Week 4 HW: Protein Design
Homework 4
Protein Design Part I — amino acids, protein structure, helices, and β-sheets.
📋 Parts
- Part A: Conceptual Questions (Shuguang Zhang) — Answer any 9 of 12 questions ✓
- Part B: Protein Analysis and Visualization (ECSOD) — ECSOD/SOD3 structure analysis ✓
- Part D: Group Brainstorm — Bacteriophage Engineering (MS2 L Lysis Protein) — stability + tunable toxicity ✓
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang (i.e. you can select two to skip).
Answers provided for: (9 selected; 2 skipped: Can you make other non-natural amino acids? Design some new amino acids. and Design a β-sheet motif that forms a well-ordered structure.)
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Answer: Meat is roughly 15–25% protein by dry weight; water content varies. For a rough estimate, assume ~500 g of meat contains ~100 g of protein (≈20%). An average amino acid has a molecular mass of ~100 Daltons (Da).
- Number of amino acids in 100 g protein ≈ 100 g / (100 × 10⁻³ kg/mol) ≈ 100 g / 0.1 kg/mol ≈ 1 mol ≈ 6 × 10²³ molecules (Avogadro’s number).
Order of magnitude: ~10²³–10²⁴ amino acid molecules per 500 g of meat.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer: Dietary proteins are digested into amino acids and small peptides before absorption. They are absorbed as monomers, not as intact proteins.
- Digestion: Stomach acid and proteases (pepsin, trypsin, chymotrypsin) hydrolyze peptide bonds.
- Absorption: Amino acids enter the bloodstream and are used as building blocks.
- Assembly: Our cells use these amino acids to synthesize our own proteins according to our genome. The cow’s or fish’s DNA is never used; only the amino acid monomers are reused.
Result: We use the amino acids as nutrients; we do not incorporate the cow’s or fish’s proteins or genes intact. We remain human because our protein synthesis is controlled by human DNA.
3. Why are there only 20 natural amino acids?
Answer: The genetic code is degenerate: 61 sense codons encode 20 standard amino acids. The number 20 reflects a balance of evolutionary and physicochemical constraints:
- Evolution: Early life likely used a smaller set of amino acids; the canonical 20 were added over time as biosynthesis pathways evolved.
- Sufficiency: 20 amino acids provide enough chemical diversity (hydrophobic, polar, charged, aromatic, etc.) to build proteins with diverse structures and functions.
- Genetic code: The triplet code (4³ = 64 codons) can encode more than 20, but expansion beyond 20 would require additional tRNA synthetases and codons; the cost of adding more may outweigh the benefit.
- Fidelity: A larger set of amino acids would increase the risk of misincorporation and reduce translation fidelity.
Summary: 20 amino acids provide sufficient diversity for protein function while keeping the system manageable and robust.
4. Where did amino acids come from before enzymes that make them, and before life started?
Answer: Abiotic (prebiotic) synthesis.
- Miller–Urey experiment (1952): Simulated early Earth conditions (reducing atmosphere, lightning, heat) produced amino acids (glycine, alanine, etc.) from simple precursors (H₂O, CH₄, NH₃, H₂).
- Extraterrestrial sources: Amino acids (e.g., glycine) are found in meteorites (e.g., Murchison) and comets; they may have been delivered to early Earth.
- Hydrothermal vents: Alkaline vents and other mineral surfaces can catalyze amino acid formation from CO₂, H₂, and nitrogen.
- Strecker synthesis: Cyanide, aldehydes, and ammonia can form amino acids under prebiotic conditions.
Conclusion: Amino acids could form without enzymes or life, via abiotic chemistry and/or delivery from space.
5. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer: Left-handed (M-type) helix.
- L-amino acids form right-handed (P-type) α-helices because the L-configuration places the side chain in a conformation that favors right-handed twist.
- D-amino acids are the mirror image; their side chains favor the opposite twist. A D-amino acid α-helix is therefore left-handed.
Summary: D-amino acid α-helix → left-handed; L-amino acid α-helix → right-handed.
6. Can you discover additional helices in proteins?
Answer: Yes. Beyond the canonical α-helix (3.6 residues/turn), other helices exist:
- 3₁₀ helix: ~3 residues/turn; tighter, shorter hydrogen bonds; often at helix termini.
- π-helix: ~4.4 residues/turn; rare; energetically less favorable.
- Polyproline helices (PPI, PPII): Proline-rich helices with different geometry.
- Collagen-like structures: Triple helical motifs.
- Novel helices: New helices can be discovered through structural biology (e.g., X-ray crystallography, cryo-EM) or designed de novo.
Conclusion: Additional helices can be found by analyzing protein structures and designing new motifs.
7. Why are most molecular helices right-handed?
Answer: Several factors favor right-handed helices:
- Chirality of L-amino acids: All natural proteins use L-amino acids. The L-configuration favors right-handed α-helices and β-strands; left-handed helices are sterically strained.
- DNA: Double helix is right-handed (B-form).
- RNA: RNA helices are typically right-handed.
- Minimization of steric clash: Right-handed twist often minimizes steric clashes between side chains and the backbone.
- Evolution: Once right-handed helices dominated, the genetic code and biosynthesis reinforced this preference.
Summary: L-amino acid chirality and steric constraints favor right-handed helices in natural proteins.
8. Why do β-sheets tend to aggregate?
Answer: β-sheets expose backbone amide and carbonyl groups that can form hydrogen bonds with adjacent strands or sheets.
- Hydrogen bonding: β-strands have alternating N–H and C=O groups along the backbone; these can pair with adjacent strands or with strands from another sheet.
- Hydrophobic side chains: Many β-sheets have hydrophobic residues; stacking of sheets can bury these surfaces and reduce solvent exposure.
- Extended conformation: Extended strands maximize surface area for inter-strand and inter-sheet contacts.
- Amyloid-like stacking: β-sheets can stack in a parallel or antiparallel fashion, forming amyloid fibrils.
Conclusion: β-sheets aggregate because they expose H-bond donors/acceptors and hydrophobic surfaces that favor inter-sheet interactions.
9. What is the driving force for β-sheet aggregation?
Answer: Main driving forces:
- Hydrogen bonding: Backbone–backbone H-bonds between strands from different molecules or sheets.
- Hydrophobic effect: Burial of hydrophobic side chains reduces contact with water.
- Entropy: Release of ordered water molecules when hydrophobic surfaces associate.
- π–π stacking: Aromatic side chains (e.g., Phe, Tyr) can stack between sheets.
- Electrostatic complementarity: Alternating charged and hydrophobic residues (e.g., in ionic self-complementary peptides) can drive ordered assembly.
Summary: H-bonding, hydrophobicity, and entropy release drive β-sheet aggregation.
10. Why do many amyloid diseases form β-sheets?
Answer: Many disease-associated proteins aggregate into amyloid fibrils rich in β-sheet structure:
- Misfolding: Proteins that are normally α-helical or disordered can misfold into β-sheet-rich conformations under stress (e.g., pH, temperature, mutations).
- Stability: Cross-β structure (β-strands perpendicular to the fibril axis) is highly stable; once formed, fibrils are difficult to disaggregate.
- Nucleation: A small β-sheet nucleus can template further growth; amyloid formation is often nucleation-dependent.
- Examples: Aβ (Alzheimer’s), α-synuclein (Parkinson’s), prion (PrP), huntingtin (Huntington’s).
Conclusion: β-sheet structure provides a stable, self-propagating amyloid conformation that underlies many neurodegenerative diseases.
11. Can you use amyloid β-sheets as materials?
Answer: Yes. Amyloid-like β-sheet structures are used as materials:
- Self-assembling peptides: Shuguang Zhang’s ionic self-complementary peptides form stable β-sheet nanofibers and scaffolds for tissue engineering, drug delivery, and 3D cell culture.
- Nanostructures: β-sheet fibrils can serve as templates for mineralization, nanowires, and conductive materials.
- Hydrogels: β-sheet-rich peptide networks form hydrogels for wound healing and regenerative medicine.
- Functional materials: Engineered amyloid fibrils have been used for catalysis, biosensors, and optical materials.
Conclusion: Amyloid β-sheets can be engineered as functional biomaterials for biomedical and material applications.
Part B. Protein Analysis and Visualization
Protein Selected
| Field | Value |
|---|---|
| Name | Extracellular superoxide dismutase [Cu-Zn] |
| Gene | SOD3 (aka ECSOD) |
| Organism | Homo sapiens (human) |
| Chosen Structure (RCSB PDB) | 2JLP |
| Classification (RCSB) | OXIDOREDUCTASE |
Why I selected it (brief):
ECSOD is a secreted antioxidant enzyme that detoxifies superoxide radicals in the extracellular space, helping protect tissues from oxidative stress. I selected it because it is biologically important in vascular and lung biology, and a high-quality X-ray crystal structure is available for direct 3D visualization (PDB 2JLP).
1) Identify the amino acid sequence of the protein
Canonical protein sequence source: UniProt (Entry: P08294)
IMPORTANT NOTE ABOUT SEQUENCE VS STRUCTURE:
The UniProt canonical protein is the biological sequence. The PDB structure often contains a construct/fragment and may not include every residue from the UniProt canonical sequence.
How to obtain the sequence (recommended workflow):
- A) UniProt canonical sequence (P08294): Go to UniProt entry P08294 → Download the FASTA (canonical sequence)
- B) PDB construct sequence (2JLP): Go to the RCSB page for 2JLP → Download FASTA Sequence
UniProt FASTA (P08294)
PDB FASTA (2JLP)
Download from RCSB 2JLP → Sequence tab → FASTA. The PDB chain in 2JLP is 222 aa per chain (Chains A, B, C, D).
2) How long is it? What is the most frequent amino acid?
| Length type | Value |
|---|---|
| UniProt canonical length | 251 amino acids (from UniProt P08294) |
| PDB (2JLP) chain length | 222 amino acids (each chain A–D in 2JLP) |
Most frequent amino acid (using the provided Colab notebook):
- Input sequence used: UniProt canonical (P08294)
- Most frequent amino acid: Alanine (A)
- Count: 33
- Note: Frequency depends on whether the canonical full-length or the crystallized construct sequence is used.
3) How many protein sequence homologs are there? (UniProt BLAST)
Tool: UniProt BLAST
Procedure: Paste the FASTA sequence (UniProt canonical P08294) → Run BLAST with default settings.
Results to record:
- Total hits/homologs: (run BLAST to fill)
- Example organisms among top hits: (e.g., vertebrate species)
- Typical identity range of strong hits: (e.g., 70–100%)
Write-up sentence:
“Using UniProt BLAST, ECSOD (SOD3) returned ______ homologous sequences under the selected parameters, with strong matches across vertebrate species.”
4) Does the protein belong to any protein family?
Yes.
- Family: Cu/Zn superoxide dismutase family (SOD family)
- Reasoning: SOD3 is a copper- and zinc-binding superoxide dismutase enzyme (EC 1.15.1.1) and is classified as a Cu/Zn SOD.
5) Identify the structure page in RCSB
| Field | Value |
|---|---|
| PDB ID | 2JLP |
| Title | Crystal structure of human extracellular copper-zinc superoxide dismutase. |
| Link | RCSB PDB — 2JLP |
6) When was the structure solved? Is it a good quality structure?
| Field | Value |
|---|---|
| Deposited | 2008-09-14 |
| Released | 2009-03-17 |
| Experimental method | X-RAY DIFFRACTION |
| Resolution | 1.70 Å |
| R-work | 0.150 |
| R-free | 0.185 |
Quality statement:
This is a good quality structure because its resolution (1.70 Å) is better than 2.70 Å (smaller Å = higher resolution detail).
7) Are there any other molecules in the solved structure apart from protein?
Yes.
Small-molecule ligands listed for 2JLP (3 unique):
| Ligand | Description |
|---|---|
| CU | Copper (II) ion |
| ZN | Zinc ion |
| SCN | Thiocyanate ion |
Also present: Solvent water molecules (HOH) are included in the crystal structure.
Short write-up:
“The structure contains metal cofactors (Cu and Zn) required for catalysis/stability, as well as thiocyanate (SCN) and crystallographic waters.”
8) Does the protein belong to any structure classification family?
Yes.
- SCOPe / fold-level description: “Cu,Zn superoxide dismutase-like” fold/superfamily (structure classification consistent with Cu/Zn SOD enzymes)
Write-up sentence:
“Structurally, ECSOD adopts the conserved Cu/Zn superoxide dismutase fold, consistent with other Cu/Zn SOD family proteins.”
9) Open the structure in PyMOL + required visualizations
Recommended PyMOL command checklist
Load:
A) Cartoon:
B) Ribbon:
C) Ball and stick:

D) Color by secondary structure (helices vs sheets):
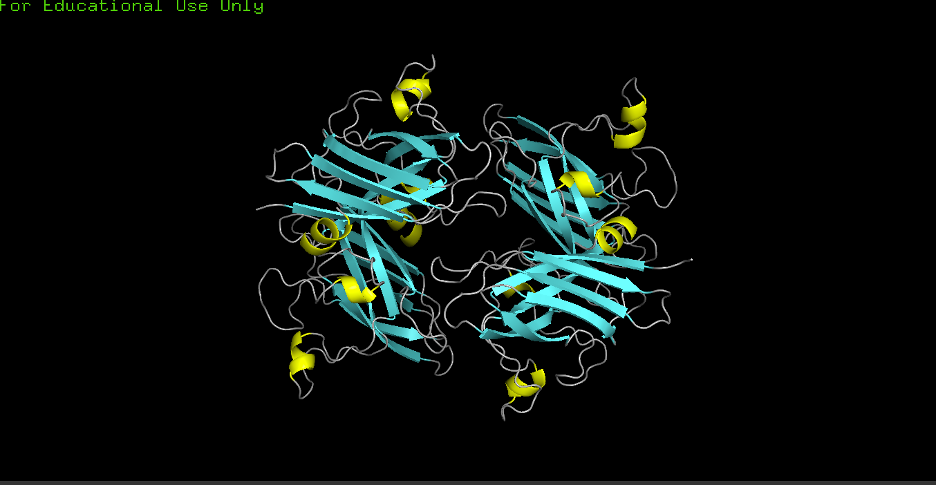
- Observation: More helices or sheets? More sheets. Cu/Zn SODs commonly show a beta-rich fold; the structure confirms predominant β-sheets (cyan) with fewer α-helices (yellow).
E) Color by residue type (hydrophobic vs hydrophilic distribution):
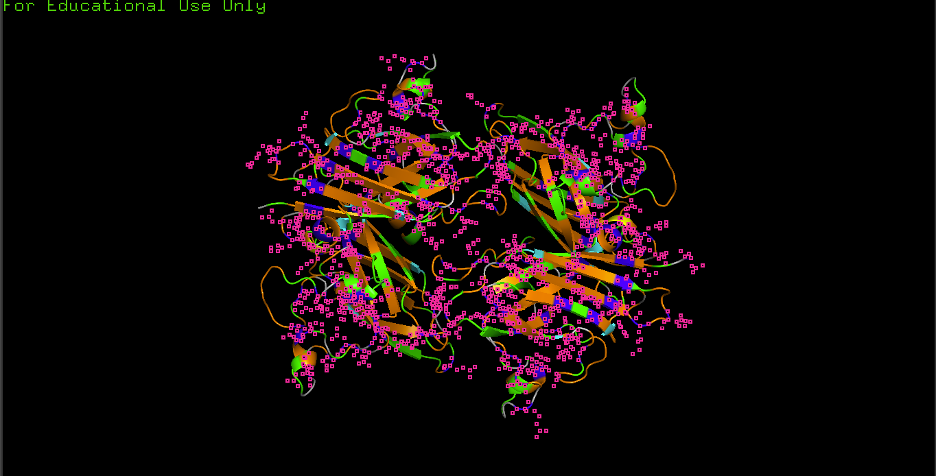
- Observation: Hydrophobics mostly: CORE
- Observation: Hydrophilics mostly: SURFACE
- Interpretation: “Hydrophobic residues tend to cluster in the core, while polar/charged residues tend to be more surface exposed (typical of soluble proteins).”
F) Surface visualization + pockets/holes:
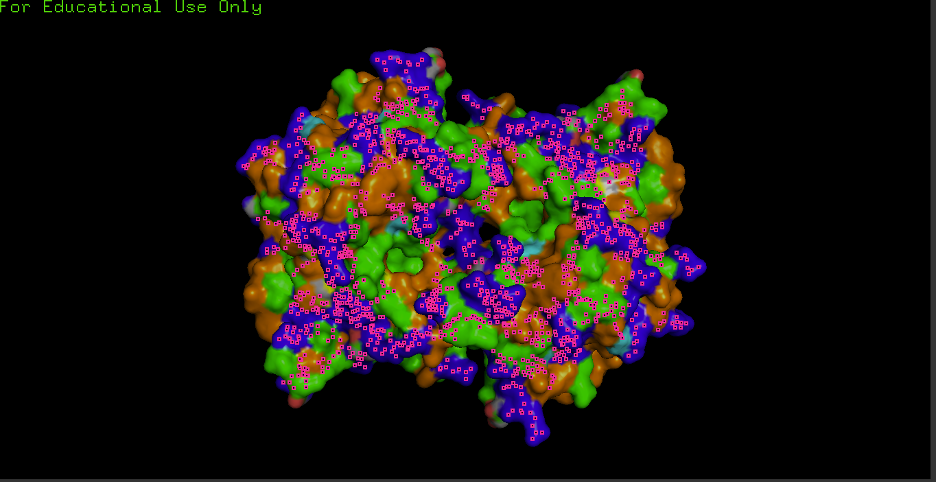
- Observation: Any grooves/holes/binding pockets visible? Yes.
- Where? Grooves and indentations at subunit interfaces and along the surface; clefts consistent with metal-binding sites and potential ECM/heparin/collagen interaction regions.
- Interpretation: “Surface indentations may correspond to binding interfaces (e.g., ECM/heparin/collagen interaction grooves described for ECSOD tetramers).”
Part D. Group Brainstorm — Bacteriophage Engineering
Computational engineering plan for the MS2 L Lysis Protein (group of ~3–4 students).
1. Executive Summary
- Goals chosen: (1) Increased stability (easiest); (2) Tunable toxicity — design a panel of L variants with graded lysis strength (attenuated → wild-type → enhanced) for predictable, dose-dependent control (hard).
- Approach: Use Protein Language Models (e.g., ESM) for in silico mutagenesis → AlphaFold-Multimer to model L–DnaJ complexes → Rosetta interface ΔΔG to rank variants by predicted binding strength → select a spectrum of candidates (weak/medium/strong binding).
- Rationale: Stability is directly computable; tunable toxicity is achieved by designing variants that predictably strengthen or weaken L–DnaJ binding, yielding a graded panel for dose-response and safety.
2. Scope and Assumptions
- Scope: MS2 L protein (75 aa); focus on single-point and small combinatorial mutations at the L–DnaJ interface.
- Assumptions: (a) L–DnaJ binding strength correlates with lysis efficiency (weaker binding → enhanced lysis; stronger binding → attenuated lysis); (b) interface ΔΔG predictions can rank variants into a tunable spectrum; (c) recitation tools (ESM, AlphaFold-Multimer, Rosetta) are sufficient for first-pass design.
- Potential pitfalls:
- Limited training data on phage–bacteria interactions — models may not generalize well to L–DnaJ or other host targets.
- Overlapping gene constraints — the lys gene overlaps coat and replicase; mutations must preserve frameshift and avoid disrupting adjacent genes.
- Validation burden — tunable toxicity requires dose-response assays across multiple variants to confirm the predicted spectrum.
3. Target Engineering Goals
| Goal | Strategy | Tools |
|---|---|---|
| Increased stability | Identify stabilizing mutations (core packing, H-bonds) | ESM mutagenesis, Rosetta ΔΔG |
| Tunable toxicity | Design variants with graded L–DnaJ binding strength: attenuated (stronger binding) → wild-type → enhanced (weaker binding) | AlphaFold-Multimer (L + DnaJ), Rosetta interface ΔΔG |
4. Proposed Pipeline Schematic
5. Tools and Rationale
| Tool | Why it helps |
|---|---|
| ESM / Protein LMs | Learn evolutionary constraints; predict tolerated vs. destabilizing mutations; generate interface-focused variants. |
| AlphaFold-Multimer | Model L–DnaJ complex structure; identify interface residues for tunable design (strengthen or weaken binding). |
| Rosetta ΔΔG | (a) Stability: filter destabilizing variants; (b) Interface: rank variants by L–DnaJ binding strength to build a graded panel (attenuated → enhanced). |
| Part | Content |
|---|---|
| Part A | 9 conceptual questions (Shuguang Zhang) — amino acids, helices, β-sheets |
| Part B | ECSOD/SOD3 (PDB 2JLP) — sequence, structure, PyMOL visualization |
| Part D | MS2 L Lysis Protein — group computational engineering proposal (stability + tunable toxicity) |
Summary (EOF)
| Question | Topic |
|---|---|
| 1 | Amino acid molecules in 500 g meat (~10²³ molecules) |
| 2 | Digestion vs. incorporation — humans don’t become cow/fish |
| 3 | Why only 20 natural amino acids |
| 4 | Prebiotic amino acid synthesis |
| 5 | D-amino acid α-helix → left-handed |
| 6 | Additional helices beyond α (3₁₀, π, etc.) |
| 7 | Right-handed helices due to L-amino acid chirality |
| 8 | β-sheet aggregation — H-bonding, hydrophobicity |
| 9 | Driving force — H-bonds, entropy, hydrophobicity |
| 10 | Amyloid diseases — misfolding, β-sheet stability |
| 11 | Amyloid β-sheets as materials (e.g., Zhang’s peptides) |
Skipped: Can you make other non-natural amino acids? Design some new amino acids. | Design a β-sheet motif that forms a well-ordered structure.