Week 4 HW: Protein Design Part 1
PART A: CONCEPTUAL QUESTIONS
1.How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To calculate this we have to make some equivalences. 1 aminoacid = 100 Da = 100g/mol
Meat= 20% protein
Look only for the protein mass
500g of meat x 0.20 = 100g of protein
Moles of aminoacid
100g/mol100g=1 mol amino acids
Number of molecules
1 mol= 6.022 x 10^23 molecules
ANSWER: In 500g of meat considerign it only having around 20% of protein would have around 6x10^23 aminoacid molecules
2.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This does not occur, because we eat the meat of either animal but it gets digested in the stomach, which is breaking down into amino acids. These amino acids are reused to create our own human proteins with the instructions of our DNA.
3.Why are there only 20 natural amino acids?
We got only 20 amino acids because of how the genetic code and translation machinery evolved. The genetic code is based on the one of codons to encode the amino acids, but even if there are 64 codons we only map 20 amino acids and the stop signals. As amino acids need trna and ribosomes to be decoded into proteins, if there would be a higher amount than the natural 20, it would require evolving new enzymes and recognition systems. With the 20 amino acids we have, we also cover all our chemical diversity needs to allow the very complex structures.
4.Can you make other non-natural amino acids? Design some new amino acids.
Yes, you could make other non-natural amino acids, exchanging R groups and giving new properties that can not be found in nature. We could design an amino acid that could bind to metal adding a strong ligand group like a pyridine in the structure. This modification is added to the alfa carbon as an r group, not making any change to base structure with the amino and carboxylic ends.
Bywater R. P. (2018). Why twenty amino acid residue types suffice(d) to support all living systems. PloS one, 13(10), e0204883. https://doi.org/10.1371/journal.pone.0204883
6.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Proteins are normally made up of L-aminoacids, which form a right handed α-helix. If we end up doing them with D-aminoacids, the structure would mirror the natural form, giving a left handed helix because the chirality of the amino acids determines the direction of the backbone twisting.
7.Can you discover additional helices in proteins?
Apart from the usual alpha helices in proteins, there have been discovered others like the pi-helix and the 3-10 helix. 310-Helices represent the 3rd principal secondary structure, its given by the regular and repetitive H bonding network between carbonyl group in residue and the amide nitrogen in residue. These interactions give rise to a helix with 3 residue per turn and a helical pitch, following a succession of b-turnsending in a thin and elongated helix. π-helices contribute to protein folding and are relevant as ligand-binding site contributors, given by single residue insertions between alpha helices. New helices can be found by identifying repeating H-bonding patterns and backbone angles.
(2024). Revisiting 310-helices: biological relevance, mimetics and applications. Exploration of Drug Science. https://doi.org/10.37349/eds.2024.00034
8.Why are most molecular helices right-handed?
This right handed direction comes back to the chirality of the amino acids that are making up the protein. As usual proteins are made up of l-aminoacids, their steric constraints make the backbone rotate towards the right, this way minimizes the atomic clashes, being preferred as it is more energetically stable.
9.Why do β-sheets tend to aggregate?
Beta sheets tend to aggregate because of the strong hydrogen bonding networks formed in their backbones. In this structure, we see exposed hydrogen bond donors and acceptors, the flat sheet surfaces stack easily and there´s hydrophobic side chains interacting between them. All of these factors combined, give optimal possibilities for the b-sheets to aggregate because of the overall intermolecular forces that pack the molecules.
-What is the driving force for β-sheet aggregation?
hydrogen bonding possible because of the exposed H bond acceptors and donors in backbone
Hydrophobic interactions coming from side chains between b-sheets
Van der Waals packing, optimized by the stacking capability of the structure
10.Why do many amyloid diseases form β-sheets?
Amyloid diseases are characterized by the accumulation of misfolded proteins in tissues and organs, impairing their function. This disease can form from b-sheets because this arrangement in proteins maximizes the hydrogen bonding and the possibilities to give stable aggregates, making them accumulate and damage cells.
-Can you use amyloid β-sheets as materials?
Yes, they have potential to be used as material because of its characteristics making them self assembled, rigid and chemically stable but its direct use is unsafe, so researchers mimic AB fibrils that are non toxic.
11.Design a β-sheet motif that forms a well-ordered structure.
To make a well ordered b-sheet motif its structure is given by the b-strands linked lateral to the backbone with hydrogen bond to form a pleated sheet with side chains alternating between hydrophobic and polar residues.
H-P-H-P-H (hydrophobic faces clusters, polar residues face solvents promoting the stacking)
Val/Ile (hydrophobics residues for internal packing)
Lys/Glu (charged residues for solubility and salt bridges)
Antiparallel b-sheet, is more stable than parallel strands so H bonds between the backbone of N-H and C=O adjacents.
You end the structure using repeats to increase stability, introduce sticky ends to favor the stacking and cap termini to prevent the structure from disintegrating.
PART B: PROTEIN ANALYSIS AND VISUALIZATION
1.Briefly describe the protein you selected and why you selected it.
I chose HIF1A because it plays a crucial role in how tumors adapt to low-oxygen conditions. Many cancers exploit HIF1A by stabilizing it, even when oxygen levels are normal, creating a state called pseudohypoxia. This enables tumors to promote blood vessel growth, alter metabolism, and survive in hostile environments. Investigating HIF1A can help uncover ways to disrupt this process and potentially limit tumor progression.
2.Identify the amino acid sequence of your protein.
AA SEQ:
MEGAGGANDKKKISSERRKEKSRDAARSRRSKESEVFYELAHQLPLPHNVSSHLDKASVM RLTISYLRVRKLLDAGDLDIEDDMKAQMNCFYLKALDGFVMVLTDDGDMIYISDNVNKYM GLTQFELTGHSVFDFTHPCDHEEMREMLTHRNGLVKKGKEQNTQRSFFLRMKCTLTSRGR TMNIKSATWKVLHCTGHIHVYDTNSNQPQCGYKKPPMTCLVLICEPIPHPSNIEIPLDSK TFLSRHSLDMKFSYCDERITELMGYEPEELLGRSIYEYYHALDSDHLTKTHHDMFTKGQV TTGQYRMLAKRGGYVWVETQATVIYNTKNSQPQCIVCVNYVVSGIIQHDLIFSLQQTECV LKPVESSDMKMTQLFTKVESEDTSSLFDKLKKEPDALTLLAPAAGDTIISLDFGSNDTET DDQQLEEVPLYNDVMLPSPNEKLQNINLAMSPLPTAETPKPLRSSADPALNQEVALKLEP NPESLELSFTMPQIQDQTPSPSDGSTRQSSPEPNSPSEYCFYVDSDMVNEFKLELVEKLF AEDTEAKNPFSTQDTDLDLEMLAPYIPMDDDFQLRSFDQLSPLESSSASPESASPQSTVT VFQQTQIQEPTANATTTTATTDELKTVTKDRMEDIKILIASPSPTHIHKETTSATSSPYR DTQSRTASPNRAGKGVIEQTEKSHPRSPNVLSVALSQRTTVPEEELNPKILALQNAQRKR KMEHDGSLFQAVGIGTLLQQPDDHAATTSLSWKRVKGCKSSEQNGMEQKTIILIPSDLAC RLLGQSMDESGLPQLTSYDCEVNAPIQGSRNLLQGEELLRALDQVN
https://www.uniprot.org/uniprotkb/Q16665/entry#sequences
-How long is it?
The length of the protein is: 826 amino acids.
-What is the most frequent amino acid?
The most common amino acid is: L (lysine), which appears 83 times.
https://colab.research.google.com/drive/1hTUfWcd194QJaz_IzDPkatACbLsnHMbS?usp=sharing

-How many protein sequence homologs are there for your protein?
Hint: Use Uniprot’s BLAST tool to search for homologs.
There are 250 protein sequence homologs codifying the same protein through different species.

-Does your protein belong to any protein family?
Yes, HIF-1A is part of a protein family, and is a key member of the bHLH-PAS protein family of transcription factors. The family mainly gives insight to the protein’s structure: bHLH, a basic helix loop helix domain that is gonna interact with DNA; PAS, a domain crucial for dimerization, needed to become an active transcription factor when partnered with HIF-1B.
Kunej T. (2021). Integrative Map of HIF1A Regulatory Elements and Variations. Genes, 12(10), 1526. https://doi.org/10.3390/genes12101526
3.Identify the structure page of your protein in RCSB
https://www.wwpdb.org/pdb?id=pdb_00009ofu
https://www.rcsb.org/structure/9OFU#entity-1
When was the structure solved?
The structure was solved in 2025.
Its initial deposition was on 30th of April, initially released on 11th of June, and the latest revision on 27th of August.
Is it a good quality structure?
It has a resolution of 3.90 Å. I think it is a good quality structure as it has a fair resolution when compared to the size of the sample that was used.
Are there any other molecules in the solved structure apart from protein?
As HIF-1A works in collaboration to other structures as a transcription factor, the sample (Dimer of HIF-1a-ARNT Heterodimers Complexed on 52-bp HRE/HAS) and so on the molecule, the structure was solved with its other proteins interacting, them being:
- hypoxia inducible factor a1 (hif-1a)
- aryl hydrocarbon receptor nuclear translocator (hif-1b)
- 52nt hypoxia response elements (forward and reverse)
Does your protein belong to any structure classification family?
Yes, is a key member of the bHLH-PAS protein family. It gives insight to the protein’s structure: bHLH, a basic helix loop helix domain that is gonna interact with DNA; PAS, a domain crucial for dimerization.
4.Open the structure of your protein in any 3D molecule visualization software:
-PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
PyMOL>fetch 9OFU TITLE Dimer of HIF-1a-ARNT Heterodimers Complexed on 52-bp HRE/HAS ExecutiveLoad-Detail: Detected mmCIF CmdLoad: ".\9ofu.cif" loaded as "9OFU".
-Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
CARTOON
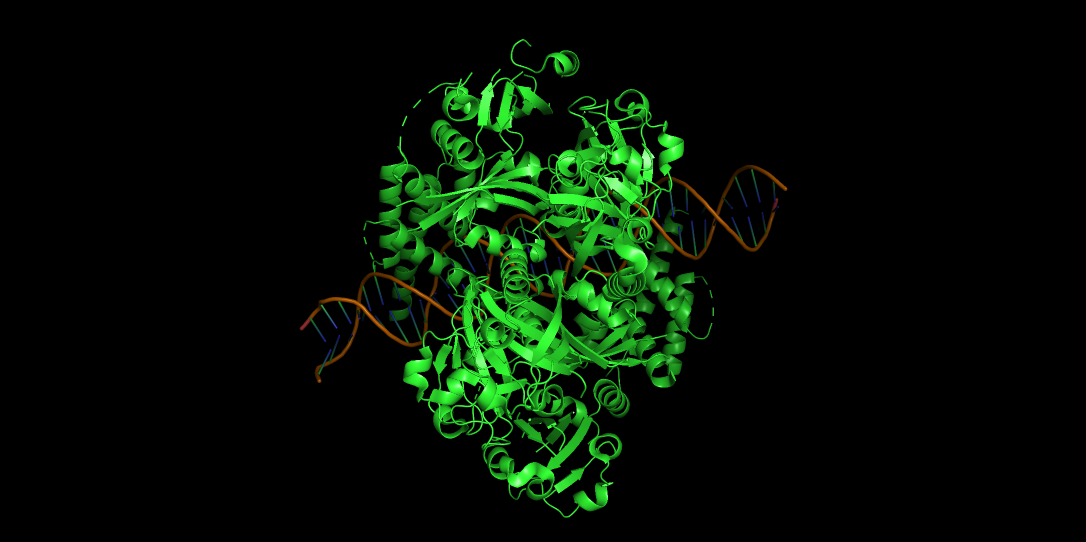
RIBBON
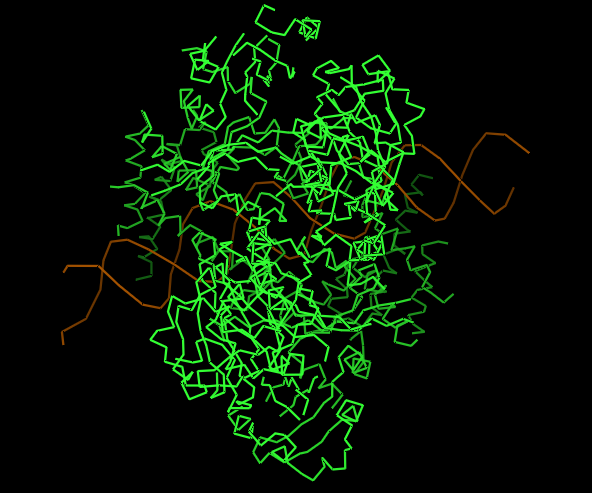
BALL AND STICK
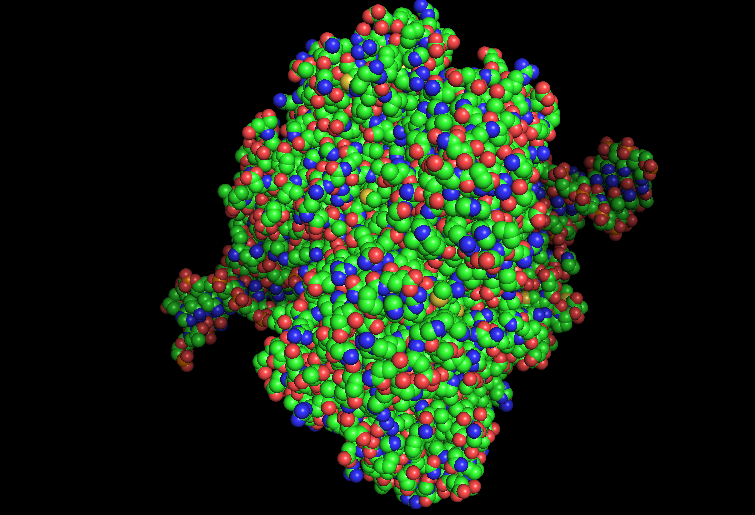
-Color the protein by secondary structure.
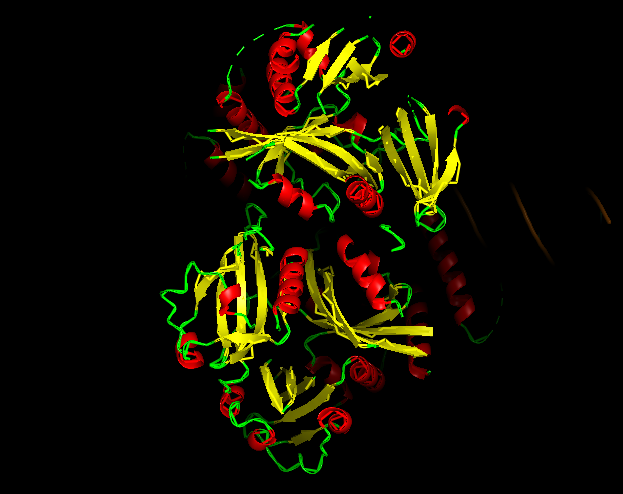
-Red → a-helices
-Yellow → b-sheets
-Green → loops
Does it have more helices or sheets?
It contains more helices than sheets, and it makes sense because HIF1A has the bHLH domain that’s helix rich to wrap around DNA to act as transcription factor.
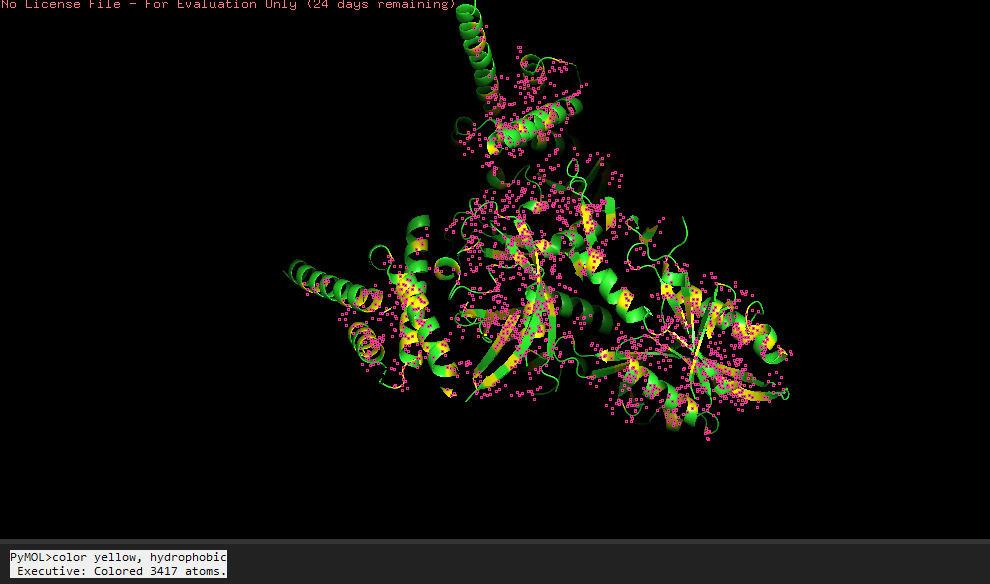
-Color the protein by residue type.
HYDROPHOBIC RESIDUES (YELLOW)
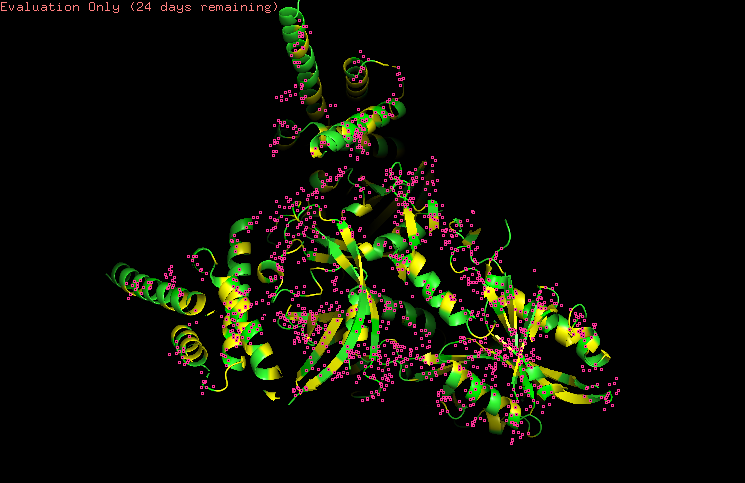
POLAR RESIDUES (GREEN)
POSITIVE RESIDUES (RED)
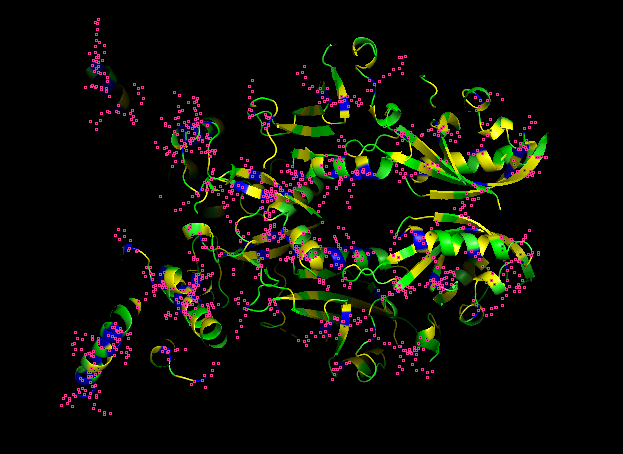
NEGATIVE RESIDUES (BLUE)
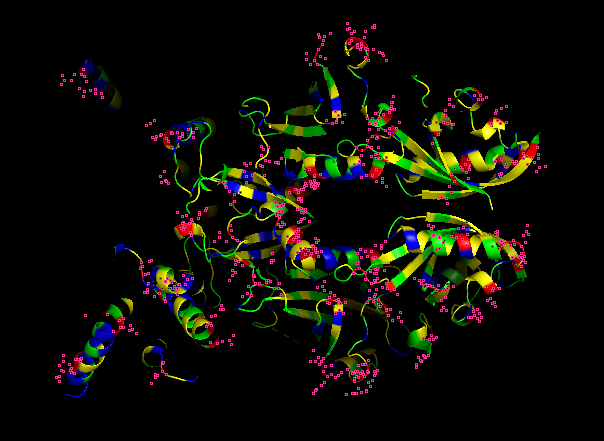
What can you tell about the distribution of hydrophobic vs hydrophilic residues?
From what it is seen in the structure after the coloring, most of the hydrophobic residues can be found inside the protein core, while the hydrophilic and differently charged residues are mainly in the surface. A positive charge can be seen near DNA.
-Visualize the surface of the protein.
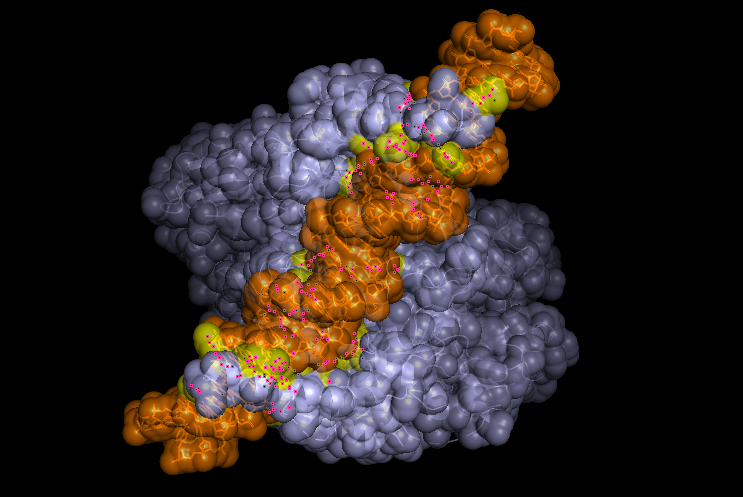 Does it have any “holes” (aka binding pockets)?
Does it have any “holes” (aka binding pockets)?
Yeah, in the surface visualization you can see grooves and small holes on the protein surface. This makes sense and this protein dimer interacts with dna for transcription. The grooves correspond to where the hif1a and arnt bind to dna, and the pockets between the proteins because of the interface that’s created between the structures making the dimer.
Part C. Using ML-Based Protein Design Tools
https://www.rcsb.org/3d-view/9OFU
As before we looked into the whole dimer structure, for the protein design tool, I chose to focus into the hif1a chain.
1.Deep Mutational Scans
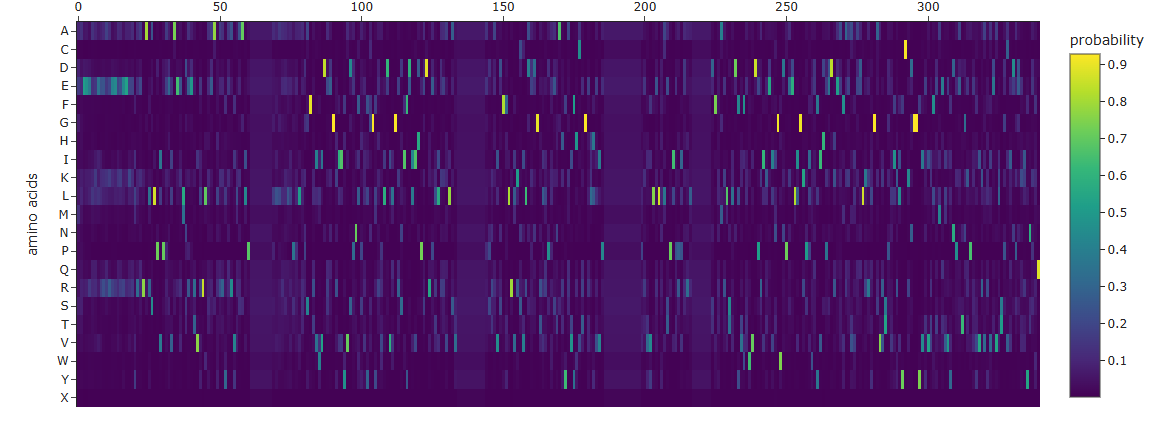
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
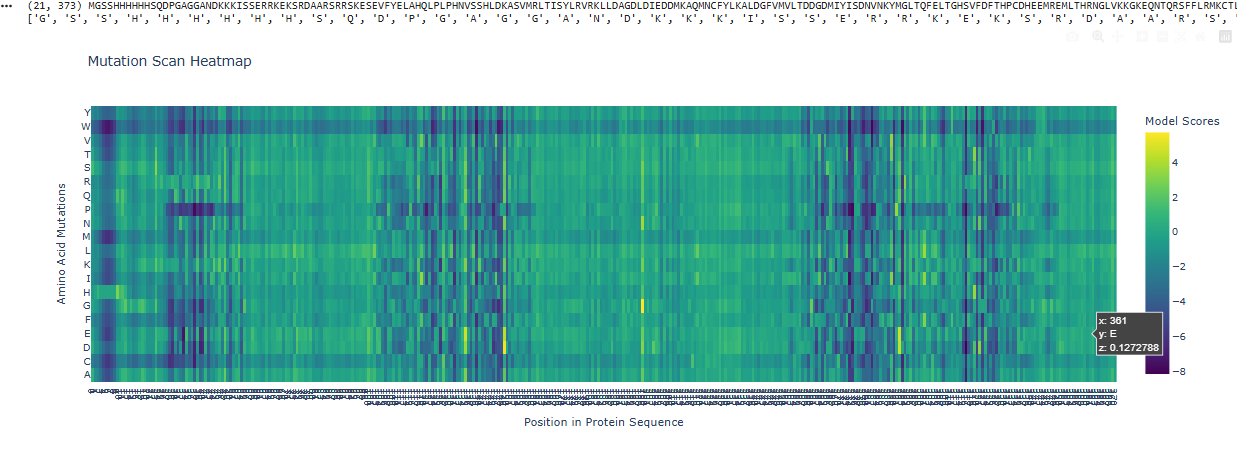 Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
I can see that overall a big part of the protein is pretty constant, not giving rise to a lot of mutations, except in those 3 spots, one and the start, middle and end.
2.Latent Space Analysis
-Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins?
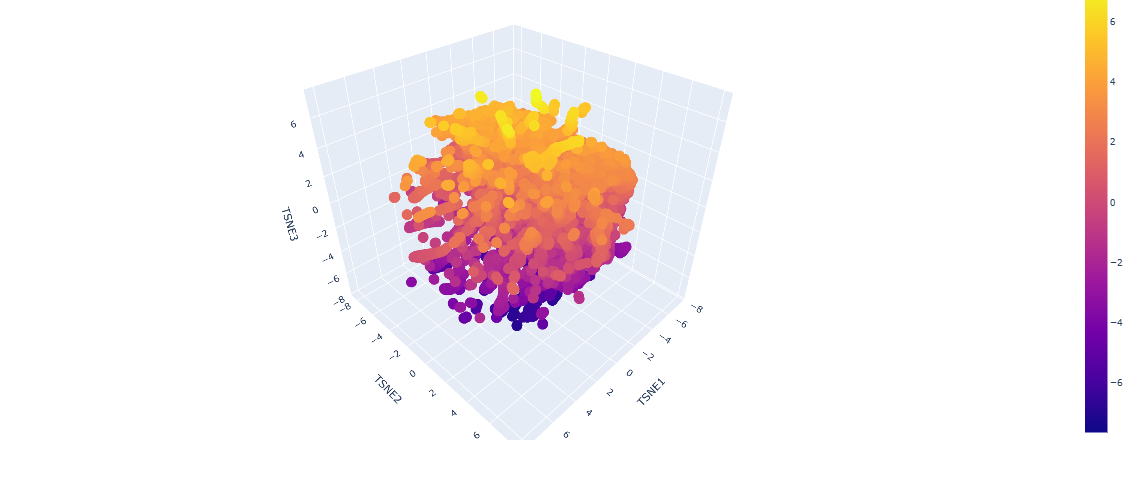
From looking at the different proteins embedded into the dataset, the proteins are organized into neighborhoods that are grouped into proteins that have similar interactions inside the host, as it can see a lot of proteins that have interactions with DNA towards negative numbers tsne2, while other proteins found in fungi or plants are upwards.
-Place your protein in the resulting map and explain its position and similarity to its neighbors.
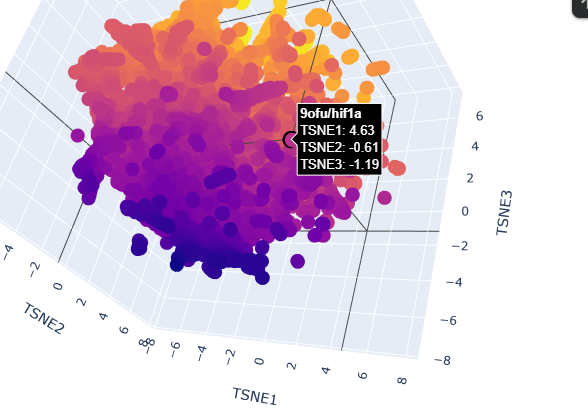
When placing my protein into the resulting map, it’s integrated into the purple side of the graph, towards the negative tsne2. I can see some similarities with the neighboring protein as they have interactions with dna or have transcription domains.
3. Protein Generation
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Predicted structure with mutations
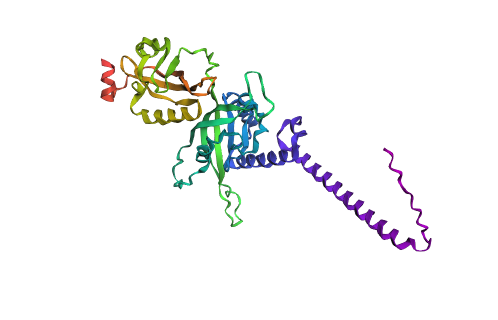 Original structure
Original structure
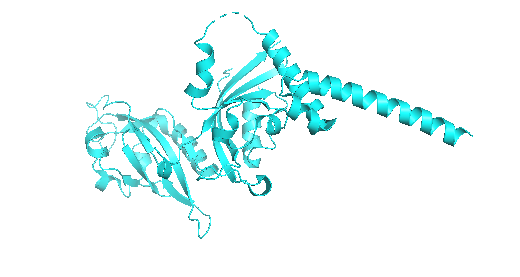
Looking at both structures there’s not a lot of rash differences, but that can be as I just did a 3 cycle of folding based on the sequence with the program. One of the main differences is that you can see a higher quantity of alpha helices and a more compacted protein compared to the predicted sequence.
Input this sequence into ESMFold and compare the predicted structure to your original.
New Sequence: SLEEILKELEEKREKEEEERYREEALSLPIPEEVAKKLDKETVKRLKEALEKYKRVKEALPXXXXXXXXLLEELLKLEKENSFKVWIDEEGKIIYVDPNIYKYAGMKVIDVLGKSIFDIIDPNDRELLKEKLSVXXXXXXXXXXEPRKVDFFLRIKSILDDDGKLIDEKNAHYKVFRAKGEILKVPXXXXXXXXXXXXXPEYVLELNLTPITDPEKEXXXXXXXTFKAKLTKDFVFEWVDPKIKEINGWNPEDLIGKPIYEFIHPEDREAFEKVLEELKKYGKVVSPEIKLYCKNGGYIIVRFTMTMKYNPETNKPEEVLAEIEVLSPCIEPEKIYNEIQ
Part D. Group Brainstorm on Bacteriophage Engineering
-Choose one or two main goals from the list that you think you can address computationally.
- Increase the stability of L protein
- Increase the lytic activity
-Write a 1-page proposal (bullet points or short paragraphs) describing:
The L protein from bacteriophage MS2, is responsible for the lysis of infected cells in E,coli. Studies suggest that this protein interacts with host proteins for this desirable effect but small mutations can strongly affect its function. L protein being small and sensitive to mutations, through computational tools we can study these possible changes and their effects on usual interactions.
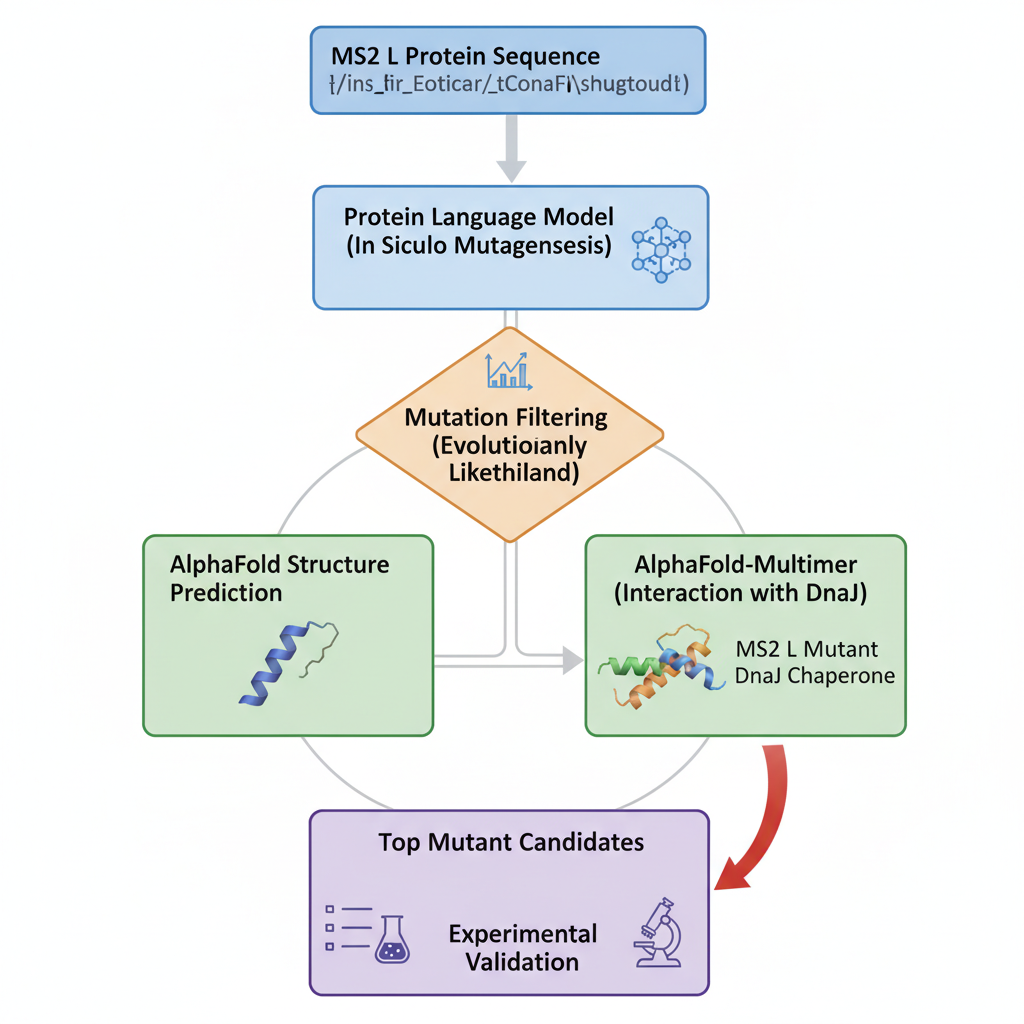
-Which tools/approaches from recitation you propose using.
We can use PLMs to perform mutagenesis of the protein, generating single aas substitutions across the protein, and score these mutations. Alphafold gives us a more visual form to look into these new sequences and the feasibility of them existing and being stable. At the same time, we can look into host proteins interactions to make it possible to try and have more insight into the way this protein interacts to be effective.
-Why do you think those tools might help solve your chosen sub-problem?
Protein language models like,smn2, lets us capture patterns in protein sequences, making it possible to identify mutations that are structurally feasible.
Alphafold, lets us predict structures based on sequence, letting us evaluate how mutations might destabilize the protein folds and let us predict how it interacts with other protein complexes.
-Name one or two potential pitfalls.
We have limited understanding of the exact mechanism of MS2L protein causing the lysis of bacteria. This makes the mutations a bit random as we dont know if they are gonna be beneficial for stability but decrease its lytic effect.