Week 5 HW: Protein Design Part 2
SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
-Design short peptides that bind mutant SOD1.
-Then decide which ones are worth advancing toward therapy.
**Part 1: Generate Binders with PepMLM**
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Sequence:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
MUTATION A4V:
MATKVVCCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
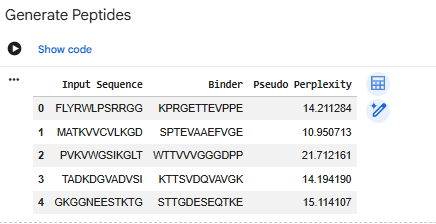
-Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
- SPTEVAAEFVGE
- WTTVVVGGGDPP
- KTTSVDQVAVGK
- STTGDESEQRKE
- CONTROL: FLYRWLPSRRGG
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
**Part 2: Evaluate Binders with AlphaFold3**
- Navigate to the AlphaFold Server: alphafoldserver.com
- For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
- Record the ipTM score and briefly describe where the peptide appears to bind.
CONTROL PEPTIDE
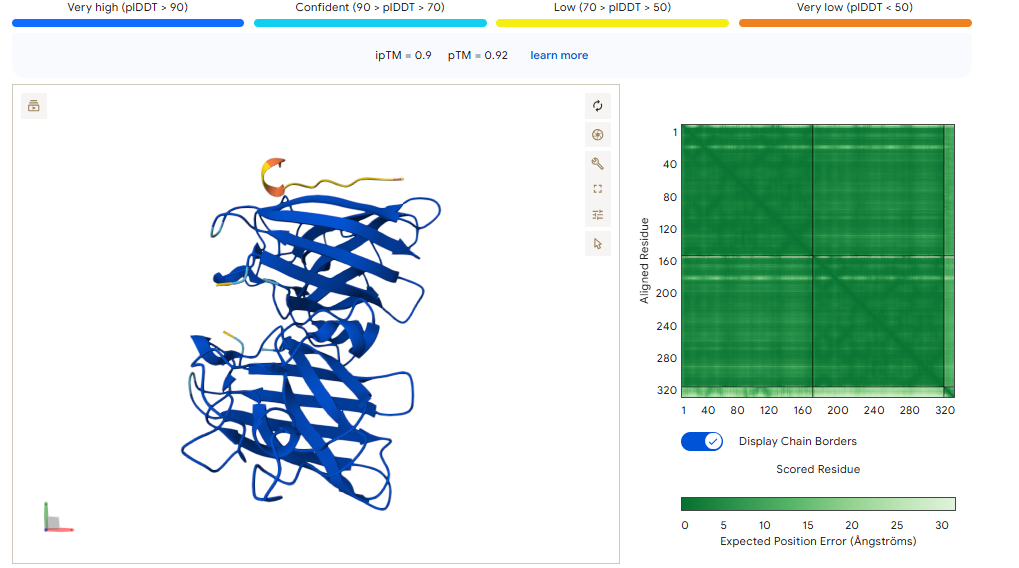
The control shows a ipTM of 0.90, and it can be seen engaging to the surface of the b-barrel.
A PEPTIDE
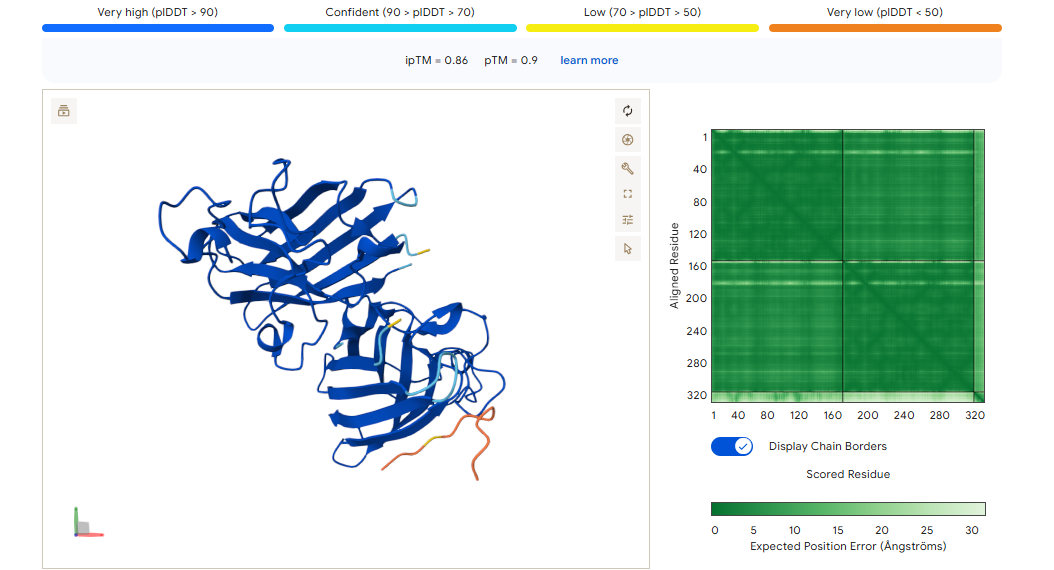
ipTM of 0.86 It shows a high confidence and specialized binding mode. It is near the n terminus where the a4v mutations is, looking a bit buried into where the mutant valine is.
B PEPTIDE
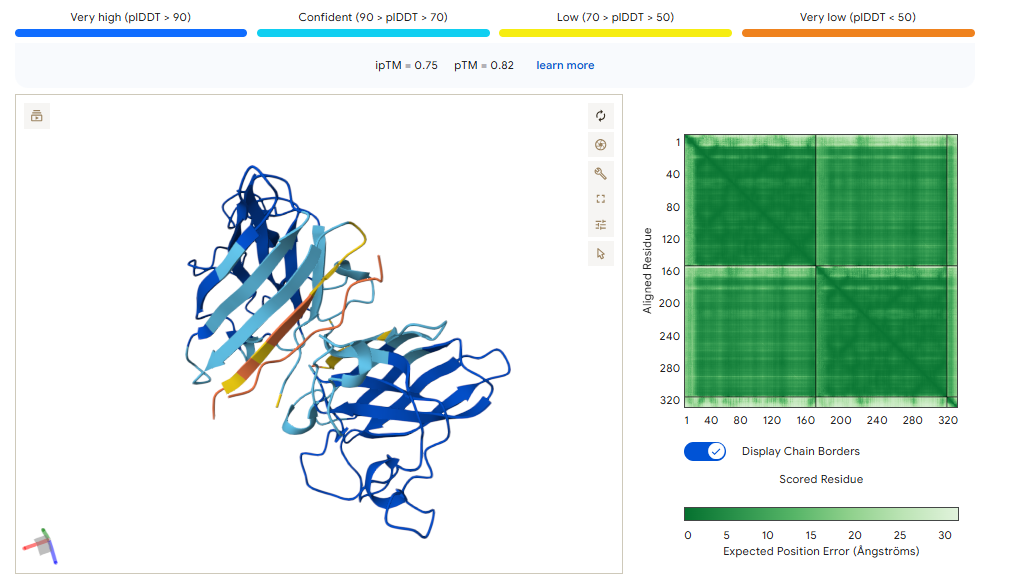
ipTM of 0.75This is the lowest score of the group. It shows flexible groups indicated by the yellow/orange colors and has more of a loop region rather than the b-barrel. Seems mostly surface bounded.
C PEPTIDE
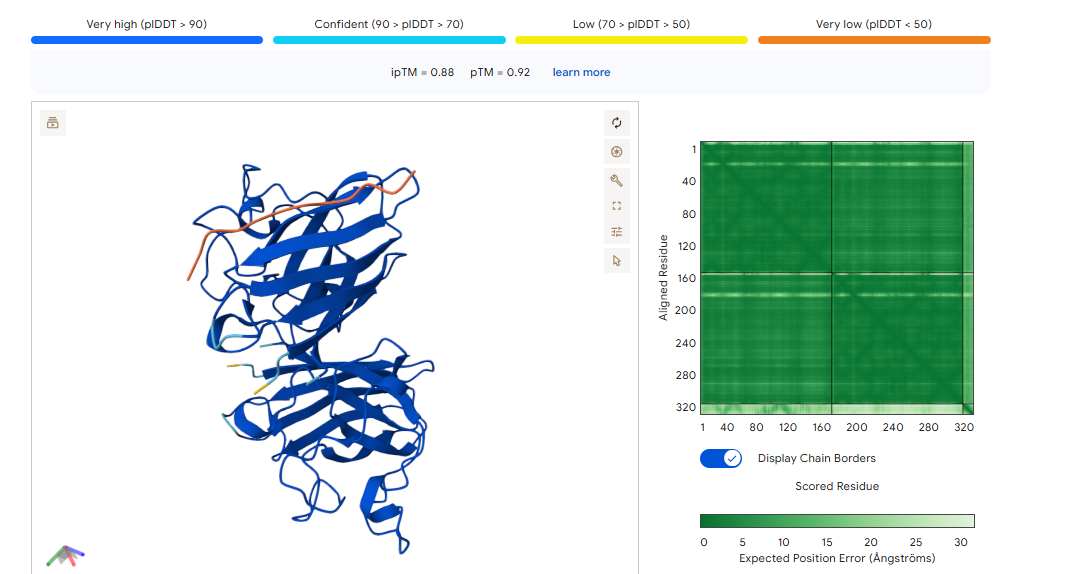
ipTM of 0.88, and looks like it approaches the dimer interface. In the PAE, there are cross chain signals, which means the peptide is acting as a bridge between the SOD1 monomers, where it is partially buried.
D PEPTIDE
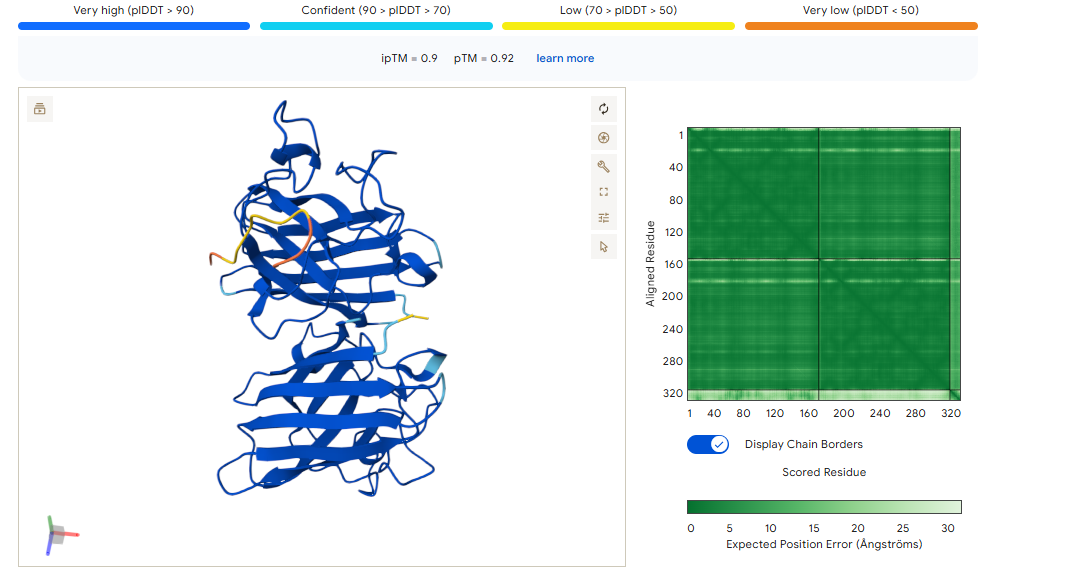
ipTM of 0.90, this score makes this peptide stand out as it has the same score as the known binder. It gives a full capture to the target area, interacting with the b-barrel and dimer interface.
**Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse**
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide!
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see.
- Do peptides with higher ipTM also show stronger predicted affinity?
- Are any strong binders predicted to be hemolytic or poorly soluble?
- Which peptide best balances predicted binding and therapeutic properties?
A PEPTIDE
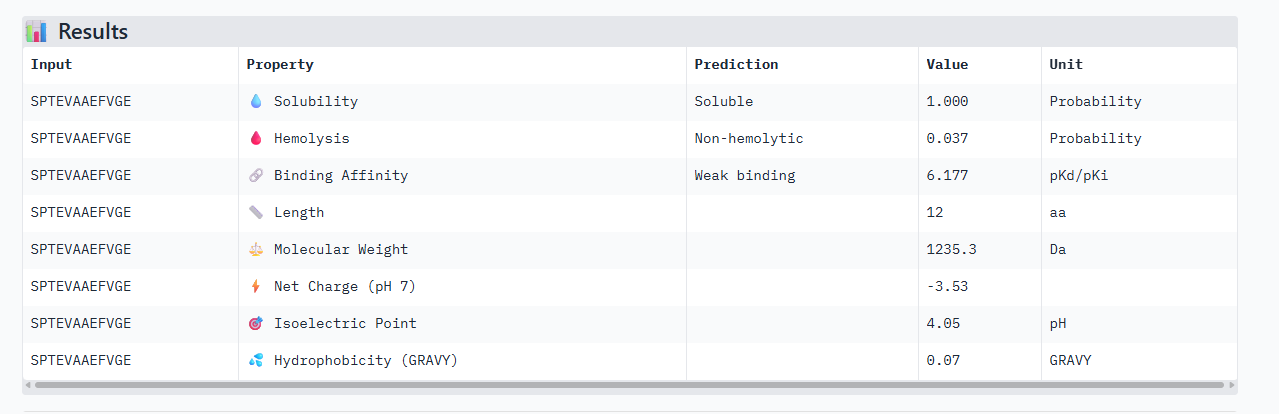
In the Alphafold3, it shows a high confidence and the structure suggests a tight fit to the SOD1 pocket. In the Peptiverse analysis, it confirms A, as the strongest binder with an affinity of 6.177,
B PEPTIDE
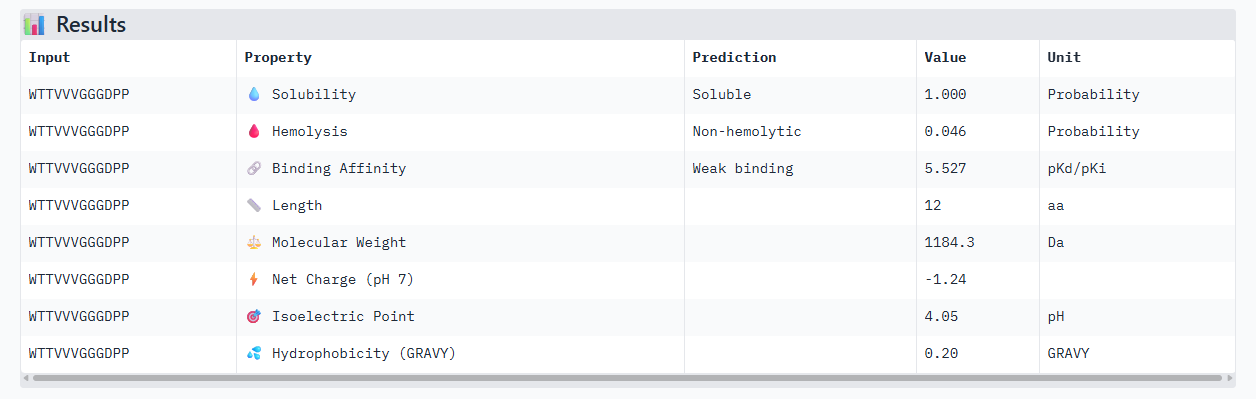
Alphafold3, gives us a confidence of 0.75, maybe because the triple valine in the sequence given arises is pretty flexible and harder to place. In the peptiverse, even though we have this bit of uncertainty in the structure, it shows a decent affinity.
C PEPTIDE
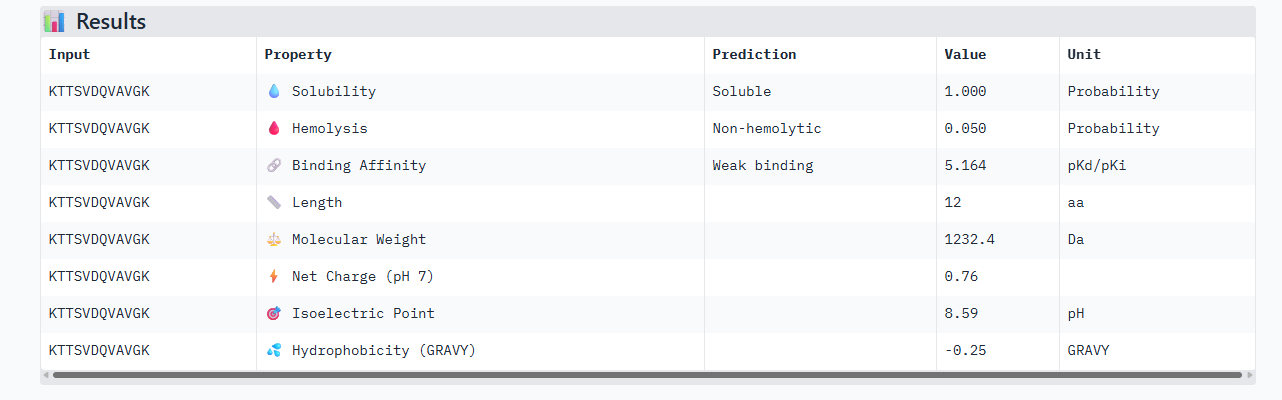
Has a high confidence of 0.88 in Alphafold3, given a structure that interacts between the dimer interface. In the peptiverse, it’s a bit shocking to see it as the peptide with the lowest affinity of 5.164. This score lets us know that even if the peptide is structurally sound, it is chemically weak.
D PEPTIDE
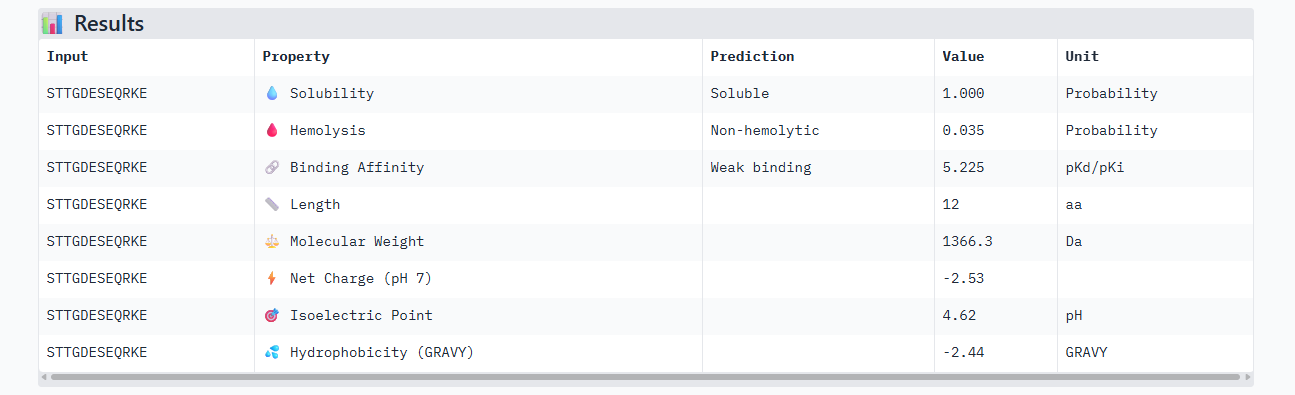
This peptide has the highest score in Alphafold3, having the same 0.90 ipTM as our control. In the peptiverse, it does not show the best affinity with 5.225, however it has the lowest hemolysis score. It could be the most certain design but could lack the chemically to be a potent drug.
Choose one peptide you would advance and justify your decision briefly.
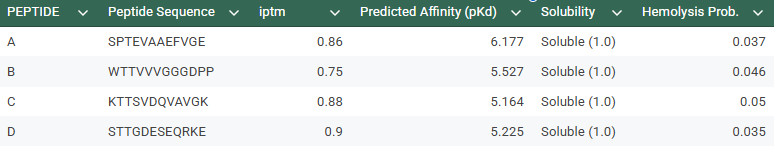
Based on the overall analysis the best choice would be PEPTIDE A. Even if it doesn’t have the best ipTM score compared to the other peptides, it still holds a high fidelity. In the peptiverse, this high possibility of interaction is confirmed with a good affinity score, and also it being safe with having a low score in the hemolysis probability.
**Part 4: Generate Optimized Peptides with moPPIt**
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available).
- Generate peptides.
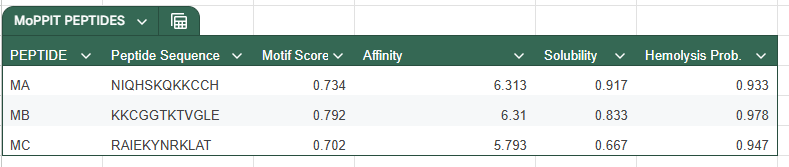
With this model we successfully pushed the predictability affinity higher, specially wiTH designs MA and MB; but all three have a toxicity problem because of their high hemolysis probability. Overall based on the scores, the best peptide would be MA.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
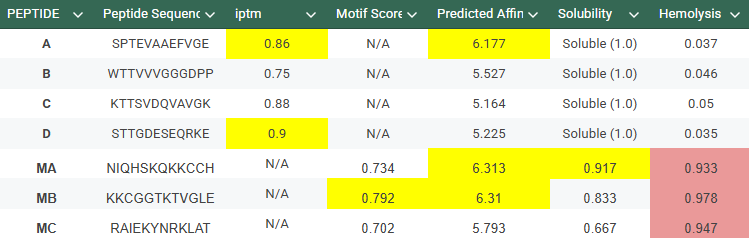
When using MoPPit, to create new peptide designs, we enable motif and affinity guidance. This guidance can be seen in the de novo peptides made by the model, as all 3 of the structures have a high affinity and motif score compared to the PEPMLM peptides,which sequencing was taken from random sites of the mutated SOD1. While these new peptides are theoretically better at interacting with our target, if we were to advance with any of them for a clinical study, they would be toxic from their predicted hemolysis score. If we had to choose the safest peptide between all the ones created, peptides from PEPMLM are better. Peptide A from PEPMLM because it maintains a very respectable affinity score (6.18) and high structural confidence (0.86) while remaining non-toxic and fully soluble.
Details
BOLTZ Has not been done as troubles have been present at creating the Boltz account.
Part C: Final Porject: L- Protein Mutants
[L-Protein Mutants] link: https://pages.htgaa.org/2026a/jamilet-ordonez-villacis/labs/week-05-lab-protein-design-part-ii/index.html