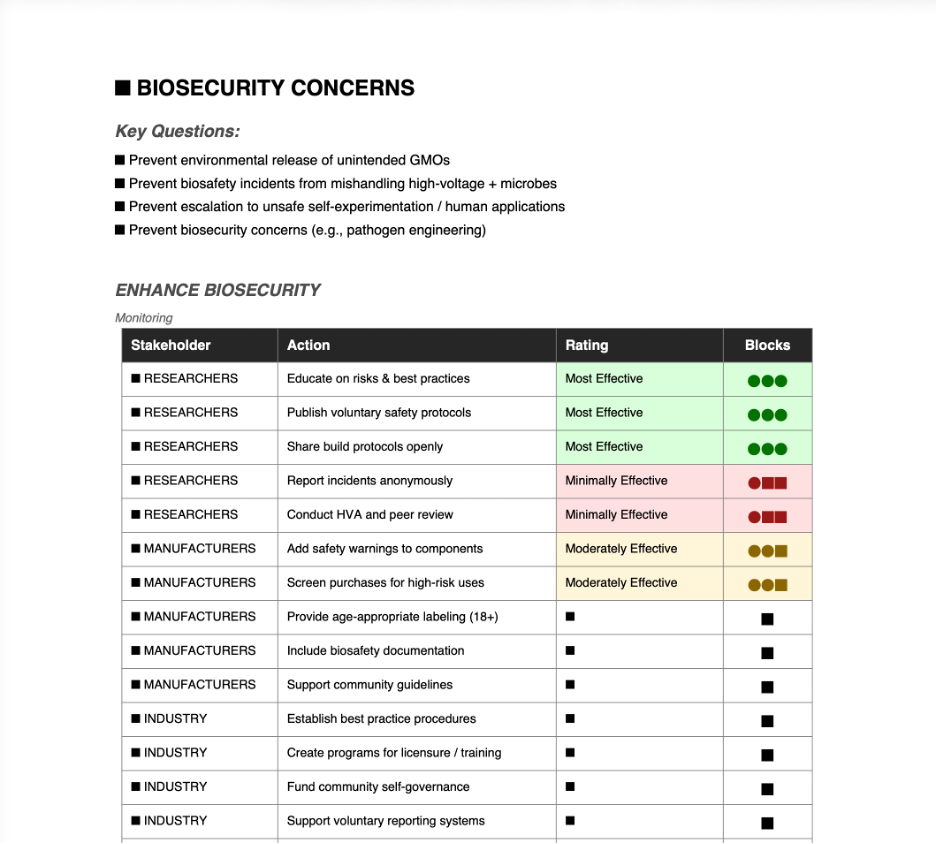
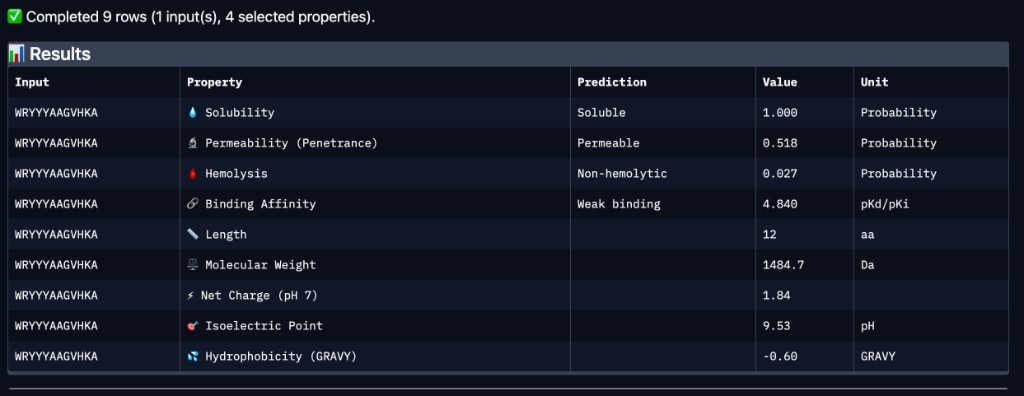
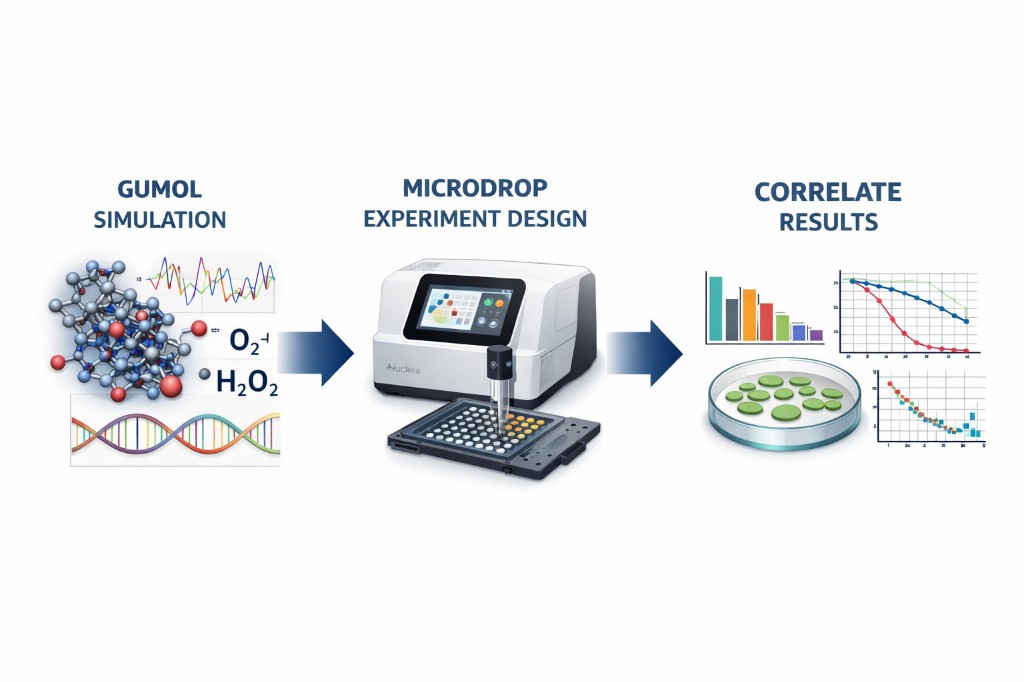
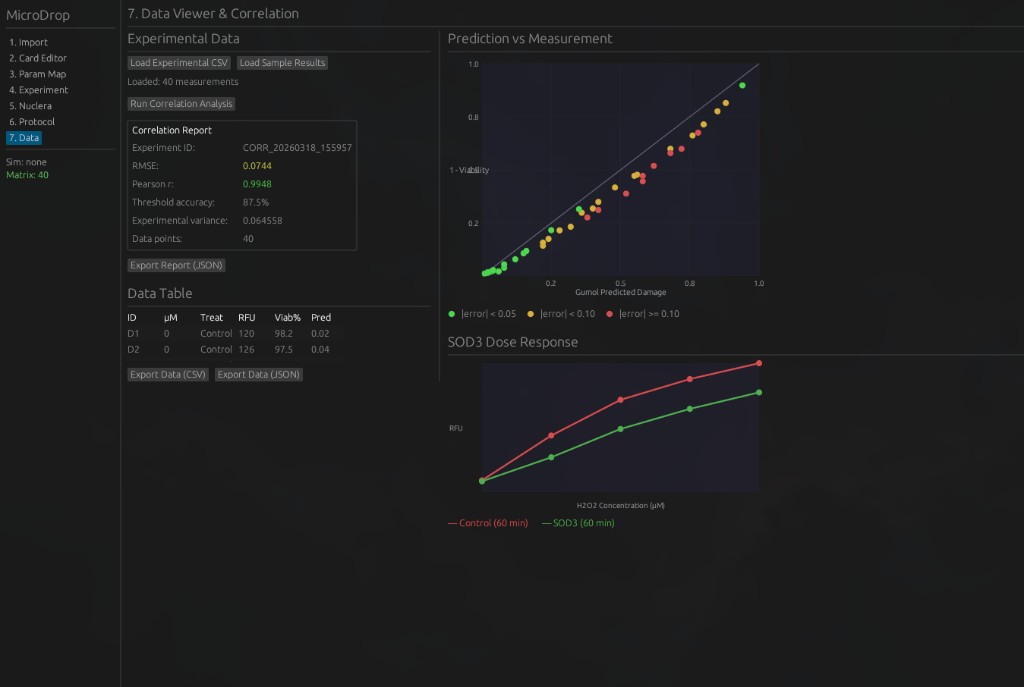
Python protocol for the Opentrons OT-2 that deposits fluorescent bacteria to reproduce the T-Rex QR code on a black agar plate. Black pixels in the 33×33 grid are mapped to plate coordinates; one color (Green) is used for the monochrome design.
Copy this into the first code block of the HTGAA26 Opentrons Colab notebook (after the prerequisite setup):
from opentrons import types
metadata = {
'author': 'James Utley',
'protocolName': 'T-Rex QR Code Bio-Art',
'description': 'Deposits fluorescent E. coli to create a scannable QR code with T-Rex on black agar',
'source': 'HTGAA 2026 Opentrons Lab',
'apiLevel': '2.20'
}
##############################################################################
### Robot deck setup constants - don't change these
##############################################################################
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
well_colors = {
'A1': 'Red',
'B1': 'Green',
'C1': 'Orange'
}
# 33x33 QR code pixel grid (1 = black/deposit, 0 = white/skip)
# Extracted from T-Rex QR code image
QR_PIXELS = [
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0],
[0, 0, 0, 1, 0, 1, 0, 1, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 1, 1, 0, 1, 0, 0, 0, 1, 0, 1, 0, 1, 0, 0, 0],
[0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 1, 1, 0, 0, 1, 1, 0, 0, 1, 1, 0, 1, 0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0],
[0, 0, 0, 1, 0, 1, 0, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 1, 0, 1, 0, 1, 0, 0, 0],
[0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 1, 0, 0, 1, 0, 0, 1, 0, 0, 1, 0, 0, 1, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 1, 1, 1, 1, 1, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 1, 1, 0, 1, 0, 1, 0, 0, 1, 0, 0, 1, 1, 0, 1, 0, 1, 0, 0, 0],
[0, 0, 0, 1, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 1, 0, 1, 0, 0, 1, 0, 1, 1, 1, 1, 0, 1, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 1, 0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0],
[0, 0, 0, 0, 1, 1, 1, 0, 1, 0, 1, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 0, 0, 0, 1, 1, 0, 1, 1, 1, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 1, 1, 1, 1, 0, 0, 1, 1, 0, 0, 0, 1, 1, 1, 0, 0, 0, 1, 1, 0, 1, 1, 1, 0, 1, 1, 0, 0, 0],
[0, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 1, 0, 0, 1, 1, 1, 1, 0, 0, 0, 1, 0, 0, 1, 1, 1, 0, 0, 1, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 0, 1, 0, 1, 1, 0, 1, 0, 0, 1, 1, 0, 0, 0, 0, 0, 0, 1, 0, 0, 1, 1, 1, 1, 0, 1, 0, 0, 0],
[0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 0, 1, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0, 1, 0, 1, 1, 1, 1, 1, 0, 0, 0, 1, 1, 0, 0, 1, 1, 0, 0, 1, 0, 0, 0],
[0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 0, 1, 0, 1, 0, 0, 0, 0, 1, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 1, 1, 1, 1, 1, 0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 1, 0, 0, 1, 0, 0, 1, 0, 1, 0, 0, 1, 0, 0, 0],
[0, 0, 0, 1, 0, 0, 0, 1, 0, 0, 1, 1, 0, 1, 1, 0, 1, 0, 0, 0, 0, 0, 0, 0, 1, 1, 0, 0, 0, 1, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
[0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0],
]
def run(protocol):
##############################################################################
### Load labware, modules and pipettes
##############################################################################
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
temperature_module = protocol.load_module('temperature module gen2', COLORS_DECK_SLOT)
temperature_plate = temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul', 'Cold Plate')
color_plate = temperature_plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
##############################################################################
### Helper functions
##############################################################################
def location_of_color(color_string):
for well, color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
def dispense_and_detach(pipette, volume, location):
above_location = location.move(types.Point(z=location.point.z + 5))
pipette.move_to(above_location)
pipette.dispense(volume, location)
pipette.move_to(above_location)
##############################################################################
### T-Rex QR Code patterning
##############################################################################
PIXEL_SIZE_MM = 2.0 # spacing between pixels (mm)
VOLUME_UL = 0.5 # 0.5 uL per drop (~117 uL total, fits in 200uL PCR strip well)
COLOR = 'Green'
# Collect (row, col) for all black pixels
black_pixels = []
for row in range(len(QR_PIXELS)):
for col in range(len(QR_PIXELS[row])):
if QR_PIXELS[row][col] == 1:
black_pixels.append((row, col))
# One tip, one color - aspirate in batches of 20uL (max pipette volume)
pipette_20ul.pick_up_tip()
source = location_of_color(COLOR)
for i in range(0, len(black_pixels), 20):
batch = black_pixels[i:i+20]
vol = min(len(batch) * VOLUME_UL, 20)
pipette_20ul.aspirate(vol, source)
for row, col in batch:
# Map pixel (row,col) to plate coords: center at (16,16), 2mm per pixel
x_mm = (col - 16) * PIXEL_SIZE_MM
y_mm = (row - 16) * PIXEL_SIZE_MM
loc = center_location.move(types.Point(x=x_mm, y=y_mm))
dispense_and_detach(pipette_20ul, VOLUME_UL, loc)
pipette_20ul.drop_tip()