Week 6 HW: Genetic Circuits Part I

PART A: DNA Assembly
The Phusion Flash High-Fidelity PCR Master Mix is a ready made reagent kit for fast and accurate PCR, used to select specific DNA sequences and amplify them for cloning, genotyping, sequencing, and pathogen detection.
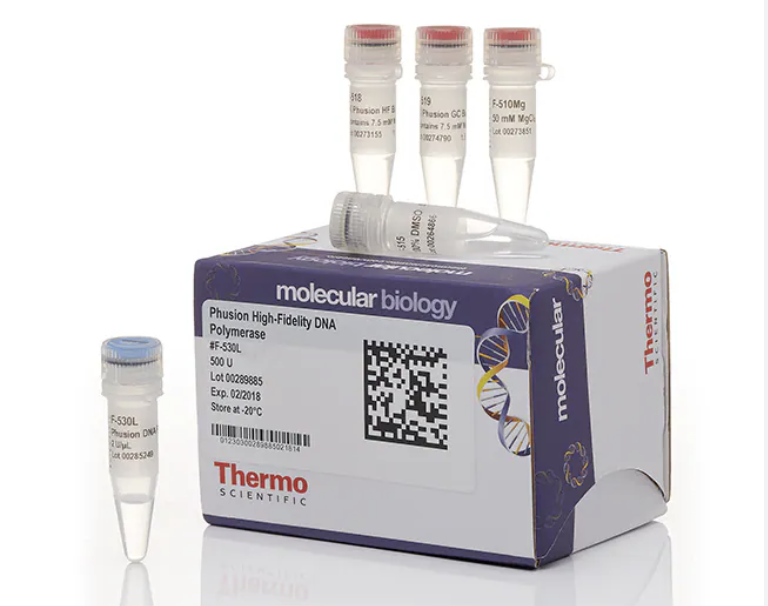
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA Polymerase:
This is the enzyme that synthesises the new complementary strands of DNA during the extension step of a PCR by proofreading the nucleotide sequence and adding complementary nucleotides in a 5’ to 3’ direction, starting from the 3’ end of a primer to form a complementary strand to those split during denaturation. The Phusion technology enhances the enzymes ability to bind to the DNA strand and also has 3’to 5’ exonuclease activity so that if the wrong base is added it can be removed and replaced with the correct base.
dNTP (deoxynucleoside triphosphates):
These are the nucleotides, the building blocks of the DNA that the DNA Polymerase used to build the new complimentary strand of DNA from.
HF reaction buffer (high fidelity):
This provides the ideal chemical environment (salts, pH) for the Phusion enzyme. This buffer specifically is optimised to prioritise the accuracy of the polymerase to ensure the lowest possible error rate.
MgCl2:
Is a catalytic co-factor that binds to the active site of the polymerase and facilitate the biochemical reaction.
100% DMSO (Dimethyl Sulphoxide):
DMSO is a chemical additive that can be introduced for difficult PCRs. It acts as a denaturant that lowers the melting temperature of the DNA if needed for a PCR with DNA templates that are GC rich or have complex secondary structures.
What are some factors that determine primer annealing temperature during PCR?
The annealing temperature for the Phusion mix is slightly higher than ordinary PCR polymerase so it is recommended to us the Tm calculator for Thermo fisher DNA polymerase.
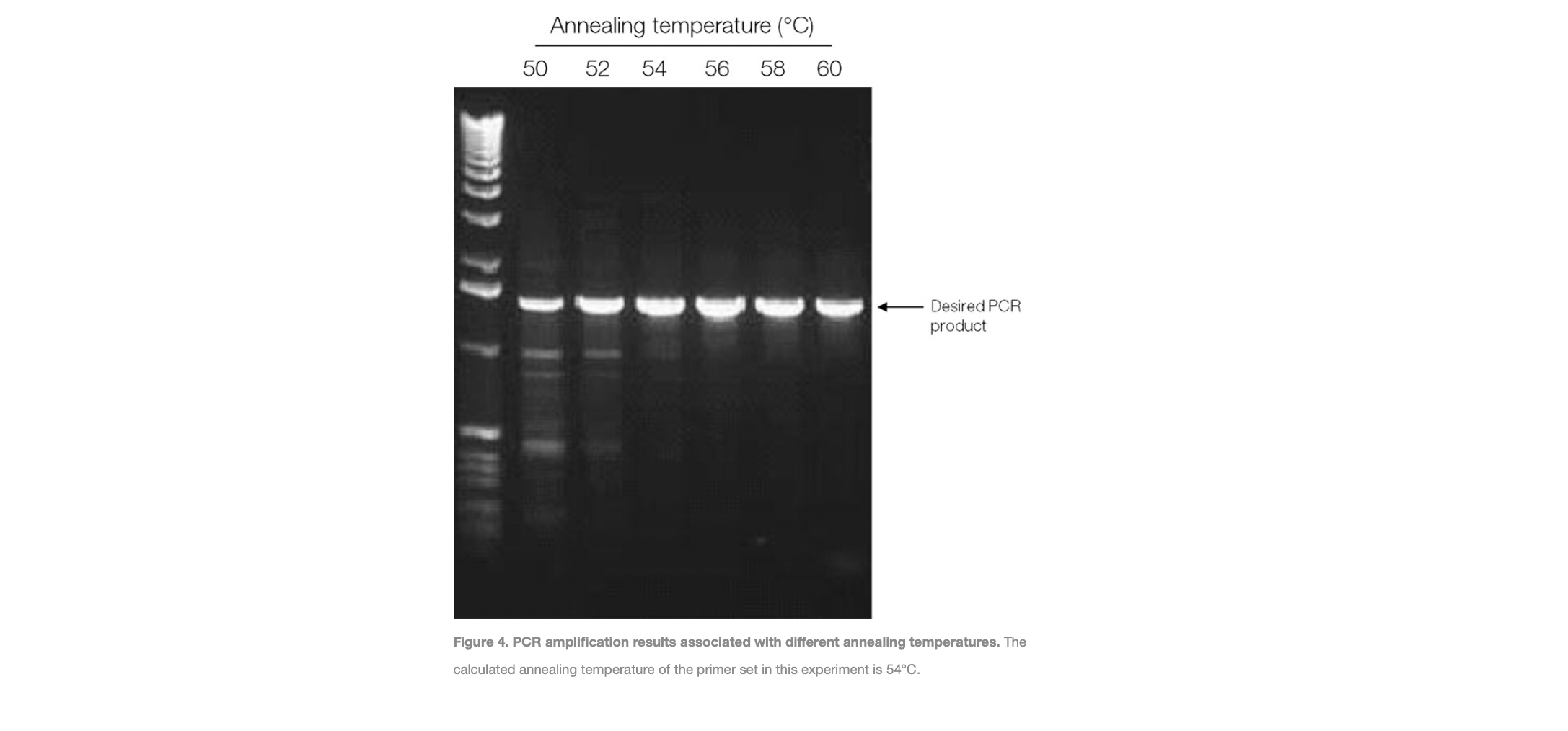
Figure from Thermofisher: https://www.thermofisher.com/uk/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html
The Melting Temperature of the primers (Tm)
This is defined as the temperature at which 50% of the primer form a duplex with the target DNA.
This is determined by the primers:
Base Composition: The number of Hydrogen bonds. Since G-C pairs have three bonds and A-T pairs have two, a higher GC content increases the Tm.
Primer Length: Longer primers generally have higher thermodynamic stability and require a higher Tm.
The Buffer Environment
The reaction buffer components significantly affect annealing temperature:
Salt Concentration (Na+ or K+): Cations neutralise the negative charge of the DNA backbone, increasing the stability of the primer-template duplex and raising the required Tm.
PCR Additives, co-solvents and modified nucleotides lower the Tm e.g DMSO, using 7-deaza-dGTP instead of dGTP
Specialized Buffers: some buffers contain isostabilizing components designed to allow a universal annealing temperature (regardless of the specific primer sequence.)
Experimental Optimisation
The final annealing temperature used in the thermal cycler is often adjusted based on the observed experimental results based on PCR yield.
If there is no or low amplification the temperature is lowered (in 2–3∘C increments) to encourage binding.
If non-specific PCR products appear, the temperature is raised to enhance specificity.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
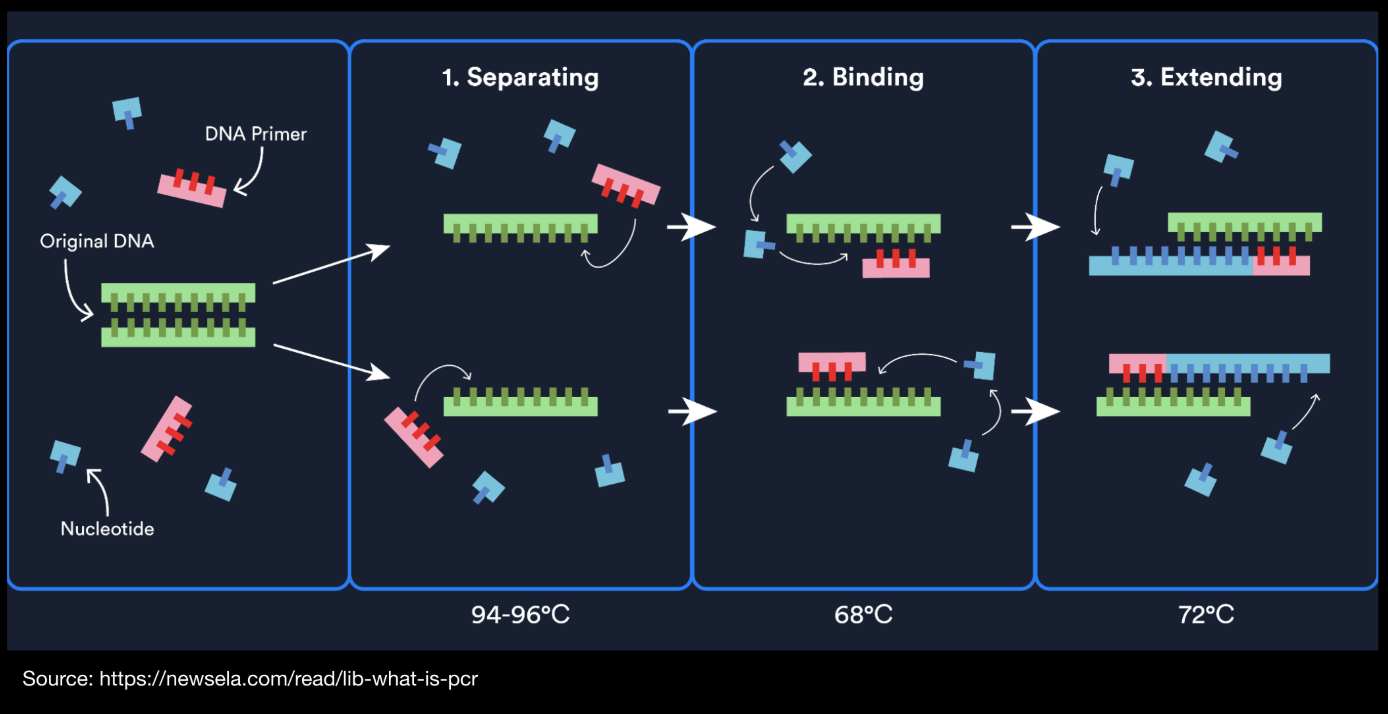
Figure from Newsela: https://app.newsela.com/view/ck9nooouh094h0iqjb6jgexg5/
PCR
PCR is an amplification technique which uses primers to targets a specific DNA sequences of interest from a template and creating many linear copies through a cycle of denaturation, annealing and extension via DNA polymerase.
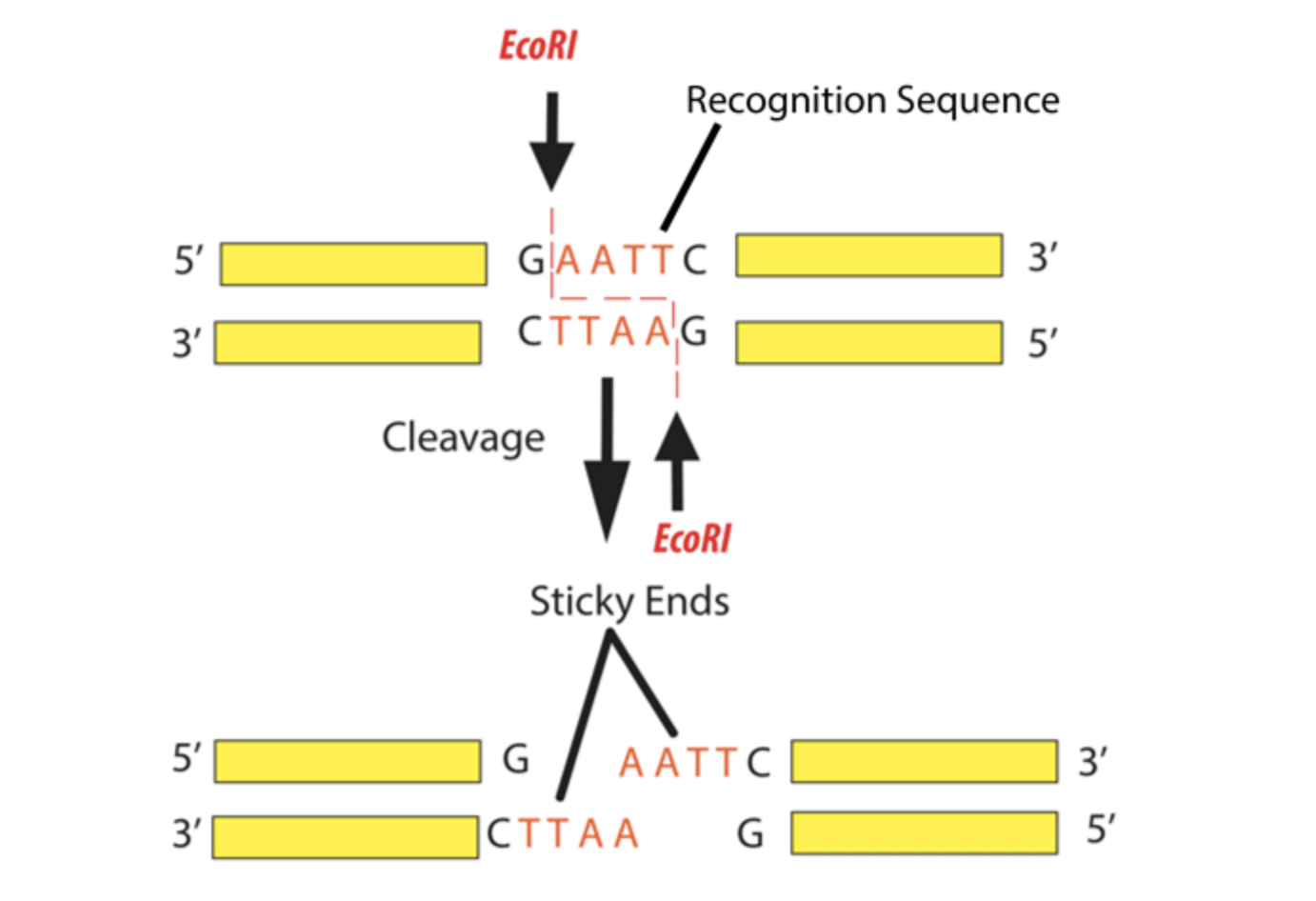
Figure from Addgene: https://www.addgene.org/protocols/restriction-digest/
Restriction Enzyme Digest
In contrast, a restriction enzyme digest is a cleavage technique where restriction endonucleases recognise and cut the phosphodiester bonds at specific recognition sites within an existing DNA molecule.
Compare and Contrast
| Properties | PCR | Restriction Enzyme Digest |
|---|---|---|
| Action | Amplification (Building new DNA) | Cleavage (Cutting existing DNA) |
| Components Requirements | Template DNA (not much needed), Primers, dNTPs, MgCl2, reaction buffer | Larger quantities of DNA (with specific recognition sequence), restriction enzymes, reaction buffer, water |
| Targeting | customisable: primers define the specific boundaries of the target DNA, allowing for the amplification of any sequence of interest provided the flanking sequences are known. | Fixed: dna fragments are defined by the location and frequency of specific palindromic recognition sites within the existing DNA molecule |
| Temperature | dynamic cycles of 95°C, 60°C, 72°C | Usually stable 37°C incubation |
| Output | High yield of copies of a specific DNA segment | Cleaved fragments of existing molecule |
| End Type | Often blunt | Often sticky |
| Mutation | Not possible | Possible |
PCR is the preferred technique when you have a small amount of starting template, need to define the exact boundaries of a fragment where no natural cut sites exist or intend to perform precise engineering by introducing mutations or adding overlaps for Gibson Assembly.
In contrast, a restriction digest is preferable for diagnostic applications, linearising plasmids and traditional sub-cloning where the DNA sequence must be perfectly preserved. Since it is a cleavage method that does not involve synthesis, it is highly predictable and stable. It is the simpler, more reliable choice when suitable recognition sites are already located in the DNA and it is excellent for creating compatible sticky ends for ligation. Additionally, because it only requires a single incubation temperature in a stable buffer, it is often a faster and more cost-effective option for routine tasks.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Verify that the insert sequence and backbone have long, matching overlaps of 20–22 base pairs of sequence identity between adjoining fragments and that a high fidelity polymerase like Phusion is used in PCR to to ensure that the overlap sequences are accurate.
Eliminate the template DNA with a Dpnl digest. In the lab, this specifically targets and digests methylated GATC sequences found in the template. Since the amplified fragments are unmethylated, they remain intact. This ensures that the only circular DNA formed during the 50°C incubation comes from the new assembly.
Purify the DNA with the Zymo DNA Clean & Concentrator kit. This remove the Phusion enzyme, dNTPs and salts from the PCR reaction because they can interfere with the Gibson Master Mix enzymes (the exonuclease and ligase).
Diagnostic Gel Electrophoresis. Run the samples on an agarose gel to confirm that the backbone and insert are the expected band sizes, the DNA is linearised and that there are no non-specific contaminants that might compete for binding during the assembly.
DNA Quantification. Use a Nanodrop or Qubit to identify the exact DNA concentration in the sample (ng/μL). Gibson Assembly depends on a specific molar ratio (lab states generally 2:1 Insert to Vector). Accurate quantification ensures that the correct volume of each fragment is added so that the overlapping ends can find each other and anneal efficiently.
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmids are introduced into the E.coli cells via transformation (transfection in mammalian cells). These processes create pores in bacterial cell walls though heat shock, electroporation, chemical or physical transformation, sonication or micro shockwaves.
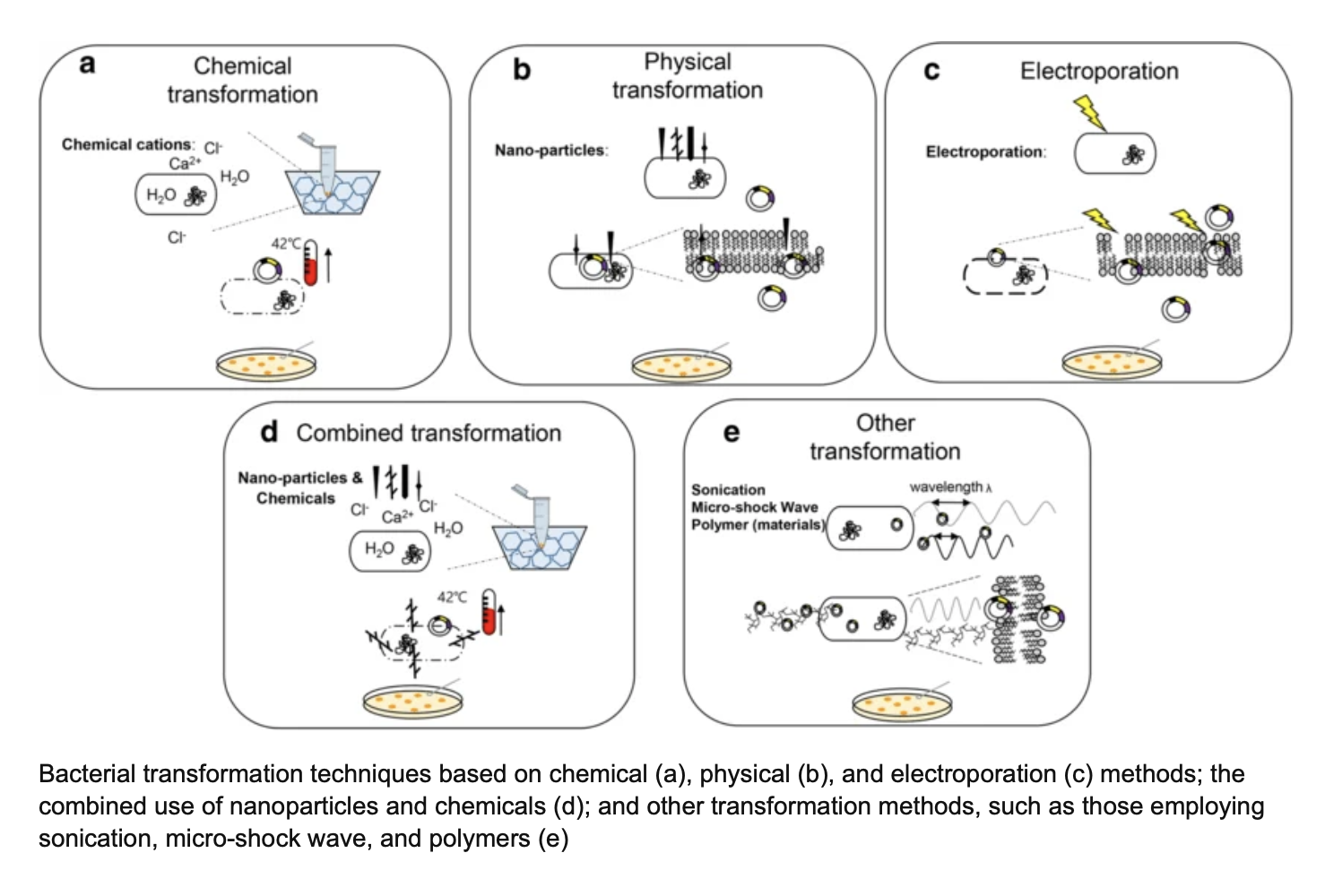
Figure from Research Gate: https://www.researchgate.net/figure/Bacterial-transformation-techniques-based-on-chemical-a-physical-b-and_fig1_336797847
In the lab heat shock is used. The sudden temperature change of moving the cells from ice to a 42°C water bath causes the bacterial cell wall and membrane to open up by generating temporary pores. The plasmid can then enter the E.coli cells from the surrounding liquid through diffusion.
Immediately after the 45-second heat shock, the cells are transferred back to ice for 5 mins to stabilise the membranes.
The cells are then added to SOC growth media and incubated at 37°C for 60 minutes. This recovery period allows the pores to close and gives the bacteria time to start multiplying and expressing the antibiotic resistance gene before being placed on selective agar plates.
Only cells that had successfully received the plasmid will survive the antibiotics and grow.
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate assembly is a cloning technique that allows for a seamless and ordered assembly of multiple DNA fragments in a single reaction.
- Type IIS Restriction Enzyme Digest
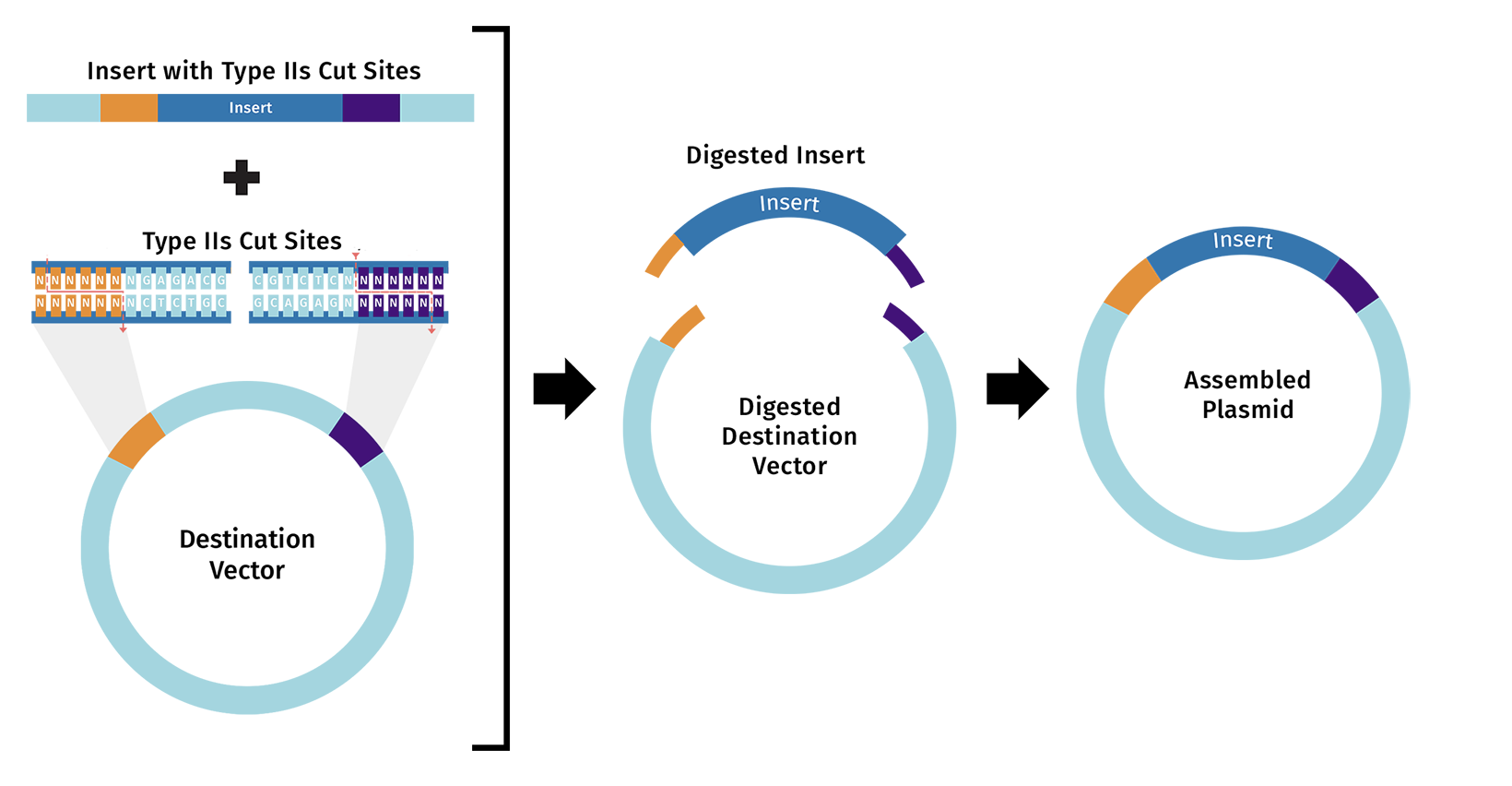
Figure from Snap Gene :https://www.snapgene.com/guides/golden-gate-assembly
The method first uses Type IIS restriction enzymes (e.g BsaI) to perform a digest. These enzymes are unique as they recognise non-palindromic sequences and cleave the DNA at a shifted site outside of the recognition sequence. This shifted cleavage creates variable sticky ends (fusion sites).
- DNA Ligation
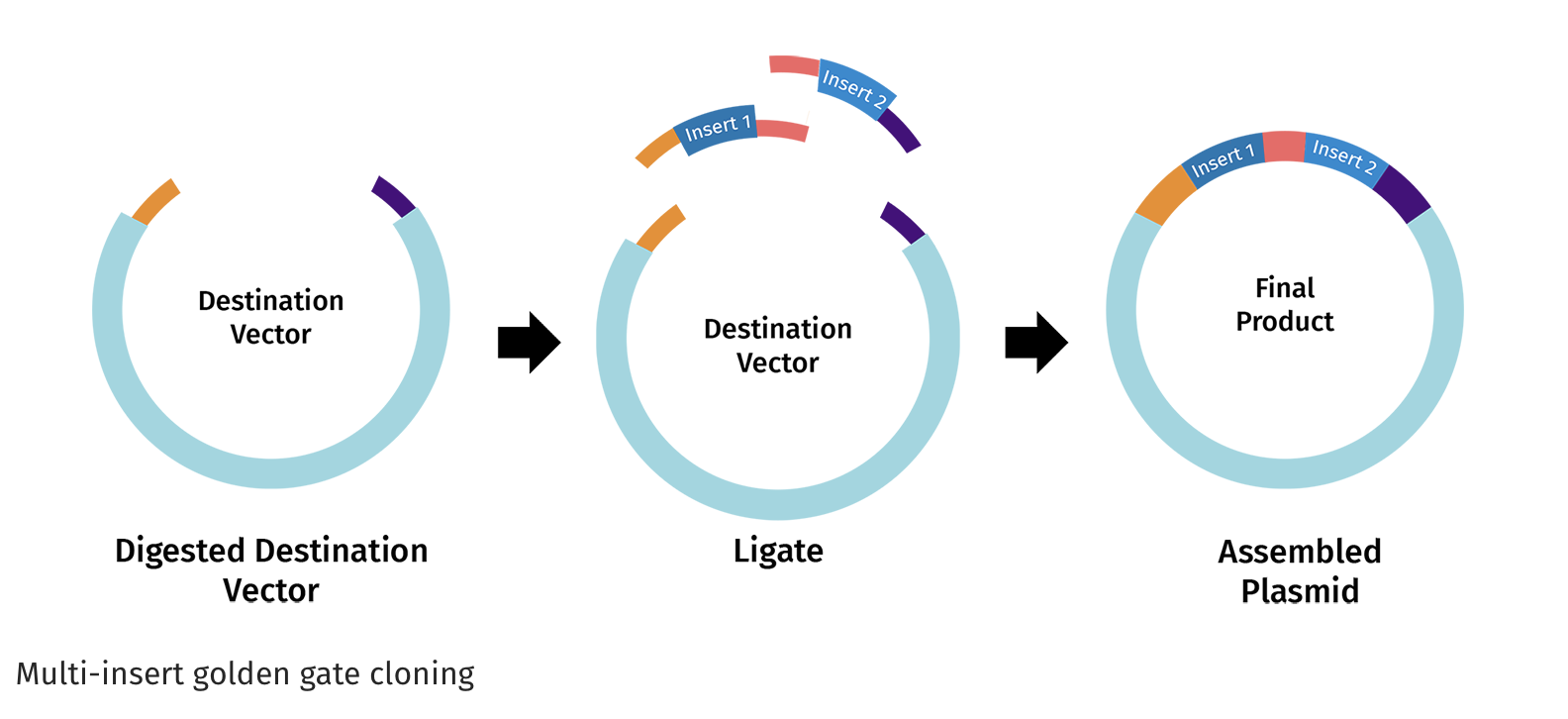
Figure from Snap Gene :https://www.snapgene.com/guides/golden-gate-assembly
Once the vector and DNA insert are digested, the complementary overhangs are joined together by DNA ligase to create the assembled plasmid. The process is seamless because the restriction sites are eliminated in the final construct.
Model this assembly method with Benchling!
To model a simple Golden Gate Assembly in Benchling:
I first prepared my vector and insert. I downloaded a empty pET-28a(+) vector from Snapgene and found a standard GFP sequence from the Registry of Standard Biological Parts.
In Benchling I screened both for exisiting BsaI cut sites and luckily we were all clear:
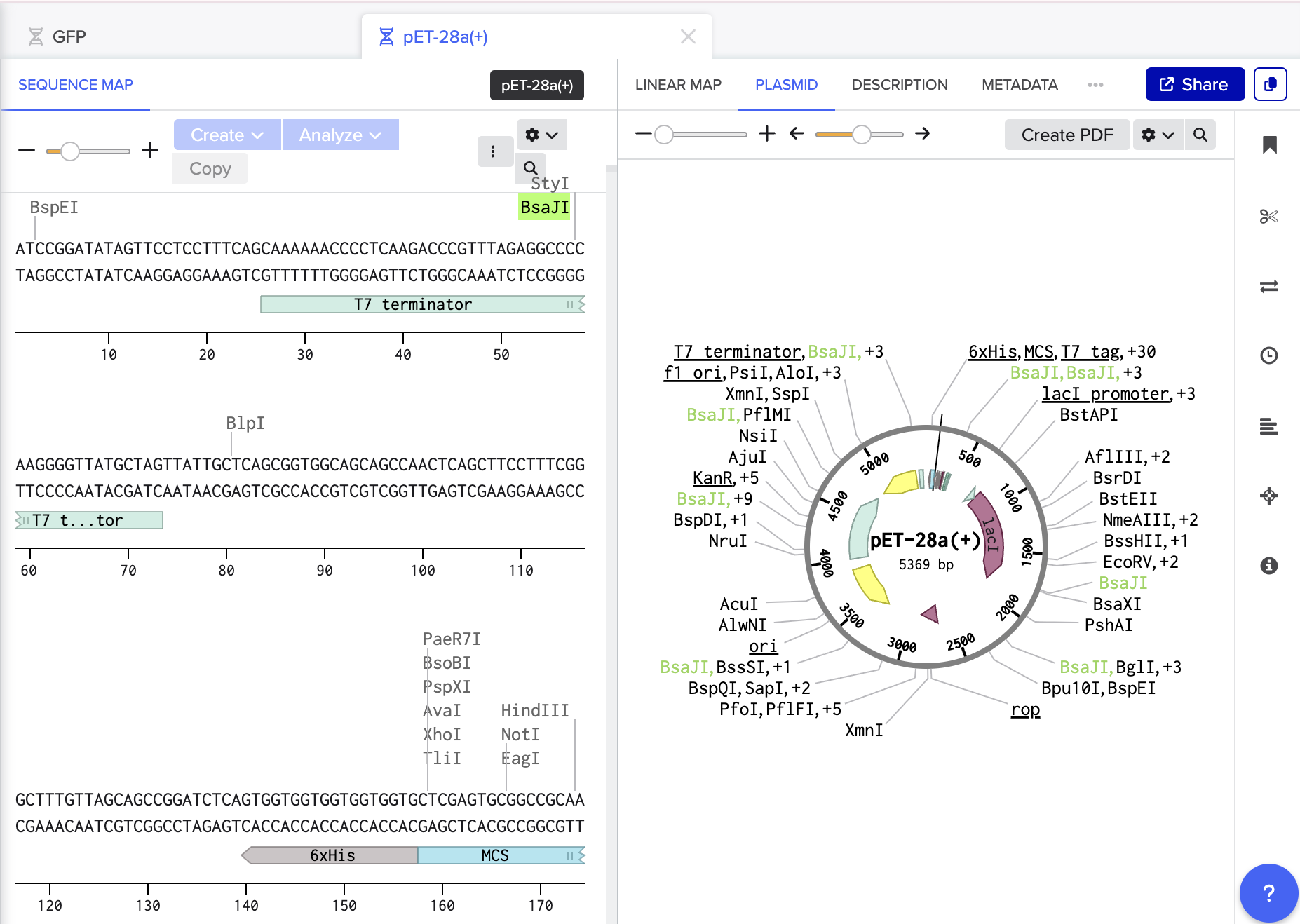
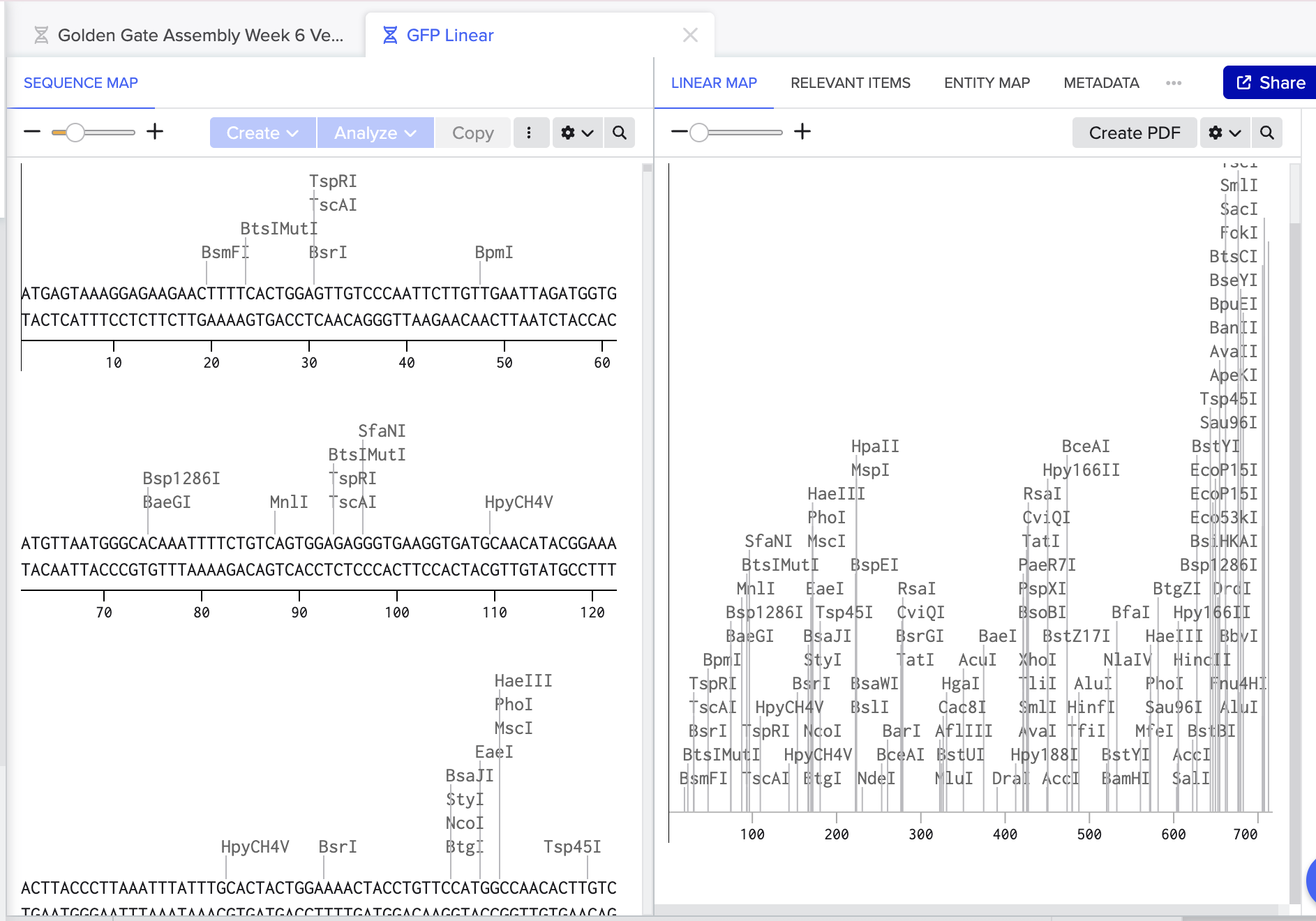
- I then prepared my BsaI restriction sites and overhangs for both the vector and insert ensuring correct directionality.
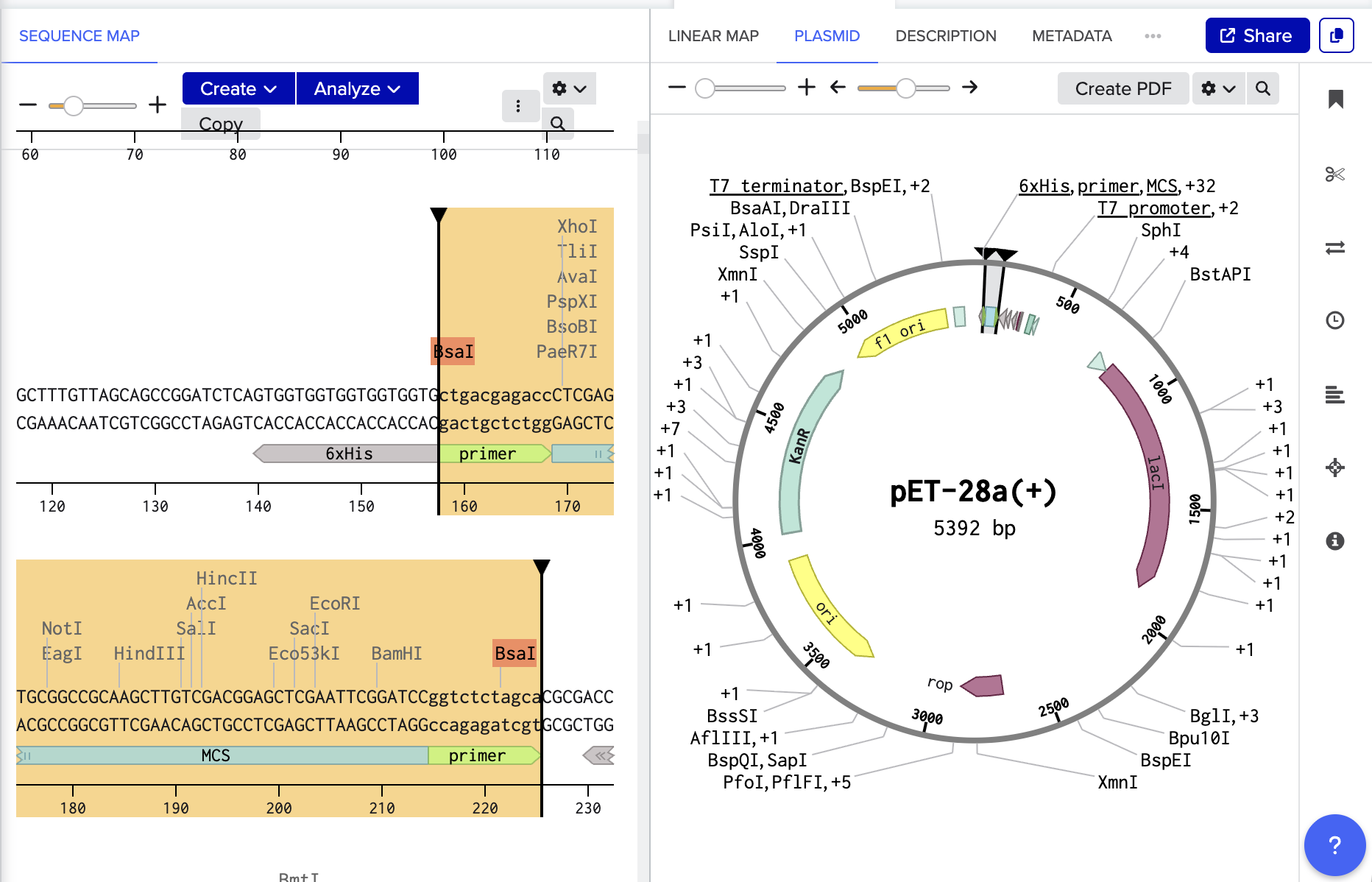
For my Vector we are cutting out the MCS so:
OVERHANG - RESTRICTION SITE - MCS - RESTRICTION SITE - OVERHANG
CTGAC GAGACC MCS GGTCTC TAGCA
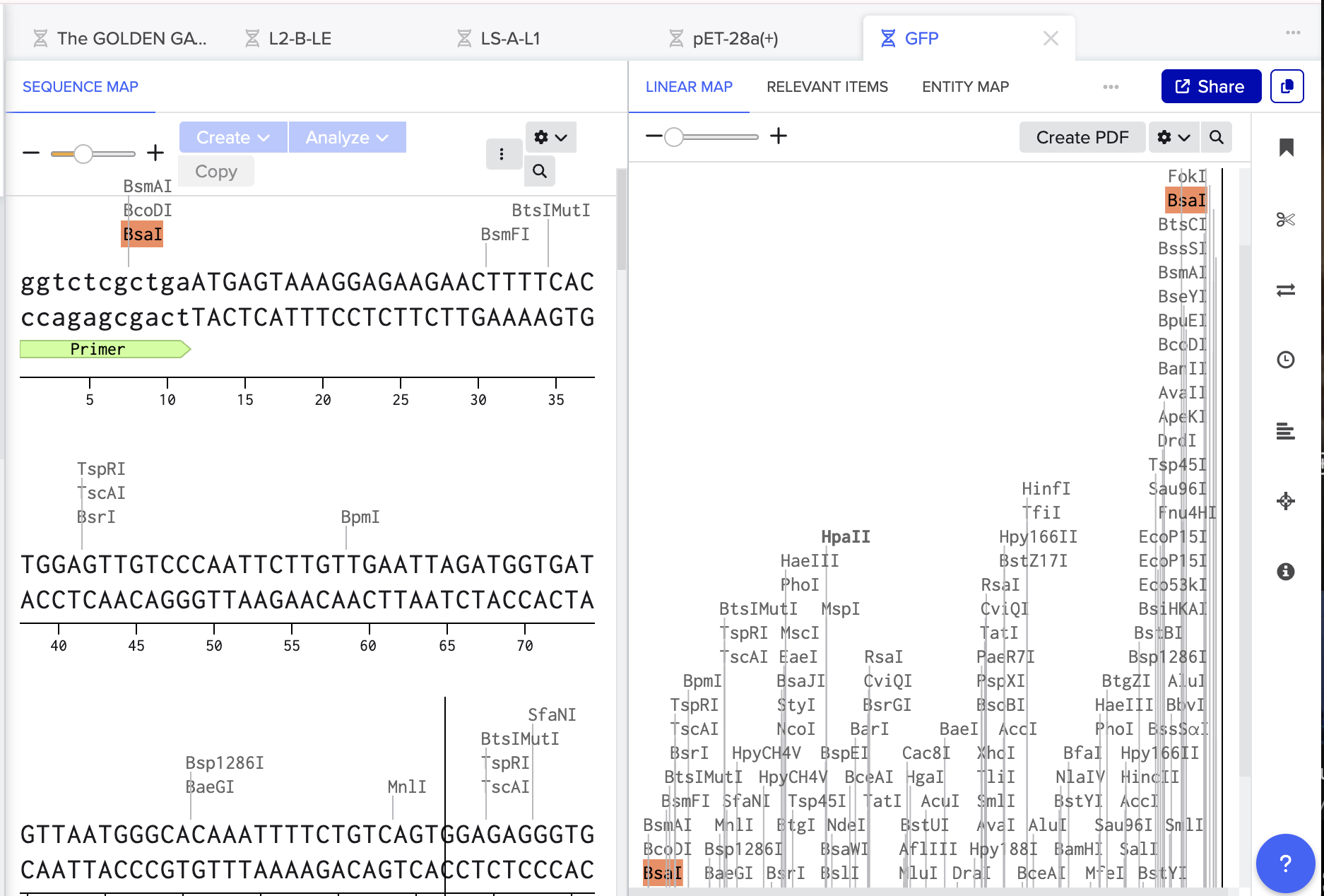
For my GFP Insert we are creating complimentry sticky end so:
RESTRICTION SITE - OVERHANG - GFP - OVERHANG - RESTRICTION SITE
GGTCTC GCTGA GFP AGCAC GAGACC
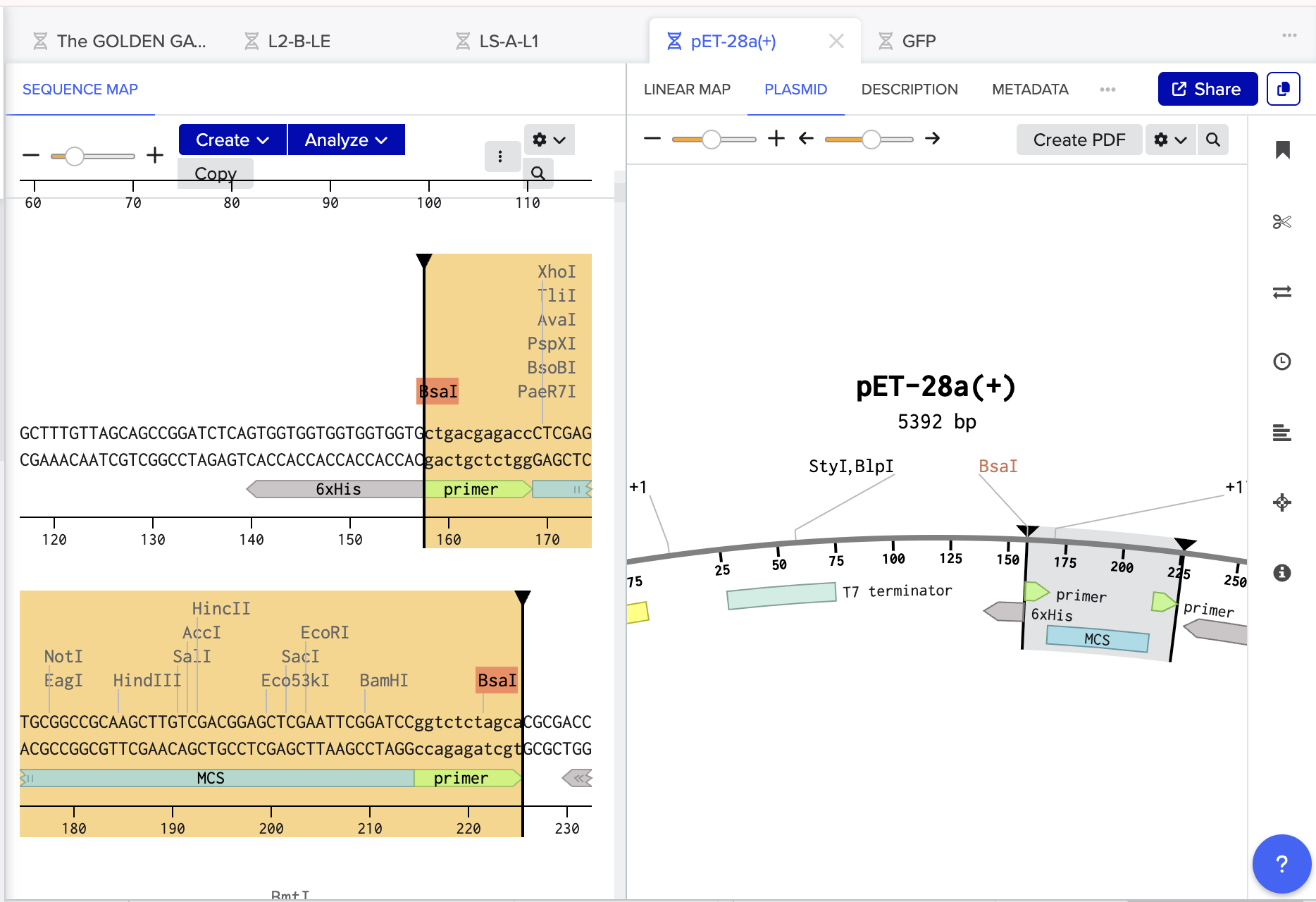
- Then I opened the Benchling Assembly Tool and selected Golden Gate. I added my vector as the backbone, making sure to select existing BsaI cut sites. I then added my GFP as the insert.
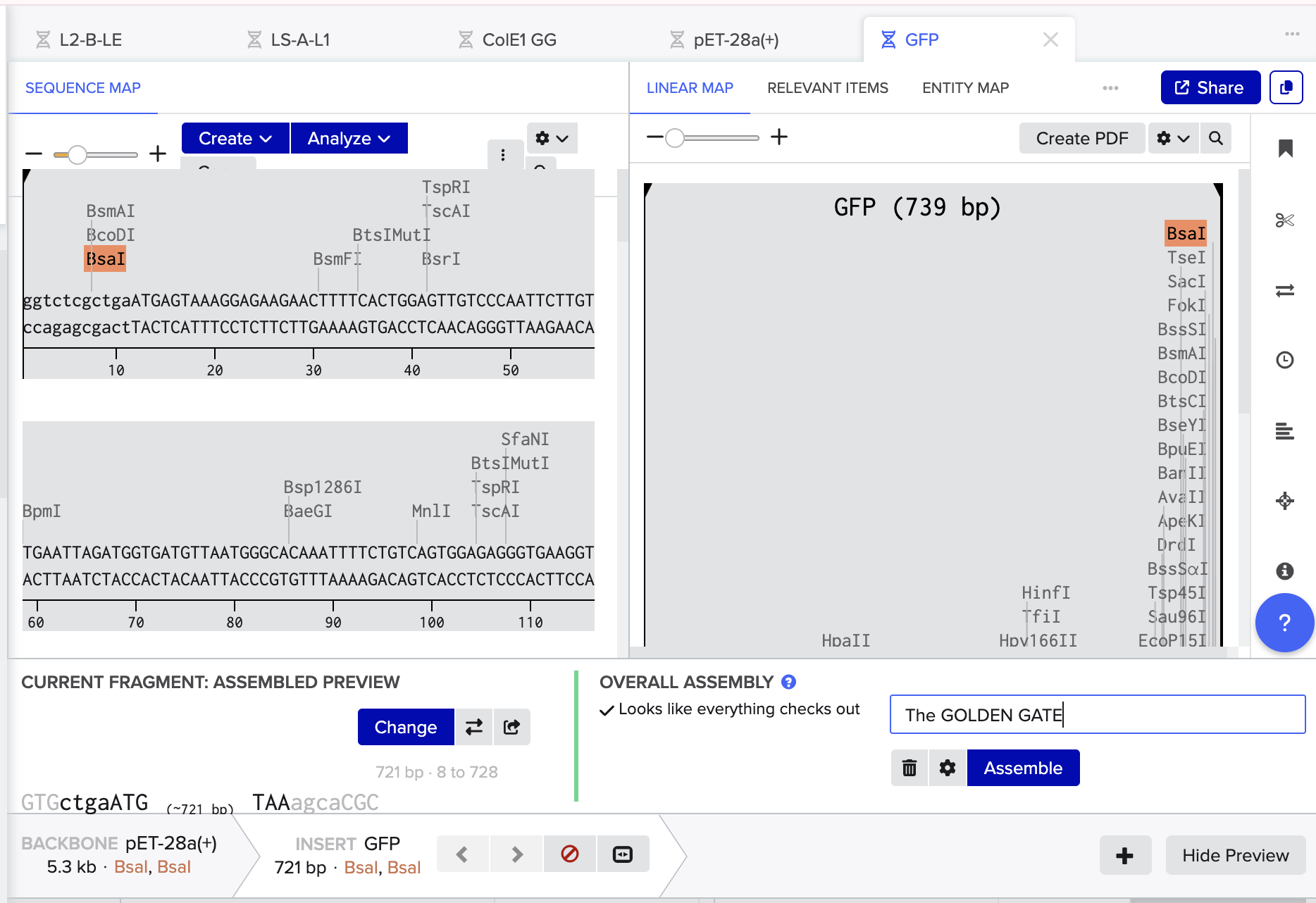
- Mercifully, this was accepted by Benchling and everything was complimentary and happy so I pressed assemble! And here we have the recombinant plasmid pET-28a(+)/ GFP:
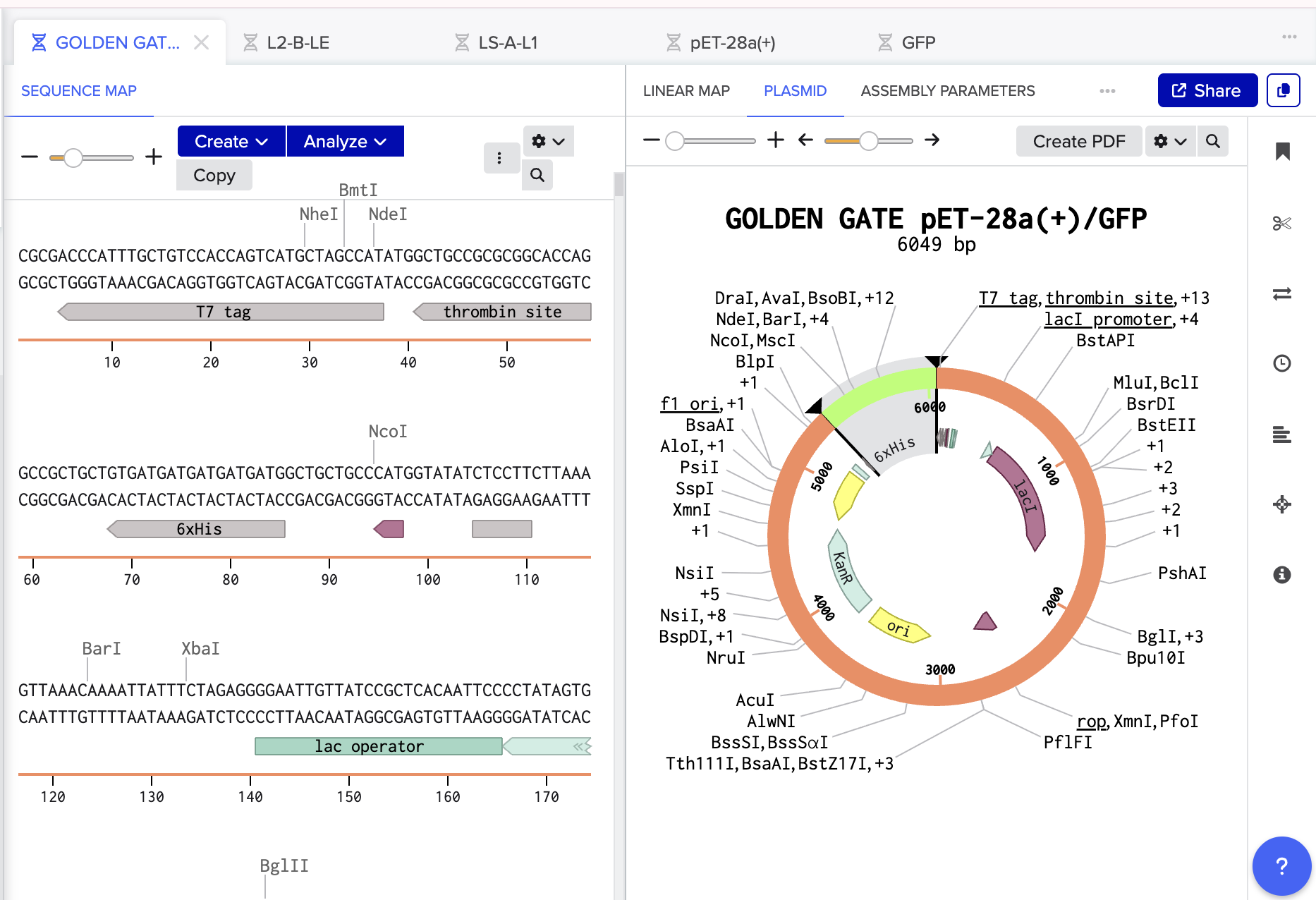
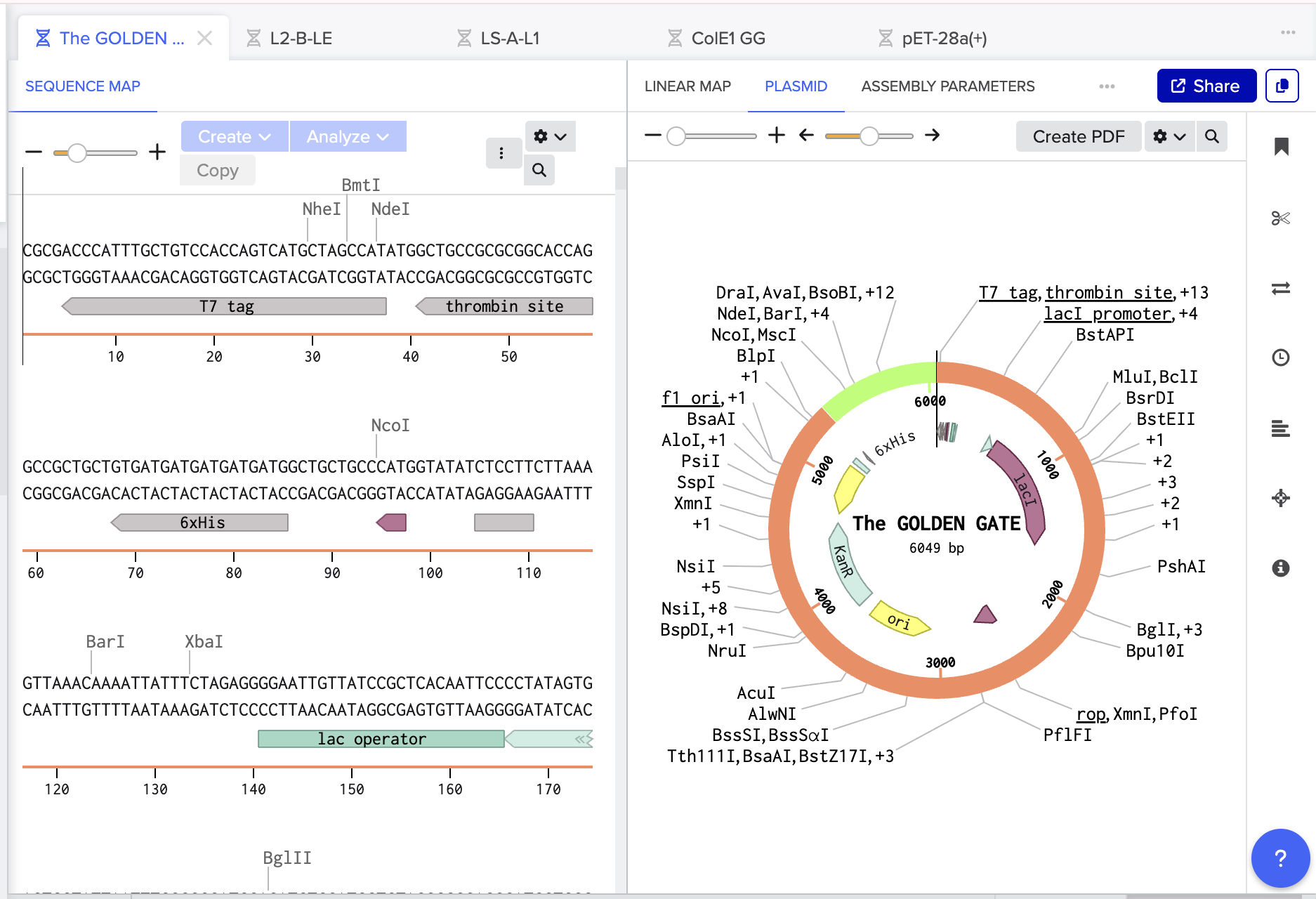
The Benchling Links are below:
Recombinant Plasmid: https://benchling.com/s/seq-kne6FeCThT1SY8yJEm6Y?m=slm-OTGqNj7vDDl0cWvWRb6E
Vector: https://benchling.com/s/seq-Sk6djpIs6kgEsF0KKI6H?m=slm-SvPeLVKrZAmtwOunSb27
Insert: https://benchling.com/s/seq-I2fzIiisOFvixW8peO3l?m=slm-uqvbBCJ0iNJbn08LPcx0
- Following Ice’s Benchling Basics Tutorial I also simulated a multifragment Golden Gate:
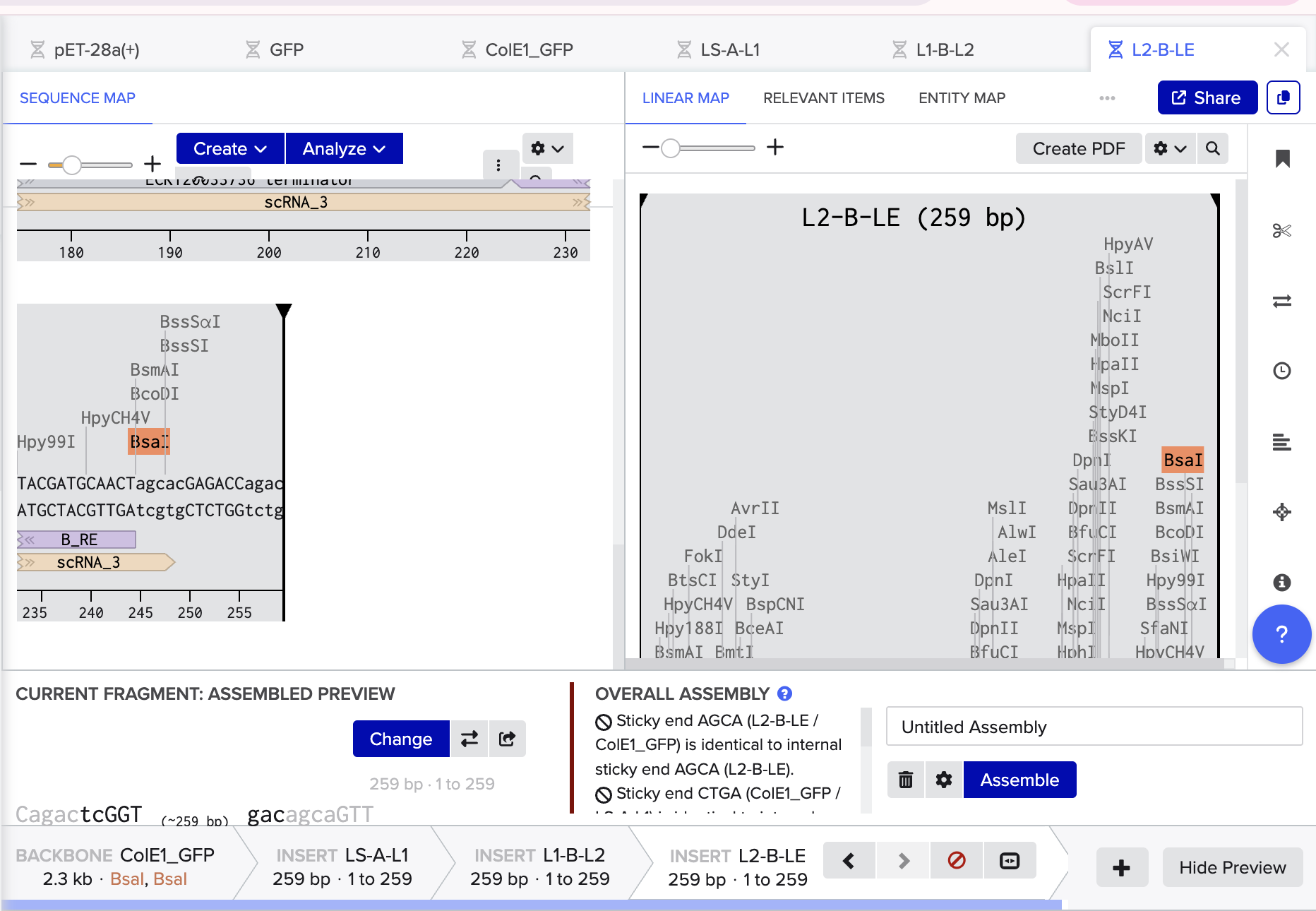
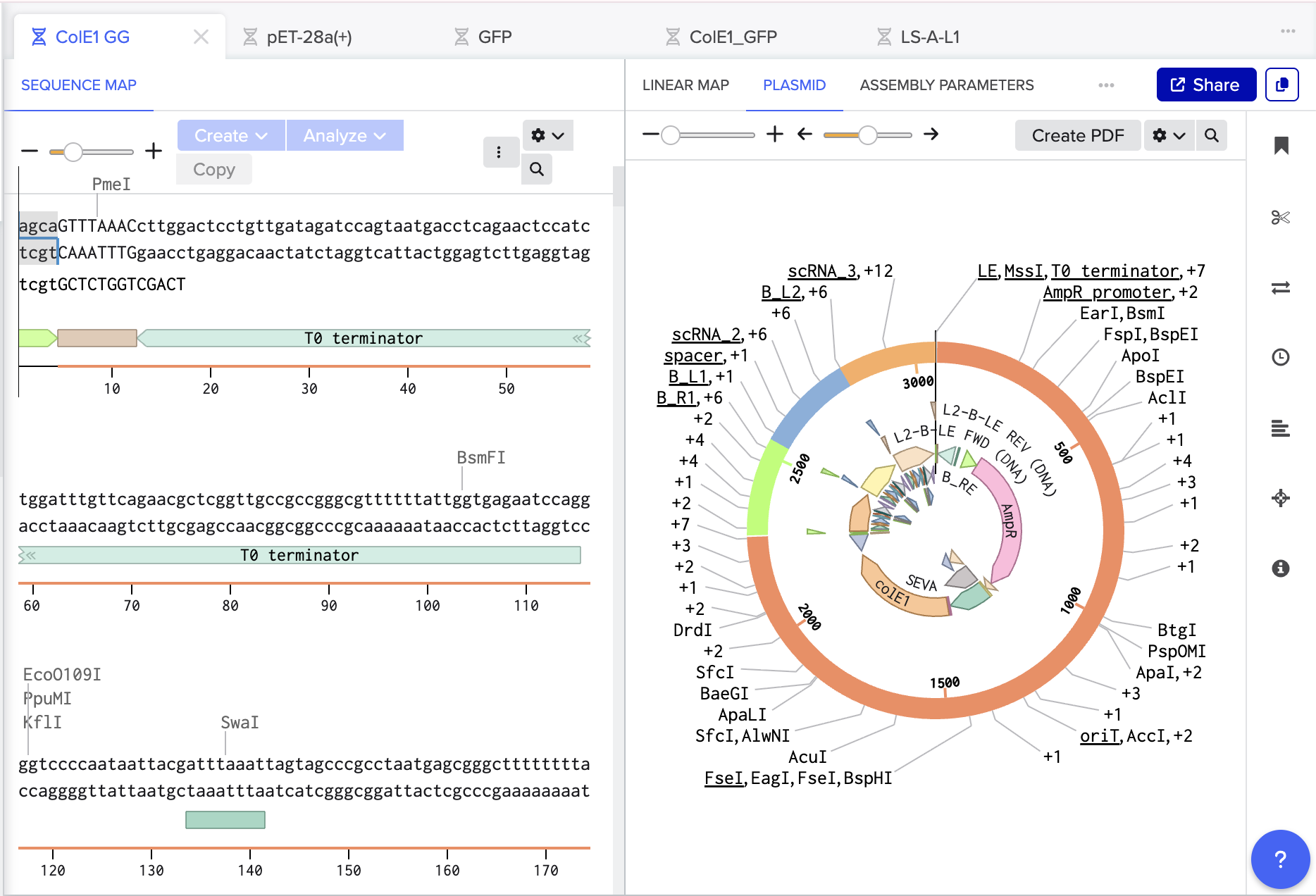
The Benchling Links is below:
PART B: Asimov Kernel not possible to complete without liscence access.
References:
Addgene. (n.d.). PCR protocol (thermal cycling). https://www.addgene.org/protocols/pcr/
Bloch, K. D., & Grossmann, B. (2001). Digestion of DNA with restriction endonucleases. Current Protocols in Molecular Biology, Chapter 3, Unit 3.1. https://doi.org/10.1002/0471142727.mb0301s31
Massachusetts Institute of Technology. (2015). Molecular cloning using the Gibson Assembly cloning kit (NEB E5510S). 7.15 Experimental Molecular Genetics. MIT OpenCourseWare. https://ocw.mit.edu/courses/7-15-experimental-molecular-genetics-spring-2015/857fcd5fb6b6b392ab478e8167337b8f_MIT7_15S15_Molecular.pdf
SnapGene. (n.d.). Gibson Assembly guide. https://www.snapgene.com/guides/gibson-assembly SnapGene. (n.d.). Golden Gate Assembly guide. https://www.snapgene.com/guides/golden-gate-assembly
Thermo Fisher Scientific. (n.d.). PCR cycling considerations—Time and temperature. https://www.thermofisher.com/uk/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-cycling-considerations.html
Thermo Fisher Scientific. (n.d.). Phusion High-Fidelity DNA Polymerase (F531L). https://www.thermofisher.com/order/catalog/product/F531L