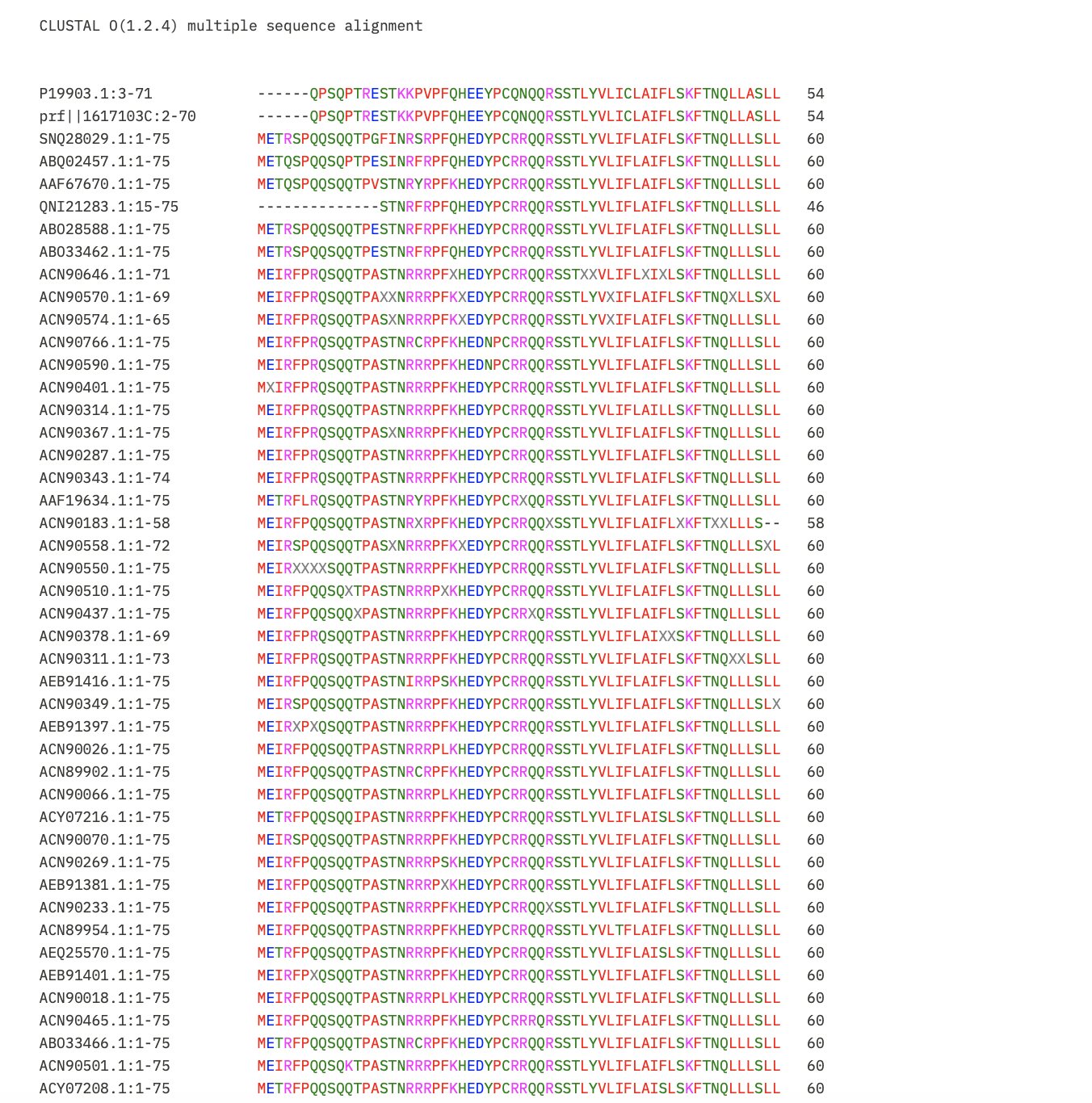
Selected Mutations
I have selected five mutations by prioritising those with high positive LLR scores while cross-referencing experimental data. I avoided mutations in conserved regions by analyzing the multiple sequence alignment generated by Clustal Omega using pBLAST results.
| Mutation | Region | LLR Score | Rational |
|---|---|---|---|
| K50L | Transmembrane | 2.56 | This is the highest LLR score in the entire dataset, indicating high confidence in the mutation. The Clustal Omega alignment shows this position is variable, meaning it is a safe candidate for functional enhancement. |
| N53L | Transmembrane | 1.86 | A top LLR score. The alignment shows that while the surrounding motif is conserved, position 53 itself is not strictly conserved suggesting it is a safe site for substitution. |
| S9Q | Soluble | 2.01 | This is the highest-scoring mutation in the N-terminal soluble region (1–17). The clustal omega alignment confirms this site is in a highly variable spot in the N-terminal tail. |
| F5Q | Soluble | 1.79 | Position 5 is located at the N-terminus, which the Clustal alignment identifies as the most variable part of the protein. By choosing a high-scoring mutation in this flexible region it can minimise the risk of negatively impacting protein stability. |
| Y39L | Transmembrane | 2.24 | This mutation sits at the boundary of the transmembrane region. It was selected because it is the highest-scoring remaining variant that is not at a conserved site. Using a Leucine substitution here aligns with the hydrophobic nature of the membrane entry domain. |
Check in AlphaFold-Multimer
I will proceed to the last step with mutation K50L. ESM says the mutation is high confidence (LLR 2.56) and pBLAST/Clustal Omega says the position is flexible and safe to change. Finally, I will check with AlphaFold-Multimer to see if the mutation allows the protein to assemble into a stable, 8-unit pore that can perforate a membrane.
I chose to try a homooctamer (8 chains), this is because it is suggested that the protein functions by assembling to make a perforation in the bacterial membrane.
Query Sequence for K50L:
METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT:METRSPQQSQQTPGFINRSRPFQHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLDAVIRTVETLRQLLT
Results:
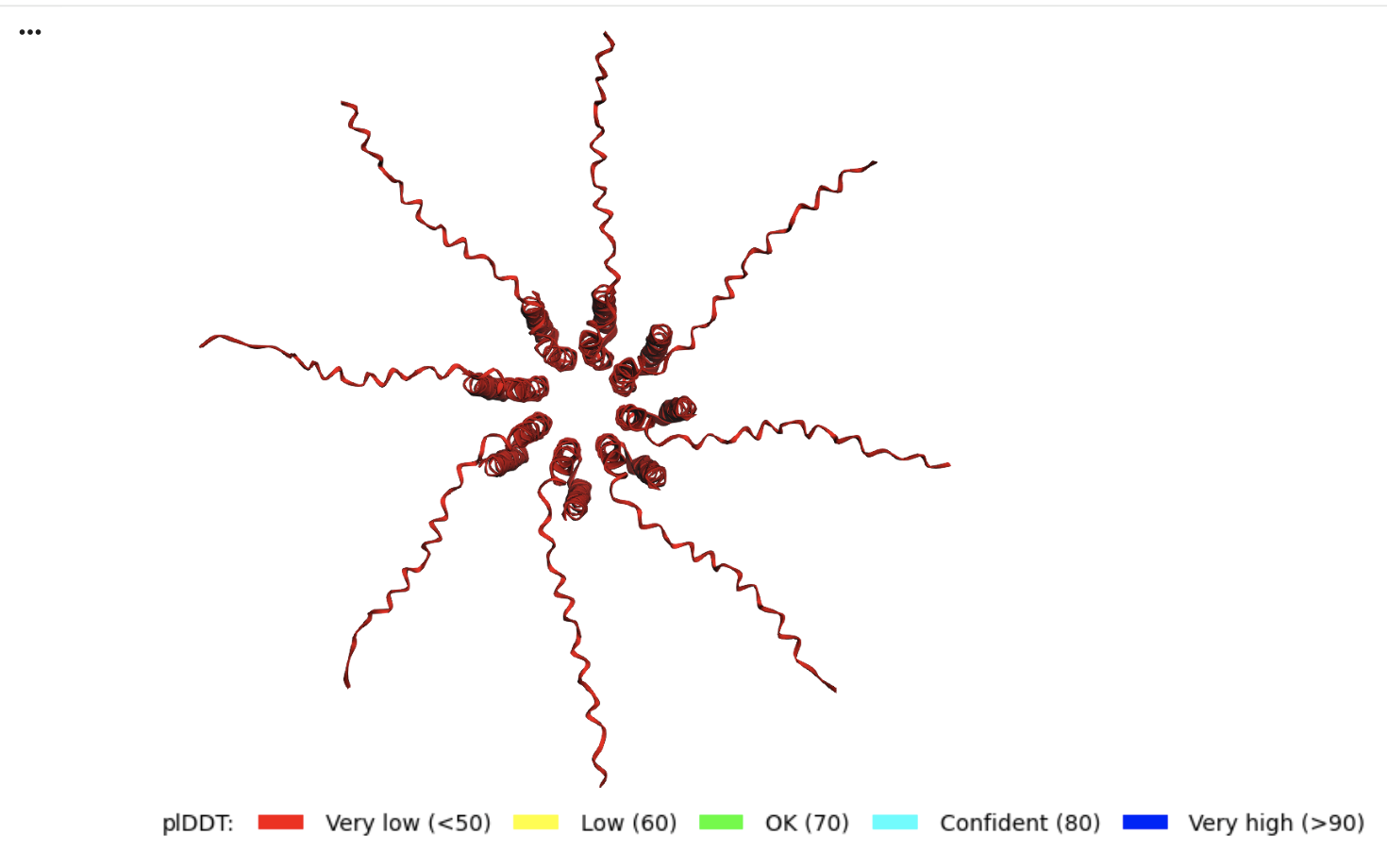
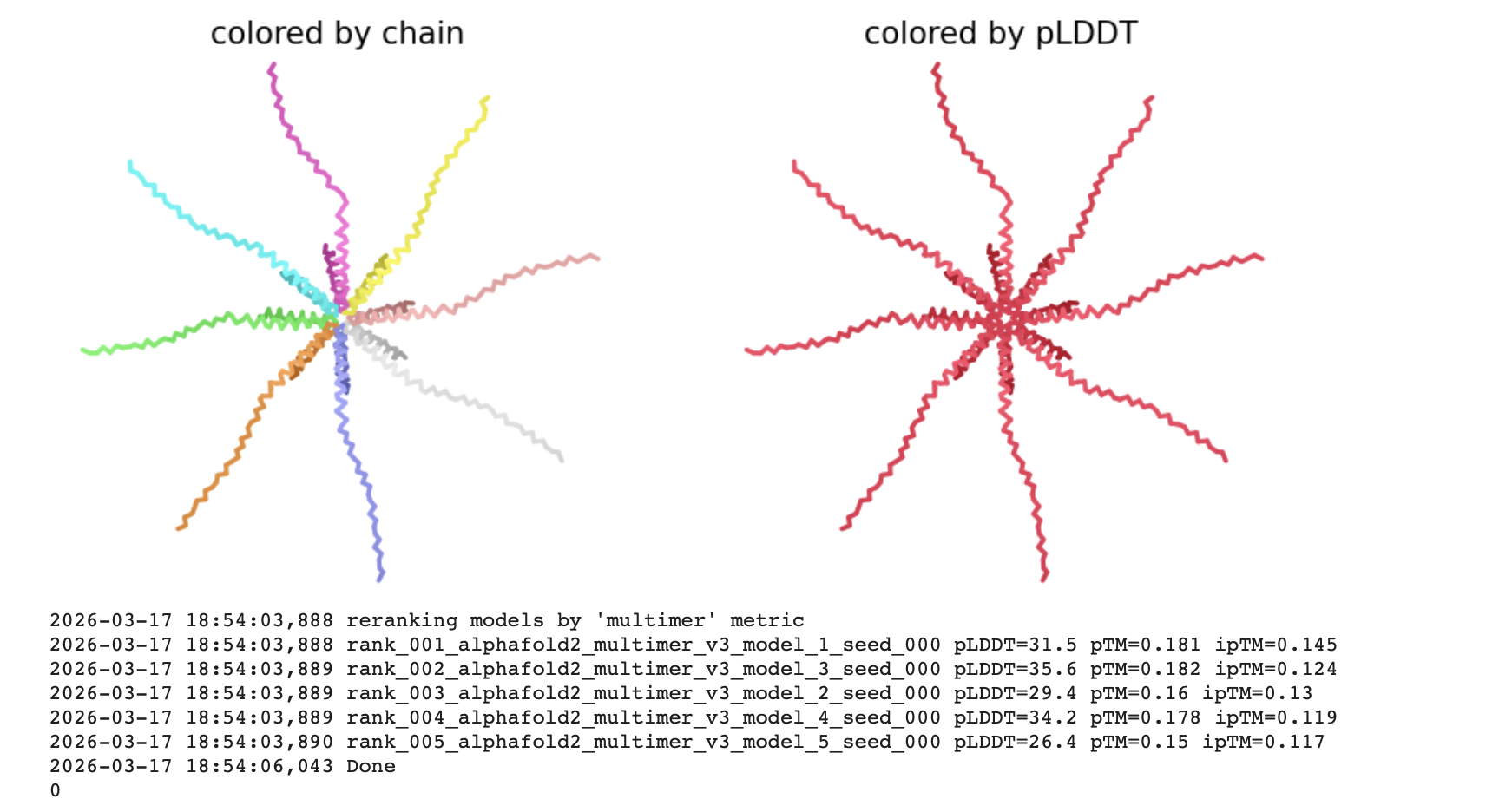


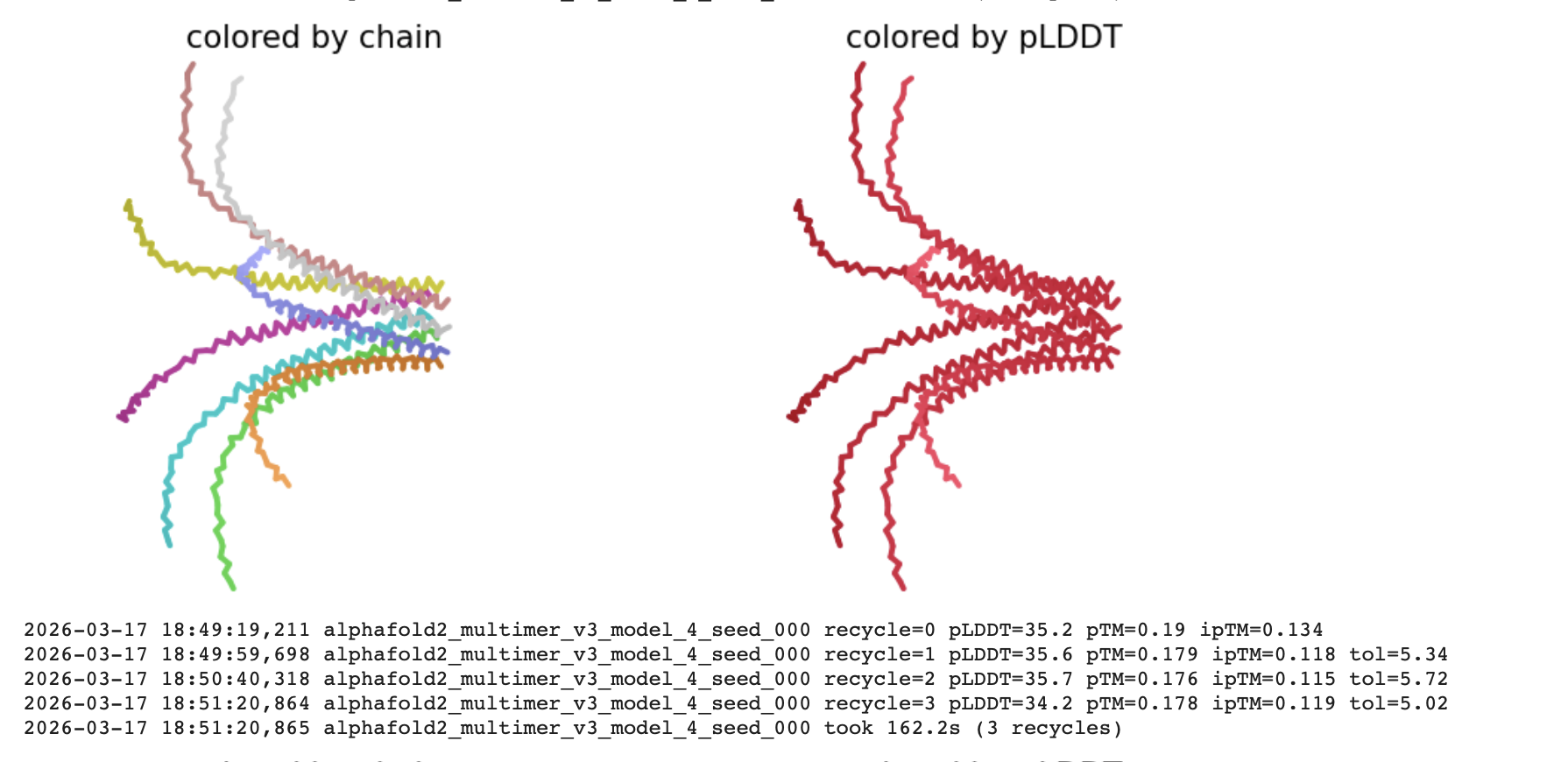
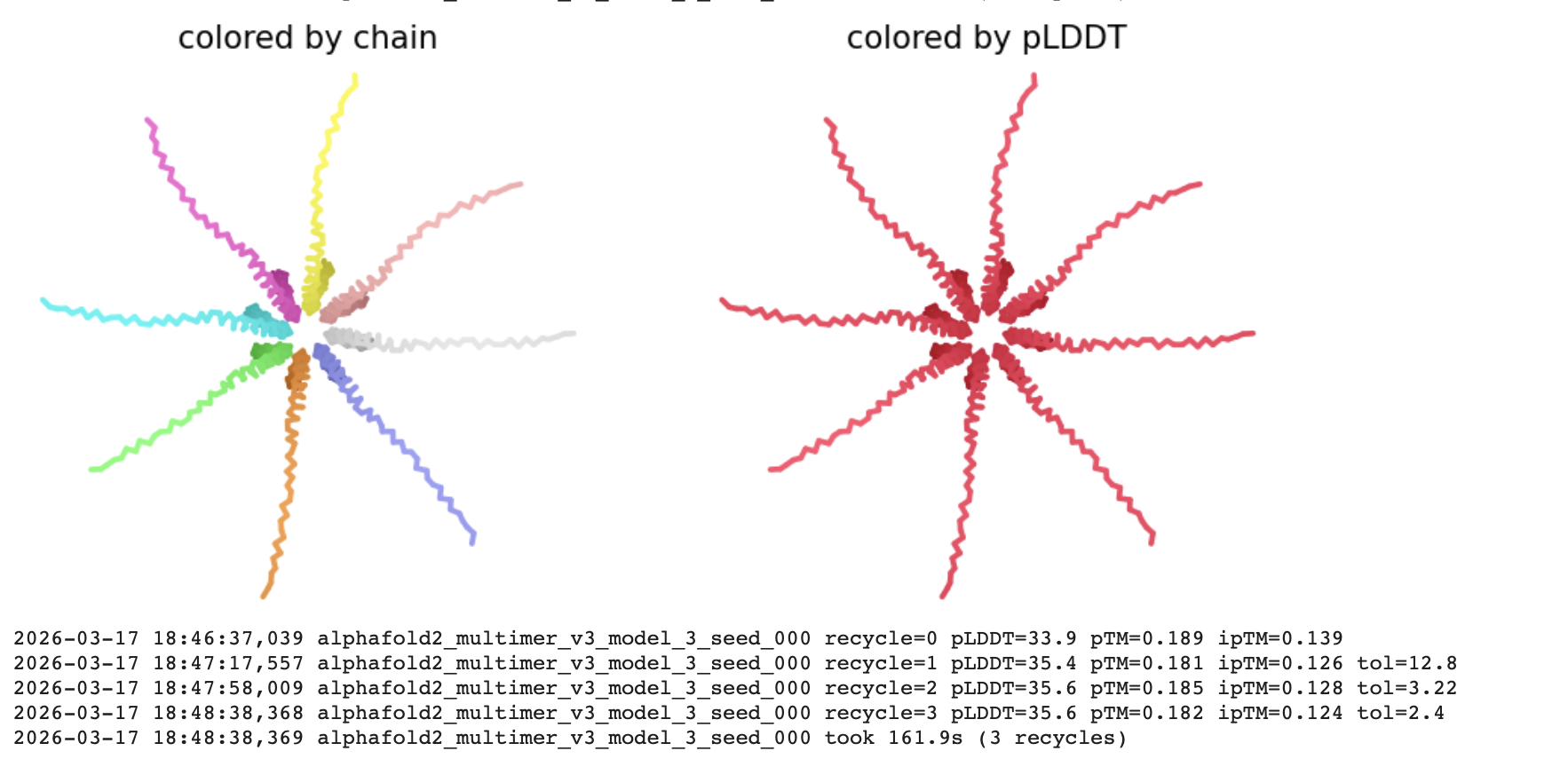
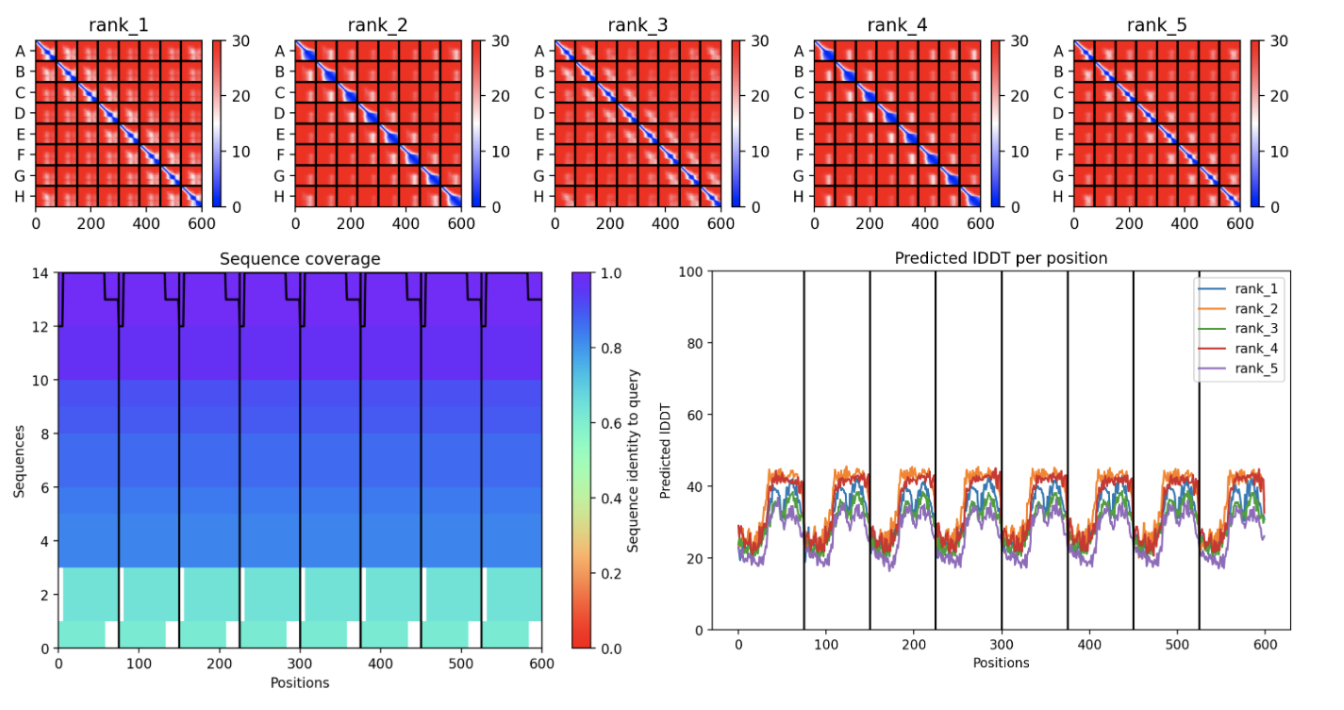
Interpretation
The pLDDT (Predicted Local Distance Difference Test)
The all red structure suggests the pLDDT is very low (below 50). This is a strong indicator that the Alphafold has very low confidence in the predicted 3D structure.
Predicted Aligned Error (PAE) Plot:
The ‘blue diagonal’ across rank 1-5 suggests that the model is generally confident about the relative positions of residues within each individual chain. However, large proportion of red implies low confidence in the relative positioning of residues between different chains. This indicates that while the internal structure of each monomer might be reasonably predicted, the way these monomers assemble into the octamer might be highly uncertain.
The MSA Sequence Coverage Plot:
The predominantly purple and dark blue’ areas signify a strong and diverse Multiple Sequence Alignment. This means the low prediction confidence (seen in the PAE and pLDDT) is likely not due to insufficient evolutionary information but inherent challenges in the modelling such a large and complex octamer.