Complete end-to-end project documentation including governance assessment and interactive Python application.
/\_/\
( o.o )
> ^ <
/| |\
(_| |_)
"Meow! Check out both sections above!"
BioVolt Governance Assessment — Policy Options Comparison - I filled this out anyways but the project is located in the Sub as the Cat suggested- Meow
The table below compares three governance approaches for the BioVolt DIY electroporation device across multiple criteria. Scoring: 3 = Best, 2 = Moderate, 1 = Worst.
Criteria
Option 1: Community Self-Governance
Option 2: Safety Warnings & Labels
Option 3: Regulatory Licensing
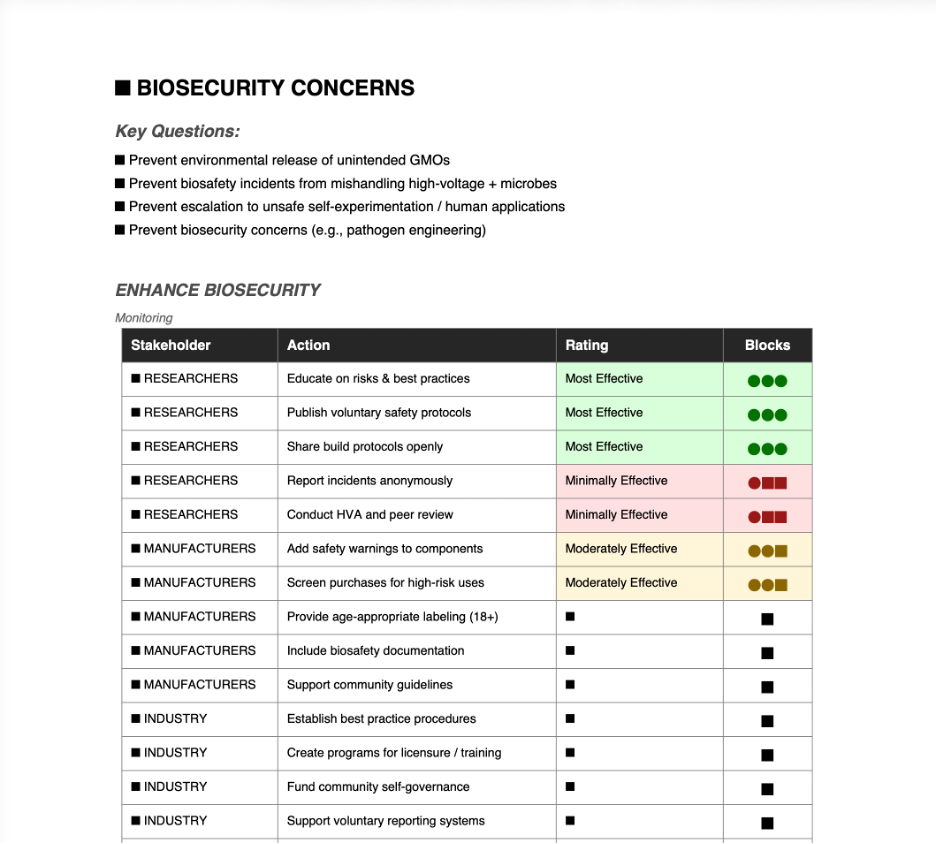
ENHANCE BIOSECURITY
• By preventing incidents
2
2
3
• By helping respond
3
1
2
FOSTER LAB SAFETY
• By preventing incident
2
2
3
• By helping respond
3
1
3
PROTECT THE ENVIRONMENT
• By preventing incidents
2
2
3
• By helping respond
3
1
2
OTHER CONSIDERATIONS
• Minimizing costs and burdens
3
2
1
• Feasibility?
3
3
1
• Not impede research
3
2
1
• Promote constructive applications
3
2
1
Option Summaries
Option 1 — Community-Led Self-Governance: ✓ Best for: response capacity, feasibility, minimizing burdens, not impeding research ✗ Weaker on: prevention (relies on voluntary participation; rogue actors may ignore)
Option 2 — Targeted Product Restrictions (Safety Warnings/Labels): ✓ Best for: feasibility, moderate prevention without bans ✗ Weaker on: response capacity (warnings don’t help after incidents), limited impact on determined bad actors
Option 3 — Regulatory Classification (Licensing/HVA Review): ✓ Best for: prevention (permits, training, HVA peer review blocks worst misuse) ✗ Weaker on: costs, feasibility, impedes DIY research, harms global equity
Recommendation: Prioritize Option 1 (community self-governance) as primary, combine with Option 2 (warnings) as secondary safeguard. Avoid Option 3 unless clear evidence of high-risk proliferation emerges.
Subsections of Week 1 HW: Principles and Practices
DIY Electroporation Project: BioVolt - First rolled out at DEFCON 32- Now revisted from END to END
Biological engineering application/tool to develop: BioVolt is a portable, ultra-low-cost DIY electroporation device (~$10-20 in parts) that uses a piezoelectric crystal from a barbecue lighter to generate ~2,000 V pulses for temporary cell membrane permeabilization. This enables DNA/RNA uptake in bacteria (e.g., E. coli), yeast, plant protoplasts, or even stem cells for genetic transformation. Inspired by the DEFCON 32 talk “You got a lighter I need to do some Electroporation” (presented by Dr. James Utley (Me), Phil Rhodes, and Josh Hill from Viva Securus/Syndicate Laboratories), it builds on frugal biohacking principles: piezoelectric trigger pulsing, custom microfluidic cuvettes from aluminum tape/magnets/glass slides, and simple high-voltage testing.
DEFCON 32 Presentation — Where It Started for me
At DEFCON 32 the talk I presented focused on the device itself — proving that a barbecue lighter’s piezoelectric crystal could generate sufficient voltage to temporarily permeabilize cell membranes for DNA uptake. The talk covered design details, demos, troubleshooting (e.g., arc gap tuning with Post-it notes), and the biohacking ethos behind building a ~$10 electroporator.
Key highlights from the talk: ~2,000 V pulses via lighter clicks, high cell mortality (50-70%) but viable transformants, GFP reporter demos, open protocols encouraged.
Next Phase: End-to-End Pipeline with Efficiency Focus
The next phase of BioVolt moves beyond the device and brings the entire workflow end to end, with a focus on efficiency and frugal validation. The goal: take a piezoelectric electroporator built from a barbecue lighter and prove — through a full pipeline — that it actually works. The pipeline includes:
Plasmid amplification via thermal cycling — Before electroporation, the initial plasmid source will be amplified using the MJ Research PTC-100 thermal cycler (Peltier-effect programmable controller) available in the lab. This ensures sufficient plasmid DNA concentration for transformation.
DNA concentration measurement — Using the Rodeo open colorimeter (visible light version for OD600 cell density measurements) and, if possible, the UV version for DNA concentration quantification. This provides pre- and post-transformation metrics.
Electroporation — Transformation of cells with the amplified plasmid DNA using the BioVolt piezoelectric device, followed by recovery and plating.
Post-transformation PCR verification — For good measure, PCR will be run after transformation using the same thermal cycler to check whether the insert is present in the recovered cells. This triangulates and correlates with plating results to provide a hasty “close enough” frugal validation.
Gel electrophoresis confirmation — Agarose gel electrophoresis to visualise PCR products and verify successful transformation (e.g., presence of reporter genes like GFP via band patterns under UV).
The aim is to triangulate multiple data points — plasmid amplification, colorimetric/UV measurement, transformation plating, and post-transformation PCR — to build confidence that the piezo electroporator from a lighter actually delivers. Fingers crossed, this provides a credible, frugal, end-to-end validation of a DIY electroporation workflow.
This democratizes synthetic biology for education, citizen science, and personal biohacking in resource-limited settings.
Lab Setup & Tools in Action - You can see I got some goods to work with!
My biohacker lab integrates the device with the full verification pipeline.
On to the assignement - Interactive Governance Assessment Form
An interactive Python application (app.py) is provided to assess governance and risk mitigation strategies for the BioVolt project. The form uses a block-based rating scale where more filled blocks mean more effective:
Blocks
Rating
Meaning
●○○
Minimally Effective
Low impact — unlikely to achieve the goal
●●○
Moderately Effective
Moderate impact — partial success likely
●●●
Most Effective
High impact — highly likely to achieve goal
Project File Structure
BioVolt_week_01_hw_principles_and_practices/
├── _index.md # This file — project documentation (Hugo page)
├── app.py # Interactive governance assessment application
├── requirements.txt # Python dependencies
├── Biohacker_Lab.jpeg # Lab overview photo
├── in_da_lab.jpeg # Working in the lab photo
├── Volt_Test.jpeg # High-voltage testing with insulation tester
├── rodeo-colorimeter.png # IO Rodeo open colorimeter
├── BioVolt_govern_UI.png # Screenshot of the application UI
└── Biovolt_Govern_Report.png # Screenshot of the PDF report output
Prerequisites
Python 3.x installed on your system
tkinter (usually included with Python; on Linux you may need python3-tk)
Installation
Navigate to the project directory:
cd BioVolt_week_01_hw_principles_and_practices
Install required dependencies:
pip install -r requirements.txt
Running the Application
python app.py
How to Use the Form
Launch — The application opens a dark-themed window with the assessment matrix.
Read the instructions — System instructions are displayed at the top of the form explaining the block-based rating system.
Review each concern category — Three categories are presented, each with context questions:
Equity Concerns — access, regulation, educational barriers, global equity
Environmental Concerns — microbial activity, non-human organisms, public concerns
Rate each action — For every action under each stakeholder (Researchers, Manufacturers, Industry, Organizations), click one of three block-rating buttons:
The selected button stays highlighted with its rating colour
A status indicator appears to the right showing your selection
Other buttons in the same row reset to their default state
Export to PDF — Click the “EXPORT TO PDF” button to generate a report containing:
Cover page with assessment date and completion count
Rating scale legend with colour-coded descriptions
Full assessment tables for each concern category
Colour-coded rows: green tint for Most Effective, amber for Moderate, red for Minimal
Block indicators (●●● / ●●○ / ●○○) printed in every row
Summary page with counts and percentages for each rating level
Reset — Click “RESET MATRIX” to clear all selections and start over.
Application Features
Block-based rating scale — intuitive system where more blocks = more effective (no ambiguity)
Dark theme UI — dark background with neon accent colours for readability
Persistent button state — selected buttons remain highlighted with their rating colour
Status indicators — each row shows the current selection in text beside the buttons
Scrollable interface — mouse wheel support for navigating the full assessment matrix
Neon accent bars — left-side accent bars on each concern card for visual hierarchy
Colour-coded PDF output — rating cells are tinted to match their effectiveness level
Summary statistics — PDF includes a final page with counts and percentages
Empty export protection — warns you if no ratings are selected before exporting
Form reset — one-click reset with confirmation dialog
Screenshots
Application UI — Dark-themed interface with block-based rating buttons and colour-coded status indicators:
PDF Report Output — Exported assessment with colour-coded rows, block indicators, and stakeholder ratings:
Governance / Policy Goals (Preventing Harm)
Focus on non-tool-function risks: Prevent environmental release of unintended GMOs, biosafety incidents from mishandling high-voltage + microbes, escalation to unsafe self-experimentation/human applications, or biosecurity concerns (e.g., pathogen engineering). Core aims: Minimize biosafety/biosecurity harms, promote responsible use, avoid stifling innovation with heavy regulation, encourage informed DIYbio practices, and address public/environmental concerns.
Three Potential Governance/Policy Actions
Action 1: Community-Led Self-Governance with Voluntary Guidelines and Reporting
Goal: Foster peer accountability and safe practices through DIYbio networks, reducing risks via shared norms without external mandates.
Design:
Opt-in: DIYbio communities, forums (e.g., Discord, Reddit, The ODIN users), and makerspaces.
Fund: Crowdfunding, donations, or volunteer time.
Approve: Community-elected moderators or biosafety working groups.
Implement: Publish voluntary guidelines (e.g., “BioVolt Safety Protocol” on protocols.io or GitHub), require protocol sharing for builds, anonymous incident reporting (expand “Ask a Biosafety Expert” services).
Risks / What could go wrong (incorrect assumptions, uncertainties): Assumes broad ethical participation - rogue actors may ignore; self-reporting misses hidden issues; low adoption if seen as “extra work.”
Assumptions, “Success” and “Failure” rubric:
Success (best - 1): High adoption -> fewer accidents, strong norms against risky uses (e.g., no human trials), community self-corrects.
Mid (2): Partial uptake -> safety improvements in visible projects, but gaps remain.
Failure (worst - 3): Guidelines ignored -> no risk reduction, or “forbidden fruit” effect increases experimentation.
Action 2: Targeted Product Restrictions (e.g., Safety Warnings / Age Limits on Kits & Components)
Goal: Reduce impulsive or uninformed misuse by requiring clear hazard labels on high-voltage components (e.g., piezoelectric lighters, capacitors) or full kits, without banning access.
Approve: Consumer safety agencies or state-level consumer protection (e.g., modeled on CRISPR kit labeling laws).
Implement: Mandatory labels (“Not for human use; biological hazard when combined with genetic material; 18+ recommended”).
Risks / What could go wrong: Warnings may not deter determined users (parts sourced separately); patchy enforcement online/global; could increase black-market activity.
Mid (2): Labels added but often ignored by experienced users.
Failure (worst - 3): Little impact on bad actors; adds cost/delays for legitimate builders.
Unintended consequences: Drives activity underground, reducing community visibility/oversight.
Action 3: Treat as if it has a Regulatory Classification as Restricted Biotech Equipment (e.g., Licensing for High-Voltage Builds) Pledge reporting and Safe use.
Goal: Treat advanced DIY electroporators like controlled lab tools - require permits/training for >1,000 V devices to prevent proliferation to high-risk genetic work.
Design:
Opt-in: Individual builders/users via registration.
Fund: User fees.
Approve: Government agencies (e.g., expanding CDC/NIH biosafety rules or local health depts).
Implement: Permits, training requirements, inspections for community labs/shared spaces.
Hazard Vulnerability Assessment (HVA) and Peer Review: Conduct a comprehensive HVA and require peer review through a pseudo-IRB-like entity - a multidisciplinary and independent review board focusing on environmental and human safety. This entity would evaluate proposed uses, assess risks, and provide guidance on safe protocols before high-voltage builds are deployed.
Risks / What could go wrong: Hard to define safe thresholds; bureaucracy kills accessibility; overreach chills innovation globally.
Unintended consequences: Harms global equity/education; favors institutional labs only.
Overall Tradeoffs & Prioritization
Prioritize Action 1 (community self-governance) as primary: Lowest overregulation risk, aligns with DIY ethos, adaptable to low current misuse evidence, leverages community goodwill.
Combine with Action 2 (targeted warnings) as secondary: Adds minimal external safeguard for public health, deters casual risks without bans.
Avoid/minimize Action 3 unless clear evidence of high-risk proliferation: Highest chance of killing accessibility and innovation, poor fit for low-harm tool like BioVolt.
Key uncertainties (misuse rates, community response, enforcement feasibility) favor lighter interventions. Monitor via voluntary reporting; escalate only if serious incidents arise. This balances empowerment with responsible governance for biosafety and preventing broader DIY genetic risks.
Made with love and the AI Slop is from Cursor-GLM 4.7
Week 1: Professor Questions
Answers organized by instructor, please click the question to reveal the answer!
Instructions: Click the triangle (▶) or question text to expand and view the full answer.
[SECTION 1] Questions from Professor Jacobson
Source: Lecture 2 slides
▶ Question 1: Nature's machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
Answer
Executive Summary: DNA polymerase intrinsic error rate (~10⁻⁷) would cause ~320 errors per human genome replication (3.2 × 10⁹ bp). Biology employs multilayer error correction (proofreading, mismatch repair, excision repair) to achieve final fidelity of ~10⁻⁹ to 10⁻¹⁰ errors per base per division, yielding 0.3-3 errors per replication in normal somatic cells.
Error Rate of DNA Polymerase
DNA polymerase has an intrinsic error rate of approximately 1 error per 10⁷ nucleotides during DNA synthesis. With integrated 3’ to 5’ exonuclease proofreading activity, this improves to approximately 1 error per 10⁸-10⁹ nucleotides.
Comparison to Human Genome Length
The human genome contains approximately 3.2 × 10⁹ base pairs.
Without proofreading:
Error rate: ~10⁻⁷ per nucleotide
Expected errors per replication: ~320 errors per genome copy
With proofreading:
Error rate: ~10⁻⁸ to 10⁻⁹ per nucleotide
Expected errors per replication: ~3-32 errors per genome copy
How Biology Deals with This Discrepancy
Biology employs multiple layers of error correction that act sequentially:
Proofreading (3’ → 5’ exonuclease activity)
DNA polymerase detects incorrect base pairing via geometric distortion
Removes mismatched nucleotide immediately
Reduces error rate by approximately 100-1000-fold
Mismatch Repair (MMR) System
Post-replication surveillance mechanism
In bacteria (E. coli): MutS, MutL, and MutH proteins
In eukaryotes: MSH (MutS homolog), MLH (MutL homolog), and PMS protein families
System identifies mismatched base pairs, excises incorrect strand segment, and resynthesizes
Further reduces error rate by approximately 100-1000-fold
Nucleotide Excision Repair (NER)
Repairs bulky DNA lesions (UV-induced thymine dimers, chemical adducts)
Removes damaged nucleotide segments (20-30 nt patches)
Base Excision Repair (BER)
Corrects small base modifications (deamination, oxidation, alkylation)
DNA glycosylases remove damaged bases; AP endonucleases process abasic sites
Result: The combined fidelity of replication in eukaryotic somatic cells typically achieves ~10⁻⁹ to 10⁻¹⁰ errors per base per cell division, depending on organism, cell type, and proliferation status. This ensures 0.3-3 errors per genome replication under normal physiological conditions.
Note: Fidelity varies by context. Cancer cells with MMR defects exhibit 100-1000× higher mutation rates. Germline cells employ additional proofreading mechanisms. Some DNA polymerases (e.g., Pol η, translesion synthesis polymerases) have lower fidelity by design for specialized repair functions.
▶ Question 2: How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don't work to code for the protein of interest?
Answer
Executive Summary: For a typical 400-residue protein, the number of synonymous DNA sequences (due to codon degeneracy) is astronomically large—on the order of 10¹⁰⁰ or more, calculated as the product of synonymous codon counts across all positions. In practice, most sequences fail due to codon usage bias, mRNA secondary structure, RNA instability, splicing interference, cryptic regulatory elements, and synthesis/cloning constraints.
Number of Different Ways to Code for a Protein
The genetic code is degenerate—61 sense codons encode 20 standard amino acids plus start/stop signals. Each amino acid (except Met and Trp) has multiple synonymous codons:
Leucine, Serine, Arginine: 6 codons each
Isoleucine: 3 codons
Methionine, Tryptophan: 1 codon each
For an average human protein (~400 amino acids):
The total number of synonymous DNA sequences is the product of synonymous codon counts across all positions:
N = ∏(i=1 to 400) n_i
where n_i = number of synonymous codons for amino acid i.
Rough estimate:
Average degeneracy per amino acid ≈ 3 codons (weighted by frequency)
Total combinations ≈ 3⁴⁰⁰ ≈ 10¹⁹⁰ possible DNA sequences
Even conservative estimates (e.g., leucine-rich proteins) yield 10¹⁰⁰+ combinations.
Why All These Different Codes Don’t Work in Practice
Even though multiple sequences encode the same amino acid sequence, the vast majority fail to express functional protein due to:
1. Codon Usage Bias
Each organism has preferred codons reflecting tRNA abundance (Plotkin & Kudla 2011)
E. coli prefers different codons than humans (e.g., AGG/AGA rare in bacteria, common in mammals)
Rare codons → ribosome stalling → may alter co-translational folding kinetics
Using non-optimal codons can reduce expression 10-1000-fold (Gustafsson et al. 2004)
2. mRNA Secondary Structure
Certain nucleotide sequences form stem-loops or hairpins
Strong secondary structures can:
Block ribosome binding
Stall translation
Trigger mRNA degradation
3. RNA Stability
AU-rich sequences → rapid mRNA degradation
GC-rich sequences → more stable mRNA
Wrong codon choice can drastically reduce mRNA half-life
4. Splicing Interference
Certain sequences create cryptic splice sites
Can cause exon skipping or intron retention
Results in truncated or non-functional protein
5. Ribosome Binding Sites (RBS) Interference
Shine-Dalgarno sequences (prokaryotes) or Kozak sequences (eukaryotes)
Internal RBS-like sequences can cause premature translation initiation
Results in truncated proteins
6. Restriction Enzyme Sites
Cloning often requires avoiding certain restriction sites
Limits sequence choices for practical molecular biology
7. Repetitive Sequences
Long homopolymer runs (e.g., AAAAAA) cause synthesis/sequencing errors
Can trigger recombination or replication errors
Quantitative Example:
For a 10-amino acid peptide (assuming average 3-fold degeneracy), there are theoretically 3¹⁰ ≈ 59,000 synonymous sequences. However, accounting for all the constraints listed above, only an estimated 10²-10³ sequences (~1-2%) would be practically functional.
[SECTION 2] Questions from Dr. LeProust
Source: Lecture 2 slides
▶ Question 3: What's the most commonly used method for oligo synthesis currently?
Answer
Executive Summary: Phosphoramidite chemistry on solid-phase support (Caruthers method, 1981) is the current industry standard, with typical coupling efficiency of 98.5-99.5% per cycle and practical length ceiling of 150-200 nucleotides.
Phosphoramidite Chemistry (Solid-Phase Synthesis)
The phosphoramidite method on solid support is the dominant technology for oligonucleotide synthesis worldwide.
Key Features:
Invented: Marvin Caruthers and colleagues (1981)
Platform: Solid-phase synthesis on controlled-pore glass (CPG) or polystyrene beads
Direction: 3’ → 5’ synthesis (chain grows from 3’-OH to 5’ end)
Cycle efficiency: Typically 98.5-99.5% per nucleotide addition
Practical length limit: 150-200 nucleotides for routine synthesis
Four-Step Cycle:
Detritylation (acid treatment)
Removes DMT (dimethoxytrityl) protecting group from 5’-OH
Converts unstable phosphite (P³⁺) to stable phosphate (P⁵⁺)
Forms phosphate backbone
Advantages:
High throughput (96-384 well formats)
Automated
Scalable (nmol to µmol scale)
Well-established chemistry
Current Platforms: Commercial platforms include BioAutomation and ABI/Applied Biosystems synthesizers for traditional column-based synthesis. Newer high-throughput approaches include Twist Bioscience (silicon-based microarray synthesis) and Custom Array (electrochemical synthesis on chips).
▶ Question 4: Why is it difficult to make oligos longer than 200nt via direct synthesis?
Answer
Executive Summary: Cumulative coupling inefficiency (even at 99% per cycle) yields only ~13% full-length product at 200 nt. Dominant failure modes are deletion sequences from incomplete coupling, depurination during detritylation, and increasing purification difficulty as n-1, n-2… products accumulate.
Cumulative Coupling Errors and Deletion Sequences
The primary challenge is imperfect coupling efficiency in each phosphoramidite addition cycle.
The Mathematics of Error Accumulation:
Coupling efficiency per cycle: typically 98.5-99.5%
Stepwise failure rate: 0.5-1.5% per cycle
Yield of full-length product = (coupling efficiency)^n where n = oligo length
Yield Calculation:
Length
Coupling Efficiency
Full-Length Yield
50 nt
99%
60%
100 nt
99%
37%
150 nt
99%
22%
200 nt
99%
13%
300 nt
99%
5%
At 200 nucleotides with 99% efficiency:
Only 13% of molecules are full-length correct sequence
87% are deletion products (n-1, n-2, n-3… truncations)
Specific Problems Beyond 200nt (in order of impact):
Deletion Sequences from Incomplete Coupling
Failed coupling at position i → all subsequent additions build on truncated chain
Creates heterogeneous mixture of n-1, n-2, n-3… products
Capping step blocks these from extending, but they remain in final pool
Depurination During Acid Treatment
Detritylation uses trichloroacetic acid or dichloroacetic acid
Causes glycosidic bond cleavage at purines (A, G)
Cumulative damage over 200+ cycles
Results in abasic sites and chain breaks
Purification Difficulty
Full-length (200 nt) vs. n-1 (199 nt) differ by <0.5% in mass
HPLC and PAGE separation becomes marginal
Impure product affects downstream applications
Secondary Structure Formation
Long single-stranded oligos form intramolecular hairpins during synthesis
Blocks reagent access to growing 3’-OH end (on solid support, growing from 3’ end)
Reduces effective coupling efficiency in later cycles
Practical Solutions: Modern approaches avoid direct synthesis beyond 200 nt by using gene assembly from overlapping 60-80 nt oligos (polymerase cycling assembly, Gibson assembly), column-based assembly methods (e.g., Twist Bioscience chip synthesis followed by assembly), or emerging enzymatic synthesis using terminal deoxynucleotidyl transferase-based methods.
▶ Question 5: Why can't you make a 2000bp gene via direct oligo synthesis?
Answer
Executive Summary: Direct phosphoramidite synthesis of 2000 nt is practically infeasible due to vanishingly low yields (0.99^2000 ≈ 10⁻⁹), prohibitive synthesis time (~2-3 weeks continuous), cumulative depurination, and insurmountable purification challenges. Modern gene synthesis uses hierarchical assembly of 60-80 nt oligos into fragments, then full-length genes.
Practical Infeasibility with Current Phosphoramidite Chemistry
Making a 2000 bp gene via direct oligonucleotide synthesis is practically infeasible with standard phosphoramidite chemistry due to insurmountable yield, time, and purification barriers.
Yield Barriers:
At 99% coupling efficiency (best-case scenario):
Yield = 0.99^2000 ≈ 2 × 10⁻⁹ (0.0000002%)
To obtain 1 picomole of full-length product requires ~0.5 moles of starting material
Equivalent to ~660 grams of protected nucleotide phosphoramidites
Material cost alone: ~$500,000 - $1,000,000
Even at 99.5% efficiency (exceptional, rarely achieved):
Yield = 0.995^2000 ≈ 5 × 10⁻⁵ (0.005%)
Still economically and practically prohibitive
Physical/Chemical Barriers:
Synthesis Time
Typical cycle time: 10-15 minutes per nucleotide addition
2000 cycles = 20,000-30,000 minutes = 14-21 days continuous synthesis
Reagent degradation over extended periods
Instrument reliability over multi-week runs
Cumulative Depurination
2000 acid detritylation steps
Each cycle causes low-frequency glycosidic bond cleavage at purines
Accumulates to extensive abasic sites and strand breaks
Secondary Structure Collapse
Long single-stranded DNA forms extensive intramolecular structure
Hairpins and G-quadruplexes block reagent access
Synthesis typically stalls beyond 300-400 nt even with optimized conditions
Solubility and Handling
Very long oligos can precipitate on solid support
Reduced accessibility to coupling reagents
Cleavage and deprotection become inefficient
Practical Solution: Hierarchical Gene Assembly
Modern commercial gene synthesis uses multi-step assembly:
Step 1: Oligo Synthesis
Synthesize 30-50 oligonucleotides (60-80 nt each, with 20-40 nt overlaps)
Yield per oligo: 60-95% (high quality)
Step 2: Fragment Assembly
Assemble oligos into 4-6 intermediate fragments (400-600 bp each)
Methods: Polymerase cycling assembly (PCA), Gibson assembly, Golden Gate
Yield per fragment: 70-90%
Step 3: Final Assembly
Combine fragments into full 2000 bp gene
Gibson assembly or restriction enzyme-based methods
Final yield: 60-85% overall
Example for 2000 bp gene:
40 oligos × 70 nt average = 2800 nt synthesized capacity
Assemble into 5 fragments (~400 bp each)
Final Gibson assembly into 2000 bp construct
Overall yield: ~70% (vs. 10⁻⁹% for direct synthesis)
Commercial Gene Synthesis: Major vendors (Twist Bioscience, IDT, GenScript, Thermo Fisher) offer typical academic pricing of $0.07-0.20/bp, though this is highly variable depending on sequence complexity (GC content, repeats, secondary structure), turnaround time (5-10 days standard, 2-3 days expedited), and order volume. Standard turnaround is 5-10 days with rush options of 2-3 days.
[SECTION 3] Question from Professor George Church
Source: Lecture 2 slides
▶ Question 6: [Using Google & Prof. Church's slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the "Lysine Contingency"?
(I chose this question from the three options)
Answer
Executive Summary: The commonly listed essential amino acids in vertebrates include His, Ile, Leu, Lys, Met, Phe, Thr, Trp, Val, and conditionally Arg. The “Lysine Contingency” from Jurassic Park is scientifically flawed because lysine is already naturally essential in all vertebrates—the genetic modification provides zero additional biocontainment. Moreover, lysine is abundant in all natural food sources, and deficiency takes months to years to be lethal.
The Commonly Listed Essential Amino Acids in Vertebrates
Essential amino acids cannot be synthesized de novo by vertebrate metabolism and must be obtained from diet. The standard list for humans and most vertebrates includes: Histidine (His, H), Isoleucine (Ile, I), Leucine (Leu, L), Lysine (Lys, K) [focus of Jurassic Park scenario], Methionine (Met, M), Phenylalanine (Phe, F), Threonine (Thr, T), Tryptophan (Trp, W), Valine (Val, V), and Arginine (Arg, R), which is conditionally essential—essential in juveniles, young/growing animals, and during illness, though adults can synthesize limited amounts via the urea cycle.
Mnemonic:“PVT TIM HALL” (Phe, Val, Thr, Trp, Ile, Met, His, Arg, Leu, Lys)
Note: The classification varies slightly by species and life stage. Arginine is typically considered semi-essential or conditionally essential in adult mammals.
The “Lysine Contingency” from Jurassic Park
In Jurassic Park (Michael Crichton, 1990), InGen implemented a “Lysine Contingency” as a biocontainment measure. The plan involved genetically engineering dinosaurs unable to synthesize lysine, making them dependent on lysine supplements in their food. The theory was that if they escaped, they would die from lysine deficiency. As Dr. Wu stated: “The lysine contingency is intended to prevent the spread of the animals is case they ever got off the island.”
Why the Lysine Contingency is Scientifically Flawed
Critical Problem: ALL ANIMALS ALREADY REQUIRE DIETARY LYSINE
1. Lysine is Naturally Essential in All Vertebrates
Humans, dinosaurs, birds, and mammals cannot synthesize lysine de novo. Animals lost the lysine biosynthesis pathway approximately 500 million years ago during early vertebrate evolution. The dinosaurs would have required dietary lysine regardless of any genetic modification. Therefore, the “contingency” provides zero additional biocontainment—it is entirely redundant.
2. Lysine is Abundant in Natural Food Sources
Based on USDA nutritional databases, lysine is widespread in both plant and animal food sources. Plant sources include legumes (soybeans, lentils, beans) containing 1-2% lysine by dry weight, seeds and grains with 0.2-0.8% lysine, and grasses and leafy vegetation with 0.3-0.6% lysine. Animal sources are even richer: insects contain approximately 2-3% lysine by dry weight, while vertebrate muscle tissue, fish, and eggs contain 1.5-2.5% lysine by weight.
Estimated lysine intake for large theropods (carnivorous dinosaurs):
Note: The following are rough extrapolations from modern vertebrate nutritional requirements and are not based on direct measurements of dinosaur metabolism. Assuming an estimated daily food intake of 50-100 kg meat (scaled from modern large carnivores) and lysine content of meat at approximately 1.5-2.0 g/100g, the estimated daily lysine intake would be 750-2000 g. Compared to an estimated lysine requirement of approximately 10-50 g/day (scaled from mammals, though highly uncertain), even conservative estimates suggest 10-100× excess lysine intake.
Estimated lysine intake for herbivorous dinosaurs:
Assuming estimated daily vegetation consumption of hundreds of kg for sauropods and lysine content in plant matter of 0.3-1.0% dry weight, the estimated daily lysine intake would be hundreds of grams. This substantially exceeds the likely requirement of 50-200 g/day when scaled from large herbivorous mammals.
Key Point: Even consuming exclusively grass, leaves, or insects would likely provide sufficient lysine to meet metabolic needs, assuming dinosaur requirements scaled similarly to modern vertebrates.
3. Timescale of Lysine Deficiency is Impractical
Lysine deficiency symptoms develop slowly: immune system impairment occurs over weeks to months, growth retardation takes months, and muscle wasting progresses over months to years. Lethality from severe deficiency requires months to years. A dinosaur escaping into the wild would eat naturally available food and immediately obtain sufficient lysine, never developing deficiency symptoms. The timescale mismatch is fatal to the strategy: containment must occur in minutes to hours (the escape window), while lysine deficiency lethality takes months to years. The result is a completely ineffective biocontainment strategy.
4. Better Biocontainment Strategies
If the goal is preventing escaped dinosaurs from surviving or reproducing, several approaches would be more effective than the lysine contingency.
Metabolic Dependencies: Creating auxotrophy for synthetic amino acids not found in nature (such as D-amino acids or unnatural amino acids requiring continuous supplementation), nucleotide auxotrophy (e.g., thymine requirement), or vitamin/cofactor dependencies (e.g., engineered B12 requirement) would provide genuine containment.
Genetic Kill Switches: Conditional lethality genes requiring antidote molecules, thermosensitive essential genes that allow survival only at controlled temperatures, or light-dependent survival mechanisms requiring specific UV or wavelength exposure offer programmed containment.
Reproductive Control: All-female populations (as attempted in Jurassic Park), meiotic drive systems ensuring sterility, or genetic incompatibility with wild relatives would prevent population establishment.
Environmental Dependencies: Temperature-sensitive phenotypes surviving only in controlled climates or organisms requiring specific atmospheric pressure or composition would restrict habitat range.
Conclusion: How This Affects My View of the Lysine Contingency
The Lysine Contingency is scientifically flawed as a biocontainment strategy and represents a misunderstanding of vertebrate nutritional biochemistry. The strategy fails on four fundamental levels: (1) it is not a contingency since lysine is already naturally essential in all vertebrates, making the modification redundant; (2) it is not limiting since lysine is abundant in nearly all natural food sources; (3) it is not fast-acting since lysine deficiency takes months to years to be lethal in large vertebrates; and (4) it provides no additional biocontainment barrier beyond natural biology.
From a biosafety perspective, the lysine contingency demonstrates the risk of “security theater” in synthetic biology—creating the appearance of control without meaningful containment. Real biocontainment requires dependencies on synthetic or artificial inputs not present in natural ecosystems. Modern synthetic biology approaches include unnatural amino acid dependencies (e.g., amber suppressor systems with synthetic tRNAs), genetic kill switches (toxin-antitoxin modules, essential gene knockout with complementation), orthogonal genetic systems (expanded genetic code, xenobiology with XNA), and metabolic dependencies on synthetic nutrients or specific light wavelengths.
Narrative function in Jurassic Park: The flawed lysine contingency serves as a plot device illustrating InGen’s overconfidence and foreshadows that all their control measures will fail (“Life finds a way”). It highlights the dangers of inadequate risk assessment and overconfidence in genetic engineering safeguards.
Lessons for modern synthetic biology: Biological containment is extremely difficult and requires multiple redundant safeguards. Single-point dependencies, especially on naturally occurring molecules, are inadequate. Rigorous testing and evolutionary escape rate measurements are essential for any containment strategy.
[REFERENCES]
Primary Literature and Resources
DNA Replication Fidelity (Q1):
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. 6th edition. Garland Science, 2014. Chapter 5: DNA Replication, Repair, and Recombination.
Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497-529. doi:10.1146/annurev.biochem.69.1.497
Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106(2):302-323. doi:10.1021/cr0404794
Genetic Code and Translation (Q2):
Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 2011;12(1):32-42. doi:10.1038/nrg2899
Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22(7):346-353. doi:10.1016/j.tibtech.2004.04.006
Tuller T, Carmi A, Vestsigian K, et al. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141(2):344-354. doi:10.1016/j.cell.2010.03.031
Oligonucleotide Synthesis (Q3-Q5):
Caruthers MH. Gene synthesis machines: DNA chemistry and its uses. Science. 1985;230(4723):281-285. doi:10.1126/science.3863253
Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014;11(5):499-507. doi:10.1038/nmeth.2918
Hughes RA, Ellington AD. Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb Perspect Biol. 2017;9(1):a023812. doi:10.1101/cshperspect.a023812
Amino Acid Nutrition and Biosafety (Q6):
Reeds PJ. Dispensable and indispensable amino acids for humans. J Nutr. 2000;130(7):1835S-1840S. doi:10.1093/jn/130.7.1835S
WHO/FAO/UNU Expert Consultation. Protein and amino acid requirements in human nutrition. WHO Technical Report Series 935. Geneva: World Health Organization; 2007.
USDA National Nutrient Database for Standard Reference (Release 28). Agricultural Research Service, U.S. Department of Agriculture. 2015.
Crichton M. Jurassic Park. New York: Alfred A. Knopf; 1990.
Mandell DJ, Lajoie MJ, Mee MT, et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature. 2015;518(7537):55-60. doi:10.1038/nature14121 [Modern unnatural amino acid containment systems]
Document created: February 10, 2026 Author: James Utley, PhD Affiliation: Syndicate Laboratories, Panama City, Panama Course: HTGAA 2026 Spring — Week 1 Homework