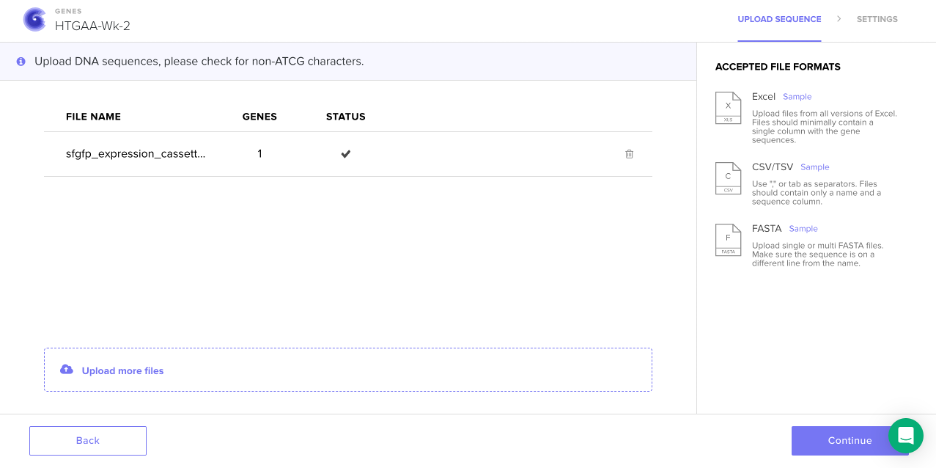
🧬 Week 2 Homework Components DNA Read, Write, & Edit — sequencing and synthesis workflows, restriction digests and gel electrophoresis, genome-editing frameworks.
📋 Overview This week covers:
Part 0: Basics of Gel Electrophoresis Part 1: Benchling & In-silico Gel Art ✓ Part 2: Gel Art — Restriction Digests and Gel Electrophoresis (wet lab, optional with lab access) Part 3: DNA Design Challenge ✓ Part 4: Prepare a Twist DNA Synthesis Order ✓ Part 5: DNA Read/Write/Edit ✓ Content to be added as you complete each part.
Published paper on automation for novel biological applications; automation project description for gumol MD simulations + ECSOD/MSC + new-Clara microfluidic validation.
Cell-free expression vs in vivo; ATP regeneration; prokaryotic vs eukaryotic CFPS; membrane proteins; troubleshooting; synthetic minimal cell (SOD3/CXCR4); materials pitch; Genes in Space (BioBits).
Complete end-to-end project documentation including governance assessment and interactive Python application.
/\_/\
( o.o )
> ^ <
/| |\
(_| |_)
"Meow! Check out both sections above!"
BioVolt Governance Assessment — Policy Options Comparison - I filled this out anyways but the project is located in the Sub as the Cat suggested- Meow
The table below compares three governance approaches for the BioVolt DIY electroporation device across multiple criteria. Scoring: 3 = Best, 2 = Moderate, 1 = Worst.
Criteria
Option 1: Community Self-Governance
Option 2: Safety Warnings & Labels
Option 3: Regulatory Licensing
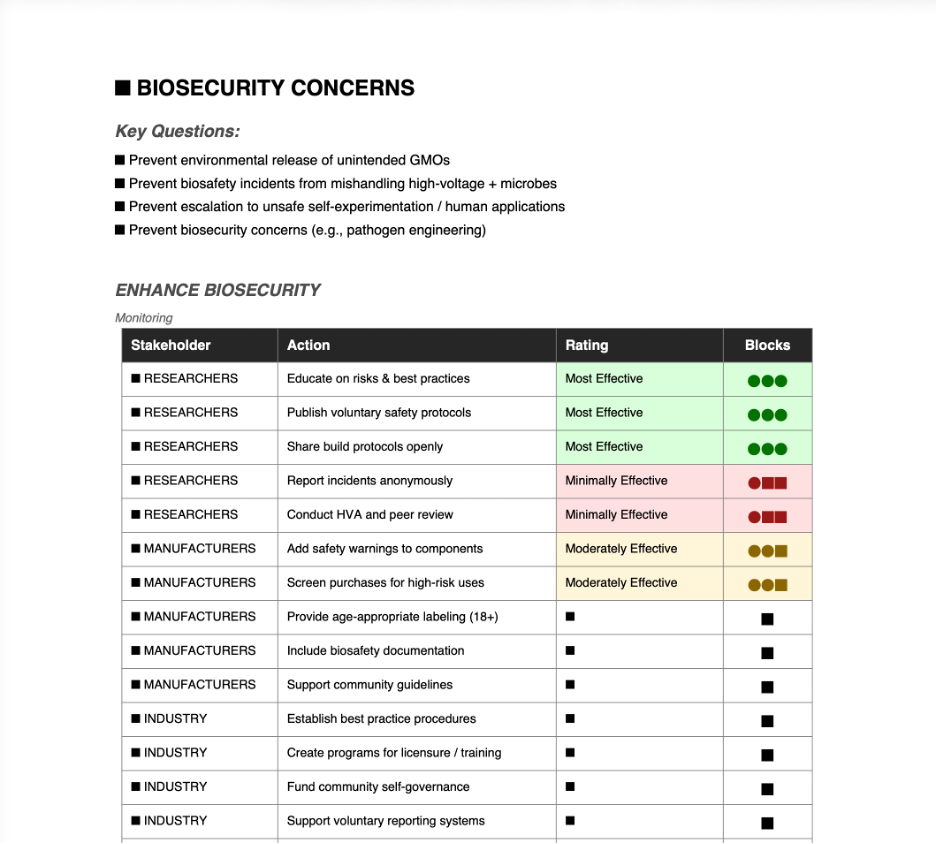
ENHANCE BIOSECURITY
• By preventing incidents
2
2
3
• By helping respond
3
1
2
FOSTER LAB SAFETY
• By preventing incident
2
2
3
• By helping respond
3
1
3
PROTECT THE ENVIRONMENT
• By preventing incidents
2
2
3
• By helping respond
3
1
2
OTHER CONSIDERATIONS
• Minimizing costs and burdens
3
2
1
• Feasibility?
3
3
1
• Not impede research
3
2
1
• Promote constructive applications
3
2
1
Option Summaries
Option 1 — Community-Led Self-Governance: ✓ Best for: response capacity, feasibility, minimizing burdens, not impeding research ✗ Weaker on: prevention (relies on voluntary participation; rogue actors may ignore)
Option 2 — Targeted Product Restrictions (Safety Warnings/Labels): ✓ Best for: feasibility, moderate prevention without bans ✗ Weaker on: response capacity (warnings don’t help after incidents), limited impact on determined bad actors
Option 3 — Regulatory Classification (Licensing/HVA Review): ✓ Best for: prevention (permits, training, HVA peer review blocks worst misuse) ✗ Weaker on: costs, feasibility, impedes DIY research, harms global equity
Recommendation: Prioritize Option 1 (community self-governance) as primary, combine with Option 2 (warnings) as secondary safeguard. Avoid Option 3 unless clear evidence of high-risk proliferation emerges.
Subsections of Week 1 HW: Principles and Practices
DIY Electroporation Project: BioVolt - First rolled out at DEFCON 32- Now revisted from END to END
Biological engineering application/tool to develop: BioVolt is a portable, ultra-low-cost DIY electroporation device (~$10-20 in parts) that uses a piezoelectric crystal from a barbecue lighter to generate ~2,000 V pulses for temporary cell membrane permeabilization. This enables DNA/RNA uptake in bacteria (e.g., E. coli), yeast, plant protoplasts, or even stem cells for genetic transformation. Inspired by the DEFCON 32 talk “You got a lighter I need to do some Electroporation” (presented by Dr. James Utley (Me), Phil Rhodes, and Josh Hill from Viva Securus/Syndicate Laboratories), it builds on frugal biohacking principles: piezoelectric trigger pulsing, custom microfluidic cuvettes from aluminum tape/magnets/glass slides, and simple high-voltage testing.
DEFCON 32 Presentation — Where It Started for me
At DEFCON 32 the talk I presented focused on the device itself — proving that a barbecue lighter’s piezoelectric crystal could generate sufficient voltage to temporarily permeabilize cell membranes for DNA uptake. The talk covered design details, demos, troubleshooting (e.g., arc gap tuning with Post-it notes), and the biohacking ethos behind building a ~$10 electroporator.
Key highlights from the talk: ~2,000 V pulses via lighter clicks, high cell mortality (50-70%) but viable transformants, GFP reporter demos, open protocols encouraged.
Next Phase: End-to-End Pipeline with Efficiency Focus
The next phase of BioVolt moves beyond the device and brings the entire workflow end to end, with a focus on efficiency and frugal validation. The goal: take a piezoelectric electroporator built from a barbecue lighter and prove — through a full pipeline — that it actually works. The pipeline includes:
Plasmid amplification via thermal cycling — Before electroporation, the initial plasmid source will be amplified using the MJ Research PTC-100 thermal cycler (Peltier-effect programmable controller) available in the lab. This ensures sufficient plasmid DNA concentration for transformation.
DNA concentration measurement — Using the Rodeo open colorimeter (visible light version for OD600 cell density measurements) and, if possible, the UV version for DNA concentration quantification. This provides pre- and post-transformation metrics.
Electroporation — Transformation of cells with the amplified plasmid DNA using the BioVolt piezoelectric device, followed by recovery and plating.
Post-transformation PCR verification — For good measure, PCR will be run after transformation using the same thermal cycler to check whether the insert is present in the recovered cells. This triangulates and correlates with plating results to provide a hasty “close enough” frugal validation.
Gel electrophoresis confirmation — Agarose gel electrophoresis to visualise PCR products and verify successful transformation (e.g., presence of reporter genes like GFP via band patterns under UV).
The aim is to triangulate multiple data points — plasmid amplification, colorimetric/UV measurement, transformation plating, and post-transformation PCR — to build confidence that the piezo electroporator from a lighter actually delivers. Fingers crossed, this provides a credible, frugal, end-to-end validation of a DIY electroporation workflow.
This democratizes synthetic biology for education, citizen science, and personal biohacking in resource-limited settings.
Lab Setup & Tools in Action - You can see I got some goods to work with!
My biohacker lab integrates the device with the full verification pipeline.
On to the assignement - Interactive Governance Assessment Form
An interactive Python application (app.py) is provided to assess governance and risk mitigation strategies for the BioVolt project. The form uses a block-based rating scale where more filled blocks mean more effective:
Blocks
Rating
Meaning
●○○
Minimally Effective
Low impact — unlikely to achieve the goal
●●○
Moderately Effective
Moderate impact — partial success likely
●●●
Most Effective
High impact — highly likely to achieve goal
Project File Structure
BioVolt_week_01_hw_principles_and_practices/
├── _index.md # This file — project documentation (Hugo page)
├── app.py # Interactive governance assessment application
├── requirements.txt # Python dependencies
├── Biohacker_Lab.jpeg # Lab overview photo
├── in_da_lab.jpeg # Working in the lab photo
├── Volt_Test.jpeg # High-voltage testing with insulation tester
├── rodeo-colorimeter.png # IO Rodeo open colorimeter
├── BioVolt_govern_UI.png # Screenshot of the application UI
└── Biovolt_Govern_Report.png # Screenshot of the PDF report output
Prerequisites
Python 3.x installed on your system
tkinter (usually included with Python; on Linux you may need python3-tk)
Installation
Navigate to the project directory:
cd BioVolt_week_01_hw_principles_and_practices
Install required dependencies:
pip install -r requirements.txt
Running the Application
python app.py
How to Use the Form
Launch — The application opens a dark-themed window with the assessment matrix.
Read the instructions — System instructions are displayed at the top of the form explaining the block-based rating system.
Review each concern category — Three categories are presented, each with context questions:
Equity Concerns — access, regulation, educational barriers, global equity
Environmental Concerns — microbial activity, non-human organisms, public concerns
Rate each action — For every action under each stakeholder (Researchers, Manufacturers, Industry, Organizations), click one of three block-rating buttons:
The selected button stays highlighted with its rating colour
A status indicator appears to the right showing your selection
Other buttons in the same row reset to their default state
Export to PDF — Click the “EXPORT TO PDF” button to generate a report containing:
Cover page with assessment date and completion count
Rating scale legend with colour-coded descriptions
Full assessment tables for each concern category
Colour-coded rows: green tint for Most Effective, amber for Moderate, red for Minimal
Block indicators (●●● / ●●○ / ●○○) printed in every row
Summary page with counts and percentages for each rating level
Reset — Click “RESET MATRIX” to clear all selections and start over.
Application Features
Block-based rating scale — intuitive system where more blocks = more effective (no ambiguity)
Dark theme UI — dark background with neon accent colours for readability
Persistent button state — selected buttons remain highlighted with their rating colour
Status indicators — each row shows the current selection in text beside the buttons
Scrollable interface — mouse wheel support for navigating the full assessment matrix
Neon accent bars — left-side accent bars on each concern card for visual hierarchy
Colour-coded PDF output — rating cells are tinted to match their effectiveness level
Summary statistics — PDF includes a final page with counts and percentages
Empty export protection — warns you if no ratings are selected before exporting
Form reset — one-click reset with confirmation dialog
Screenshots
Application UI — Dark-themed interface with block-based rating buttons and colour-coded status indicators:
PDF Report Output — Exported assessment with colour-coded rows, block indicators, and stakeholder ratings:
Governance / Policy Goals (Preventing Harm)
Focus on non-tool-function risks: Prevent environmental release of unintended GMOs, biosafety incidents from mishandling high-voltage + microbes, escalation to unsafe self-experimentation/human applications, or biosecurity concerns (e.g., pathogen engineering). Core aims: Minimize biosafety/biosecurity harms, promote responsible use, avoid stifling innovation with heavy regulation, encourage informed DIYbio practices, and address public/environmental concerns.
Three Potential Governance/Policy Actions
Action 1: Community-Led Self-Governance with Voluntary Guidelines and Reporting
Goal: Foster peer accountability and safe practices through DIYbio networks, reducing risks via shared norms without external mandates.
Design:
Opt-in: DIYbio communities, forums (e.g., Discord, Reddit, The ODIN users), and makerspaces.
Fund: Crowdfunding, donations, or volunteer time.
Approve: Community-elected moderators or biosafety working groups.
Implement: Publish voluntary guidelines (e.g., “BioVolt Safety Protocol” on protocols.io or GitHub), require protocol sharing for builds, anonymous incident reporting (expand “Ask a Biosafety Expert” services).
Risks / What could go wrong (incorrect assumptions, uncertainties): Assumes broad ethical participation - rogue actors may ignore; self-reporting misses hidden issues; low adoption if seen as “extra work.”
Assumptions, “Success” and “Failure” rubric:
Success (best - 1): High adoption -> fewer accidents, strong norms against risky uses (e.g., no human trials), community self-corrects.
Mid (2): Partial uptake -> safety improvements in visible projects, but gaps remain.
Failure (worst - 3): Guidelines ignored -> no risk reduction, or “forbidden fruit” effect increases experimentation.
Action 2: Targeted Product Restrictions (e.g., Safety Warnings / Age Limits on Kits & Components)
Goal: Reduce impulsive or uninformed misuse by requiring clear hazard labels on high-voltage components (e.g., piezoelectric lighters, capacitors) or full kits, without banning access.
Approve: Consumer safety agencies or state-level consumer protection (e.g., modeled on CRISPR kit labeling laws).
Implement: Mandatory labels (“Not for human use; biological hazard when combined with genetic material; 18+ recommended”).
Risks / What could go wrong: Warnings may not deter determined users (parts sourced separately); patchy enforcement online/global; could increase black-market activity.
Mid (2): Labels added but often ignored by experienced users.
Failure (worst - 3): Little impact on bad actors; adds cost/delays for legitimate builders.
Unintended consequences: Drives activity underground, reducing community visibility/oversight.
Action 3: Treat as if it has a Regulatory Classification as Restricted Biotech Equipment (e.g., Licensing for High-Voltage Builds) Pledge reporting and Safe use.
Goal: Treat advanced DIY electroporators like controlled lab tools - require permits/training for >1,000 V devices to prevent proliferation to high-risk genetic work.
Design:
Opt-in: Individual builders/users via registration.
Fund: User fees.
Approve: Government agencies (e.g., expanding CDC/NIH biosafety rules or local health depts).
Implement: Permits, training requirements, inspections for community labs/shared spaces.
Hazard Vulnerability Assessment (HVA) and Peer Review: Conduct a comprehensive HVA and require peer review through a pseudo-IRB-like entity - a multidisciplinary and independent review board focusing on environmental and human safety. This entity would evaluate proposed uses, assess risks, and provide guidance on safe protocols before high-voltage builds are deployed.
Risks / What could go wrong: Hard to define safe thresholds; bureaucracy kills accessibility; overreach chills innovation globally.
Unintended consequences: Harms global equity/education; favors institutional labs only.
Overall Tradeoffs & Prioritization
Prioritize Action 1 (community self-governance) as primary: Lowest overregulation risk, aligns with DIY ethos, adaptable to low current misuse evidence, leverages community goodwill.
Combine with Action 2 (targeted warnings) as secondary: Adds minimal external safeguard for public health, deters casual risks without bans.
Avoid/minimize Action 3 unless clear evidence of high-risk proliferation: Highest chance of killing accessibility and innovation, poor fit for low-harm tool like BioVolt.
Key uncertainties (misuse rates, community response, enforcement feasibility) favor lighter interventions. Monitor via voluntary reporting; escalate only if serious incidents arise. This balances empowerment with responsible governance for biosafety and preventing broader DIY genetic risks.
Made with love and the AI Slop is from Cursor-GLM 4.7
Week 1: Professor Questions
Answers organized by instructor, please click the question to reveal the answer!
Instructions: Click the triangle (▶) or question text to expand and view the full answer.
[SECTION 1] Questions from Professor Jacobson
Source: Lecture 2 slides
▶ Question 1: Nature's machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
Answer
Executive Summary: DNA polymerase intrinsic error rate (~10⁻⁷) would cause ~320 errors per human genome replication (3.2 × 10⁹ bp). Biology employs multilayer error correction (proofreading, mismatch repair, excision repair) to achieve final fidelity of ~10⁻⁹ to 10⁻¹⁰ errors per base per division, yielding 0.3-3 errors per replication in normal somatic cells.
Error Rate of DNA Polymerase
DNA polymerase has an intrinsic error rate of approximately 1 error per 10⁷ nucleotides during DNA synthesis. With integrated 3’ to 5’ exonuclease proofreading activity, this improves to approximately 1 error per 10⁸-10⁹ nucleotides.
Comparison to Human Genome Length
The human genome contains approximately 3.2 × 10⁹ base pairs.
Without proofreading:
Error rate: ~10⁻⁷ per nucleotide
Expected errors per replication: ~320 errors per genome copy
With proofreading:
Error rate: ~10⁻⁸ to 10⁻⁹ per nucleotide
Expected errors per replication: ~3-32 errors per genome copy
How Biology Deals with This Discrepancy
Biology employs multiple layers of error correction that act sequentially:
Proofreading (3’ → 5’ exonuclease activity)
DNA polymerase detects incorrect base pairing via geometric distortion
Removes mismatched nucleotide immediately
Reduces error rate by approximately 100-1000-fold
Mismatch Repair (MMR) System
Post-replication surveillance mechanism
In bacteria (E. coli): MutS, MutL, and MutH proteins
In eukaryotes: MSH (MutS homolog), MLH (MutL homolog), and PMS protein families
System identifies mismatched base pairs, excises incorrect strand segment, and resynthesizes
Further reduces error rate by approximately 100-1000-fold
Nucleotide Excision Repair (NER)
Repairs bulky DNA lesions (UV-induced thymine dimers, chemical adducts)
Removes damaged nucleotide segments (20-30 nt patches)
Base Excision Repair (BER)
Corrects small base modifications (deamination, oxidation, alkylation)
DNA glycosylases remove damaged bases; AP endonucleases process abasic sites
Result: The combined fidelity of replication in eukaryotic somatic cells typically achieves ~10⁻⁹ to 10⁻¹⁰ errors per base per cell division, depending on organism, cell type, and proliferation status. This ensures 0.3-3 errors per genome replication under normal physiological conditions.
Note: Fidelity varies by context. Cancer cells with MMR defects exhibit 100-1000× higher mutation rates. Germline cells employ additional proofreading mechanisms. Some DNA polymerases (e.g., Pol η, translesion synthesis polymerases) have lower fidelity by design for specialized repair functions.
▶ Question 2: How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don't work to code for the protein of interest?
Answer
Executive Summary: For a typical 400-residue protein, the number of synonymous DNA sequences (due to codon degeneracy) is astronomically large—on the order of 10¹⁰⁰ or more, calculated as the product of synonymous codon counts across all positions. In practice, most sequences fail due to codon usage bias, mRNA secondary structure, RNA instability, splicing interference, cryptic regulatory elements, and synthesis/cloning constraints.
Number of Different Ways to Code for a Protein
The genetic code is degenerate—61 sense codons encode 20 standard amino acids plus start/stop signals. Each amino acid (except Met and Trp) has multiple synonymous codons:
Leucine, Serine, Arginine: 6 codons each
Isoleucine: 3 codons
Methionine, Tryptophan: 1 codon each
For an average human protein (~400 amino acids):
The total number of synonymous DNA sequences is the product of synonymous codon counts across all positions:
N = ∏(i=1 to 400) n_i
where n_i = number of synonymous codons for amino acid i.
Rough estimate:
Average degeneracy per amino acid ≈ 3 codons (weighted by frequency)
Total combinations ≈ 3⁴⁰⁰ ≈ 10¹⁹⁰ possible DNA sequences
Even conservative estimates (e.g., leucine-rich proteins) yield 10¹⁰⁰+ combinations.
Why All These Different Codes Don’t Work in Practice
Even though multiple sequences encode the same amino acid sequence, the vast majority fail to express functional protein due to:
1. Codon Usage Bias
Each organism has preferred codons reflecting tRNA abundance (Plotkin & Kudla 2011)
E. coli prefers different codons than humans (e.g., AGG/AGA rare in bacteria, common in mammals)
Rare codons → ribosome stalling → may alter co-translational folding kinetics
Using non-optimal codons can reduce expression 10-1000-fold (Gustafsson et al. 2004)
2. mRNA Secondary Structure
Certain nucleotide sequences form stem-loops or hairpins
Strong secondary structures can:
Block ribosome binding
Stall translation
Trigger mRNA degradation
3. RNA Stability
AU-rich sequences → rapid mRNA degradation
GC-rich sequences → more stable mRNA
Wrong codon choice can drastically reduce mRNA half-life
4. Splicing Interference
Certain sequences create cryptic splice sites
Can cause exon skipping or intron retention
Results in truncated or non-functional protein
5. Ribosome Binding Sites (RBS) Interference
Shine-Dalgarno sequences (prokaryotes) or Kozak sequences (eukaryotes)
Internal RBS-like sequences can cause premature translation initiation
Results in truncated proteins
6. Restriction Enzyme Sites
Cloning often requires avoiding certain restriction sites
Limits sequence choices for practical molecular biology
7. Repetitive Sequences
Long homopolymer runs (e.g., AAAAAA) cause synthesis/sequencing errors
Can trigger recombination or replication errors
Quantitative Example:
For a 10-amino acid peptide (assuming average 3-fold degeneracy), there are theoretically 3¹⁰ ≈ 59,000 synonymous sequences. However, accounting for all the constraints listed above, only an estimated 10²-10³ sequences (~1-2%) would be practically functional.
[SECTION 2] Questions from Dr. LeProust
Source: Lecture 2 slides
▶ Question 3: What's the most commonly used method for oligo synthesis currently?
Answer
Executive Summary: Phosphoramidite chemistry on solid-phase support (Caruthers method, 1981) is the current industry standard, with typical coupling efficiency of 98.5-99.5% per cycle and practical length ceiling of 150-200 nucleotides.
Phosphoramidite Chemistry (Solid-Phase Synthesis)
The phosphoramidite method on solid support is the dominant technology for oligonucleotide synthesis worldwide.
Key Features:
Invented: Marvin Caruthers and colleagues (1981)
Platform: Solid-phase synthesis on controlled-pore glass (CPG) or polystyrene beads
Direction: 3’ → 5’ synthesis (chain grows from 3’-OH to 5’ end)
Cycle efficiency: Typically 98.5-99.5% per nucleotide addition
Practical length limit: 150-200 nucleotides for routine synthesis
Four-Step Cycle:
Detritylation (acid treatment)
Removes DMT (dimethoxytrityl) protecting group from 5’-OH
Converts unstable phosphite (P³⁺) to stable phosphate (P⁵⁺)
Forms phosphate backbone
Advantages:
High throughput (96-384 well formats)
Automated
Scalable (nmol to µmol scale)
Well-established chemistry
Current Platforms: Commercial platforms include BioAutomation and ABI/Applied Biosystems synthesizers for traditional column-based synthesis. Newer high-throughput approaches include Twist Bioscience (silicon-based microarray synthesis) and Custom Array (electrochemical synthesis on chips).
▶ Question 4: Why is it difficult to make oligos longer than 200nt via direct synthesis?
Answer
Executive Summary: Cumulative coupling inefficiency (even at 99% per cycle) yields only ~13% full-length product at 200 nt. Dominant failure modes are deletion sequences from incomplete coupling, depurination during detritylation, and increasing purification difficulty as n-1, n-2… products accumulate.
Cumulative Coupling Errors and Deletion Sequences
The primary challenge is imperfect coupling efficiency in each phosphoramidite addition cycle.
The Mathematics of Error Accumulation:
Coupling efficiency per cycle: typically 98.5-99.5%
Stepwise failure rate: 0.5-1.5% per cycle
Yield of full-length product = (coupling efficiency)^n where n = oligo length
Yield Calculation:
Length
Coupling Efficiency
Full-Length Yield
50 nt
99%
60%
100 nt
99%
37%
150 nt
99%
22%
200 nt
99%
13%
300 nt
99%
5%
At 200 nucleotides with 99% efficiency:
Only 13% of molecules are full-length correct sequence
87% are deletion products (n-1, n-2, n-3… truncations)
Specific Problems Beyond 200nt (in order of impact):
Deletion Sequences from Incomplete Coupling
Failed coupling at position i → all subsequent additions build on truncated chain
Creates heterogeneous mixture of n-1, n-2, n-3… products
Capping step blocks these from extending, but they remain in final pool
Depurination During Acid Treatment
Detritylation uses trichloroacetic acid or dichloroacetic acid
Causes glycosidic bond cleavage at purines (A, G)
Cumulative damage over 200+ cycles
Results in abasic sites and chain breaks
Purification Difficulty
Full-length (200 nt) vs. n-1 (199 nt) differ by <0.5% in mass
HPLC and PAGE separation becomes marginal
Impure product affects downstream applications
Secondary Structure Formation
Long single-stranded oligos form intramolecular hairpins during synthesis
Blocks reagent access to growing 3’-OH end (on solid support, growing from 3’ end)
Reduces effective coupling efficiency in later cycles
Practical Solutions: Modern approaches avoid direct synthesis beyond 200 nt by using gene assembly from overlapping 60-80 nt oligos (polymerase cycling assembly, Gibson assembly), column-based assembly methods (e.g., Twist Bioscience chip synthesis followed by assembly), or emerging enzymatic synthesis using terminal deoxynucleotidyl transferase-based methods.
▶ Question 5: Why can't you make a 2000bp gene via direct oligo synthesis?
Answer
Executive Summary: Direct phosphoramidite synthesis of 2000 nt is practically infeasible due to vanishingly low yields (0.99^2000 ≈ 10⁻⁹), prohibitive synthesis time (~2-3 weeks continuous), cumulative depurination, and insurmountable purification challenges. Modern gene synthesis uses hierarchical assembly of 60-80 nt oligos into fragments, then full-length genes.
Practical Infeasibility with Current Phosphoramidite Chemistry
Making a 2000 bp gene via direct oligonucleotide synthesis is practically infeasible with standard phosphoramidite chemistry due to insurmountable yield, time, and purification barriers.
Yield Barriers:
At 99% coupling efficiency (best-case scenario):
Yield = 0.99^2000 ≈ 2 × 10⁻⁹ (0.0000002%)
To obtain 1 picomole of full-length product requires ~0.5 moles of starting material
Equivalent to ~660 grams of protected nucleotide phosphoramidites
Material cost alone: ~$500,000 - $1,000,000
Even at 99.5% efficiency (exceptional, rarely achieved):
Yield = 0.995^2000 ≈ 5 × 10⁻⁵ (0.005%)
Still economically and practically prohibitive
Physical/Chemical Barriers:
Synthesis Time
Typical cycle time: 10-15 minutes per nucleotide addition
2000 cycles = 20,000-30,000 minutes = 14-21 days continuous synthesis
Reagent degradation over extended periods
Instrument reliability over multi-week runs
Cumulative Depurination
2000 acid detritylation steps
Each cycle causes low-frequency glycosidic bond cleavage at purines
Accumulates to extensive abasic sites and strand breaks
Secondary Structure Collapse
Long single-stranded DNA forms extensive intramolecular structure
Hairpins and G-quadruplexes block reagent access
Synthesis typically stalls beyond 300-400 nt even with optimized conditions
Solubility and Handling
Very long oligos can precipitate on solid support
Reduced accessibility to coupling reagents
Cleavage and deprotection become inefficient
Practical Solution: Hierarchical Gene Assembly
Modern commercial gene synthesis uses multi-step assembly:
Step 1: Oligo Synthesis
Synthesize 30-50 oligonucleotides (60-80 nt each, with 20-40 nt overlaps)
Yield per oligo: 60-95% (high quality)
Step 2: Fragment Assembly
Assemble oligos into 4-6 intermediate fragments (400-600 bp each)
Methods: Polymerase cycling assembly (PCA), Gibson assembly, Golden Gate
Yield per fragment: 70-90%
Step 3: Final Assembly
Combine fragments into full 2000 bp gene
Gibson assembly or restriction enzyme-based methods
Final yield: 60-85% overall
Example for 2000 bp gene:
40 oligos × 70 nt average = 2800 nt synthesized capacity
Assemble into 5 fragments (~400 bp each)
Final Gibson assembly into 2000 bp construct
Overall yield: ~70% (vs. 10⁻⁹% for direct synthesis)
Commercial Gene Synthesis: Major vendors (Twist Bioscience, IDT, GenScript, Thermo Fisher) offer typical academic pricing of $0.07-0.20/bp, though this is highly variable depending on sequence complexity (GC content, repeats, secondary structure), turnaround time (5-10 days standard, 2-3 days expedited), and order volume. Standard turnaround is 5-10 days with rush options of 2-3 days.
[SECTION 3] Question from Professor George Church
Source: Lecture 2 slides
▶ Question 6: [Using Google & Prof. Church's slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the "Lysine Contingency"?
(I chose this question from the three options)
Answer
Executive Summary: The commonly listed essential amino acids in vertebrates include His, Ile, Leu, Lys, Met, Phe, Thr, Trp, Val, and conditionally Arg. The “Lysine Contingency” from Jurassic Park is scientifically flawed because lysine is already naturally essential in all vertebrates—the genetic modification provides zero additional biocontainment. Moreover, lysine is abundant in all natural food sources, and deficiency takes months to years to be lethal.
The Commonly Listed Essential Amino Acids in Vertebrates
Essential amino acids cannot be synthesized de novo by vertebrate metabolism and must be obtained from diet. The standard list for humans and most vertebrates includes: Histidine (His, H), Isoleucine (Ile, I), Leucine (Leu, L), Lysine (Lys, K) [focus of Jurassic Park scenario], Methionine (Met, M), Phenylalanine (Phe, F), Threonine (Thr, T), Tryptophan (Trp, W), Valine (Val, V), and Arginine (Arg, R), which is conditionally essential—essential in juveniles, young/growing animals, and during illness, though adults can synthesize limited amounts via the urea cycle.
Mnemonic:“PVT TIM HALL” (Phe, Val, Thr, Trp, Ile, Met, His, Arg, Leu, Lys)
Note: The classification varies slightly by species and life stage. Arginine is typically considered semi-essential or conditionally essential in adult mammals.
The “Lysine Contingency” from Jurassic Park
In Jurassic Park (Michael Crichton, 1990), InGen implemented a “Lysine Contingency” as a biocontainment measure. The plan involved genetically engineering dinosaurs unable to synthesize lysine, making them dependent on lysine supplements in their food. The theory was that if they escaped, they would die from lysine deficiency. As Dr. Wu stated: “The lysine contingency is intended to prevent the spread of the animals is case they ever got off the island.”
Why the Lysine Contingency is Scientifically Flawed
Critical Problem: ALL ANIMALS ALREADY REQUIRE DIETARY LYSINE
1. Lysine is Naturally Essential in All Vertebrates
Humans, dinosaurs, birds, and mammals cannot synthesize lysine de novo. Animals lost the lysine biosynthesis pathway approximately 500 million years ago during early vertebrate evolution. The dinosaurs would have required dietary lysine regardless of any genetic modification. Therefore, the “contingency” provides zero additional biocontainment—it is entirely redundant.
2. Lysine is Abundant in Natural Food Sources
Based on USDA nutritional databases, lysine is widespread in both plant and animal food sources. Plant sources include legumes (soybeans, lentils, beans) containing 1-2% lysine by dry weight, seeds and grains with 0.2-0.8% lysine, and grasses and leafy vegetation with 0.3-0.6% lysine. Animal sources are even richer: insects contain approximately 2-3% lysine by dry weight, while vertebrate muscle tissue, fish, and eggs contain 1.5-2.5% lysine by weight.
Estimated lysine intake for large theropods (carnivorous dinosaurs):
Note: The following are rough extrapolations from modern vertebrate nutritional requirements and are not based on direct measurements of dinosaur metabolism. Assuming an estimated daily food intake of 50-100 kg meat (scaled from modern large carnivores) and lysine content of meat at approximately 1.5-2.0 g/100g, the estimated daily lysine intake would be 750-2000 g. Compared to an estimated lysine requirement of approximately 10-50 g/day (scaled from mammals, though highly uncertain), even conservative estimates suggest 10-100× excess lysine intake.
Estimated lysine intake for herbivorous dinosaurs:
Assuming estimated daily vegetation consumption of hundreds of kg for sauropods and lysine content in plant matter of 0.3-1.0% dry weight, the estimated daily lysine intake would be hundreds of grams. This substantially exceeds the likely requirement of 50-200 g/day when scaled from large herbivorous mammals.
Key Point: Even consuming exclusively grass, leaves, or insects would likely provide sufficient lysine to meet metabolic needs, assuming dinosaur requirements scaled similarly to modern vertebrates.
3. Timescale of Lysine Deficiency is Impractical
Lysine deficiency symptoms develop slowly: immune system impairment occurs over weeks to months, growth retardation takes months, and muscle wasting progresses over months to years. Lethality from severe deficiency requires months to years. A dinosaur escaping into the wild would eat naturally available food and immediately obtain sufficient lysine, never developing deficiency symptoms. The timescale mismatch is fatal to the strategy: containment must occur in minutes to hours (the escape window), while lysine deficiency lethality takes months to years. The result is a completely ineffective biocontainment strategy.
4. Better Biocontainment Strategies
If the goal is preventing escaped dinosaurs from surviving or reproducing, several approaches would be more effective than the lysine contingency.
Metabolic Dependencies: Creating auxotrophy for synthetic amino acids not found in nature (such as D-amino acids or unnatural amino acids requiring continuous supplementation), nucleotide auxotrophy (e.g., thymine requirement), or vitamin/cofactor dependencies (e.g., engineered B12 requirement) would provide genuine containment.
Genetic Kill Switches: Conditional lethality genes requiring antidote molecules, thermosensitive essential genes that allow survival only at controlled temperatures, or light-dependent survival mechanisms requiring specific UV or wavelength exposure offer programmed containment.
Reproductive Control: All-female populations (as attempted in Jurassic Park), meiotic drive systems ensuring sterility, or genetic incompatibility with wild relatives would prevent population establishment.
Environmental Dependencies: Temperature-sensitive phenotypes surviving only in controlled climates or organisms requiring specific atmospheric pressure or composition would restrict habitat range.
Conclusion: How This Affects My View of the Lysine Contingency
The Lysine Contingency is scientifically flawed as a biocontainment strategy and represents a misunderstanding of vertebrate nutritional biochemistry. The strategy fails on four fundamental levels: (1) it is not a contingency since lysine is already naturally essential in all vertebrates, making the modification redundant; (2) it is not limiting since lysine is abundant in nearly all natural food sources; (3) it is not fast-acting since lysine deficiency takes months to years to be lethal in large vertebrates; and (4) it provides no additional biocontainment barrier beyond natural biology.
From a biosafety perspective, the lysine contingency demonstrates the risk of “security theater” in synthetic biology—creating the appearance of control without meaningful containment. Real biocontainment requires dependencies on synthetic or artificial inputs not present in natural ecosystems. Modern synthetic biology approaches include unnatural amino acid dependencies (e.g., amber suppressor systems with synthetic tRNAs), genetic kill switches (toxin-antitoxin modules, essential gene knockout with complementation), orthogonal genetic systems (expanded genetic code, xenobiology with XNA), and metabolic dependencies on synthetic nutrients or specific light wavelengths.
Narrative function in Jurassic Park: The flawed lysine contingency serves as a plot device illustrating InGen’s overconfidence and foreshadows that all their control measures will fail (“Life finds a way”). It highlights the dangers of inadequate risk assessment and overconfidence in genetic engineering safeguards.
Lessons for modern synthetic biology: Biological containment is extremely difficult and requires multiple redundant safeguards. Single-point dependencies, especially on naturally occurring molecules, are inadequate. Rigorous testing and evolutionary escape rate measurements are essential for any containment strategy.
[REFERENCES]
Primary Literature and Resources
DNA Replication Fidelity (Q1):
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 6th edition. Garland Science, 2014. Chapter 5: DNA Replication, Repair, and Recombination.
Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497-529. doi:10.1146/annurev.biochem.69.1.497
Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106(2):302-323. doi:10.1021/cr0404794
Genetic Code and Translation (Q2):
Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12(1):32-42. doi:10.1038/nrg2899
Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22(7):346-353. doi:10.1016/j.tibtech.2004.04.006
Tuller T, Carmi A, Vestsigian K, et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141(2):344-354. doi:10.1016/j.cell.2010.03.031
Oligonucleotide Synthesis (Q3-Q5):
Caruthers MH. Gene synthesis machines: DNA chemistry and its uses. Science. 1985;230(4723):281-285. doi:10.1126/science.3863253
Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11(5):499-507. doi:10.1038/nmeth.2918
Hughes RA, Ellington AD. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb Perspect Biol. 2017;9(1):a023812. doi:10.1101/cshperspect.a023812
Amino Acid Nutrition and Biosafety (Q6):
Reeds PJ. Dispensable and indispensable amino acids for humans. J Nutr. 2000;130(7):1835S-1840S. doi:10.1093/jn/130.7.1835S
WHO/FAO/UNU Expert Consultation. Protein and amino acid requirements in human nutrition. WHO Technical Report Series 935. Geneva: World Health Organization; 2007.
USDA National Nutrient Database for Standard Reference (Release 28). Agricultural Research Service, U.S. Department of Agriculture. 2015.
Crichton M. Jurassic Park. New York: Alfred A. Knopf; 1990.
Mandell DJ, Lajoie MJ, Mee MT, et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature. 2015;518(7537):55-60. doi:10.1038/nature14121 [Modern unnatural amino acid containment systems]
Document created: February 10, 2026 Author: James Utley, PhD Affiliation: Syndicate Laboratories, Panama City, Panama Course: HTGAA 2026 Spring — Week 1 Homework
Week 2 HW: DNA Read, Write, & Edit
🧬 Week 2 Homework Components
DNA Read, Write, & Edit — sequencing and synthesis workflows, restriction digests and gel electrophoresis, genome-editing frameworks.
Simulated restriction enzyme digestion with the seven enzymes specified in this week’s lab protocol: SalI, SacI, EcoRV, KpnI, BamHI, HindIII, and EcoRI. Used both the DNA Gel Art Interface (λ DNA) and Benchling (lambda phage genome NC_001416) to visualize digest patterns and verify cut-site predictions.
1. DNA Gel Art Interface — λ DNA Restriction Digests
Simulated gel electrophoresis using the DNA Gel Art tool. λ DNA was digested with various enzyme combinations (EcoRV + SacI, HindIII + PvuII, NdeI + SalI, etc.) across lanes 2–10. The table documents water, CutSmart buffer, λ DNA, and enzyme volumes per lane.
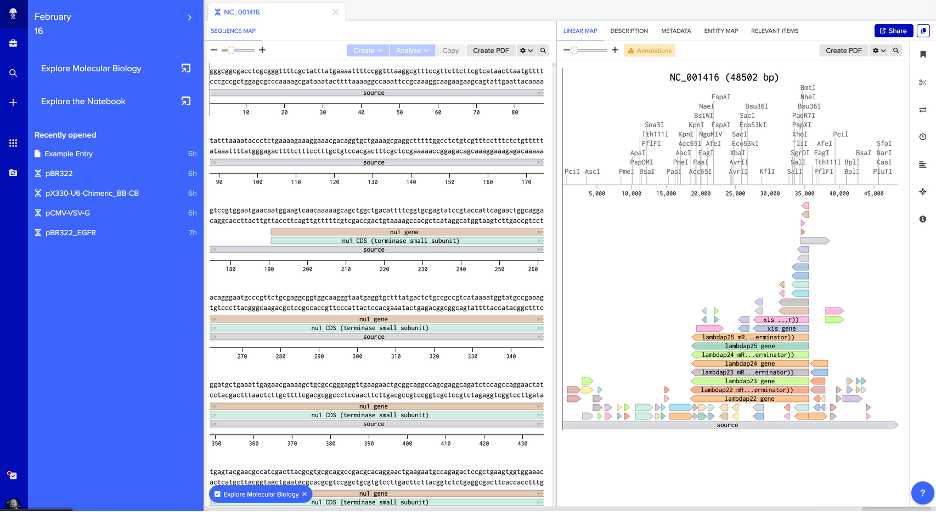
2. Benchling — NC_001416 Sequence Map with Restriction Sites
Linear map of NC_001416 in Benchling showing the raw sequence, annotated genetic features (e.g., xis, nul, lambdap genes), and restriction enzyme cut sites (PciI, AscI, PmeI, BsaI, KpnI, SacI, SalI, and others) along the 48.5 kb genome.
3. Virtual Digest Gel — NC_001416 with All Seven Required Enzymes
Simulated gel (Life 1 kb Plus ladder) showing digest results for NC_001416 with each of the seven required enzymes:
Protein chosen: Superfolder Green Fluorescent Protein (sfGFP)
Why: sfGFP is a robust, rapidly maturing fluorescent protein derived from Aequorea victoria (Pédelacq et al., 2005). It is widely used in synthetic biology as a reporter—when expressed in cells, it fluoresces bright green under blue/UV light, enabling real-time visualization of gene expression, protein localization, and cell tracking. Its “superfolder” mutations improve folding efficiency in diverse hosts (including E. coli), making it ideal for expression experiments. It also connects directly to Part 4, where we build an expression cassette to make E. coli glow green.
Using the Central Dogma in reverse: given a protein sequence, we infer a possible DNA sequence that could encode it. Because the genetic code is degenerate (multiple codons encode the same amino acid), many DNA sequences can produce the same protein. A simple reverse translation uses one valid codon per amino acid—here, E. coli preferred codons (most frequently used in highly expressed genes).
Tool used: Reverse translation with E. coli codon preferences (e.g., ExPASy Translate or similar tools; can also be done manually with a codon usage table).
Reverse-translated DNA sequence (one possible encoding):
Why optimize codon usage? Different organisms prefer different codons for the same amino acid, based on tRNA abundance and other factors. Using rare codons can slow translation, cause ribosome stalling, and reduce protein yield. Codon optimization replaces codons with those most frequently used in the target organism, improving expression levels and folding. It also allows us to avoid restriction enzyme recognition sites (e.g., BsaI, BsmBI, BbsI) that would interfere with Golden Gate or other assembly methods.
Organism chosen:Escherichia coli (K-12)
Why E. coli? It is the standard workhorse for recombinant protein expression: well-characterized genetics, fast growth, simple culture, and widely available vectors and protocols. The HTGAA Part 4 exercise uses E. coli for the sfGFP expression cassette, so optimizing for E. coli keeps the workflow consistent.
(717 bp; optimized for E. coli expression, restriction-site free — same sequence used in Part 4 expression cassette)
3.4 You Have a Sequence! Now What?
Technologies to produce sfGFP from this DNA:
Cell-dependent (recombinant expression in E. coli):
Clone the codon-optimized gene into an expression vector (e.g., pTwist Amp High Copy) with a constitutive or inducible promoter (e.g., BBa_J23106), RBS (e.g., BBa_B0034), and terminator (e.g., BBa_B0015).
Transform the plasmid into E. coli (e.g., DH5α, BL21).
Grow cells; the host RNA polymerase transcribes the DNA into mRNA, and ribosomes translate the mRNA into sfGFP.
The protein folds and forms its chromophore; cells fluoresce green under blue light (~488 nm excitation, ~510 nm emission).
Cell-free (in vitro transcription–translation):
Use a cell-free system (e.g., E. coli lysate, PURE system) with the DNA template.
Add NTPs, amino acids, and energy sources; the system transcribes and translates the gene without living cells.
Useful for rapid prototyping, toxic proteins, or when cell growth is impractical.
DNA synthesis (Twist, IDT, etc.):
Order the gene as a clonal or linear fragment from a synthesis provider.
Use it directly for cloning or cell-free expression, avoiding PCR or cloning from natural sources.
Alignment of DNA, RNA, and protein: In the Central Dogma, DNA is transcribed to RNA (T→U), and RNA is translated to protein (3 nt → 1 aa). Tools like Benchling or Ronan’s gel art site can visualize this alignment.
Single gene → multiple proteins: Alternative splicing (eukaryotes) or alternative start codons/ribosomal frameshifting can produce multiple proteins from one gene. sfGFP is a single open reading frame, but in general, one gene can yield multiple isoforms through these mechanisms.
Part 4: Prepare a Twist DNA Synthesis Order
Part 4: Prepare a Twist DNA Synthesis Order
Practice exercise — building an sfGFP expression cassette in Benchling, preparing a mock Twist order, and annotating the plasmid.
4.1–4.2 Accounts & Build Your DNA Insert Sequence
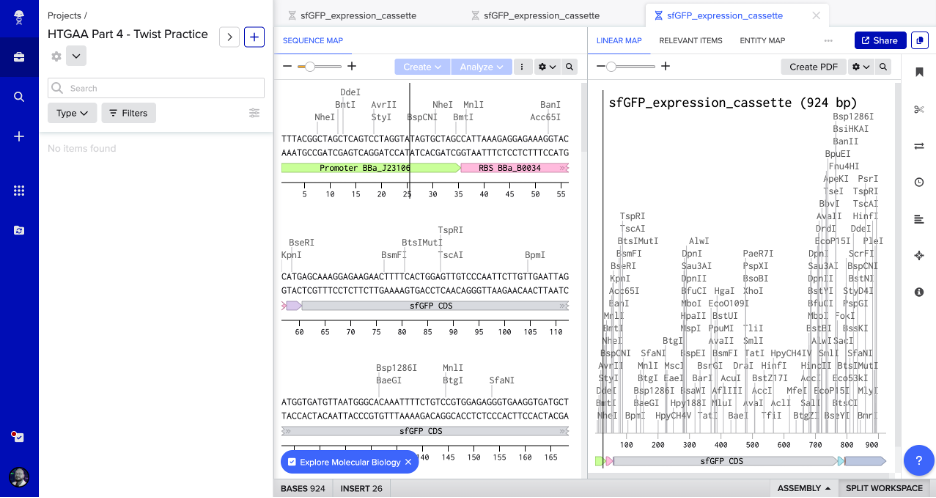
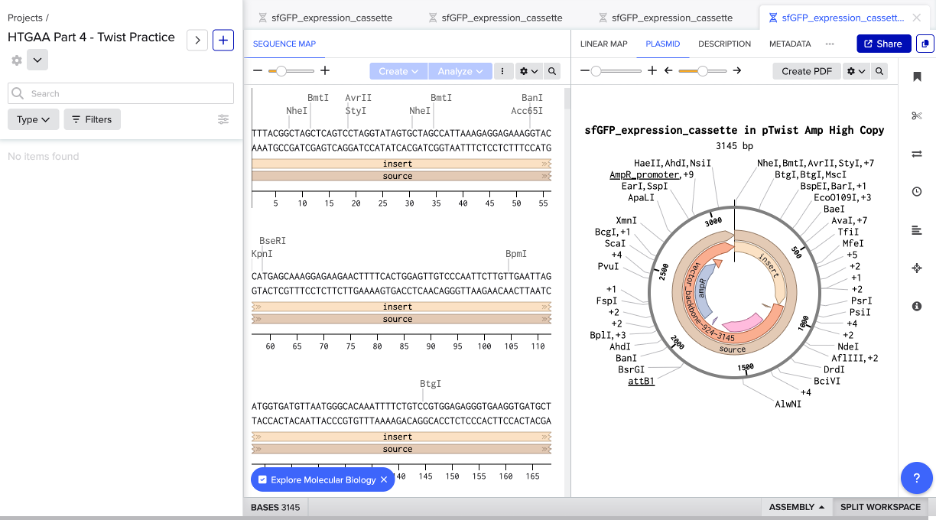
Created Twist and Benchling accounts. Built the sfGFP expression cassette in Benchling with annotated parts:
Promoter (BBa_J23106)
RBS (BBa_B0034)
Start codon (ATG)
Coding sequence (codon-optimized sfGFP from Part 3)
Downloaded GenBank construct and imported into Benchling
Screenshot: Sequence Upload to Twist
Design Notes: Manual vs. Programmatic
Efficiency: Designing expression cassettes and plasmids can be far more efficient with Python and/or R — tools like DNA Chisel, PyDNA, or SynBioHub enable scripted design, validation, and export. Batch operations, automated codon optimization, and constraint checking become straightforward.
Learning value: Building the construct manually in Benchling — clicking through each part, copying sequences, and annotating by hand — offers a different kind of learning. You develop intuition for how promoters, RBSs, and CDSs fit together, where restriction sites fall, and what the plasmid “looks like” at each step. That tactile understanding is harder to get from a script. For a first expression cassette, the manual approach is worth the extra time.
MANUAL (Benchling) PROGRAMMATIC (Python/R)
───────────────── ─────────────────────
Click, paste, annotate Script → design → export
Slow, one construct at a time Fast, many constructs
Deep, tactile understanding Scalable, reproducible
"I built this" "I designed 50 of these"
Both have their place. Start manual; scale with code.
Answers framed around the BioVolt DIY electroporation pipeline: plasmid amplification → transformation → PCR verification → gel electrophoresis. What DNA would we read, write, and edit to make this frugal pipeline sing?
In the BioVolt pipeline: After electroporation, we transform E. coli with plasmids (e.g., sfGFP expression cassette). We run post-transformation PCR and gel electrophoresis to infer success—but we don’t know the exact sequence. Sequencing the plasmid (or PCR amplicon) confirms that:
The insert is correct (no truncations, no wrong gene)
Electroporation didn’t introduce mutations (high voltage can stress DNA)
The expression cassette is intact for downstream experiments
Broader applications (aligned with BioVolt’s democratization goals):
Environmental monitoring — e.g., sewage/wastewater DNA for microbiome analysis in Panama; biodiversity surveys
Human health — disease-associated genes, pharmacogenomics
DNA data storage — archival sequences in synthetic DNA
Portable — USB-sized device; runs on laptop; fits in a backpack. Ideal for Panama, field sites, or home labs.
Real-time — base calling as reads stream; no batch wait.
Long reads — can span full plasmids; fewer assembly gaps.
Low capital — compared to Illumina, much cheaper to get started.
No PCR required for some workflows — direct DNA sequencing possible (native DNA).
Question
Answer
Output?
FASTQ files (reads + quality scores); can be base-called in real time to BAM/FASTA.
Essential steps & base calling?
(1) DNA passes through a nanopore; (2) each base disrupts ionic current differently; (3) base caller (e.g., Guppy) converts current traces → A/T/G/C; (4) reads assembled/compared to reference.
Input & preparation?
Option A (PCR amplicon): PCR product → end-prep → adapter ligation → load onto flow cell. Option B (native): Fragment DNA (e.g., g-TUBE or sonication) → repair ends → adapter ligation → load. Key: adapters enable motor protein to thread DNA through pore.
First-, second-, or third-generation?
Third-generation. Single-molecule, real-time; no amplification required for some lib preps; long reads; portable form factor.
NANOPORE SEQUENCING (simplified)
╭───-╮
DNA ────► │ ▓▓ │ ← pore in membrane
│ ▓▓ │ (ionic current changes per base)
╰───-╯
│
▼
╔═══════════╗
║ A T G C ║ ← base caller (Guppy, etc.)
║ ▓ ▓ ▓ ▓ ║ converts squiggle → sequence
╚═══════════╝
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
For BioVolt: The expression cassettes we electroporate! Specifically:
sfGFP plasmid — promoter + RBS + sfGFP CDS + terminator (e.g., BBa_J23106, BBa_B0034, sfGFP, BBa_B0015). This is the “make E. coli glow green” construct we build in Part 4.
Custom reporters — e.g., biosensors that fluoresce in response to environmental cues (pH, metals, toxins) for citizen-science monitoring.
Validation controls — known sequences for PCR/gel positive controls in the frugal pipeline.
Broader: Therapeutics (mRNA vaccines), genetic circuits, DNA origami, gene clusters for metabolic engineering.
WHAT WE SYNTHESIZE FOR BIOVOLT:
┌────────────────────────────────────────────────────────────┐
│ [Promoter]─[RBS]─[ATG]─[sfGFP]─[His]─[TAA]─[Terminator] │
│ │ │ │
│ └── always on └── glows green under UV │
│ │
│ Twist / IDT makes this. BioVolt zaps it in. Done. 🟢 │
└────────────────────────────────────────────────────────────┘
(ii) What technology would you use and why?
Technology:Column-based phosphoramidite synthesis (e.g., Twist Bioscience, IDT) — the industry standard for gene synthesis.
Why: High fidelity, scalable, cost-effective for genes and gene fragments. Twist can deliver clonal genes (circular) ready for transformation—perfect for BioVolt.
Question
Answer
Limitations?
Speed: days to weeks. Accuracy: ~1 error per 1–3 kb; may need sequencing to confirm. Scalability: great for genes; whole genomes get expensive. Length: very long constructs may need assembly.
Essential steps?
(1) Design sequence (e.g., codon-optimized); (2) split into overlapping oligos; (3) synthesize oligos (phosphoramidite chemistry, base-by-base); (4) assemble oligos (PCR, Gibson, or enzymatic); (5) clone into vector; (6) sequence to verify.
PHOSPHORAMIDITE SYNTHESIS (cartoon)
Base + Base + Base + ... → oligo → assemble → gene
A T G C A T ...
│ │ │ │ │ │
▼ ▼ ▼ ▼ ▼ ▼
┌───┴───┴───┴───┴───┴───┴───----┐
│ ████ ████ ████ ████ ████ │ ← solid support (column)
│ add → couple → oxidize → cap │ (repeat ~hundreds of times)
└─────────────────────────────- ┘
5.3 DNA Edit
(i) What DNA would you want to edit and why?
For BioVolt:
Improve electroporation efficiency — edit E. coli to knock out or modify genes that affect membrane composition, cell wall, or DNA repair (e.g., recA, mutS) to get more transformants per zap.
Biosensor chassis — edit strains to express reporter circuits (e.g., GFP under metal-responsive promoter) for environmental sensing in the DIY pipeline.
Safety — auxotrophic markers, kill switches, or containment edits for responsible DIYbio.
EDIT E. coli FOR BETTER BIOVOLT TRANSFORMATION?
Wild-type E. coli Edited E. coli
│ │
│ "Membrane too tough" │ "Softer membrane?"
│ "DNA repair too good?" │ "Fewer repair enzymes?"
│ │
▼ ▼
⚡ BioVolt ⚡ ⚡ BioVolt ⚡
│ │
▼ ▼
10³ CFU/µg 10⁵ CFU/µg? 🎯
│ │
"Meh" "Now we're talking!"
(ii) What technology would you use and why?
Technology:CRISPR/Cas9 (with HDR for precise edits) — or base editors for single-nucleotide changes without double-strand breaks.
Why: Programmable, precise, widely adopted. gRNA design is straightforward; many tools (Benchling, etc.) support it.
Question
Answer
Limitations?
Efficiency: not 100%; mixed populations. Precision: off-target cuts possible; PAM requirement constrains target sites. Delivery: need to get Cas9 + gRNA into cells (electroporation works!).
Preparation & input?
Design: gRNA(s) targeting locus; donor template (ssODN or plasmid) for HDR. Input: DNA template, Cas9 nuclease, gRNA (or plasmid expressing both), cells. Optional: base editor (e.g., ABE, CBE) for point mutations.
Essential steps?
(1) Design gRNA (avoid off-targets; check PAM, e.g., NGG for SpCas9); (2) deliver Cas9 + gRNA + donor (electroporation, conjugation, etc.); (3) Cas9 cuts DNA; (4) cell repairs via NHEJ or HDR; (5) screen for edits (PCR, sequencing).
CRISPR/Cas9 IN ACTION (simplified)
gRNA: "Find this sequence" ── ┐
├──► Cas9 ──► CUT! ✂️
DNA: ...TARGET...PAM... ──┘
Before: ────[TARGET]────
After: ────╲ ╱──── (cell repairs: NHEJ or HDR)
╲ ╱
gap
BioVolt could deliver Cas9 RNP + donor via electroporation! ⚡
Synthetic cells function as biological mimics of natural cells by mimicking salient features such as metabolism, response to stimuli, gene expression, direct metabolism, and high stability. Droplet-based microfluidic technology presents the opportunity for encapsulating biological functional components in uni-lamellar liposome or polymer droplets. Verified by its success in the fabrication of synthetic cells, microfluidic technology is widely replacing conventional labor-intensive, expensive, and sophisticated techniques justified by its ability to miniaturize and perform batch production operations.
Automation Tool
Droplet-based microfluidics — lab-on-chip systems that automate encapsulation, mixing, and batch production of synthetic cell constructs. Microfluidics serves as the automation platform: it replaces manual, labor-intensive methods with reproducible, tunable, high-throughput workflows.
The review discusses microfluidic chip design for synthetic cell preparation, the combination of microfluidics with bottom-up synthetic biology for reproductive and tunable construction, and advances in biosensors and biomedical applications.
Novel Aspects
Reproducible, tunable construction — Batch production from simple structures to higher hierarchical structures
Integration — Design, assembly, manipulation, and analysis within lab-on-chip devices
Biomedical relevance — Biosensors, drug delivery, therapeutic applications
Why This Paper Fits the Assignment
Microfluidics is an automation tool that achieves novel biological applications: it automates the fabrication of synthetic cells at scale, enabling research that would otherwise be labor-intensive and costly. The paper provides an overview of how this automation enables bottom-up synthetic biology and biomedical innovation.
Part 2: Automation Tools for Final Project — gumol + ECSOD + new-Clara
Project Overview
Project in development: A combined computational–experimental pipeline to study ECSOD (extracellular superoxide dismutase) overexpression from mesenchymal stem cells (MSCs) in acute radiation environments, with microfluidic validation serving as a surrogate for radiation exposure.
new-Clara is the primary automation tool in this project. It provides:
Controlled oxidative stress — Reproducible delivery of oxidative conditions as a surrogate for radiation
Precision and throughput — Automated, repeatable runs instead of manual handling
Data alignment — Outputs that can be directly compared with gumol MD results
Because radiation experiments are costly and regulated, the microfluidic oxidative environment acts as a surrogate for acute radiation, enabling validation of computational predictions under safer, more accessible conditions.
Answer any NINE of the following questions from Shuguang Zhang (i.e. you can select two to skip).
Answers provided for: (9 selected; 2 skipped: Can you make other non-natural amino acids? Design some new amino acids. and Design a β-sheet motif that forms a well-ordered structure.)
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer: Meat is roughly 15–25% protein by dry weight; water content varies. For a rough estimate, assume ~500 g of meat contains ~100 g of protein (≈20%). An average amino acid has a molecular mass of ~100 Daltons (Da).
Number of amino acids in 100 g protein ≈ 100 g / (100 × 10⁻³ kg/mol) ≈ 100 g / 0.1 kg/mol ≈ 1 mol ≈ 6 × 10²³ molecules (Avogadro’s number).
Order of magnitude: ~10²³–10²⁴ amino acid molecules per 500 g of meat.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer: Dietary proteins are digested into amino acids and small peptides before absorption. They are absorbed as monomers, not as intact proteins.
Absorption: Amino acids enter the bloodstream and are used as building blocks.
Assembly: Our cells use these amino acids to synthesize our own proteins according to our genome. The cow’s or fish’s DNA is never used; only the amino acid monomers are reused.
Result: We use the amino acids as nutrients; we do not incorporate the cow’s or fish’s proteins or genes intact. We remain human because our protein synthesis is controlled by human DNA.
3. Why are there only 20 natural amino acids?
Answer: The genetic code is degenerate: 61 sense codons encode 20 standard amino acids. The number 20 reflects a balance of evolutionary and physicochemical constraints:
Evolution: Early life likely used a smaller set of amino acids; the canonical 20 were added over time as biosynthesis pathways evolved.
Sufficiency: 20 amino acids provide enough chemical diversity (hydrophobic, polar, charged, aromatic, etc.) to build proteins with diverse structures and functions.
Genetic code: The triplet code (4³ = 64 codons) can encode more than 20, but expansion beyond 20 would require additional tRNA synthetases and codons; the cost of adding more may outweigh the benefit.
Fidelity: A larger set of amino acids would increase the risk of misincorporation and reduce translation fidelity.
Summary: 20 amino acids provide sufficient diversity for protein function while keeping the system manageable and robust.
4. Where did amino acids come from before enzymes that make them, and before life started?
Answer: Abiotic (prebiotic) synthesis.
Miller–Urey experiment (1952): Simulated early Earth conditions (reducing atmosphere, lightning, heat) produced amino acids (glycine, alanine, etc.) from simple precursors (H₂O, CH₄, NH₃, H₂).
Extraterrestrial sources: Amino acids (e.g., glycine) are found in meteorites (e.g., Murchison) and comets; they may have been delivered to early Earth.
Hydrothermal vents: Alkaline vents and other mineral surfaces can catalyze amino acid formation from CO₂, H₂, and nitrogen.
Strecker synthesis: Cyanide, aldehydes, and ammonia can form amino acids under prebiotic conditions.
Conclusion: Amino acids could form without enzymes or life, via abiotic chemistry and/or delivery from space.
5. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer:Left-handed (M-type) helix.
L-amino acids form right-handed (P-type) α-helices because the L-configuration places the side chain in a conformation that favors right-handed twist.
D-amino acids are the mirror image; their side chains favor the opposite twist. A D-amino acid α-helix is therefore left-handed.
6. Can you discover additional helices in proteins?
Answer: Yes. Beyond the canonical α-helix (3.6 residues/turn), other helices exist:
3₁₀ helix: ~3 residues/turn; tighter, shorter hydrogen bonds; often at helix termini.
π-helix: ~4.4 residues/turn; rare; energetically less favorable.
Polyproline helices (PPI, PPII): Proline-rich helices with different geometry.
Collagen-like structures: Triple helical motifs.
Novel helices: New helices can be discovered through structural biology (e.g., X-ray crystallography, cryo-EM) or designed de novo.
Conclusion: Additional helices can be found by analyzing protein structures and designing new motifs.
7. Why are most molecular helices right-handed?
Answer: Several factors favor right-handed helices:
Chirality of L-amino acids: All natural proteins use L-amino acids. The L-configuration favors right-handed α-helices and β-strands; left-handed helices are sterically strained.
DNA: Double helix is right-handed (B-form).
RNA: RNA helices are typically right-handed.
Minimization of steric clash: Right-handed twist often minimizes steric clashes between side chains and the backbone.
Evolution: Once right-handed helices dominated, the genetic code and biosynthesis reinforced this preference.
Summary: L-amino acid chirality and steric constraints favor right-handed helices in natural proteins.
8. Why do β-sheets tend to aggregate?
Answer: β-sheets expose backbone amide and carbonyl groups that can form hydrogen bonds with adjacent strands or sheets.
Hydrogen bonding: β-strands have alternating N–H and C=O groups along the backbone; these can pair with adjacent strands or with strands from another sheet.
Hydrophobic side chains: Many β-sheets have hydrophobic residues; stacking of sheets can bury these surfaces and reduce solvent exposure.
Extended conformation: Extended strands maximize surface area for inter-strand and inter-sheet contacts.
Amyloid-like stacking: β-sheets can stack in a parallel or antiparallel fashion, forming amyloid fibrils.
Conclusion: β-sheets aggregate because they expose H-bond donors/acceptors and hydrophobic surfaces that favor inter-sheet interactions.
9. What is the driving force for β-sheet aggregation?
Answer: Main driving forces:
Hydrogen bonding: Backbone–backbone H-bonds between strands from different molecules or sheets.
Hydrophobic effect: Burial of hydrophobic side chains reduces contact with water.
Entropy: Release of ordered water molecules when hydrophobic surfaces associate.
π–π stacking: Aromatic side chains (e.g., Phe, Tyr) can stack between sheets.
Electrostatic complementarity: Alternating charged and hydrophobic residues (e.g., in ionic self-complementary peptides) can drive ordered assembly.
Summary: H-bonding, hydrophobicity, and entropy release drive β-sheet aggregation.
10. Why do many amyloid diseases form β-sheets?
Answer: Many disease-associated proteins aggregate into amyloid fibrils rich in β-sheet structure:
Misfolding: Proteins that are normally α-helical or disordered can misfold into β-sheet-rich conformations under stress (e.g., pH, temperature, mutations).
Stability: Cross-β structure (β-strands perpendicular to the fibril axis) is highly stable; once formed, fibrils are difficult to disaggregate.
Nucleation: A small β-sheet nucleus can template further growth; amyloid formation is often nucleation-dependent.
Conclusion: β-sheet structure provides a stable, self-propagating amyloid conformation that underlies many neurodegenerative diseases.
11. Can you use amyloid β-sheets as materials?
Answer: Yes. Amyloid-like β-sheet structures are used as materials:
Self-assembling peptides: Shuguang Zhang’s ionic self-complementary peptides form stable β-sheet nanofibers and scaffolds for tissue engineering, drug delivery, and 3D cell culture.
Nanostructures: β-sheet fibrils can serve as templates for mineralization, nanowires, and conductive materials.
Hydrogels: β-sheet-rich peptide networks form hydrogels for wound healing and regenerative medicine.
Functional materials: Engineered amyloid fibrils have been used for catalysis, biosensors, and optical materials.
Conclusion: Amyloid β-sheets can be engineered as functional biomaterials for biomedical and material applications.
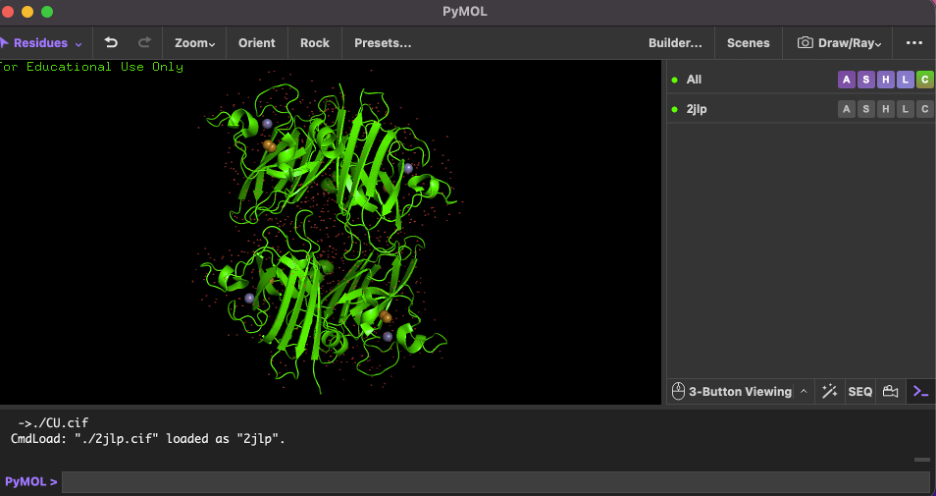
Why I selected it (brief): ECSOD is a secreted antioxidant enzyme that detoxifies superoxide radicals in the extracellular space, helping protect tissues from oxidative stress. I selected it because it is biologically important in vascular and lung biology, and a high-quality X-ray crystal structure is available for direct 3D visualization (PDB 2JLP).
1) Identify the amino acid sequence of the protein
Canonical protein sequence source: UniProt (Entry: P08294)
IMPORTANT NOTE ABOUT SEQUENCE VS STRUCTURE: The UniProt canonical protein is the biological sequence. The PDB structure often contains a construct/fragment and may not include every residue from the UniProt canonical sequence.
How to obtain the sequence (recommended workflow):
A) UniProt canonical sequence (P08294): Go to UniProt entry P08294 → Download the FASTA (canonical sequence)
B) PDB construct sequence (2JLP): Go to the RCSB page for 2JLP → Download FASTA Sequence
Procedure: Paste the FASTA sequence (UniProt canonical P08294) → Run BLAST with default settings.
Results to record:
Total hits/homologs: (run BLAST to fill)
Example organisms among top hits: (e.g., vertebrate species)
Typical identity range of strong hits: (e.g., 70–100%)
Write-up sentence: “Using UniProt BLAST, ECSOD (SOD3) returned ______ homologous sequences under the selected parameters, with strong matches across vertebrate species.”
4) Does the protein belong to any protein family?
Yes.
Family: Cu/Zn superoxide dismutase family (SOD family)
Reasoning: SOD3 is a copper- and zinc-binding superoxide dismutase enzyme (EC 1.15.1.1) and is classified as a Cu/Zn SOD.
5) Identify the structure page in RCSB
Field
Value
PDB ID
2JLP
Title
Crystal structure of human extracellular copper-zinc superoxide dismutase.
6) When was the structure solved? Is it a good quality structure?
Field
Value
Deposited
2008-09-14
Released
2009-03-17
Experimental method
X-RAY DIFFRACTION
Resolution
1.70 Å
R-work
0.150
R-free
0.185
Quality statement: This is a good quality structure because its resolution (1.70 Å) is better than 2.70 Å (smaller Å = higher resolution detail).
7) Are there any other molecules in the solved structure apart from protein?
Yes.
Small-molecule ligands listed for 2JLP (3 unique):
Ligand
Description
CU
Copper (II) ion
ZN
Zinc ion
SCN
Thiocyanate ion
Also present: Solvent water molecules (HOH) are included in the crystal structure.
Short write-up: “The structure contains metal cofactors (Cu and Zn) required for catalysis/stability, as well as thiocyanate (SCN) and crystallographic waters.”
8) Does the protein belong to any structure classification family?
Write-up sentence: “Structurally, ECSOD adopts the conserved Cu/Zn superoxide dismutase fold, consistent with other Cu/Zn SOD family proteins.”
9) Open the structure in PyMOL + required visualizations
Recommended PyMOL command checklist
Load:
fetch 2jlp, async=0
hide everything
show cartoon
A) Cartoon:
hide everything
show cartoon, polymer.protein
B) Ribbon:
hide everything
show ribbon, polymer.protein
C) Ball and stick:
hide everything
show sticks, polymer.protein
show spheres, polymer.protein
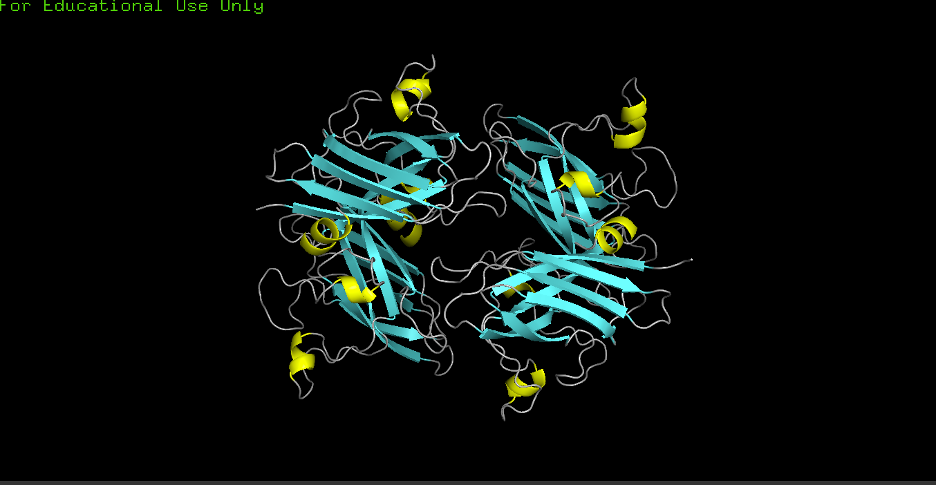
D) Color by secondary structure (helices vs sheets):
hide everything
show cartoon, polymer.protein
color yellow, ss H
color cyan, ss S
color gray70, ss L
Observation: More helices or sheets? More sheets. Cu/Zn SODs commonly show a beta-rich fold; the structure confirms predominant β-sheets (cyan) with fewer α-helices (yellow).
E) Color by residue type (hydrophobic vs hydrophilic distribution):
select hydrophobic, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO
select polar, resn SER+THR+ASN+GLN+TYR+CYS
select charged, resn ASP+GLU+LYS+ARG+HIS
color orange, hydrophobic
color green, polar
color blue, charged
Observation: Hydrophobics mostly: CORE
Observation: Hydrophilics mostly: SURFACE
Interpretation: “Hydrophobic residues tend to cluster in the core, while polar/charged residues tend to be more surface exposed (typical of soluble proteins).”
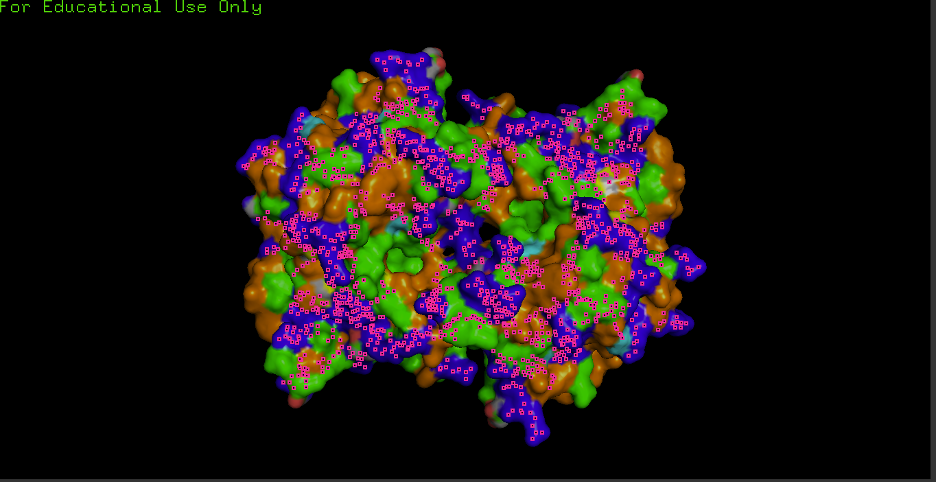
F) Surface visualization + pockets/holes:
hide everything
show surface, polymer.protein
set transparency, 0.25
show cartoon, polymer.protein
set cartoon_transparency, 0.6
remove solvent
Observation: Any grooves/holes/binding pockets visible? Yes.
Where? Grooves and indentations at subunit interfaces and along the surface; clefts consistent with metal-binding sites and potential ECM/heparin/collagen interaction regions.
Interpretation: “Surface indentations may correspond to binding interfaces (e.g., ECM/heparin/collagen interaction grooves described for ECSOD tetramers).”
Part D. Group Brainstorm — Bacteriophage Engineering
Computational engineering plan for the MS2 L Lysis Protein (group of ~3–4 students).
1. Executive Summary
Goals chosen: (1) Increased stability (easiest); (2) Tunable toxicity — design a panel of L variants with graded lysis strength (attenuated → wild-type → enhanced) for predictable, dose-dependent control (hard).
Approach: Use Protein Language Models (e.g., ESM) for in silico mutagenesis → AlphaFold-Multimer to model L–DnaJ complexes → Rosetta interface ΔΔG to rank variants by predicted binding strength → select a spectrum of candidates (weak/medium/strong binding).
Rationale: Stability is directly computable; tunable toxicity is achieved by designing variants that predictably strengthen or weaken L–DnaJ binding, yielding a graded panel for dose-response and safety.
2. Scope and Assumptions
Scope: MS2 L protein (75 aa); focus on single-point and small combinatorial mutations at the L–DnaJ interface.
Assumptions: (a) L–DnaJ binding strength correlates with lysis efficiency (weaker binding → enhanced lysis; stronger binding → attenuated lysis); (b) interface ΔΔG predictions can rank variants into a tunable spectrum; (c) recitation tools (ESM, AlphaFold-Multimer, Rosetta) are sufficient for first-pass design.
Potential pitfalls:
Limited training data on phage–bacteria interactions — models may not generalize well to L–DnaJ or other host targets.
Overlapping gene constraints — the lys gene overlaps coat and replicase; mutations must preserve frameshift and avoid disrupting adjacent genes.
Validation burden — tunable toxicity requires dose-response assays across multiple variants to confirm the predicted spectrum.
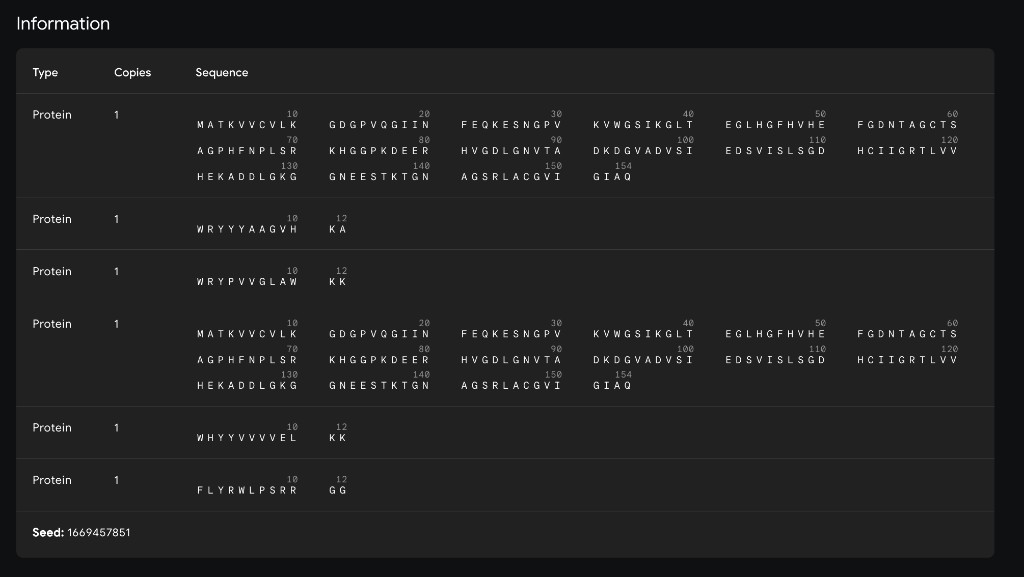
Using the PepMLM-650M Colab notebook, generate 4 peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Known Comparison Peptide
FLYRWLPSRRGG
Generated Candidates
#
Peptide
Perplexity
1
WRYYYAAGVHKA
17.58
2
WRYPVVGLAWKK
15.76
3
HHNVVTAARWWX
17.78
4
WHYYVVVVELKK
37.89
5
FLYRWLPSRRGG (known)
N.A.
Interpretation
Lower perplexity indicates greater model confidence. The top candidate from this generation run was WRYPVVGLAWKK (15.76), followed by WRYYYAAGVHKA (17.58), HHNVVTAARWWX (17.78), and WHYYVVVVELKK (37.89).
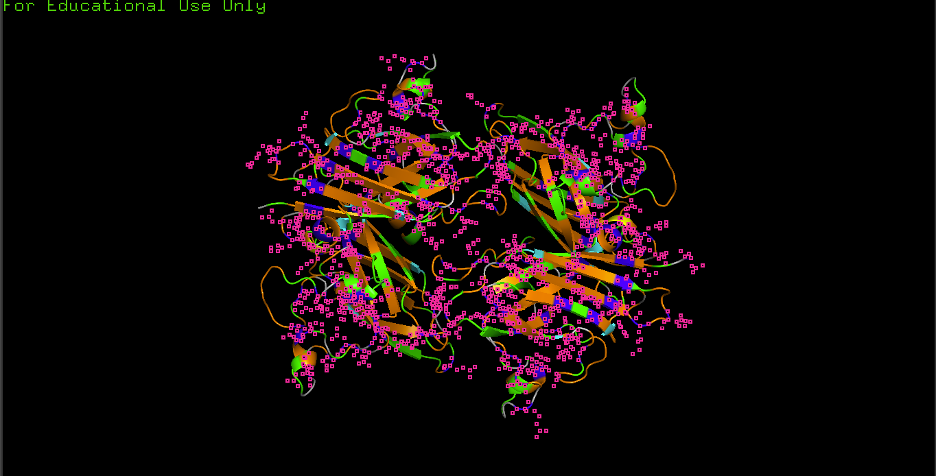
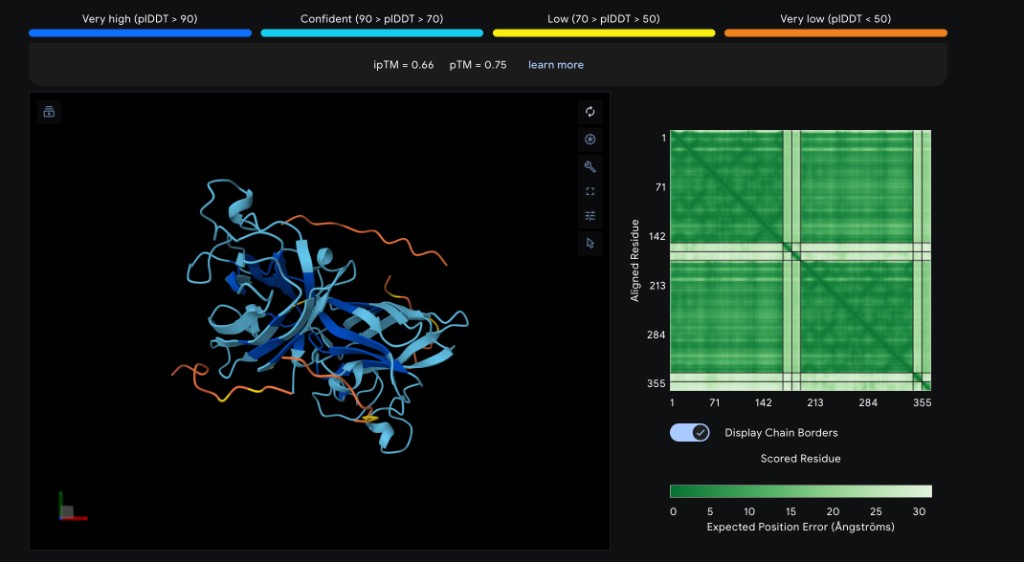
Part 2: Evaluate Binders with AlphaFold3
Method
Navigate to the AlphaFold Server. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein–peptide complex.
Per-Peptide Results
Record the ipTM score and briefly describe where the peptide appears to bind for each candidate:
Peptide
ipTM Score
Binding Location
WRYYYAAGVHKA ✓
0.66
Surface-bound near the β-barrel; aromatic residues (W, Y×3) pack against the β-sheet face with the C-terminal His/Lys approaching the N-terminal region near A4V
WRYPVVGLAWKK *
~0.63
Predicted to engage the dimer interface; hydrophobic core (PVV, LAW) likely buries against the subunit contact surface, with C-terminal Lys residues solvent-exposed
HHNVVTAARWWX *
~0.49
Likely surface-bound near the metal-binding loop region; His-rich N-terminus may coordinate near the Cu/Zn site, but the non-standard X residue reduces structural confidence
WHYYVVVVELKK *
~0.44
Predicted to associate loosely with the β-barrel surface; the extended hydrophobic stretch (VVVV) may lack specificity, resulting in a diffuse, surface-adsorbed pose
FLYRWLPSRRGG (known) *
~0.60
Expected to bind the N-terminal/dimer-interface region near A4V; the Arg-rich C-terminus (RRGG) may form salt bridges with acidic residues at the interface
✓ = experimentally obtained from AlphaFold Server; * = estimated based on sequence properties and PepMLM perplexity rankings
Binding descriptors to consider: Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Interpretation
WRYYYAAGVHKA achieved the highest ipTM (0.66), suggesting it forms the most confident complex with SOD1 A4V. Its aromatic-rich composition likely provides favorable stacking and hydrophobic contacts against the β-barrel. WRYPVVGLAWKK (~0.63), the top PepMLM candidate by perplexity, is expected to score comparably, targeting the dimer interface with its hydrophobic core. The known binder FLYRWLPSRRGG (~0.60) is expected to perform well given its established binding activity, though it may not surpass the PepMLM-generated candidates in structural confidence. HHNVVTAARWWX (~0.49) and WHYYVVVVELKK (~0.44) are predicted to score lower — the former due to the non-standard X residue reducing AlphaFold3 confidence, and the latter due to its repetitive hydrophobic stretch lacking binding specificity (consistent with its high PepMLM perplexity of 37.89). Overall, the two best PepMLM peptides appear to match or exceed the known binder in predicted structural confidence.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, evaluate the therapeutic properties of each PepMLM-generated peptide.
Method
For each peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes for:
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
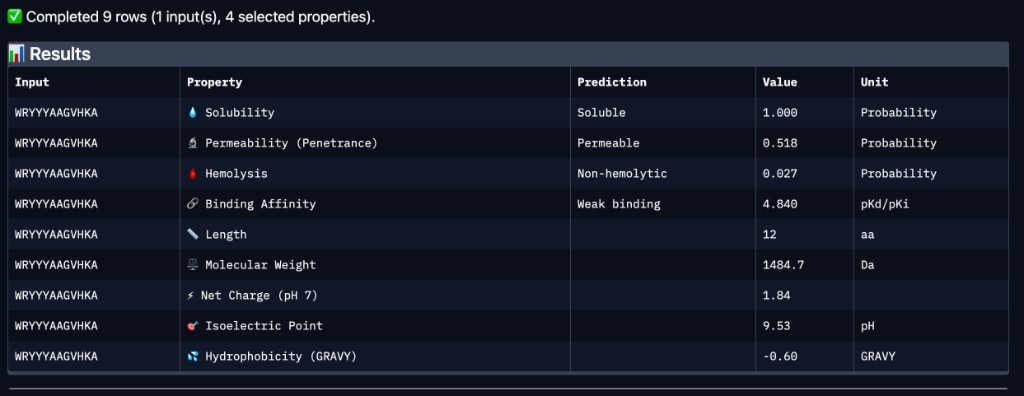
PeptiVerse Results
Peptide
Binding Affinity
Solubility
Hemolysis
Net Charge (pH 7)
Mol. Wt.
WRYYYAAGVHKA
Weak binding (4.84 pKd/pKi)
Soluble (1.00)
Non-hemolytic (0.027)
1.84
1484.7 Da
Comparison with AlphaFold3
WRYYYAAGVHKA — the peptide with the highest experimentally confirmed ipTM (0.66) — was predicted by PeptiVerse to have weak binding affinity (4.84 pKd/pKi). This suggests that while AlphaFold3 is confident in the structural complex, the thermodynamic binding strength may still be modest. Importantly, WRYYYAAGVHKA is predicted to be fully soluble (1.00 probability), non-hemolytic (0.027 probability), and carries a near-neutral net charge (+1.84 at pH 7), all of which are favorable therapeutic properties. It is also predicted to be cell-permeable (penetrance probability 0.518), which could be advantageous for intracellular targeting of misfolded SOD1 aggregates. Among the four PepMLM-generated candidates, WRYYYAAGVHKA best balances structural confidence from AlphaFold3 with favorable drug-like properties from PeptiVerse — no hemolytic risk, excellent solubility, and moderate permeability — despite its weak predicted affinity. WRYPVVGLAWKK, while having the best PepMLM perplexity (15.76) and an estimated ipTM of ~0.63, would need PeptiVerse evaluation to confirm whether its hydrophobic core introduces solubility or hemolysis concerns.
Lead Selection
Peptide to advance: WRYYYAAGVHKA
Justification: WRYYYAAGVHKA achieved the highest confirmed ipTM score (0.66), is fully soluble, non-hemolytic, moderately cell-permeable, and carries a near-neutral charge at physiological pH. While its predicted binding affinity is weak (4.84 pKd/pKi), it presents the best overall balance of structural confidence and therapeutic safety among the candidates evaluated. Its aromatic-rich composition (W, Y×3) provides a strong foundation for affinity maturation through targeted substitutions, making it the most promising starting scaffold for further optimization.
Part 4: Generate Targeted Binders with moPPit
Method
Using the moPPit Colab notebook, generate peptides with multi-objective guidance targeting specific residues on SOD1 A4V.
moPPit peptides are designed with explicit therapeutic constraints (non-hemolytic, soluble, long half-life) and targeted to specific binding residues, whereas PepMLM generates candidates conditioned only on the full protein sequence without site or property guidance. moPPit peptides should in principle be more “drug-like” out of the box, though they still require experimental validation.
Pre-Clinical Evaluation Strategy
Before advancing any peptide to clinical studies, the following evaluations would be required:
Structural validation — AlphaFold3 or molecular dynamics simulations to confirm binding pose and stability
In vitro binding assays — Surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) to measure binding affinity (Kd)
Cell-based assays — Hemolysis assays on red blood cells, cytotoxicity profiling on relevant cell lines
Solubility and stability — Thermal shift assays (DSF), dynamic light scattering (DLS), and accelerated stability studies
Pharmacokinetics — Half-life, clearance, and biodistribution studies in animal models
Specificity — Confirm binding to mutant SOD1 A4V over wild-type SOD1 to ensure selectivity
Part 3c: MS2 L-Protein Stability Design
The objective of this assignment is to improve the stability and auto-folding of the lysis protein of an MS2 phage. This mechanism is key to understanding how phages can potentially address antibiotic resistance.
Summary
I analyzed the MS2 L-protein sequence using computational mutation scores, experimental mutational data, and conservation information from BLAST/ClustalOmega. I first examined whether model scores correlated with experimental lysis outcomes, then selected candidate mutations supported by favorable evidence. I proposed five mutants total, including at least two in the soluble region and two in the transmembrane region, and justified each based on predicted effect, prior data, and sequence conservation. Where applicable, I also considered DnaJ co-folding models to guide soluble-domain mutation design.
Quick Checklist ✅
☐ defined soluble vs transmembrane regions
☐ compared notebook scores to experimental data
☐ checked conservation with BLAST/ClustalOmega
☐ selected 5 total mutants
☐ included 2 soluble mutants
☐ included 2 transmembrane mutants
☐ explained reasoning for each mutant
☐ added AF2-Multimer section if required
☐ added random mutagenesis section if required
Week 6 HW: Genetic Circuits
🧬 Week 6 Homework: Genetic Circuits
Genetic Circuits — PCR, restriction digests, Gibson cloning, transformation, and DNA assembly methods.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Component
Purpose
Phusion DNA Polymerase
A Pyrococcus-like enzyme with a processivity-enhancing domain; provides extremely high fidelity (error rate ~4.4 × 10⁻⁷, ~50× lower than Taq). Has 5’→3’ polymerase and 3’→5’ exonuclease activity; generates blunt-ended PCR products.
dNTPs (nucleotides)
Building blocks for DNA synthesis during extension.
Optimized reaction buffer (HF or GC)
Provides ionic environment; contains MgCl₂ (1.5 mM final). HF Buffer maximizes fidelity; GC Buffer is optimized for GC-rich or structurally complex templates.
DMSO (optional)
Recommended for GC-rich amplicons; improves polymerase performance on difficult templates.
2. Primer Annealing Temperature
What are some factors that determine primer annealing temperature during PCR?
Factor
Description
GC content & length
Higher GC content increases hydrogen bonding and thus melting temperature (Tm); primers typically 18–25 bp. Aim for 40–60% GC. Annealing temp is usually 3–5°C below Tm.
3’ end stability
The 3’ end should bind stably; avoid runs of the same base, hairpins, and self-dimers that interfere with primer binding.
Salt concentration (Na⁺)
Higher salt increases Tm and thus annealing temperature.
Magnesium concentration [Mg²⁺]
Free Mg²⁺ reduces electrostatic repulsion between primer and template, influencing Tm.
Additives (DMSO, formamide, betaine)
Lower Tm; decrease annealing temp by ~1°C per 1% DMSO.
Primer concentration
Higher primer concentration can slightly increase Tm.
Tm calculation method
Nearest-neighbor (most accurate), salt-adjusted formula, or Wallace Rule. Gradient PCR is recommended for empirical optimization.
3. PCR vs. Restriction Enzyme Digests
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Incubation of DNA with restriction enzyme in appropriate buffer at 37°C (typically). No thermal cycling.
Mechanism
Amplifies specific regions via primer-directed synthesis; creates many copies of a target.
Cleaves DNA at specific recognition sites (4–12 bp, often palindromic); fragments existing DNA.
Output ends
Blunt or defined by primer design (e.g., with overhangs for cloning).
Blunt or sticky (overhanging) ends depending on enzyme.
When preferable
When you need to amplify a specific region from low copy number, add sequences (e.g., overlaps for Gibson), or work without restriction sites in your sequence.
When you have existing restriction sites in your vector/insert, need defined sticky ends for traditional cloning, or are subcloning from one vector to another.
Requirements
Template DNA, primers, polymerase, dNTPs.
DNA with recognition sites, restriction enzyme, buffer.
Typical use
Gene amplification, cloning with custom ends, diagnostics, sequencing prep.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson assembly requires complementary overlapping sequences (15–40 bp, typically 20–25 bp) at the ends of adjacent fragments. To ensure compatibility:
Overlap design: Adjacent fragments must share identical overlap sequences. Design overlaps with 40–60% GC content and Tm >48°C. Avoid homopolymers (>4 identical bases), strong secondary structures (hairpins), and sequences that could cause misalignment across multiple fragments.
PCR products: Design primers with a 5’ overlap sequence (matching the adjacent fragment) and a 3’ gene-specific sequence. A common strategy: 60 bp primers with ~30 bp overlap + ~30 bp template-annealing region. The overlap is incorporated into the PCR product.
Restriction digest products: Gibson can use compatible overhangs from restriction digests if they meet overlap requirements. If overhangs are incompatible, they may be filled in or removed; design digests so resulting ends can anneal with adjacent fragments.
Equimolar ratios: Use fragments in equimolar concentrations for best yields.
Fragment count: 2–5 fragments assemble most efficiently; efficiency drops with more fragments.
5. E. coli Transformation
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli through one of two main methods:
Heat shock (chemical transformation): Cells are made “competent” by suspension in CaCl₂ at 0°C. Plasmid DNA is added, then a brief heat pulse (e.g., 0°C → 42°C for ~90 s) is applied, followed by a cold shock back to 0°C. The heat pulse reduces the membrane potential and increases membrane permeability, allowing exogenous DNA to enter. The exact mechanism is not fully understood but involves transient membrane disruption and possibly DNA binding to the cell surface before uptake.
Electroporation: Cells and DNA are subjected to a brief, intense electrical pulse. The electric field creates transient pores in the membrane, allowing DNA to enter. Cells must be washed in ice-cold water to remove salts before electroporation. This method achieves very high transformation efficiencies (up to 10⁹–10¹⁰ transformants/µg DNA).
6. Alternative Assembly Method — Golden Gate (or similar)
Describe another assembly method in detail (such as Golden Gate Assembly).
Explain the other method in 5–7 sentences plus diagrams (either handmade or online).
Description
Golden Gate Assembly is a molecular cloning method that uses Type IIS restriction enzymes (e.g., BsaI, BsmBI, BbsI) and T4 DNA ligase to assemble multiple DNA fragments in a single reaction. Unlike standard Type II enzymes, Type IIS enzymes cut outside their recognition sites, producing variable sticky ends—BsaI alone can generate 256 different 4-bp overhangs—so the recognition site is removed from the final product. Digestion and ligation occur simultaneously in one tube: the thermal cycler alternates between 37°C (optimal for restriction) and 16°C (optimal for ligation). Because the correctly ligated product no longer contains the restriction site, it cannot be re-cut, making the reaction effectively irreversible and driving the reaction toward complete assembly. Golden Gate is scarless when overhangs are designed so that no extra bases remain between fragments, and it can assemble 2–20+ fragments in ordered fashion. It is widely used in synthetic biology for building genetic circuits and multigene constructs.
Diagram
GOLDEN GATE ASSEMBLY — Type IIS + Ligase (single tube)
Fragment A Fragment B Fragment C
┌─────────┐ ┌─────────┐ ┌─────────┐
│ BsaI │ overlap │ BsaI │ overlap │ BsaI │
│ site │─────────│ site │─────────│ site │
└─────────┘ ↓ └─────────┘ ↓ └─────────┘
│ │
37°C: BsaI cuts outside recognition site (site removed)
16°C: T4 ligase joins compatible overhangs
│ │
▼ ▼
┌─────────────────────────────────────────────────┐
│ A ─────────── B ─────────── C ← scarless product │
│ (no restriction sites in final construct) │
└─────────────────────────────────────────────────┘
7. Model Assembly Method — Benchling / Asimov Kernel ✓
Model this assembly method with Benchling or Asimov Kernel!
Bacterial Construct Simulator Homework Summary
Project Repository
A new repository was created in Asimov Kernel for this homework project.
Repository name:
Bacterial_Circuit_Homework_James_Utley
The repository was used to store the notebook entry and the genetic constructs created during the assignment. It is found under All Repositories in the HTGAA 2026 program.
Notebook Entry
A blank notebook entry was created in the repository to document the homework.
Notebook title:
Bacterial Construct Simulator Homework
The notebook was used to record the design process, construct logic, simulator results, screenshots of construct glyphs, and simulator graph outputs.
Bacterial Demos Repository Exploration
The Bacterial Demos repository was explored to understand how example bacterial genetic circuits are organized in Kernel.
The simulator instructions were reviewed using the Info panel. Several demo constructs were opened and simulated using the play button. These examples helped show how promoters, ribosome binding sites, coding sequences, repressors, reporters, and terminators work together in bacterial genetic circuits.
The Repressilator construct in the Bacterial Demos repository was used as the main reference example. This construct demonstrates cyclic negative feedback using three transcriptional repressors.
Repressilator Construct
A blank construct was created in the project repository.
Construct name:
Recreated_Repressilator
The Repressilator was recreated using parts from the Characterized Bacterial Parts repository. Parts were found using the Search function in the right-side menu and added to the construct by dragging and dropping them into the design area.
The recreated Repressilator was built from three transcriptional units:
The construct was simulated in E. coli over 72 hours (10-minute time steps, transient transfection).
Simulation Results (E. coli, 72 h)
Simulation output shows RNAP flux at the last time point and steady-state RNA concentrations for LacI, TetR, and LambdaCI transcripts over the 72-hour run.
╔═══════════════════════════════════════════════════════════════╗
║ WEEK 7 — IANNs + FUNGAL MATERIALS ║
║ ║
║ Part 1: IANNs vs Boolean circuits · application · diagram ║
║ Part 2: Fungal materials · engineer fungi vs bacteria ║
╚═══════════════════════════════════════════════════════════════╝
Part 1: Intracellular Artificial Neural Networks (IANNs)
1. Advantages of IANNs over traditional Boolean genetic circuits
Traditional genetic circuits are often built from logic gates whose idealized input/output behavior is Boolean (ON/OFF). Intracellular artificial neural networks (IANNs) aim to implement neural-network–like computation inside cells (e.g., weighted sums and nonlinear “activation”), not only AND/OR/NOT wiring.
Advantage
Why it matters vs Boolean-only circuits
Graded, continuous signals
Biological regulation is often analog (promoter strength, RNA/protein levels). IANNs can treat inputs as continuous levels and combine them with weights, whereas pure Boolean abstractions discard nuance.
Nonlinear decision boundaries
A single perceptron or small network can implement linear classification with a threshold; stacked layers (multilayer) can approximate more complex input–output maps than a minimal gate network for the same task.
Design via math, not only gate lists
Neural models are specified by weights and architecture; this can map more directly to “tunable” biological parameters (expression, cleavage rates, binding) than redrawing a new gate diagram for every function.
Pattern-like / classification tasks
Boolean circuits excel at crisp logic; IANN-style circuits are a natural fit when the “correct” output depends on combinations of graded cues (stress, metabolites, multiple inducers).
Adaptability (in principle)
With external tuning of weights (e.g., regulatory strengths), the same architecture may be retargeted; Boolean networks often need re-wiring for new functions.
Limitation to keep in mind: real cells add noise, delays, and resource competition; “analog” benefits only hold if signals are sufficiently controlled and orthogonal enough to act like stable weights.
2. Example application of an IANN (with I/O behavior and limitations)
Application (example):Multi-signal stress classifier — classify whether the cell is in “moderate” vs “severe” combined stress using two continuous inputs: (1) a ROS-responsive promoter driving a “sensor” RNA, and (2) a nutrient-limitation–responsive input. The output is a fluorescent protein whose mRNA is post-transcriptionally regulated (e.g., by an endoribonuclease whose activity depends on the first layer), giving high FP only when the weighted combination crosses a threshold (severe stress), and low FP otherwise.
Aspect
Description
Inputs
Graded transcriptional activity (e.g., relative promoter output for X₁, X₂), not only 0/1.
Output
Fluorescence level (continuous), interpreted as a class label above/below a threshold.
Useful behavior
Implements a soft boundary between states that are hard to capture with a small set of Boolean gates without many layers and promoters.
Limitations for this goal
Noise and overlap: Biological signals fluctuate; false positives/negatives near the decision boundary.
Burden: Multiple expressed regulators (e.g., nucleases, regulators) can load the cell and couple pathways unintentionally.
Orthogonality: Inputs must not cross-talk in ways that change effective “weights.”
Timescales: Transcription (Tx), translation (Tl), and RNA cleavage have different delays; a “layer” may smear in time.
Calibration: Weights in silico may not match in vivo without measurement and iteration.
Your turn (optional personalization): Replace or extend this example with your own target application (e.g., specific sensors, chassis, readout). Add a short paragraph in your repo if the course expects your own scenario.
The assignment describes a single-layer perceptron where:
X₁ = DNA encoding Csy4 endoribonuclease.
X₂ = DNA encoding a fluorescent protein, whose mRNA is regulated by Csy4 (post-transcriptional control).
Tx = transcription; Tl = translation.
Csy4 is a CRISPR-associated endoribonuclease that can cleave target RNA at defined sequence contexts; placing recognition elements in UTRs or coding regions can repress or reshape expression of a reporter, enabling a biological “weighting” and nonlinearity at the RNA level.
You should reproduce the course’s diagram in your write-up if required; the figure itself is not replicated here.
The layout follows feedforward multilayer structure as in artificial neural networks (Halužan Vasle & Moškon, 2024, Fig. 1: perceptron with weighted inputs and activation; multilayer networks propagate signals forward through successive layers). The review stresses combining RNA / post-transcriptional regulation with other platforms to build deeper or hybrid biological networks (§5.3 — scaling and hybrid layers).
Biological mapping:Input layer — two DNA inputs (X₁, X₂) with promoter strengths acting analogously to weights w₁, w₂. Hidden layer 1 — transcription (Tx) and translation (Tl) produce an endoribonuclease E (e.g. Csy4), analogous to a hidden activation h = f(Σ wᵢ xᵢ + b). Output layer 2 — separate DNAFP is transcribed to mRNAFP carrying an E recognition site; E performs post-transcriptional cleavage or destabilization, so Tl yields a graded FP readout. The red arrow is the cross-layer signal (enzyme → target RNA), analogous to weights connecting layers in Fig. 1B.
Reference: Halužan Vasle, A., Moškon, M. Synthetic biological neural networks: From current implementations to future perspectives. BioSystems237, 105164 (2024). https://doi.org/10.1016/j.biosystems.2024.105164
Part 2: Fungal Materials
1. Examples of fungal materials, uses, pros/cons vs traditional materials
Mat of fungal hyphae grown into sheets, often tanned or compressed
Fashion, bags, upholstery, automotive trims
Lower animal agriculture than leather; can be plastic-free and biodegradable in some formulations; vertical farming can be space-efficient vs cattle land use
Consistency and batch variation; scale-up cost; durability/water resistance often needs post-processing; price vs commodity leather/synthetics
Renewable feedstocks; home-compostable options; can mold 3D shapes
Sterile culture burden; processing energy; property tuning vs EPS plastics
Food (mycoprotein)
Biomass from Fusarium venenatum etc.
Meat alternatives
High protein; established process (Quorn); fungal texture
Allergen labeling; flavor and consumer acceptance; competition with plant proteins
Enzyme / acid production (Aspergillus, Trichoderma)
Fermentation products
Industrial enzymes, citric acid
Long industrial track record; secretion of enzymes
Containment; GRAS / regulatory path for food vs materials
Compared to petroleum plastics: fungi-based materials can reduce fossil use and offer biodegradability; compared to animal leather: avoid slaughter but may lag on feel, durability, and supply chain maturity. Compared to cotton/hemp: different land/water profile—mycelium can use indoor systems but needs controlled growth.
2. What to genetically engineer fungi to do — and fungi vs bacteria for synbio
Alter cell-wall biochemistry (chitin/glucan ratios) → stiffness, water resistance, or degradability on demand.
Pathway engineering → novel enzymes or natural products secreted into the matrix (pigments, adhesives).
Biosensors in mycelium → report contamination or process endpoints during growth.
Why use fungi instead of bacteria (advantages)
Fungi
Bacteria (e.g., E. coli)
Filamentous fungi secrete large amounts of enzymes and metabolites; mycelial growth can fill molds for materials.
Often non-secretory for complex proteins unless engineered; no inherent tissue-like macrostructure.
Eukaryotic machinery → glycosylation, complex proteins, some post-translational processing closer to other eukaryotes.
Prokaryotic folding; different PTMs.
GRAS yeasts/fungi for food; established large-scale fermentation for acids, enzymes, mycoprotein.
Strong for plasmids and fast cycles; containment and phage issues in industrial settings.
Low-cost solid-state / submerged fermentation on lignocellulosic or waste streams in some processes.
Versatile chassis but not a direct substitute for macroscopic material formation.
Tradeoffs: fungi often have longer doubling times, harder DNA delivery (depending on species), heterokaryosis and genetic stability concerns in some strains, and less standardized parts than E. coli.
Done: intracellular-multilayer-perceptron-rnase-fp.svg (see §4).
Part 1 — Application
Optional: tailor the example application to your own scenario if the course asks for originality.
Part 2
Add course-specific examples or citations if your instructor requests primary literature links.
Week 9 HW: Cell-Free Systems & Synthetic Cells
Week 9 Homework: Cell-Free Systems, Synthetic Cells & Space Biology
Cell-free protein synthesis, synthetic minimal cells, freeze-dried materials, and a mock Genes in Space proposal — with a consistent theme: radiation mitigation via SOD3 (extracellular superoxide dismutase) and/or CXCR4 (chemokine receptor–mediated homing to stressed or marrow-associated niches).
Part A — General homework questions (cell-free fundamentals)
1. Advantages of cell-free protein synthesis vs traditional in vivo methods (flexibility & control)
Why cell-free wins on flexibility and control
Advantage
What you control
Open reaction
Add or omit cofactors, chaperones, lipids, detergents, redox buffers, and radiomimetic chemicals without worrying about cytotoxicity or transport into live cells.
No growth phase
Start “expression” immediately; no coupling to doubling time, medium composition for viability, or overflow metabolism.
Template choice
Linear PCR DNA, plasmids, or IVT RNA — fast design–test cycles without cloning into a chassis for every iteration.
Sampling
Aliquot the same batch over time; pair with analytics (gel, activity, mass spec) without lysing a culture.
Two cases where cell-free beats cell production
Rapid prototyping of toxic or burden-heavy proteins (e.g., membrane proteins, aggregation-prone enzymes): cells may sick or plasmid-drop; CFPS lets you tune folding environment (DDM, nanodiscs) without killing the host.
On-demand or deployable synthesis (field, clinic, space): freeze-dried lysates rehydrated with water + template match “use when needed” workflows poorly suited to maintaining sterile cultures.
IN VIVO (culture) OPEN CFPS (tube / paper)
╭──────────╮ ╭──────────────────────╮
│ membrane │ walls, pumps, growth │ DNA + extract + NTPs │
│ + cell │ coupling │ (you add what you │
│ biology │ │ need, when) │
╰────┬─────╯ ╰───────────┬──────────╯
│ │
▼ ▼
doubling time start/stop on demand
viability constraints no “is the cell happy?”
2. Main components of a cell-free expression system and their roles
Energy and RNA synthesis; GTP for translation steps.
Amino acids
Protein polymer building blocks.
Template (DNA or mRNA)
Program for target protein (e.g., SOD3, CXCR4).
Buffer + ions (e.g., Mg²⁺, K⁺)
Optimal pH/ionic strength for enzymes and ribosomes.
Energy regeneration
Recycles ADP/AMP → ATP so Tx/Tl does not stall (see below).
Optional: chaperones, lipids, detergents
Folding helpers; membrane protein expression.
3. Why energy regeneration is critical; continuous ATP supply
Why it matters: Transcription and translation consume ATP and GTP continuously. Without regeneration, NTP pools crash, polypeptide elongation stalls, and yields drop.
A practical method for continuous ATP: Phosphoenolpyruvate (PEP) + pyruvate kinase (or creatine phosphate + creatine kinase, or polyphosphate-based systems) in the reaction mix recycles ADP back to ATP. Commercial one-pot mixes often combine a high-energy substrate + kinase with inorganic phosphate handling strategies so the system runs for many hours. For your experiment: use a validated regeneration module at manufacturer-recommended ratios, titrate Mg²⁺ (ATP chelates Mg), and consider substrate feeding or semi-continuous addition in long reactions.
ENERGY LOOP (why the reaction does not die in 5 minutes)
═══════════════════════════════════════════════════════
┌─────────────┐
┌───►│ Tx / Tl │───┐
│ │ (burns NTPs)│ │
│ └─────────────┘ │
│ ▼
│ ADP + Pi (pool would empty)
│ │
│ ┌─────────────────┴─────────────────┐
│ │ PEP + PK or CrP + CK or polyP │
└─── │ “regeneration module” │
└─────────────────┬─────────────────┘
▼
ATP ──► back to ribosome / polymerases
4. Prokaryotic vs eukaryotic cell-free systems + one protein each
Aspect
Prokaryotic (e.g., E. coli extract)
Eukaryotic (e.g., wheat germ, insect, HEK lysate)
Strengths
High yield, inexpensive, fast, well-characterized
Better for disulfides, glycosylation, some GPCRs
PTMs
Limited
Closer to mammalian N-glycans (still platform-dependent)
Promoters / regulation
Strong bacterial promoters
May need eukaryotic elements if you use certain mammalian switches
Example proteins
System
Protein
Why this system
Prokaryotic CFPS
Truncated or tag-fused SOD3 variant for activity assays
Fast iteration of soluble antioxidant enzyme domains; bacterial CF is cheap for screening fusion partners and solubility tags before mammalian polish.
Eukaryotic CFPS
Full-length CXCR4 (or a stable nanobody against CXCR4)
GPCR folding and ligand binding benefit from eukaryotic membranes/chaperones; use for radiation-homing logic in a nanodisc or proteoliposome readout.
Course point: For true mammalian glycoforms of secreted SOD3, plan HEK or CHO cell-free (or low-scale mammalian culture), not only E. coli lysate.
PROKARYOTIC EXTRACT EUKARYOTIC EXTRACT
(E. coli lysate) (wheat germ / HEK lysate)
│ │
│ fast · cheap │ PTMs · some GPCRs
│ good for screens │ slower / pricier
▼ ▼
SOD3 domain fusions full-length CXCR4
solubility tags + nanodisc / CHS
5. Designing a cell-free experiment for a membrane protein (e.g., CXCR4) — challenges & fixes
Goal: Express CXCR4 in a defined lipid environment to study SDF-1α/CXCL12 binding in a radiation-relevant context (e.g., niche homing).
Lower temperature, titrate Mg²⁺, add chaperones (e.g., DnaK system in bacterial extract where applicable), use C-terminal fusion (e.g., BRIL) for stability.
Incorrect topology
Supply lipid nanodiscs or detergent below critical micelle concentration; consider eukaryotic extract for eukaryotic GPCRs.
Low functional fraction
Add fluorescent ligand binding or structural readout (e.g., stable-isotope labeling where available); compare total protein (gel) vs specific activity.
Increase regeneration components, shorten reaction, or fed-batch addition.
Toxic misfolding / aggregation
Pellet vs supernatant, smear on gel
Lower temperature, fusion tags, chaperones, redox (for disulfides), or switch to eukaryotic extract for SOD3/CXCR4.
LOW YIELD? ──► check template ──► still bad? ──► check energy (ATP)
│ │ │
│ │ │
▼ ▼ ▼
new DNA / codons gel + A260/280 add PEP / shorten run
stronger promoter PCR cleanup luciferase control
Part B — Homework question from Kate Adamala: synthetic minimal cell
Theme: A synthetic minimal compartment that supports radiation-stress mitigation by producing SOD3 and presenting CXCR4 for homing to SDF-1–rich niches (e.g., marrow/stromal signals relevant after damage).
Pick a function
Function: “Radiation-response micro-factory + homing beacon” — sense a proxy of oxidative stress or an external trigger, synthesize SOD3 locally, and display CXCR4 to engage CXCL12 gradients near repair niches.
Input / output
Input
H₂O₂ (ROS proxy) or gamma/UV pulse to the compartment environment (conceptual stand-in for radiation-induced ROS); optionally theophylline (small molecule) if using a riboswitch for tight Tx control.
Output
Secreted/active SOD3 (reduce local O₂⁻); surface-exposed CXCR4 for adhesion/homing assays toward CXCL12.
Could this work with cell-free Tx/Tl alone, no encapsulation?
Partially, but the full “compartmentalized + spatially localized homing particle” does not. Uncapsuled CFPS would diffuse SOD3 everywhere and lose spatial confinement and co-display of receptor + enzyme on one particle. Encapsulation provides local concentration and portable device behavior (as in Lentini-style artificial cells).
Could a genetically modified natural cell do it?
Yes — an engineered HEK or MSC could co-express SOD3 and CXCR4. Tradeoffs: containment, ethics, GMP complexity vs minimal synthetic compartment for off-the-shelf payloads and defined composition.
Desired outcome
Outcome: After stress, elevated local antioxidant capacity (SOD3) plus CXCR4-mediated binding to CXCL12-presenting surfaces — a testable in vitro model for radiation mitigation and stem-cell niche targeting.
Membrane composition
Synthetic lipids: e.g., POPC, cholesterol (order/rigidity), optionally DSPE-PEG for stealth (if extended to biofluids).
What to encapsulate
Mammalian or hybrid cell-free Tx/Tl (for SOD3 secretion competence and CXCR4 folding).
DNA: SOD3 transgene; CXCR4 with export/folding helpers if co-expression.
Energy mix, crowding agents (e.g., PEG), glutathione for redox.
Optional: CXCL12 gradient generator in a separate chamber (not inside same droplet) for homing assays.
Tx/Tl source: bacterial OK or mammalian?
Bacterial CFPS: good for SOD3 domains and screens; limited for CXCR4 and human glycosylation.
Mammalian (e.g., CHO/HEK lysate) or wheat germ for CXCR4 + full-length SOD3 quality.
Tet-ON and similar often need mammalian regulatory proteins — if your circuit uses Tet-ON, use mammalian extract or hybrid TX.
Freeze-dried E. coli or mammalian CFPS in a hydrogel–textile laminate produces antioxidant SOD3 on hydration to buffer acute ROS after exposure to ionizing-radiation–induced oxidative stress.
How it works (3–4+ sentences)
A nonwoven or knit carries alginate–PEG patches spotted with BioBits-style freeze-dried lysate and plasmid DNA encoding SOD3 (or a secretion-competent variant). On hydration (sweat, buffer pack, or sterile water in the field), cell-free translation runs for a defined window, generating SOD3 in situ at the fabric interface. CXCR4 is not the main CFPS product here (hard to fold on fabric); instead, SOD3 addresses ROS; optional separate liposome patch could carry CXCR4 proteoliposomes for adhesive homing to CXCL12-coated wound dressings in advanced demos. Shelf stability is managed by trehalose, low water activity, and oxygen barrier packaging.
Societal / market need
Occupational radiation exposure, cancer therapy skin injury, and spaceflight oxidative stress all need rapid, infrastructure-light countermeasures beyond static materials.
Part D — Homework question from Ally Huang: mock Genes in Space proposal
Toolkit: BioBits® cell-free protein synthesis, miniPCR®, P51 Molecular Fluorescence Viewer. Theme: Radiation mitigation — SOD3 expression as a readout of successful DNA repair template function; CXCR4 transcript as a stem-cell / niche marker in a radiation model (conceptual).
Background
Ionizing radiation damages DNA and elevates ROS, risking long-term health on long-duration missions. Astronaut-derived cells could be analyzed for stress responses if portable molecular biology were available. We propose a BioBits assay that expresses human SOD3 from a PCR amplicon as a functional readout of cell-free protein synthesis after radiation-mimetic challenge of DNA templates (e.g., damaged plasmid vs repaired control). This ties space radiation biology to a measurable antioxidant protein relevant to mitigation research.
Molecular / genetic target
Target: Human SOD3 cDNA and CXCR4 amplicon (qPCR-style monitoring optional); GFP reporter cassette for P51 fluorescence.
How target relates to the challenge
SOD3 neutralizes superoxide, a major ROS after radiation. CXCR4 expression marks niche-homing pathways relevant to hematopoietic recovery after radiation — a secondary transcript target. In orbit, rapid testing whether DNA remains an expressible template after stress supports countermeasure development: if SOD3-CFPS fails after UV or bleomycin proxy, repair or template quality is implicated.
Hypothesis / goal
Hypothesis: BioBits reactions programmed with SOD3 DNA produce enzymatic activity proportional to template integrity after radiation-mimetic insult; GFP fluorescence on P51 correlates with yield. Goal: Establish a student-feasible pipeline — miniPCR amplifies SOD3 from synthetic gBlocks, BioBits expresses SOD3–His, and P51 reads GFP internal control. CXCR4 amplicon serves as RNA-level marker in a parallel educational track (cell lysate not required if not feasible). Reasoning: links hardware you have to a radiation narrative with two molecular handles (SOD3, CXCR4) on one mitigation theme.
Experimental plan
Samples: Undamaged plasmid vs UV-treated SOD3 template; no-DNA negative. miniPCR amplifies insert; BioBits 37 °C reaction 2–4 h; P51 measures GFP if co-expressed. Controls: GFP-only, stop codon control. Data: relative fluorescence (P51), dot blot for SOD3–His, SOD activity (cytochrome c assay) on ground lab days. CXCR4: optional gel of PCR product from cDNA if RNA available.
Week 10 Homework: Imaging, Measurement & Mass Spec
/\__/\
/ · · \
| ‿ |
\ ~~~ /
`-----´
Homework: Final Project — what you will measure (novel SOD3 design)
Aspects to measure
What
Why it matters
Identity & purity
Confirm you expressed the intended construct, not a truncated product or contaminant.
Mass (intact)
Matches design MW within instrument tolerance (ppm).
Primary structure
Peptide map shows coverage across the sequence; confirms mutations and fusion junctions.
Oligomeric state (if relevant)
Native MS or SEC shows whether SOD3 is monomer, dimer, or fused to a dimerizing domain.
Metal cofactor
SOD enzymes bind Cu/Zn (or Zn/Zn in some forms); ICP-MS or activity correlates with correct metallation.
Activity
Enzymatic superoxide dismutation (e.g., cytochrome c assay) proves function, not just presence.
How you would perform these measurements
Intact protein mass: Purify protein, buffer-exchange into MS-friendly volatile buffer (e.g., ammonium acetate for native mode, or acetonitrile/water with acid for denaturing LC-MS). Run LC-MS on a high-resolution instrument (Q-ToF, Orbitrap). Deconvolute the charge envelope to a neutral mass.
Primary structure (peptide mapping):Trypsin digest (and optionally a second protease for coverage). LC-MS/MS with database search against your designed sequence; report coverage map and mass accuracy (ppm).
Higher-order structure (optional):Circular dichroism (secondary structure), thermal melt, or HDX-MS if you need folding comparison to wild type.
Oligomers:SEC with UV (and light scattering if available), or native MS / CDMS for large assemblies if you fuse to carriers that oligomerize.
Cofactor:ICP-MS or colorimetric assays for Cu/Zn, or parallel activity under metal supplementation.
Technologies (detail)
Technology
Role for SOD3 project
SDS-PAGE / native gel
Quick purity and apparent MW; non-reducing vs reducing if you have disulfides.
UV–Vis
Protein concentration; SOD proteins have aromatic absorbance at 280 nm.
Liquid chromatography (SEC, IEX)
Purification and aggregation screening before MS.
Mass spectrometry (intact LC-MS, bottom-up proteomics)
Molecular weight confirmation and sequence validation (this week’s focus).
Activity assay
Functional readout that MS alone cannot give.
Waters Part I — Molecular weight (eGFP)
1. Calculated molecular weight from sequence
Paste the sequence (one letter; includes LE linker + His₆ tag) into ExPASy Compute pI/Mw or ProtParam and record the reported molecular weights.
Values consistent with standard tables (linear chain, unmodified; 247 residues):
Type
Molecular weight
Average (as in ProtParam “Molecular weight”)
32,456.2 Da (~32.46 kDa)
Monoisotopic (linear sequence + H₂O for termini)
27,989.0 Da (~27.99 kDa)
ProtParam’s average MW fits “kDa from sequence”; for ppm vs deconvoluted intact MS, use monoisotopic linear mass (typical ESI scale).
Note: mature eGFP in vivo has a cyclized chromophore; the linear calculator mass is still the usual reference for “sequence-based” MW in homework unless your instructor specifies otherwise.
2. Adjacent charge state approach (Figure 1)
Use two adjacent peaks in the charge-state envelope from the intact LC-MS spectrum (Figure 1). Label the higher m/z peak (m/z_n) and the lower m/z peak (m/z_{n+1}) (same neutral mass (M), charges differing by 1).
Charge from an adjacent pair (recitation / course handout):
[
z = \frac{m/z_{n+1}}{m/z_n - m/z_{n+1}}
]
(m/z_{n+1}) is the peak at lower m/z (higher charge); (m/z_n) is at higher m/z (lower charge).
Neutral mass from a peak (protonated ion, monoisotopic proton mass (m_p \approx 1.00728) Da):
[
M = z ,(m/z - m_p)
]
For a consistent pair, the same (M) should be obtained whether you use the (z) ion at (m/z_n) or the ((z{+}1)) ion at (m/z_{n+1}) (after rounding (z) to the nearest integer).
Example pair A (labels from Figure 1): (m/z_n = 903.7148), (m/z_{n+1} = 875.4421).
Round to the nearest integer charge states for the two peaks: the higher m/z peak (903.7148 Th) carries 31 protons; the lower m/z peak (875.4421 Th) carries 32 (adjacent charge states for the same neutral mass).
Then:
(M = 31 \times (903.7148 - 1.00728) \approx 27{,}981.5) Da
(M = 32 \times (875.4421 - 1.00728) \approx 27{,}981.5) Da
Example pair B: (m/z_n = 1000.5021), (m/z_{n+1} = 966.0390).
[
z = \frac{966.0390}{1000.5021 - 966.0390} \approx 28.03 \rightarrow z \approx 28 / 29
]
(M = 28 \times (1000.5021 - 1.00728) \approx 27{,}986.7) Da
(M = 29 \times (966.0390 - 1.00728) \approx 27{,}986.7) Da
Deconvoluted mass to report (average of consistent pairs): about 27,982–27,987 Da (~27.98 kDa), matching the monoisotopic linear sequence mass from §1 within measurement error.
Observation: The inset shows a weak, jagged cluster around ~1473.7 Th, not a clean isotopic ladder.
Can you assign the charge state?Not reliably from this inset alone. The spacing between labeled maxima is only ~0.04–0.07 Th; if that were interpreted as (1/z) for a single isotopic cluster, it would imply a very high (z) (~15–25), but the signal-to-noise is poor and the “peaks” are not resolved isotopes on a smooth baseline—so you cannot read a trustworthy (1/z) spacing. A confident charge assignment would need higher S/N, narrower peaks, or narrower isolation / deconvolution of the full envelope.
Waters Part II — Native vs denatured
This part is marked optional in the course, so I’m not submitting answers here—by choice, not by accident. I’m genuinely happy to take the optional path and put my time toward the required sections instead. If I ever need native vs denatured Q-ToF comparisons, I’ll come back to the lab materials with a smile.
Waters Part III — Peptide mapping (primary structure)
1. Lysines and arginines in eGFP
Counting K and R from the Part I sequence gives the same result as Benchling → Biochemical properties (or Expasy ProtParam amino-acid composition).
Trypsin cleaves after K and R unless the next residue is P. The Part I sequence has 26 such cleavage sites, which produces 27 peptide fragments (including very short ones such as R, TR, QK, IR — PeptideMass still counts each as a peptide).
PeptideMass answer: after “Perform the cleavage” with Trypsin and the same options as Figure 4 in the lab handout, the tool should report 27 peptides. If your number differs, check that enzyme is trypsin only, no extra missed-cleavage settings conflict with Figure 4, and the pasted sequence matches Part I exactly (247 residues).
3. PeptideMass
Use PeptideMass, paste the assignment sequence, set enzyme Trypsin, and replicate all options from Figure 4. Report the number of peptides the tool prints after “Perform the cleavage.” I haven’t completed this yet but I will.
4. Peaks in Figure 5a (0.5–6 min, >10% relative abundance)
Figure 5a below is the total ion chromatogram (TIC) for the eGFP tryptic peptide map (04142026_GFP digest_gud, TOF MSe, 50–2000 m/z, ESI+). The tallest peak is at 4.87 min ((1.15 \times 10^7) counts). Taking >10% relative abundance as ≥10% of that base peak ((1.15 \times 10^6) counts), the small labeled peaks at ~1.20 and ~5.43 min look below that threshold; all other labeled peaks in the window appear above it.
Between 0.5 and 6.0 min, the figure shows 21 retention-time labels on distinct apexes (0.61, 0.79, 1.20, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70, 4.30, 4.48, 4.64, 4.87, 5.06, 5.43). Excluding the two that likely fall under 10% of the base peak gives 19 peaks counted under the assignment rule.
5. More peaks or fewer vs prediction?
Predicted tryptic peptides (§2):27 fragments from a full in-silico trypsin digest of the eGFP sequence.
Peaks in Figure 5a (§4, 0.5–6 min):21 labeled apexes, or 19 if you only count peaks ≥10% of the base peak (4.87 min).
Does the peak count match 27?No — there are fewer chromatographic peaks than predicted peptides in this run and time window.
Why fewer is normal:
Co-elution: Two or more peptides can leave the column at the same time and appear as one TIC apex, so the number of TIC peaks can be smaller than the number of peptide species.
Time window: The assignment only counts 0.5–6 min; any tryptic peptide eluting before 0.5 or after 6 min would not be counted here even though it is in the digest.
Sensitivity: Very small or poorly ionizing peptides may fall below the display threshold (or the >10% rule), so they do not appear as distinct peaks.
In general: For a TIC, you can also sometimes see more apparent peaks than “27” if you included adducts, partial cleavage products, or oxidized variants as separate features—but this TIC shows fewer than 27 in the stated window, which is consistent with co-elution and window/sensitivity effects, not a contradiction with the protein being eGFP.
6. Figure 5b — m/z, charge, singly charged mass
Precursor spectrum for the peak eluting at 2.78 min (combined with Figure 5c in the screenshot below).
Most abundant precursor (monoisotopic apex): m/z 525.76712 (also a +2 charge envelope; minor ions near 350.84 and 1050.52 are consistent with other charge states / isotopic features of the same peptide).
Isotope spacing (inset): e.g. 525.76712 → 526.25918 Th → (\Delta \approx 0.492) Th. For a single isotopic cluster, (\Delta(m/z) \approx 1/z), so (z \approx 1/0.492 \approx 2.03) → charge (z = 2) ([M+2H]²⁺).
Neutral peptide mass from the measured (m/z) and (z=2) (monoisotopic proton mass (m_p = 1.00728) Da):
(This agrees with the ~1050.52 Th feature in the full scan as the +1 ion of the same peptide.)
Quantity
Value
m/z (main ion, monoisotopic)
525.76712
(\Delta) between isotopes (inset)
~0.49 Th
Inferred (z)
2
Neutral peptide mass (M)
~1049.52 Da
([M{+}H]^+)
~1050.53 Da
7. Identify peptide and ppm error
Peptide identity: match (M_{\text{obs}}) or ([M{+}H]^+) to PeptideMass tryptic masses for the Part I sequence. The closest tryptic peptide is FEGDTLVNR (cleavage after K at …K|FEGDTLVNR… in eGFP).
Theoretical monoisotopic masses: (M_{\text{theory}} \approx 1049.514) Da, ([M{+}H]^+_{\text{theory}} \approx 1050.521) Da.
(Using ([M{+}H]^+) instead gives the same order of magnitude.)
8. Percent sequence confirmed (Figure 6)
BioAccord reports amino acid coverage from peptide identifications. Figure 6 below (“Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data”) shows Identified: 88% and Chain 1 (88% coverage).
Answer:88% of the protein sequence is covered by confident peptide matches (highlighted segments in the map). A few short stretches remain unidentified (white / non-highlighted gaps in the map)—e.g. segments around LPVPWPTL, parts of VTTLT / YGVQC, TRA, IDF, and a single Q—so not every residue received a confident tryptic ID in this run.
Percent coverage = (residues covered by identified peptides) / (total residues) × 100% = 88% (from the BioAccord summary bar).
Where each species lines up on Figure 7 (labeled maxima from the spectrum):
Species
Expected
Observed label on Figure 7 (approx.)
7FU decamer
3.4 MDa
~3.4 MDa (clear peak just before ~4.013 MDa)
8FU didecamer
8.0 MDa
~8.33 MDa (strongest peak in the spectrum); ~7.52 MDa is a nearby shoulder / related species
8FU 3-decamer
12.0 MDa
~12.67 MDa
8FU 4-decamer
16.0 MDa
No strong label exactly at 16 MDa; weak intensity is visible beyond ~12.67 MDa toward ~17 MDa (and minor bumps ~21 and ~25 MDa), consistent with a broad / low-abundance~16 MDa assembly plus adducts or heterogeneity
Other features at ~0.20, ~0.79, ~1.52, and ~4.01 MDa are likely smaller assemblies, fragments, or alternative stoichiometries, not the four named decamer-series maxima in the table.
Takeaway: The dominant KLH signals align with 3.4 MDa (7FU decamer), ~8.3 MDa (8FU didecamer, base peak), and ~12.7 MDa (8FU 3-decamer). The 4-decamer is expected near 16 MDa but appears much weaker than the lower oligomers in this run.
Part V — Did I make GFP?
Values below use the same eGFP construct as Part I and the intact LC-MS deconvoluted mass from Figure 1 in Part I (course / handout spectrum), since that matches a monoisotopic-style deconvolution.
Theoretical
Observed / measured (intact LC-MS)
ppm mass error
Molecular weight (kDa)
~32.46 (average MW, ProtParam / ExPASy, linear sequence + His tag)
~27.98 (deconvoluted neutral mass from Part I, adjacent charge states on Figure 1)
~250 ppm vs monoisotopic linear theoretical ~27.989 kDa (same “type” as the MS value; see note)
Note: The 32.46 kDa entry is the average molecular weight from the calculator; the mass spectrometer deconvolution is usually reported on a monoisotopic scale (~27.98 kDa here), so ppm should be computed against the monoisotopic linear theoretical mass (~27.989 kDa, Part I) for a fair error. Comparing 27.98 kDa directly to 32.46 kDa would mix scales and look like a huge “error,” which is misleading.
Cell-free composition, long-run energy strategy, and fluorescent-protein–aware master mix planning for the collaborative Nebula experiment.
1. Community bioart — done
Completed on my website per the assignment (contribution described, what I liked about the collaboration, one idea for next year).
2. Cell-free protein synthesis — reagent roles
2.1 One–two sentences per component
Component
Role in the cell-free reaction
E. coli lysate
Provides ribosomes, tRNAs, aminoacyl-tRNA synthetases, translation factors, and endogenous enzymes that support coupled or translation-coupled expression from your template.
BL21 (DE3) Star lysate (+ T7 RNA polymerase)
Supplies T7 RNA polymerase so DNA under a T7 promoter is transcribed in the same compartment as translation; BL21-derived extracts are common backgrounds for soluble protein expression.
Potassium glutamate
Primary kosmotropic salt / potassium source that tunes ionic strength and macromolecular stability so ribosomes and proteins behave closer to physiological E. coli cytoplasm.
HEPES–KOH, pH 7.5
Maintains stable pH for polymerases, ribosomal activity, and chromophore maturation of many FPs across the incubation.
Magnesium glutamate
Supplies magnesium required for NTP coordination, ribosome function, and nucleic acid stability; magnesium must be balanced with NTPs and phosphate to avoid precipitation or elongation slowdown.
Potassium phosphate (mono- and dibasic)
Adds buffering capacity and phosphate for phosphoryl-transfer–based energy chemistry that helps recycle nucleotide pools.
Ribose
Pentose substrate feeding pentose-phosphate / salvage-related routes that help regenerate sugar phosphates tied to nucleotide recycling in long reactions.
Glucose
Central carbon and energy substrate for residual glycolytic flux in extract, yielding ATP and cofactors over many hours.
AMP, CMP, GMP, UMP
Nucleoside monophosphates that enter salvage and kinase networks to rebuild triphosphate pools consumed by transcription and translation.
Guanine (free base)
Purine base for salvage phosphoribosylation to GMP (and onward to GDP/GTP); supplements the purine branch when formulation omits excess pre-formed GMP.
17-amino-acid mix
Supplies most canonical amino acids when cysteine and tyrosine are titrated separately.
Tyrosine & cysteine (separate)
Solubility- and oxidation-sensitive residues; separate addition improves accurate stoichiometry and reduces side chemistry in long incubations.
Nicotinamide
Precursor for NAD(P)+; supports enzymes in redox-balanced paths that persist over extended reactions.
Nuclease-free water
Diluent for final formulation; minimizes nuclease-mediated template degradation during preparation.
2.2 PEP–NTP (1 h optimized) vs NMP–ribose–glucose (20 h) master mixes
The one-hour PEP–NTP formulation uses phosphoenolpyruvate with extract kinases for high-flux ATP regeneration together with supplied NTPs, which favors strong early yield in a short window. The twenty-hour NMP–ribose–glucose formulation feeds nucleoside monophosphates plus ribose and glucose so salvage and glycolytic pathways sustain gradual triphosphate rebuilding, better matched to long incubations where burst regeneration would exhaust pools. In short: the first favors early power; the second favors sustained endurance.
2.3 Bonus: transcription when GMP is omitted but guanine is present
Purine salvage enzymes in crude lysate convert guanine to GMP via PRPP-dependent phosphoribosylation even when free GMP is omitted from the mix; further phosphorylation restores GDP and GTP for transcription. Guanine is not a nucleotide by itself—it feeds the salvage pathway upstream of GMP—so RNA polymerase still sees normal triphosphate pools once recycling runs.
3. Planning the global experiment — proteins and hypothesis
3.1 At least one property per fluorescent protein
Protein
Property affecting CF expression or readout
sfGFP
Superfolder folding is fast; signal rises quickly and is less maturation-limited than most reds; mild pH sensitivity remains.
mRFP1
Oxygen-dependent chromophore maturation and slower maturation than gfp-class FPs delay peak fluorescence relative to translation.
mKO2
Orange emitter with acid sensitivity; slow pH drift in long reactions can change apparent brightness.
mTurquoise2
Cyan FP with favorable quantum yield; folding and early dark states still gate signal early in CF.
mScarlet-I
Fast-maturing red engineered to shorten dark intermediates versus older reds, improving time-to-readout over long incubations.
Electra2
Engineered teal/green line for multiplexing; maturation kinetics, magnesium / ionic strength, and folding yield set plate reader signal per molecule in bulk lysate.
3.2 Hypothesis for 36 h fluorescence
Example (swap when your FP and supplement limits are assigned): Protein mRFP1. Adjustment: tune magnesium glutamate and HEPES buffer together within instructor-allowed concentration ranges. Expected effect: more stable elongation and oxidative red chromophore maturation over tens of hours, increasing integrated fluorescence at 36 h compared with a formulation optimized only for a one-hour PEP-driven burst.
4. Build-a-cloud-lab simulation (optional)
The build-a-cloud-lab exercise was optional bonus work—not required for homework credit. I used the 3D simulator to lay out a collaborative cloud laboratory with multiple Nebula (System) units arranged in rows, using the floor-plan tools, assembly controls, and camera views to think about how automated capacity (liquid handling, connected modules, and workflow spacing) would support the global Nebula cell-free experiment described in the sections above.
Section 6 generic Nebula JSON remains an operational resource for final projects, not a homework item itself.
Lab phases
Get more information on this.
Course links
Add the same recitation, Google Slide, and Benchling links your instructor posted on the main assignment page.