Simulated restriction enzyme digestion with the seven enzymes specified in this week’s lab protocol: SalI, SacI, EcoRV, KpnI, BamHI, HindIII, and EcoRI. Used both the DNA Gel Art Interface (λ DNA) and Benchling (lambda phage genome NC_001416) to visualize digest patterns and verify cut-site predictions.
1. DNA Gel Art Interface — λ DNA Restriction Digests
Simulated gel electrophoresis using the DNA Gel Art tool. λ DNA was digested with various enzyme combinations (EcoRV + SacI, HindIII + PvuII, NdeI + SalI, etc.) across lanes 2–10. The table documents water, CutSmart buffer, λ DNA, and enzyme volumes per lane.
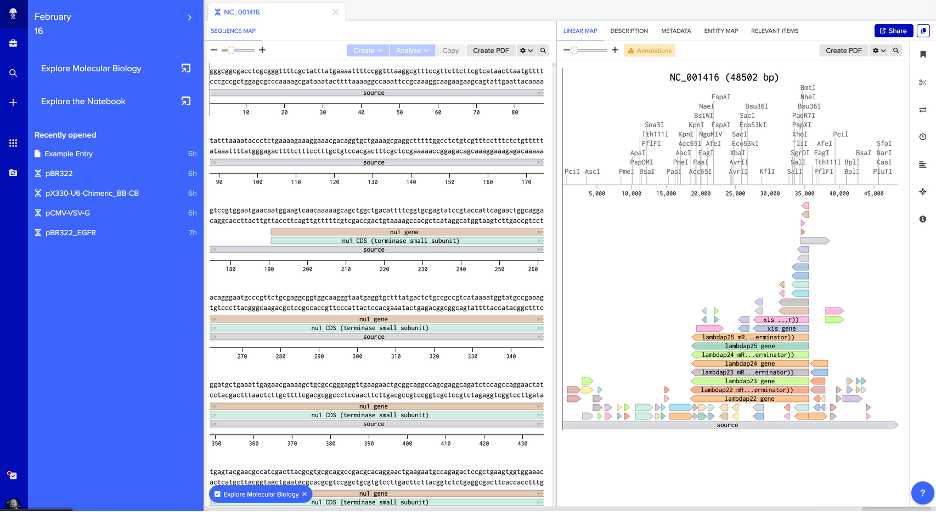
2. Benchling — NC_001416 Sequence Map with Restriction Sites
Linear map of NC_001416 in Benchling showing the raw sequence, annotated genetic features (e.g., xis, nul, lambdap genes), and restriction enzyme cut sites (PciI, AscI, PmeI, BsaI, KpnI, SacI, SalI, and others) along the 48.5 kb genome.
3. Virtual Digest Gel — NC_001416 with All Seven Required Enzymes
Simulated gel (Life 1 kb Plus ladder) showing digest results for NC_001416 with each of the seven required enzymes:
Protein chosen: Superfolder Green Fluorescent Protein (sfGFP)
Why: sfGFP is a robust, rapidly maturing fluorescent protein derived from Aequorea victoria (Pédelacq et al., 2005). It is widely used in synthetic biology as a reporter—when expressed in cells, it fluoresces bright green under blue/UV light, enabling real-time visualization of gene expression, protein localization, and cell tracking. Its “superfolder” mutations improve folding efficiency in diverse hosts (including E. coli), making it ideal for expression experiments. It also connects directly to Part 4, where we build an expression cassette to make E. coli glow green.
Using the Central Dogma in reverse: given a protein sequence, we infer a possible DNA sequence that could encode it. Because the genetic code is degenerate (multiple codons encode the same amino acid), many DNA sequences can produce the same protein. A simple reverse translation uses one valid codon per amino acid—here, E. coli preferred codons (most frequently used in highly expressed genes).
Tool used: Reverse translation with E. coli codon preferences (e.g., ExPASy Translate or similar tools; can also be done manually with a codon usage table).
Reverse-translated DNA sequence (one possible encoding):
Why optimize codon usage? Different organisms prefer different codons for the same amino acid, based on tRNA abundance and other factors. Using rare codons can slow translation, cause ribosome stalling, and reduce protein yield. Codon optimization replaces codons with those most frequently used in the target organism, improving expression levels and folding. It also allows us to avoid restriction enzyme recognition sites (e.g., BsaI, BsmBI, BbsI) that would interfere with Golden Gate or other assembly methods.
Organism chosen:Escherichia coli (K-12)
Why E. coli? It is the standard workhorse for recombinant protein expression: well-characterized genetics, fast growth, simple culture, and widely available vectors and protocols. The HTGAA Part 4 exercise uses E. coli for the sfGFP expression cassette, so optimizing for E. coli keeps the workflow consistent.
(717 bp; optimized for E. coli expression, restriction-site free — same sequence used in Part 4 expression cassette)
3.4 You Have a Sequence! Now What?
Technologies to produce sfGFP from this DNA:
Cell-dependent (recombinant expression in E. coli):
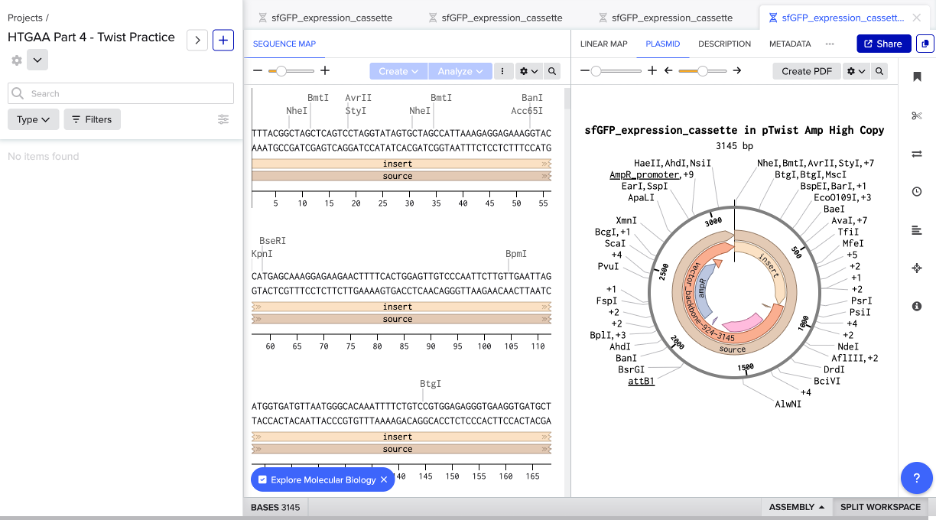
Clone the codon-optimized gene into an expression vector (e.g., pTwist Amp High Copy) with a constitutive or inducible promoter (e.g., BBa_J23106), RBS (e.g., BBa_B0034), and terminator (e.g., BBa_B0015).
Transform the plasmid into E. coli (e.g., DH5α, BL21).
Grow cells; the host RNA polymerase transcribes the DNA into mRNA, and ribosomes translate the mRNA into sfGFP.
The protein folds and forms its chromophore; cells fluoresce green under blue light (~488 nm excitation, ~510 nm emission).
Cell-free (in vitro transcription–translation):
Use a cell-free system (e.g., E. coli lysate, PURE system) with the DNA template.
Add NTPs, amino acids, and energy sources; the system transcribes and translates the gene without living cells.
Useful for rapid prototyping, toxic proteins, or when cell growth is impractical.
DNA synthesis (Twist, IDT, etc.):
Order the gene as a clonal or linear fragment from a synthesis provider.
Use it directly for cloning or cell-free expression, avoiding PCR or cloning from natural sources.
Alignment of DNA, RNA, and protein: In the Central Dogma, DNA is transcribed to RNA (T→U), and RNA is translated to protein (3 nt → 1 aa). Tools like Benchling or Ronan’s gel art site can visualize this alignment.
Single gene → multiple proteins: Alternative splicing (eukaryotes) or alternative start codons/ribosomal frameshifting can produce multiple proteins from one gene. sfGFP is a single open reading frame, but in general, one gene can yield multiple isoforms through these mechanisms.
Part 4: Prepare a Twist DNA Synthesis Order
Part 4: Prepare a Twist DNA Synthesis Order
Practice exercise — building an sfGFP expression cassette in Benchling, preparing a mock Twist order, and annotating the plasmid.
4.1–4.2 Accounts & Build Your DNA Insert Sequence
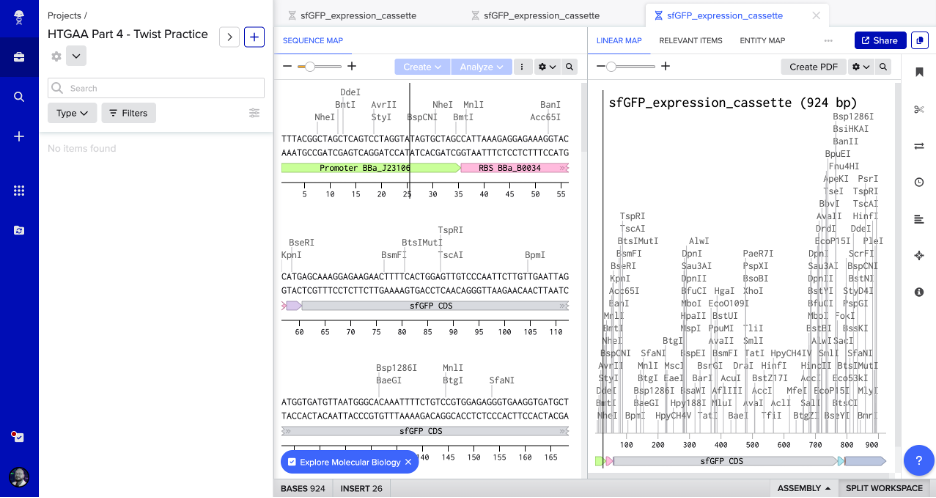
Created Twist and Benchling accounts. Built the sfGFP expression cassette in Benchling with annotated parts:
Promoter (BBa_J23106)
RBS (BBa_B0034)
Start codon (ATG)
Coding sequence (codon-optimized sfGFP from Part 3)
Downloaded GenBank construct and imported into Benchling
Screenshot: Sequence Upload to Twist
Design Notes: Manual vs. Programmatic
Efficiency: Designing expression cassettes and plasmids can be far more efficient with Python and/or R — tools like DNA Chisel, PyDNA, or SynBioHub enable scripted design, validation, and export. Batch operations, automated codon optimization, and constraint checking become straightforward.
Learning value: Building the construct manually in Benchling — clicking through each part, copying sequences, and annotating by hand — offers a different kind of learning. You develop intuition for how promoters, RBSs, and CDSs fit together, where restriction sites fall, and what the plasmid “looks like” at each step. That tactile understanding is harder to get from a script. For a first expression cassette, the manual approach is worth the extra time.
MANUAL (Benchling) PROGRAMMATIC (Python/R)
───────────────── ─────────────────────
Click, paste, annotate Script → design → export
Slow, one construct at a time Fast, many constructs
Deep, tactile understanding Scalable, reproducible
"I built this" "I designed 50 of these"
Both have their place. Start manual; scale with code.
Answers framed around the BioVolt DIY electroporation pipeline: plasmid amplification → transformation → PCR verification → gel electrophoresis. What DNA would we read, write, and edit to make this frugal pipeline sing?
In the BioVolt pipeline: After electroporation, we transform E. coli with plasmids (e.g., sfGFP expression cassette). We run post-transformation PCR and gel electrophoresis to infer success—but we don’t know the exact sequence. Sequencing the plasmid (or PCR amplicon) confirms that:
The insert is correct (no truncations, no wrong gene)
Electroporation didn’t introduce mutations (high voltage can stress DNA)
The expression cassette is intact for downstream experiments
Broader applications (aligned with BioVolt’s democratization goals):
Environmental monitoring — e.g., sewage/wastewater DNA for microbiome analysis in Panama; biodiversity surveys
Human health — disease-associated genes, pharmacogenomics
DNA data storage — archival sequences in synthetic DNA
Portable — USB-sized device; runs on laptop; fits in a backpack. Ideal for Panama, field sites, or home labs.
Real-time — base calling as reads stream; no batch wait.
Long reads — can span full plasmids; fewer assembly gaps.
Low capital — compared to Illumina, much cheaper to get started.
No PCR required for some workflows — direct DNA sequencing possible (native DNA).
Question
Answer
Output?
FASTQ files (reads + quality scores); can be base-called in real time to BAM/FASTA.
Essential steps & base calling?
(1) DNA passes through a nanopore; (2) each base disrupts ionic current differently; (3) base caller (e.g., Guppy) converts current traces → A/T/G/C; (4) reads assembled/compared to reference.
Input & preparation?
Option A (PCR amplicon): PCR product → end-prep → adapter ligation → load onto flow cell. Option B (native): Fragment DNA (e.g., g-TUBE or sonication) → repair ends → adapter ligation → load. Key: adapters enable motor protein to thread DNA through pore.
First-, second-, or third-generation?
Third-generation. Single-molecule, real-time; no amplification required for some lib preps; long reads; portable form factor.
NANOPORE SEQUENCING (simplified)
╭───-╮
DNA ────► │ ▓▓ │ ← pore in membrane
│ ▓▓ │ (ionic current changes per base)
╰───-╯
│
▼
╔═══════════╗
║ A T G C ║ ← base caller (Guppy, etc.)
║ ▓ ▓ ▓ ▓ ║ converts squiggle → sequence
╚═══════════╝
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
For BioVolt: The expression cassettes we electroporate! Specifically:
sfGFP plasmid — promoter + RBS + sfGFP CDS + terminator (e.g., BBa_J23106, BBa_B0034, sfGFP, BBa_B0015). This is the “make E. coli glow green” construct we build in Part 4.
Custom reporters — e.g., biosensors that fluoresce in response to environmental cues (pH, metals, toxins) for citizen-science monitoring.
Validation controls — known sequences for PCR/gel positive controls in the frugal pipeline.
Broader: Therapeutics (mRNA vaccines), genetic circuits, DNA origami, gene clusters for metabolic engineering.
WHAT WE SYNTHESIZE FOR BIOVOLT:
┌────────────────────────────────────────────────────────────┐
│ [Promoter]─[RBS]─[ATG]─[sfGFP]─[His]─[TAA]─[Terminator] │
│ │ │ │
│ └── always on └── glows green under UV │
│ │
│ Twist / IDT makes this. BioVolt zaps it in. Done. 🟢 │
└────────────────────────────────────────────────────────────┘
(ii) What technology would you use and why?
Technology:Column-based phosphoramidite synthesis (e.g., Twist Bioscience, IDT) — the industry standard for gene synthesis.
Why: High fidelity, scalable, cost-effective for genes and gene fragments. Twist can deliver clonal genes (circular) ready for transformation—perfect for BioVolt.
Question
Answer
Limitations?
Speed: days to weeks. Accuracy: ~1 error per 1–3 kb; may need sequencing to confirm. Scalability: great for genes; whole genomes get expensive. Length: very long constructs may need assembly.
Essential steps?
(1) Design sequence (e.g., codon-optimized); (2) split into overlapping oligos; (3) synthesize oligos (phosphoramidite chemistry, base-by-base); (4) assemble oligos (PCR, Gibson, or enzymatic); (5) clone into vector; (6) sequence to verify.
PHOSPHORAMIDITE SYNTHESIS (cartoon)
Base + Base + Base + ... → oligo → assemble → gene
A T G C A T ...
│ │ │ │ │ │
▼ ▼ ▼ ▼ ▼ ▼
┌───┴───┴───┴───┴───┴───┴───----┐
│ ████ ████ ████ ████ ████ │ ← solid support (column)
│ add → couple → oxidize → cap │ (repeat ~hundreds of times)
└─────────────────────────────- ┘
5.3 DNA Edit
(i) What DNA would you want to edit and why?
For BioVolt:
Improve electroporation efficiency — edit E. coli to knock out or modify genes that affect membrane composition, cell wall, or DNA repair (e.g., recA, mutS) to get more transformants per zap.
Biosensor chassis — edit strains to express reporter circuits (e.g., GFP under metal-responsive promoter) for environmental sensing in the DIY pipeline.
Safety — auxotrophic markers, kill switches, or containment edits for responsible DIYbio.
EDIT E. coli FOR BETTER BIOVOLT TRANSFORMATION?
Wild-type E. coli Edited E. coli
│ │
│ "Membrane too tough" │ "Softer membrane?"
│ "DNA repair too good?" │ "Fewer repair enzymes?"
│ │
▼ ▼
⚡ BioVolt ⚡ ⚡ BioVolt ⚡
│ │
▼ ▼
10³ CFU/µg 10⁵ CFU/µg? 🎯
│ │
"Meh" "Now we're talking!"
(ii) What technology would you use and why?
Technology:CRISPR/Cas9 (with HDR for precise edits) — or base editors for single-nucleotide changes without double-strand breaks.
Why: Programmable, precise, widely adopted. gRNA design is straightforward; many tools (Benchling, etc.) support it.
Question
Answer
Limitations?
Efficiency: not 100%; mixed populations. Precision: off-target cuts possible; PAM requirement constrains target sites. Delivery: need to get Cas9 + gRNA into cells (electroporation works!).
Preparation & input?
Design: gRNA(s) targeting locus; donor template (ssODN or plasmid) for HDR. Input: DNA template, Cas9 nuclease, gRNA (or plasmid expressing both), cells. Optional: base editor (e.g., ABE, CBE) for point mutations.
Essential steps?
(1) Design gRNA (avoid off-targets; check PAM, e.g., NGG for SpCas9); (2) deliver Cas9 + gRNA + donor (electroporation, conjugation, etc.); (3) Cas9 cuts DNA; (4) cell repairs via NHEJ or HDR; (5) screen for edits (PCR, sequencing).
CRISPR/Cas9 IN ACTION (simplified)
gRNA: "Find this sequence" ── ┐
├──► Cas9 ──► CUT! ✂️
DNA: ...TARGET...PAM... ──┘
Before: ────[TARGET]────
After: ────╲ ╱──── (cell repairs: NHEJ or HDR)
╲ ╱
gap
BioVolt could deliver Cas9 RNP + donor via electroporation! ⚡