Boltr — In short: targeting better speed and accuracy than AlphaFold for dynamic (flexible) regions. Rust-native biomolecular inference (Boltz2-style); LibTorch-backed; path toward Burn ML. See Final Project Selected: Boltr for full details.
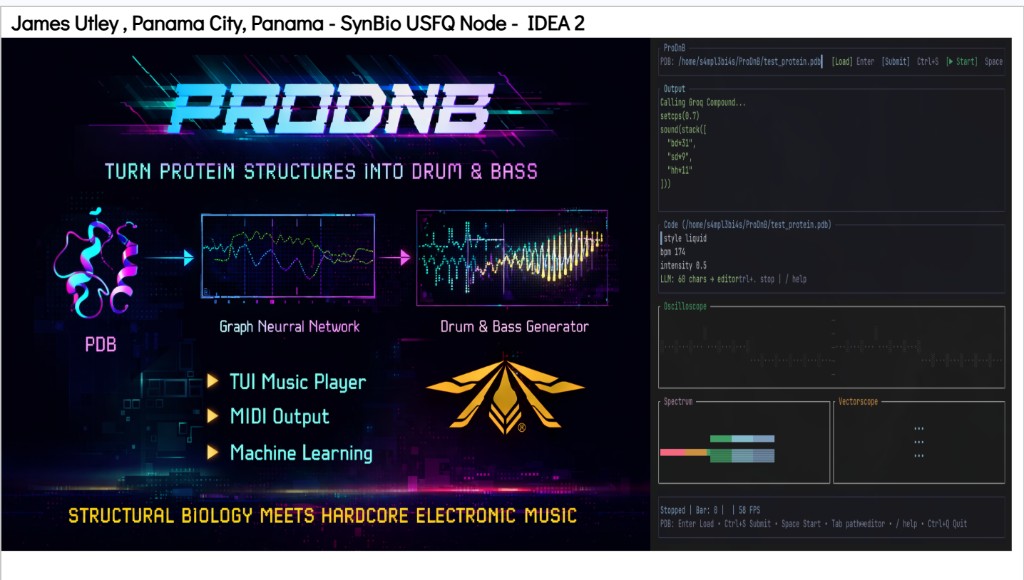
Features: TUI Music Player • MIDI Output • Machine Learning
ProDnB WebUI
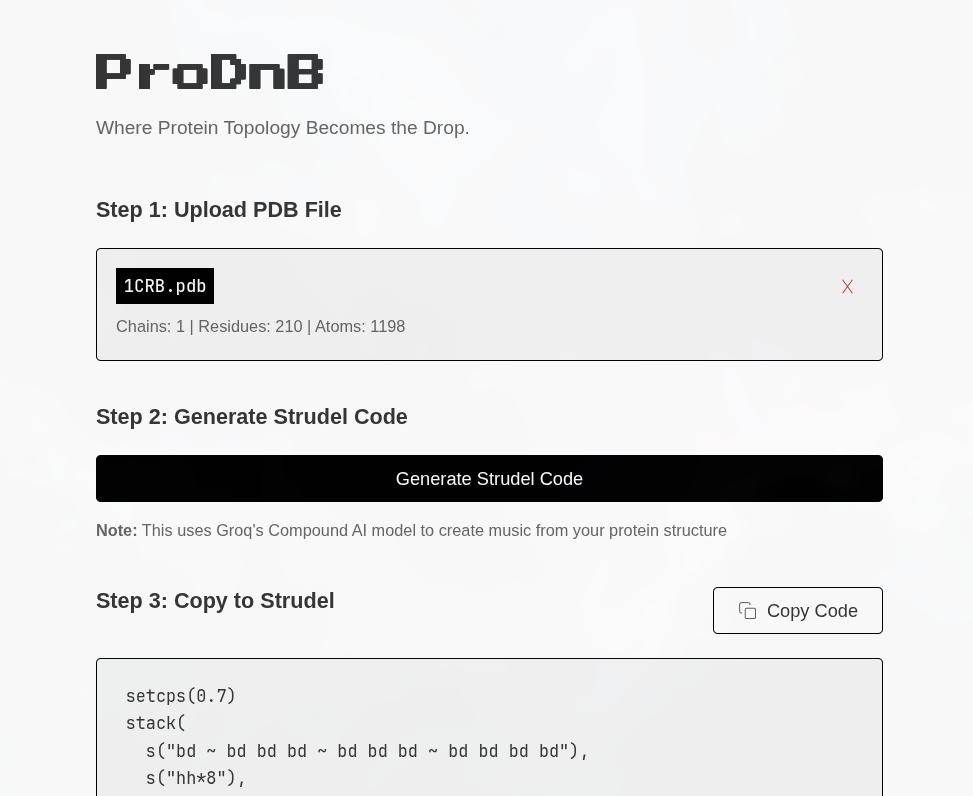
Workflow: (1) Upload PDB file → (2) Generate Strudel Code (via Groq’s Compound AI) → (3) Copy to Strudel. The WebUI parses PDB files (chains, residues, atoms) and outputs Strudel live-coding patterns for drum & bass.
IDEA 3 — BioVolt: DIY Electroporator Validation
Efficiency and frugal validation of a piezoelectric electroporator
Create a piezoelectric electroporator from a barbecue lighter and validate its functionality through a complete workflow. Triangulate multiple data points — plasmid amplification, colorimetric/UV measurement, transformation plating, and post-transformation PCR — to confirm efficacy and provide credible, frugal, end-to-end validation of a DIY electroporation workflow.
In short: I am building a structure predictor aimed at outperforming AlphaFold on both speed and accuracy in dynamic regions—loops, hinges, and other flexible or poorly ordered stretches where rigid single-state models weaken.
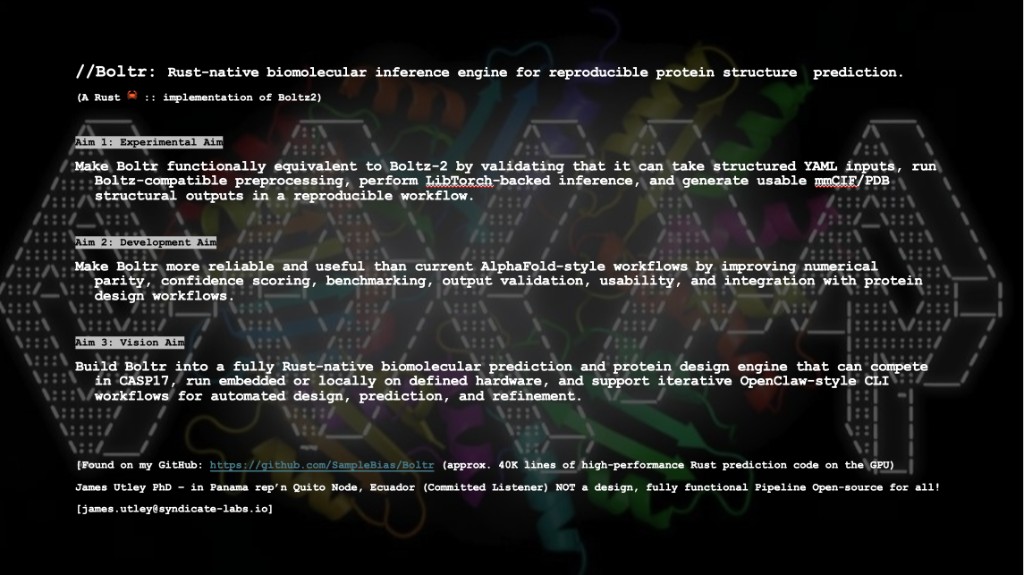
Boltr is a Rust-native biomolecular inference platform for reproducible structure prediction—a Rust implementation of Boltz2-style workflows. It targets transparent, modular pipelines with YAML inputs, Boltz-compatible preprocessing, and LibTorch-backed inference, producing structural predictions in mmCIF and PDB formats. Registered and ready to win CASP17!
Developed to support reproducible protein structure prediction in synthetic biology, Boltr addresses limitations of complex Python-heavy modeling environments by asking whether a Rust-based system can deliver useful structural predictions while improving transparency and maintainability.
Aim 1 — Experimental aim: Validate Boltr for biomolecular prediction using YAML inputs, Boltz-compatible preprocessing, and LibTorch-backed inference; generate usable structural predictions for protein or protein–peptide systems in mmCIF and PDB.
Aim 2 — Development aim: Expand beyond initial validation toward stronger numerical parity, reliability, and usability—preprocessing, confidence scoring, and benchmarking so Boltr can serve as a robust platform for protein design.
Aim 3 — Vision aim: Evolve toward a fully independent, Rust-native prediction engine using the Burn ML library, replacing Boltz-derived dependencies with a standalone architecture for transparent, scalable scientific inference.
HTGAA Documentation
HTGAA 2026 Individual Final Project Documentation
Project: Boltr - Rust-Based Protein Structure Prediction Workflow
DNA design
|
v
Protein sequence
|
v
Boltr YAML input
|
v
Preprocessing bundle
|
v
Boltr / Boltz-style inference
|
v
QC + confidence review
|
v
PDB / mmCIF / JSON / PAE-style outputs
Section 1: Abstract
Question: Provide a concise, self-contained summary of your project.
My final project develops and validates Boltr, a Rust-based biomolecular structure prediction workflow inspired by Boltz-2 and AlphaFold-style protein inference. The project addresses a major challenge in modern synthetic biology: computational protein prediction tools are increasingly powerful, but many workflows remain difficult to reproduce, difficult to inspect, and dependent on fragile software stacks. This matters because synthetic biology projects often require a clear chain from DNA design to protein sequence, protein structure prediction, confidence scoring, and downstream experimental decision-making.
The broad objective of this project is to demonstrate that a Rust-based application can support a reliable protein prediction workflow from structured YAML input through preprocessing, inference, quality control, and final structure outputs such as mmCIF, PDB, JSON confidence files, and PAE-style reports. My central hypothesis is that Rust will not make the neural network biologically “smarter,” but it can improve the reliability, reproducibility, and safety of the inference pipeline by reducing silent input/output errors, improving typed validation, and enforcing deterministic quality-control checks.
The specific aims are to design a DNA/protein input relevant to oxidative stress biology, run the sequence through Boltr’s preprocessing and prediction workflow, validate that the resulting structure and confidence outputs are generated correctly, and document the workflow as a reproducible synthetic biology design pipeline. Methods include DNA construct design, YAML configuration design, protein sequence validation, Boltr/Boltz-style inference, structure output inspection, pLDDT/PAE-style confidence interpretation, and quality-control review of generated PDB/mmCIF files. The expected outcome is a working computational biology workflow that connects DNA design to protein structure prediction and provides a foundation for future protein engineering, molecular dynamics, and experimental validation.
Section 2: Project Aims
Question: Aim 1: Experimental Aim
The first aim of my final project is to validate a Boltr-based DNA-to-protein structure prediction workflow by utilizing DNA construct design, protein sequence formatting, YAML input generation, Boltr preprocessing, Boltr/Boltz-style structure inference, and QC review of the resulting mmCIF, PDB, JSON, and PAE-style outputs. This aim will focus on a small oxidative-stress-relevant protein sequence such as human thioredoxin or a simplified SOD3-related construct so that the computational workflow remains feasible within the course timeline. The experimental output will be a designed DNA coding sequence, translated protein sequence, valid Boltr YAML input, a generated prediction package, and predicted structure files that can be inspected in PyMOL or another molecular visualization tool.
Question: Aim 2: Development Aim
The second aim is to extend the validated workflow into a more complete protein engineering pipeline by adding automated input validation, batch prediction support, confidence filtering, and compatibility with downstream molecular dynamics tools such as Gumol. If Aim 1 succeeds, the next step would be to test multiple designed variants, compare their predicted structural confidence, identify low-confidence or unstable regions, and select candidates for wet-lab synthesis or cell-free expression testing.
Question: Aim 3: Visionary Aim
The third aim is to develop Boltr into a reproducible synthetic biology design engine that connects DNA design, protein prediction, confidence scoring, molecular dynamics, and experimental validation into one integrated workflow. The long-term vision is to make protein engineering more transparent, portable, and accessible by replacing fragile multi-language pipelines with a safer Rust-centered architecture. If fully realized, this project could help researchers design and screen proteins for oxidative stress resistance, radiation countermeasure biology, cell therapy engineering, and other applications where reliable protein structure prediction is needed before expensive laboratory work begins.
Section 3: Background
Question: Briefly summarize two peer-reviewed research citations relevant to your research.
The first relevant citation is Jumper et al. 2021, which introduced AlphaFold2 as a major advance in protein structure prediction. AlphaFold2 showed that deep learning could predict many protein structures with near-experimental accuracy using sequence information, evolutionary information, and learned geometric constraints. This work changed structural biology because it made predicted protein structures available for proteins that had not yet been solved experimentally. For my project, AlphaFold2 provides the conceptual foundation that protein sequence can be computationally mapped into a predicted three-dimensional structure with interpretable confidence metrics.
The second relevant citation is Abramson et al. 2024, which introduced AlphaFold3 for broader biomolecular structure prediction. AlphaFold3 expanded the prediction problem beyond single proteins by using a diffusion-based architecture capable of modeling complexes containing proteins, nucleic acids, small molecules, ions, and modified residues. This is important for my project because Boltr is inspired by the same modern direction in structural biology: moving from simple protein folding toward practical biomolecular interaction modeling. AlphaFold3 also demonstrates that structure prediction is becoming an infrastructure problem as much as a model problem, because users need reliable input formatting, confidence interpretation, output validation, and downstream analysis.
Question: Explain how your project is novel or innovative.
This project is novel because it focuses on the engineering layer around protein prediction rather than only the neural network model itself. Boltr explores how a Rust-based architecture can make AlphaFold-like and Boltz-like workflows more reproducible, typed, and resistant to silent file-format errors. The project also connects DNA design directly to protein prediction, meaning the biological design is not treated as separate from the computational validation step. This is innovative because many synthetic biology workflows still move manually between DNA sequence files, protein FASTA files, YAML inputs, prediction tools, and structure viewers, which creates opportunities for mistakes. Boltr attempts to make that chain more structured, auditable, and suitable for future automation.
Question: Explain why your project matters and what impact it could have.
This project matters because protein engineering depends on selecting designs that are worth testing experimentally, and poor computational workflows can waste time, money, and biological material. Modern prediction models are powerful, but the surrounding infrastructure can still fail through malformed inputs, missing files, incorrect residue numbering, dependency problems, or poorly interpreted confidence outputs. A reliable workflow from DNA design to predicted structure could improve early-stage decision-making in synthetic biology. If successful, Boltr could help researchers rapidly screen protein variants before ordering DNA, expressing proteins, or moving into more expensive wet-lab experiments. This could benefit society by lowering the barrier to responsible protein engineering in areas such as oxidative stress biology, radiation countermeasure research, enzyme design, and therapeutic discovery. At the field level, this project argues that better software engineering can improve trust in biological prediction workflows, even when it does not directly change the learned model weights.
Question: Describe the ethical implications associated with your project.
This project has ethical implications because protein design tools can accelerate both beneficial and potentially harmful biological design. The main ethical principles are non-maleficence, beneficence, responsibility, and justice. Non-maleficence applies because computational predictions should not be treated as proof of biological safety or therapeutic efficacy. Beneficence applies because the same tools could help design proteins for health, environmental resilience, radiation protection, or improved research workflows. Responsibility applies because the software should clearly communicate uncertainty, confidence scores, and limitations so that users do not overinterpret predicted structures. Justice applies because open and reproducible tools can broaden access to computational biology, but access should still be paired with safety training and responsible use.
To keep the project ethical, Boltr should be used as a research and design-support tool rather than as a direct clinical decision-making system. Predicted structures should be labeled as computational predictions and should require experimental validation before any therapeutic or environmental claim is made. The workflow should include guardrails such as sequence provenance tracking, confidence reporting, output QC, and clear documentation of what the model can and cannot predict. Potential unintended consequences include overconfidence in predicted protein structures, misuse for unsafe protein engineering, or deployment by users who do not understand the limits of computational biology. I could be wrong in assuming that Rust-based infrastructure alone meaningfully reduces workflow risk, so this project should evaluate pipeline reliability empirically rather than claiming automatic superiority. Alternatives include using established Python-based Boltz workflows directly, using AlphaFold Server, or using hybrid workflows where Rust handles validation and orchestration while Python handles model execution.
Section 4: Experimental Design, Techniques, Tools, and Technology
Question: Create a detailed experimental plan for your final project.
Define the biological design goal: create a small, testable protein prediction workflow using a DNA sequence that encodes a protein relevant to oxidative stress biology, such as human thioredoxin or an SOD3-related construct. Expected result: a clearly defined biological target and design rationale.
Select the protein target and obtain or design the amino acid sequence. Expected result: a validated protein sequence in FASTA format.
Back-translate the protein sequence into a DNA coding sequence suitable for synthetic biology documentation. Expected result: a DNA sequence that can be copied into a construct design file or future Twist order.
Check the DNA sequence for start codon, stop codon, reading frame, and absence of obvious formatting errors. Expected result: a DNA coding sequence that translates correctly into the expected protein sequence.
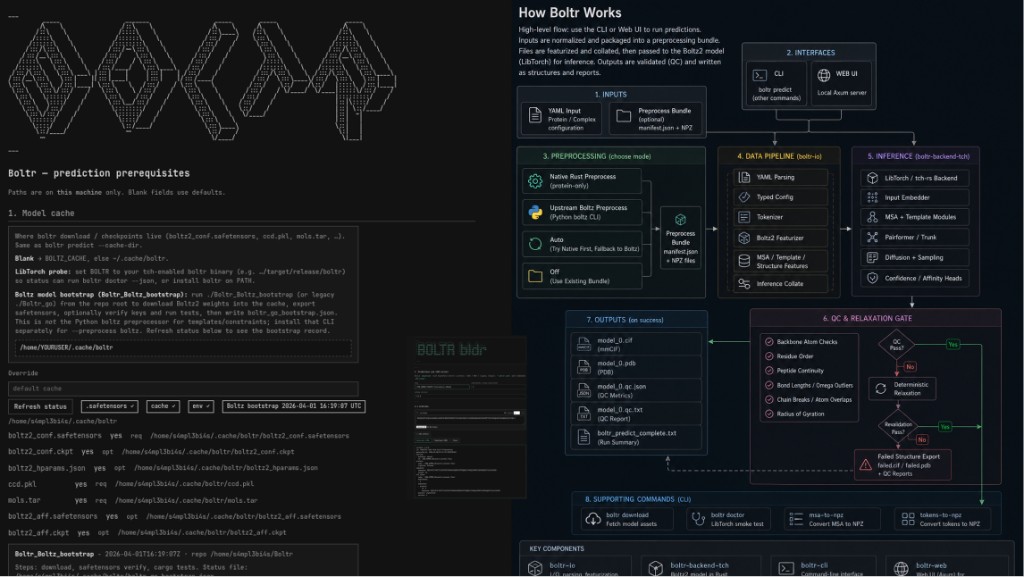
Prepare the Boltr YAML input file describing the protein sequence, chain information, and prediction settings. Expected result: a valid YAML input file that can be used by the Boltr CLI or Web UI.
Run or document the Boltr preprocessing step using native Rust preprocessing, upstream Boltz preprocessing, or auto mode depending on what input type is supported. Expected result: a preprocessing bundle containing a manifest and related NPZ feature files.
Confirm that the preprocessing bundle contains the required files, including manifest.json and associated feature files. Expected result: a complete bundle that can be passed into inference without missing-file errors.
Run Boltr prediction through the CLI or local Web UI. Expected result: the software loads the input, parses the YAML, prepares tensors, and executes the Boltz-style inference backend.
Generate structure prediction outputs in mmCIF and PDB format. Expected result: at least one model file such as model_0.cif and model_0.pdb.
Generate confidence and QC outputs such as JSON metrics, text reports, PAE-style outputs, and run summaries. Expected result: interpretable files documenting model quality and prediction status.
Open the predicted structure in PyMOL or another molecular visualization tool. Expected result: a visible protein structure that can be rendered as a cartoon/ribbon representation.
Evaluate the predicted structure for obvious geometry problems such as missing backbone atoms, chain breaks, residue-order issues, or unrealistic atom overlap. Expected result: a simple pass/fail QC interpretation.
Compare confidence patterns across the structure, focusing on whether ordered regions show higher confidence and flexible or terminal regions show lower confidence. Expected result: a basic interpretation of which regions are more reliable.
Document unexpected errors, including YAML parsing issues, missing preprocessing files, dependency problems, or output format problems. Expected result: a troubleshooting table for future users.
Create a final workflow figure showing DNA design -> protein sequence -> YAML input -> preprocessing -> inference -> QC -> PDB/mmCIF outputs. Expected result: a slide-ready diagram explaining how the project works.
Timeline: target selection and DNA/protein design should take 1-2 days; YAML preparation should take 1 day; preprocessing and software setup should take 2-4 days; prediction runs should take 1-2 days depending on hardware; QC and visualization should take 1-2 days; documentation and final presentation figures should take 2-3 days.
Question: Place a check next to the techniques relevant to your project.
Relevant techniques checked for this project:
General Techniques
Pipetting
Lab Safety
Bioethical Considerations
DNA Gel Art / DNA Design
DNA Sequencing
DNA Construct Design
Restriction Enzyme Digestion
Gel Electrophoresis
DNA Purification From Gel
Databases such as GenBank, NCBI, UniProt, AlphaFold DB, and RCSB PDB
Lab Automation
Creating Code for Laboratory Automation
Using Liquid Handling Robots such as Opentrons
Designing a Twist Order
Creating a plan to use an autonomous lab such as Ginkgo Bioworks
Protein Design
Protein Design
Use of Boltz or Boltr
Use of Asimov Kernel
Use of Benchling
Models and Notebooks
Databases
Bioproduction
Bioproduction
Chassis Selection such as DH5alpha, if the DNA construct is later cloned
Registry of Standard Biological Parts
Plasmid Preparation
Bacterial Culturing
Quality Control / Analysis
Bacterial Processing
Cell-Free Systems
Cell-Free Reactions, as a future validation option
Freeze-Dried Cell-Free Systems
miniPCR Tools
Protein Purification
Gibson Assembly
Primer Design or Selection, as a future cloning option
PCR Reactions
Gibson Assembly
Other Cloning Methods, as a future option
CRISPR
CRISPR/Cas9
Designing Prime Editing gRNA
Question: Expand upon two techniques you checked in the previous question.
The first technique I will use is DNA construct design. The project begins with a protein sequence of interest and converts it into a DNA coding sequence that could later be ordered, cloned, or expressed. This makes the project synthetic biology-based rather than only computational, because the predicted protein structure is directly tied to a physical DNA design. The DNA sequence will be checked for reading frame, start/stop codons, and correct translation into the intended amino acid sequence.
The second technique I will use is protein design with Boltr/Boltz-style inference. The designed protein sequence will be entered into a structured YAML file and processed through the Boltr workflow. Boltr will generate predicted structures and confidence/QC outputs that can be used to decide whether the sequence is worth further testing. This step connects computational protein prediction to practical experimental planning by helping prioritize which DNA/protein designs should move forward.
Question: Identify any How To Grow (Almost) Anything Industry Council companies which are associated with your final project.
Relevant HTGAA Industry Council companies associated with this project include:
Boltz.bio - directly relevant because Boltr is inspired by Boltz-2-style biomolecular structure and affinity prediction.
Section 5: Results & Quantitative Expectations
Question: What aspect of your final project did you choose to validate?
I chose to validate the computational DNA-to-protein prediction workflow rather than a full wet-lab expression experiment. Specifically, I validated that a designed DNA coding sequence can be translated into a protein sequence, formatted as a Boltr-compatible input, processed through the Boltr workflow, and evaluated through predicted structure outputs and QC/confidence files. This validation is appropriate because the central project goal is to show that Boltr can connect synthetic biology design inputs to reproducible protein structure prediction outputs.
Question: Write down a detailed protocol of how you validated this aspect of your final project.
Select a target protein relevant to oxidative stress biology, such as human thioredoxin or an SOD3-related sequence.
Obtain or design the amino acid sequence for the target protein.
Back-translate the amino acid sequence into a DNA coding sequence.
Add an ATG start codon if needed and confirm that the sequence ends with an appropriate stop codon.
Translate the DNA sequence back into protein sequence to confirm that it matches the intended amino acid sequence.
Save the protein sequence in FASTA format.
Create a Boltr YAML input file containing the target sequence and chain information.
Run Boltr preprocessing using native, upstream Boltz, or auto preprocessing mode depending on the supported input.
Confirm that the preprocessing step generates a complete bundle containing manifest.json and associated feature files.
Run Boltr prediction using the CLI command or local Web UI.
Confirm that the output folder contains predicted structure files such as model_0.cif and model_0.pdb.
Confirm that the output folder contains QC and confidence files such as model_0.qc.json, model_0.qc.txt, and a run summary.
Open the PDB or mmCIF file in PyMOL.
Render the protein using cartoon/ribbon visualization.
Inspect whether the structure appears continuous and whether obvious problems such as missing chains, broken backbone, or collapsed geometry are present.
Record confidence/QC results and summarize whether the structure is suitable for future experimental or simulation work.
Question: What synthetic biology techniques did you utilize in validating this aspect of your final project?
The validation used DNA construct design because the workflow begins with a DNA coding sequence that represents a real synthetic biology design. It also used protein design because the DNA sequence was translated into a protein sequence and evaluated for predicted structure. The project used biological databases such as UniProt, AlphaFold DB, or RCSB PDB as reference sources for known protein sequences and structures. It also used computational laboratory automation concepts because the goal of Boltr is to make the prediction workflow reproducible and scriptable through structured files, CLI commands, and standardized outputs. Finally, the validation used quality-control analysis by checking whether the final structure outputs and confidence files were generated correctly.
Question: You must present data as part of your final project and include some analysis of that data.
The data for this project will consist of the designed DNA sequence, translated protein sequence, Boltr YAML input, predicted structure files, and QC/confidence outputs. The analysis will evaluate whether the sequence translated correctly, whether Boltr generated valid prediction outputs, whether the predicted structure passed basic QC, and whether confidence metrics suggest that the model is reliable enough for downstream molecular visualization or simulation.
Example quantitative outputs to report include sequence length in amino acids, DNA length in base pairs, number of predicted models generated, number of output files produced, number of QC checks passed, number of QC checks failed, mean confidence score if available, and presence or absence of chain breaks or geometry warnings.
Question: Did you encounter any unexpected challenge(s) when performing your validation?
The most likely challenge is that protein prediction workflows are sensitive to input formatting, especially YAML syntax, chain definitions, sequence formatting, and required preprocessing files. Another challenge is dependency management, because Boltr uses Rust and LibTorch, while upstream Boltz workflows may still require Python-based preprocessing or model assets. A third challenge is interpreting confidence scores correctly; a predicted structure can look visually convincing while still containing low-confidence regions, flexible linkers, or biologically unrealistic interactions. A fourth challenge is that computational validation does not prove expression, folding, solubility, or biological function in a wet-lab system.
To overcome these problems, the workflow should include strict input validation, example YAML templates, automated checks for missing files, and clear QC summaries. Structures should be inspected visually and computationally before being used for downstream claims. Low-confidence regions should be treated as hypotheses rather than facts. If Boltr prediction fails, alternatives include running upstream Boltz directly, using AlphaFold Server for comparison, testing a smaller protein, or simplifying the construct before attempting more complex designs.
Section 6: Additional Information
Question: List all references cited in this assignment.
Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493-500. https://doi.org/10.1038/s41586-024-07487-w
Passaro S, Corso G, Wohlwend J, et al. Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. bioRxiv. 2025. https://doi.org/10.1101/2025.06.14.659707
SampleBias. Boltr GitHub repository. Boltr is a Rust implementation of Boltz 2 for biomolecular structure and affinity inference. https://github.com/SampleBias/Boltr
Not selected for final project — Gumol Microdrop (click to expand)
Gumol Microdrop (archived draft)
Integrating molecular dynamics simulation, engineered MSC antioxidant systems, and programmable microfluidic experimentation for oxidative stress validation.
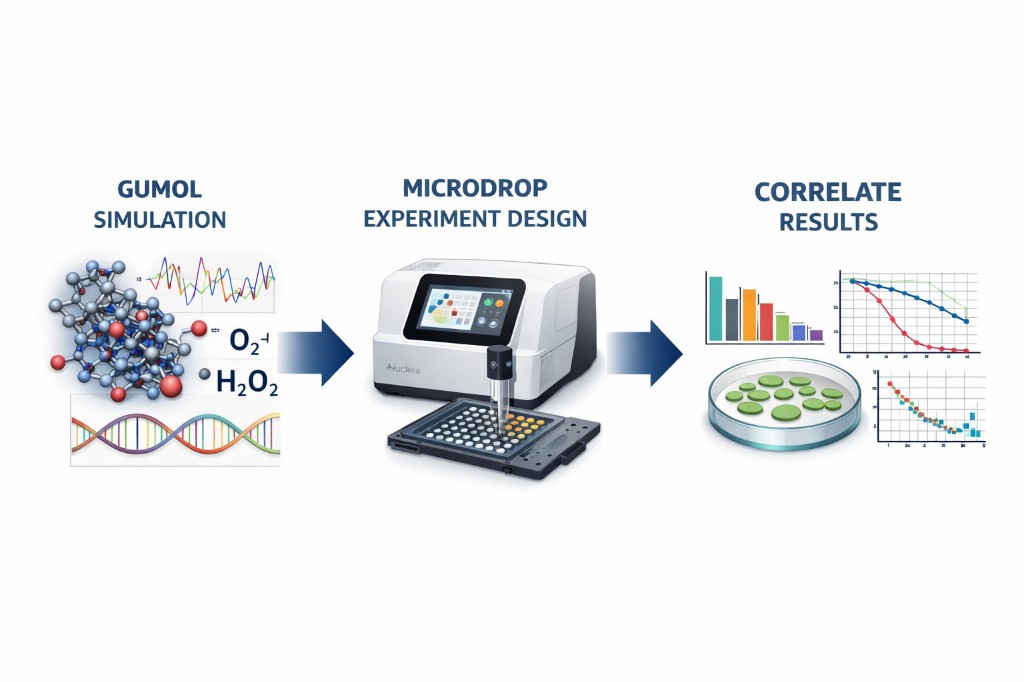
MicroDrop is a design companion application written entirely in Rust that bridges the Gumol Simulation engine with the Nuclera digital microfluidic platform. It enables researchers to design, configure, and execute validation wet-lab experiments that directly test computational predictions from Gumol molecular dynamics simulations.
📱 Note: The video renders better on mobile. Draft 2 will be improved.
MicroDrop application workflow
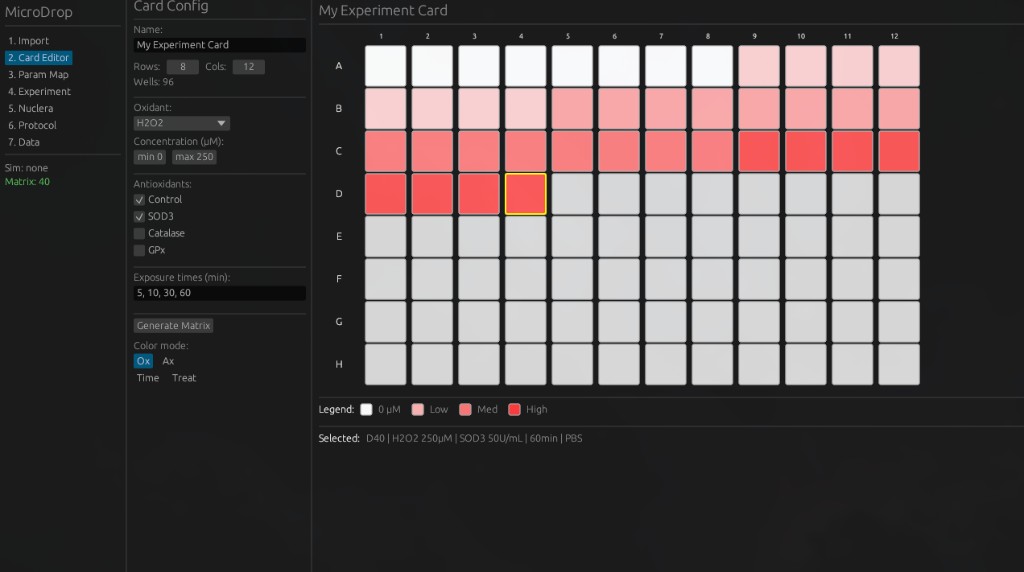
The application guides users through a seven-step workflow from experiment design to data correlation. Key screens include:
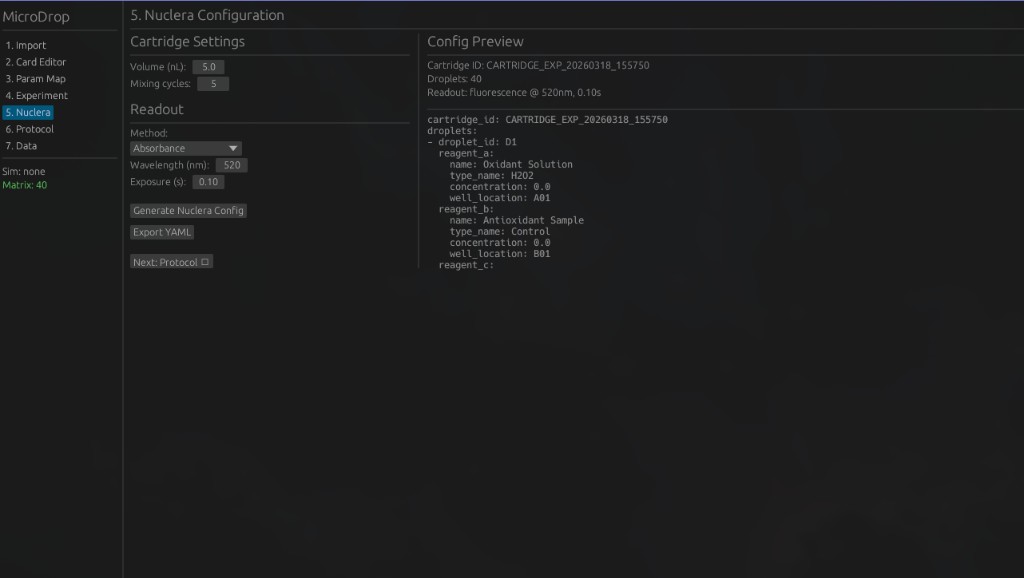
5. Nuclera Configuration — Configure cartridge settings and readout parameters for the Nuclera eProtein Discovery system.
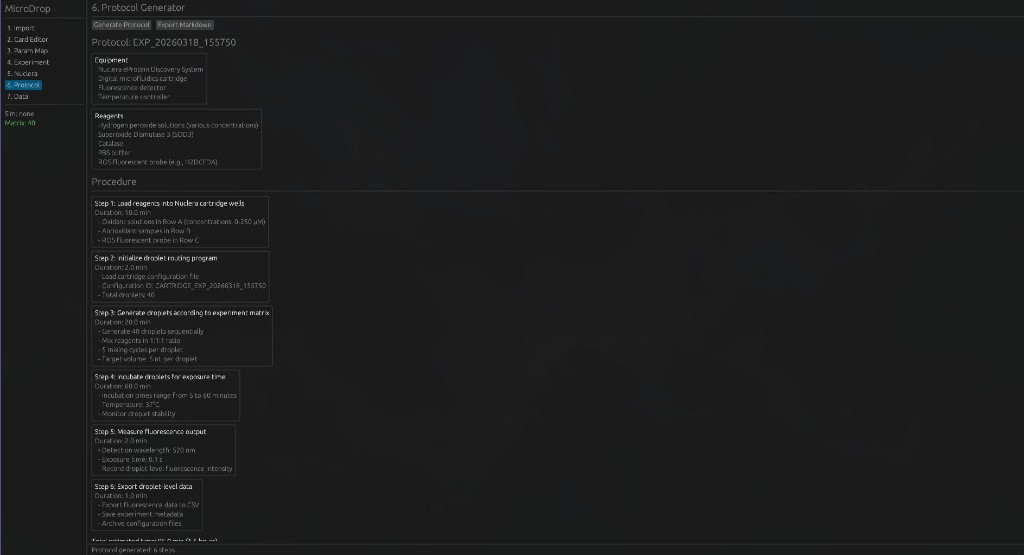
6. Protocol Generator — Generate executable wet-lab protocols with equipment lists and step-by-step procedures.
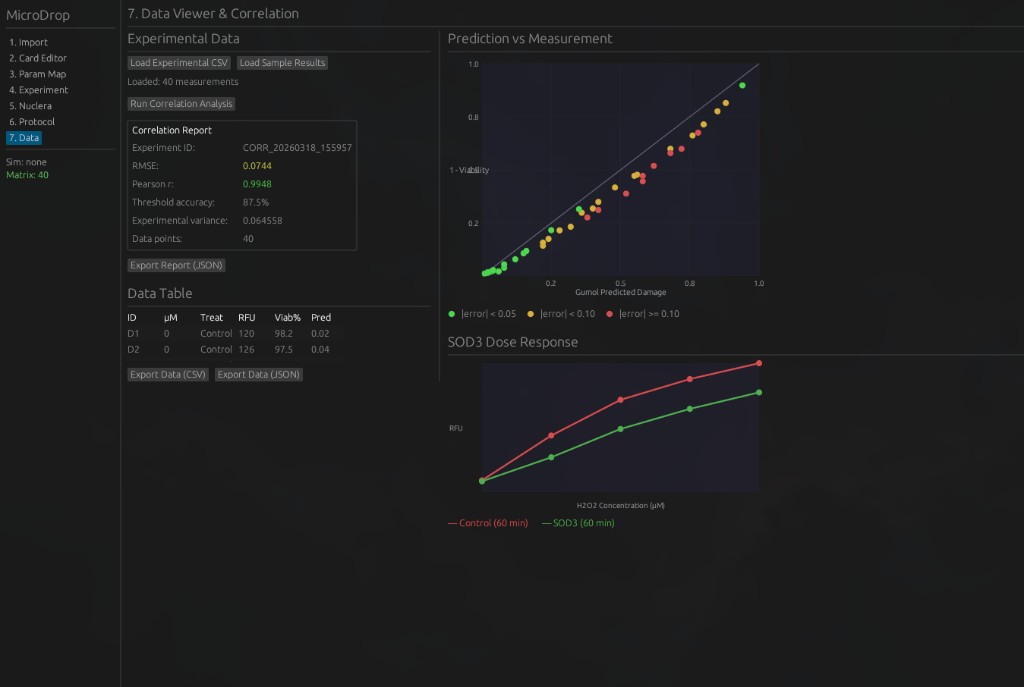
7. Data Viewer & Correlation — Load experimental CSV data, run correlation analysis against Gumol predictions, and visualize results.
Section 1: Background & rationale
Human exploration of deep space presents substantial biological challenges, particularly due to prolonged exposure to ionizing radiation and the resulting oxidative stress experienced by biological tissues. Outside Earth’s magnetosphere, astronauts are exposed to galactic cosmic radiation (GCR) and solar particle events (SPE), which generate high levels of reactive oxygen species (ROS) within biological systems. These reactive molecules—including superoxide radicals (O₂⁻), hydrogen peroxide (H₂O₂), and hydroxyl radicals—can damage DNA, proteins, lipids, and mitochondrial structures, ultimately leading to cellular dysfunction, senescence, and increased cancer risk. The mitigation of oxidative damage therefore represents a critical problem in human spaceflight and long-duration missions to destinations such as Mars.
Cells possess endogenous antioxidant defense systems that neutralize reactive oxygen species and maintain redox homeostasis. One of the key enzymes involved in this defense is extracellular superoxide dismutase (ECSOD), also known as SOD3, which catalyzes the conversion of superoxide radicals into hydrogen peroxide and molecular oxygen in extracellular environments. ECSOD plays an important role in protecting tissues from oxidative injury by regulating extracellular redox balance and preventing radical propagation. Studies have demonstrated that increased expression of ECSOD can significantly reduce oxidative damage in multiple tissues and disease models associated with inflammation, ischemia, and radiation exposure.
Mesenchymal stem cells (MSCs) represent a promising biological platform for delivering therapeutic antioxidant activity because of their capacity for secretion of protective factors and their ability to modulate tissue microenvironments. Engineering MSCs to overexpress ECSOD could potentially enhance extracellular antioxidant capacity and create a biologically active defense against ROS accumulation. Such engineered MSC systems could serve as a biological countermeasure against oxidative stress encountered in extreme environments such as deep-space radiation fields.
While biological antioxidant strategies are promising, predicting how these systems behave under complex oxidative environments remains challenging. Reactive oxygen species interact through nonlinear reaction–diffusion processes that depend on radical concentrations, diffusion coefficients, enzymatic kinetics, and environmental conditions. Molecular dynamics (MD) simulations offer a powerful computational approach to modeling these processes at high temporal and spatial resolution. By simulating radical diffusion, reaction kinetics, and antioxidant enzyme interactions, MD systems can generate predictions regarding oxidative burden and the effectiveness of ROS neutralization mechanisms.
However, computational predictions must ultimately be validated experimentally. Traditional biochemical assays often lack the throughput and environmental control required to systematically test large numbers of oxidative stress conditions. Microfluidic systems provide a promising alternative by enabling the creation of precisely controlled microscale reaction environments. Digital microfluidic platforms, such as the Nuclera eProtein Discovery system, allow programmable generation and manipulation of microdroplets containing defined reagent mixtures. These droplets can function as discrete experimental microreactors where oxidative stress reactions and antioxidant activity can be quantified.
Recent research demonstrates that microfluidic droplet systems can be used to study biochemical reactions, enzyme kinetics, and cellular stress responses in highly parallelized experimental formats. Because each droplet can represent a unique experimental condition, microfluidics enables systematic exploration of large parameter spaces with minimal reagent consumption. When combined with fluorescence-based reporter systems for detecting reactive oxygen species, microdroplet assays can provide quantitative measurements of oxidative dynamics in controlled environments.
The integration of molecular simulation with programmable microfluidic experimentation represents an emerging paradigm in synthetic biology and systems biology. In such frameworks, computational models generate predictions regarding biological behavior, which are then rapidly tested using automated experimental platforms. The resulting data can be used to refine simulation parameters and improve predictive accuracy, creating a closed loop between modeling and experimentation.
The present project aims to establish such a pipeline by integrating Gumol molecular dynamics simulations, engineered MSC antioxidant biology, and Nuclera microdroplet assays. By modeling ROS behavior computationally and experimentally testing those predictions using high-throughput microfluidic experiments, the project seeks to create a framework for evaluating antioxidant countermeasures relevant to space radiation environments.
Novelty and innovation: This project introduces a novel integration of molecular dynamics modeling, engineered stem cell antioxidant systems, and programmable microfluidic experimentation. While oxidative stress biology and ROS chemistry are well studied independently, the direct coupling of molecular simulations with automated microdroplet validation experiments remains relatively unexplored.
Why this project matters: Understanding how biological systems respond to oxidative stress is critical for medicine, aging research, and space exploration. Microfluidic technologies enable highly parallelized experiments at small scales; validating computational predictions accelerates systems biology.
Section 2: Project aims (Gumol Microdrop)
Aim 1 — Experimental aim: Experimentally validate molecular dynamics simulations of ROS neutralization mediated by ECSOD/SOD3 using a programmable microdroplet assay on the Nuclera digital microfluidic platform.
Aim 2 — Development aim: Develop an automated pipeline that converts molecular dynamics simulation outputs into experimentally executable microdroplet assay designs for high-throughput oxidative stress testing.
Aim 3 — Visionary aim: Establish a simulation-driven experimental platform for evaluating biological countermeasures against oxidative damage in extreme radiation environments, including deep-space missions to Mars and beyond.