Hey everyone!
I’m Jami, an Ecuadorian pharmacy student enrolled at Tor Vergata University in Rome. I’ve been immersed in the medical world since childhood, making me fell in love with genetics and microbiology. My interests have evolved into synthetic biology approaches for drug development, looking forward for the future of biopharmaceuticals.
1.First, describe a biological engineering application or tool you want to develop and why. As a pharmacy student, I have become increasingly interested in how drugs move from an initial idea to clinical use, and how many potential compounds fail long before they could reach patients. Drug development is an expensive and time consuming subject, and an ethically complex process, specifically in early stages. But many drugs are left behind as they fail to show strong enough effects or because the costs for further testing is too high.
Pre-Lecture HW: 1.Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of polymerase estimates to be made errors once every 10x4–10x5 nucleotides polymerized. This compared to the length of the human genome that’s approximately 3.2 billion base pairs long, that would account up to 32 thousand mutations every time a single cell divided. Biology fixes this gap with multiple systems that check for these errors. Its first system is proofreading; a function found in most polymerases. When they add a wrong base, they recognize this error, backtrack and fix it cutting the wrong base and replacing it. Another way, is mismatch repair (NMR), after polymerase is done, a second group of proteins scan the new DNA for any remaining errors.
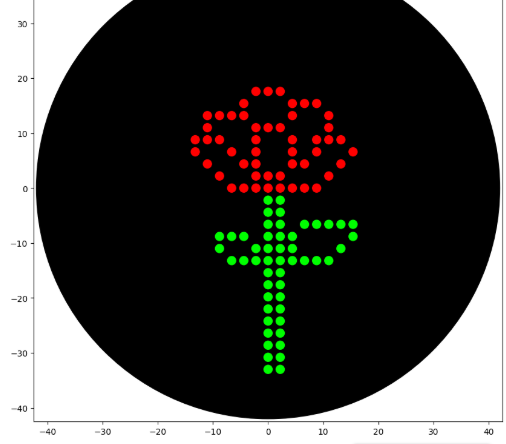
I designed a drawing with the website that was made available to us https://opentrons-art.rcdonovan.com/. It made it easier to get used to the way the points would be done inside the plate to create our art. Personally I decided to create a flower, took the exact points for each color, green (sfGFP) and red (mrFP1). I integrated the points to the base code, and customized it for the design to be possible, especially with the high quantity of points and the limit of the 20ul pipette.
PART A: CONCEPTUAL QUESTIONS
1.How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To calculate this we have to make some equivalences. 1 aminoacid = 100 Da = 100g/mol
Meat= 20% protein
Look only for the protein mass
500g of meat x 0.20 = 100g of protein
Moles of aminoacid
SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Protocol Questions Answer these questions about the protocol in this week’s lab:
-What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix with HF Buffer is a 2X master mix consisting of Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2. All that is required is the addition of template, primers and water. (New England Biolabs, 2026)
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? While Booleann circuits produce on/off outputs, IANNs generetae continuous and graded reposnes allowing us to detect subtle changes in input signals. They also can intigrate multiple inputs that are adjustable, making them more flexible and better to mimic natural cellular desicion processes.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. CFPS offers several key advantagesin felxibility and experimental control over traditional in vivo protein expression. In terms on flexibility, unlike living cells, we have an open reaction enevironment as we are not constrained by structures or the viability of the cell. We can add and remore components at any time. As we dont depend on the cell, there is no need for cloning or transformation, allowing the qucik testing of multiple gene constructs at the same time. We can express proteins that coudl be toxic or unstable in livign things because nothing is alive. Comparing the benefits for control of the experimental values, CFPS systems are superior as we cna tightly control conditions, directly manipulate the gene expression and define the environment for production.
Homework: Final Project
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Part A: Art Pixel
I was not able to complete this portion because I did not receive the email containing the project link. By the time I realized the issue, it was too late to contribute.
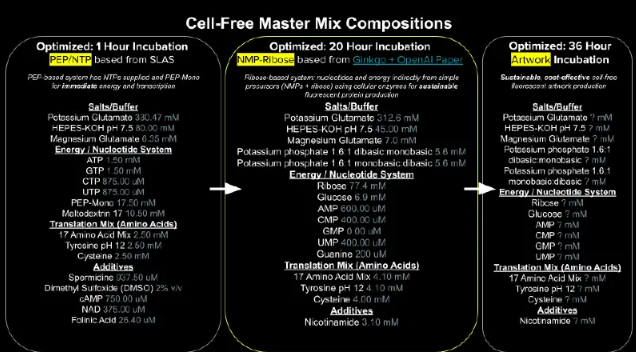
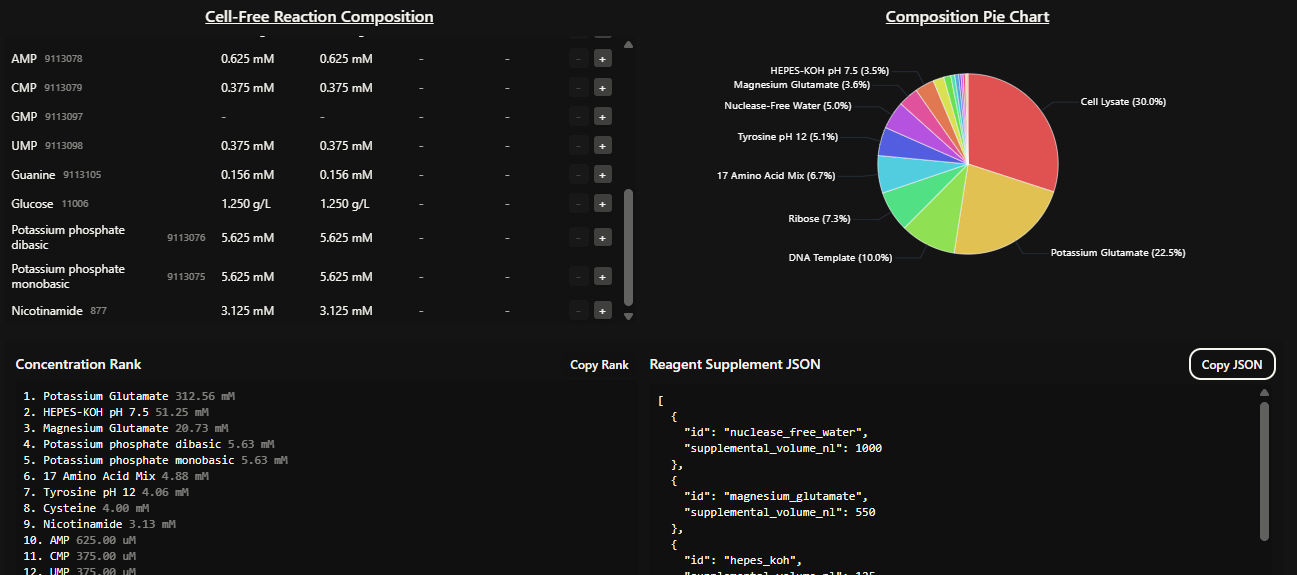
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Subsections of Homework
Week 1 HW: Principles and Practices
1.First, describe a biological engineering application or tool you want to develop and why.
As a pharmacy student, I have become increasingly interested in how drugs move from an initial idea to clinical use, and how many potential compounds fail long before they could reach patients. Drug development is an expensive and time consuming subject, and an ethically complex process, specifically in early stages. But many drugs are left behind as they fail to show strong enough effects or because the costs for further testing is too high.
This has led me to think whether there could be a more efficient way to evaluate the potential of these compounds before progressing into animal models. To address this challenge, I propose an engineered bacterial screening platform that could act as an early, low cost step in the process, especially for those drugs left behind that could be repurposed. This system could be used to report on the activation or inhibition of specific pathways, giving information on targets and if the drug should be continued to be studied before going to an animal model. It would act as an intermediate testing step making it cost effective and reducing the unnecessary animal testing.
This engineered bacterial screening could also be assembled as a living diagnostic tool for diseases that are difficult to detect using conventional methods or invasive procedures, especially when biomarkers are localized or non-specific. Engineered bacteria have already demonstrated potential as diagnostic tools because they can be programmed to detect biomarkers and respond with measurable outputs. And in this application, the engineered bacteria would work as a living biosensor, temporarily sensing for biomarkers and programs to give a clear response. Both applications would be limited to controlled clinical or research settings , allowing for the exploration through these innovative diagnostics while maintaining the commitment for patient safety.
2.Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Ensure patient safety and non-malfeasance
-Prevent uncontrolled bacterial replication, persistence, or dissemination
-Ensure engineered bacteria can be safely eliminated after use
Enhance biosecurity
-Ensure traceability and accountability in their handling and use
-Prevent repurposing of the technology for harmful or unethical applications
Promote ethical and efficient drug development
-Reduce unnecessary animal testing by strengthening early-stage screening
-Improve early decision making on drug continuation or abandonment
-Avoid discarding potentially beneficial drug candidates
Promote equity and accessibility
-Develop cost-effective screening and diagnostic tools
-Prevent socioeconomic or geographic disparities in access
-Encourage drug repurposing for neglected or rare diseases
Maintain transparency and public trust
-Clearly inform patients and research participants about the use of living biosensors
-Communicate risks, benefits, and limitations transparently
3.Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Ensure patient safety and non-malfeasance
Mandatory biosafety and containment standards for all engineered bacteria.
With this action the purpose is to prevent uncontrolled bacterial survival or harmful interactions in human hosts. When talking about genetically modified organisms (GMOs), we already have a comprehensive set of laws for them. Taking this into account, for safety it would be required rigorous assessments and protocols for the authorizations for the idea to start being used.
Enhance biosecurity and prevent misuse
Controlled access and traceability of engineered strains
-Actors: Governments, industry, research institutions
-Implementation:
Centralized strain repositories
Record-keeping of users and experiments
-Failure: Unauthorized use → biosecurity risk
The considerable attention to the control of the strains and the people using it through record keeping would help the prevention of engineered bacteria for unethical or harmful purposes. The idea of a data base is a tool, that already currently exists within EU Biosafety, that provides a unified repository of information on GMOs. We could work with them to create a specific repository for this engineering bacteria and keep records of the labs using them for drug development or as a screening tool.
Maintain transparency and public trust
Public communication campaigns and stakeholder engagement
Educate the public about risks, benefits, and uses of engineered bacteria
There should be a full transparency of the techniques and the developments of the idea. Working with the government, communicating to the public about the project could be an easy way to reach the masses and prevent miscommunication or fear of it. Italian BCH AND EU Cartagena Protocol promote the public access of biosafety information already. following in steps, public engagement programs should be used to maintain trust.
Use of engineered bacteria as a mandatory pre-animal testing step
-Actor: Regulatory agencies, pharmacological industry
-Risk: Over-reliance on bacterial models
Requiring bacterial biosensor validation before animal testing would ensure drugs engage intended pathways and reduce unnecessary animal experiments. Although this step would reduce the unnecessary use of animal models, we are assuming bacteria could be able to have the same mammalian pathways, which could give wrong results.
Promote equity and accessibility
Subsidies and open-access initiatives for bacterial screening and diagnostics
-Actors: Governments, funders, industry, NGOs
-Implementation:
Low-cost bacterial screening platforms
Partnerships to reach underserved regions
-Failure: Technology inaccessible in low-income regions
We could implement funding for low-cost bacterial screening platforms by working with public and private partnerships creating global health initiatives and open drug databases so the technology could be used globally.
Does the option:
1
2
3
4
5
Enhance Biosecurity
2
1
2
2
3
• By preventing incidents
1
1
2
2
3
• By helping respond
2
1
2
2
3
Foster Lab Safety
1
2
2
2
3
• By preventing incident
1
2
2
2
3
• By helping respond
2
2
2
2
3
Protect the environment
1
2
2
2
3
• By preventing incidents
1
2
2
2
3
• By helping respond
2
2
2
2
3
Other considerations
• Minimizing costs and burdens to stakeholders
3
3
2
2
1
• Feasibility?
1
2
1
2
2
• Not impede research
2
2
1
2
1
• Promote constructive applications
2
2
2
1
1
1:weak / 2: moderate / 3:strong
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Upon the results of the scoring, the governance actions that should be prioritized are the mandatory biosafety Nand containment standards and have control access and traceability to the engineered bacteria strains. These two goals should be at highest priority as it would be the ones preventing incidents across patient and lab safety and protection. Engineered bacteria although it used for the screening and testing tool, the consequences of failing its containment and possibilities of mutations could be severe. Existing EU Legislation don Italian biosafety measures and frameworks give a sturdy foundation for these actions, making them both feasible steps, ensuring the safeguards in our project and validated containments protocols.
Along with the need to maintain biosafety, is linked the controlled access to the engineered strains, which would be the best method to enhance the biosecurity and prevent any misuse. By maintaining repositories and detailed records of strains and ownerships and experimental uses, gives us the chance to rapidly respond to any incidents. Also, as it’s a regulatory access, the use for unethical purposes is discourage. This action builds on existing databases that share the same goals. As safety and security is assured, regulatory recognition for the engineered bacterial screening as mandatory step could be one that should give attention. This action promotes an ethical drug development by reducing unnecessary animal testing and improving early decisions. While it assumes for similar pathways and results to mammals, it reflects into the 3rs principles and offers a cost-effective way to rescue drugs that have been abandoned. There were a lot of trade offs considered when prioritizing these actions. As strong biosafety and security is needed, it becomes a new financial burden on researchers and overall, the industry. Overall, I think it’s always needed to prioritize the safety and security and ethical efficiency when it comes to creating and advancing engineered biology.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
Reflecting from this week’s work and class, I’ve realized that the importance for ethics is really needed, specially for synthetic biology. For the example of engineered bacteria, while the idea has beneficial purposes, the same properties that make them adaptable and programmable, also create concerns about misuse and unintended consequences. This made we more a ware of how closely this innovation is tied to responsibility and oversight overall. Another ethical issues that caught mya attention is reducing animal testing and ensuring patient safety., although using engineered bacteria as a pre animal testing step would minimize the used of animal models, it introduces uncertainties in how well these systems could predict humans’ responses. The ideal of heavily simplified the models leads to further skewed decisions in developments that ultimately affects patients
Public trust and transparency are also really important necessity. New technologies can face resistance and we as scientist should be able to include the people into understanding them and relieve those doubts and fear.
Ethical concerns should always be address, creating appropriated governance actions that create a safe and controlled environment, with clear regulatory limits but also the transparency to the people. All of this combined creates a place where innovation is ensured, while maintaining safety and trust.
Bibliography
-European GMO Authorisation Database (EUGinius). (n.d.). GMO authorisation index. European GMO Initiative for a Unified Database System. Retrieved February 2026, from https://euginius.eu/euginius/pages/authorisation_index.jsf (euginius.eu)
-Cartagena Protocol on Biosafety – Biosafety Clearing-House (BCH). (n.d.). Cartagena Protocol on Biosafety. Secretariat of the Convention on Biological Diversity. Retrieved February 2026, from https://bch.cbd.int/en/ (pmc.ncbi.nlm.nih.gov)
-Pant, A., & Das, B. (2022). Microbiome-based therapeutics: Opportunity and challenges. Progress in Molecular Biology and Translational Science, 191(1), 229–262. https://doi.org/10.1016/bs.pmbts.2022.07.006 (review of microbiome-related therapeutic strategies) (PubMed)
-Zhou, Y. (2022). Engineered bacteria as drug delivery vehicles: Principles and prospects. Frontiers in Bioengineering and Biotechnology (review). Retrieved from https://pmc.ncbi.nlm.nih.gov/articles/PMC11611002/ (review of engineering bacteria for therapeutic delivery) (pmc.ncbi.nlm.nih.gov)
-Kulkarni, V. S., Alagarsamy, V., Solomon, V. R., Jose, P. A., & Murugesan, S. (2023). Drug repurposing: an effective tool in modern drug discovery. Russian Journal of Bioorganic Chemistry, 49, 157–166. Retrieved from https://pmc.ncbi.nlm.nih.gov/articles/PMC9945820/ (overview of drug repurposing approaches) (pmc.ncbi.nlm.nih.gov)
Week 2 HW: DNA- Read, Write and Edit
Pre-Lecture HW:
1.Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of polymerase estimates to be made errors once every 10x4–10x5 nucleotides polymerized. This compared to the length of the human genome that’s approximately 3.2 billion base pairs long, that would account up to 32 thousand mutations every time a single cell divided. Biology fixes this gap with multiple systems that check for these errors. Its first system is proofreading; a function found in most polymerases. When they add a wrong base, they recognize this error, backtrack and fix it cutting the wrong base and replacing it. Another way, is mismatch repair (NMR), after polymerase is done, a second group of proteins scan the new DNA for any remaining errors.
2.How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Proteins are made of Animo acids, macromolecules that are mostly synthetized by humans. We can synthetize 20 standard Animo acids. They are coded by codons (a group of 3 DNA bps) of which we have 64 possible combinations. As there are more than 1 way to produce the same amino acids, we refer to this code degenerative. Because of this system, in theory, you could swap a codon for another a produce the same amino acids as product and the protein should be the same but there’s several reasons this could fail. Sometimes during the transcription and translation process, things can occur, like ribosomes slowing down to let the protein fold or just certain combination of bps make the mRNA unstable degrading before it can be translated. As the protein synthesis relies of multiples factors the degenerative code isn’t always foul proof.
1.What’s the most commonly used method for oligo synthesis currently?
Currently the most commonly used method for oligo synthesis is by using solid-phase phosphonamidite synthesis. The process adds 1 nucleotide at a time 3’-5’ thought 4 repeating steps.
2.Why is it difficult to make oligos longer than 200nt via direct synthesis?
It is difficult because of the cumulative yield and chemical side reactions. Even is the machine being in optimum state teres still some loss happening with every single base addition. By time u wan to reach 200nt, the majority of the material is going to be failure sequences rather than the target.
3.Why can’t you make a 2000bp gene via direct oligo synthesis?
This isn’t possible because the yield becomes essentially zero, as there would be no way to find target in a solution where practically all is incorrect fragments. Also, the purifications are impossible as it is too big to reliably separate all perfect 2000bps.
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Essential Aminoacids
1.Phenylalanine
2.Valine
3.Threonine
4.Tryptophan
5.Isoleucine
6.Methionine
7.Histidine
8.Arginine
9.Leucine
10.Lysine
When you look at the fact that this lysine contingency was the only fail safe for the dinosaurs, believing that its inability to produce lysine would be able to contain a scape seems a bit redundant and negligent. We all need certain amino acids that we aren’t able to produce, that’s why we make nutritious diets for us, as its abundant in nature. If we can survive without producing it on its own by resourcing to get it throughout our diet, it’s a bit irresponsible to thing this only failsafe was going to work, as we are proof of it not being limiting.
Bibliography
-Drake, J. W., Charlesworth, B., Charlesworth, D., & Crow, J. F. (1998). Rates of Spontaneous Mutation. Genetics, 148(4), 1667–1686. [Referenced via ScienceDirect: “Rates of Spontaneous Mutation” regarding replicative fidelity and evolutionary efficiency].
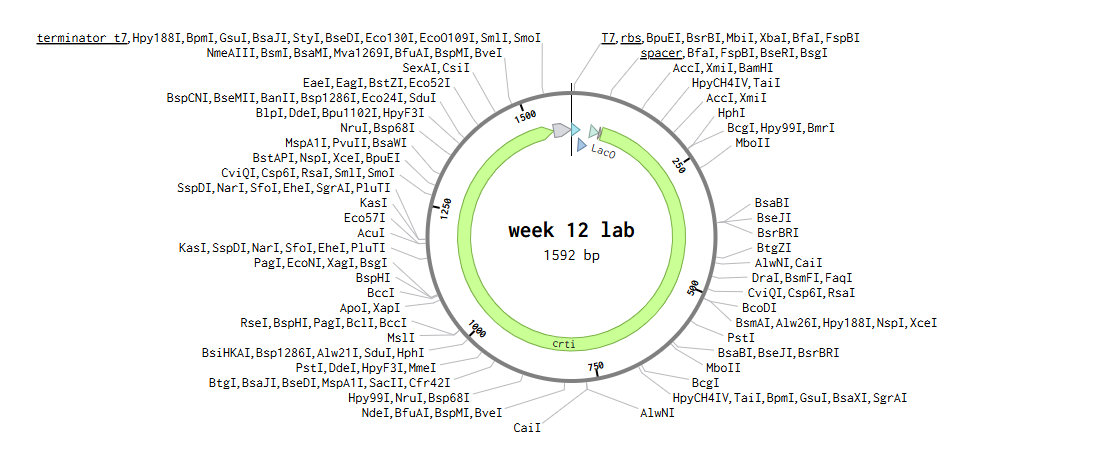
-Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks
While looking the patterns with the different restriction enzymes, I try to create the top part of a cats siluette, basically just its ears.
I used the patterns of the digestion of enzymes: Pvull, Hindll, kpnl, SalI.
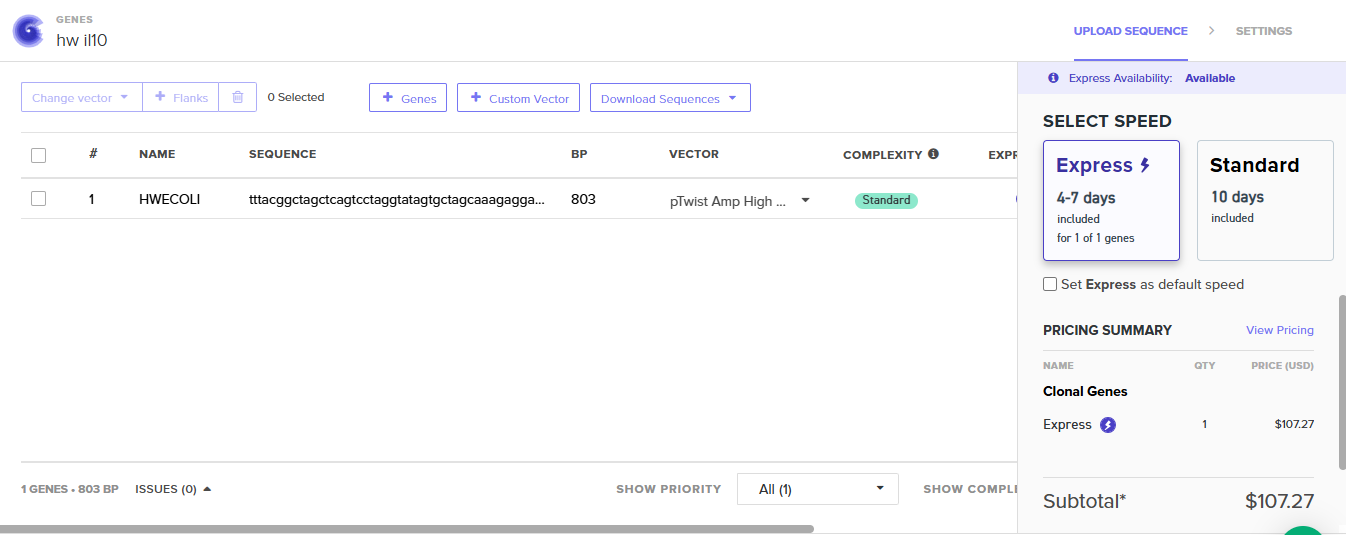
While looking into the possibilites of the use of bacteria as drug delivery systems, I saw the concept of designing bacteria with the capability of secreting anti-inflammatory proteins. I chose Interleukin-10, as its a anti-inflammatory cytokine that usually is secreted by immune cells and has a role in reducing the inflammation in tissues. I used this week assignment as a way to explore the possiblity of engineering bacteria to deliver this therapeutic protein, choosing this protein sequence and optimizing it to E.coli as a trial to see the efficacy of this concept.
In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
The genetic code is degenerate, which means most aminoacids could be encoded by more than one codon. This makes it possible for organisms to have preferences with codon for the expression of aminoacids, creating a bias. To maximaize the protein production, we need to work according to what the host prefers, improving the trnaslation efficiency and reducing errors. I chose to directly optimized for E.coli, as sequence is more a trial to see if the expression of the protein through out the bacteria is possible. Choosing E.coli simplifies the design and clonign workflow, letting us to conceptially see if the therapeutic delivery is possible.
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Some tehcnologies we could use to optain the protein from the DNA, is cell dependent expression. With this technique the IL-10 gene optimized for E.coli, gets used by the bacterial transcription machinery, producing mRNA that gets bound and translated giving us the end product of IL-10.
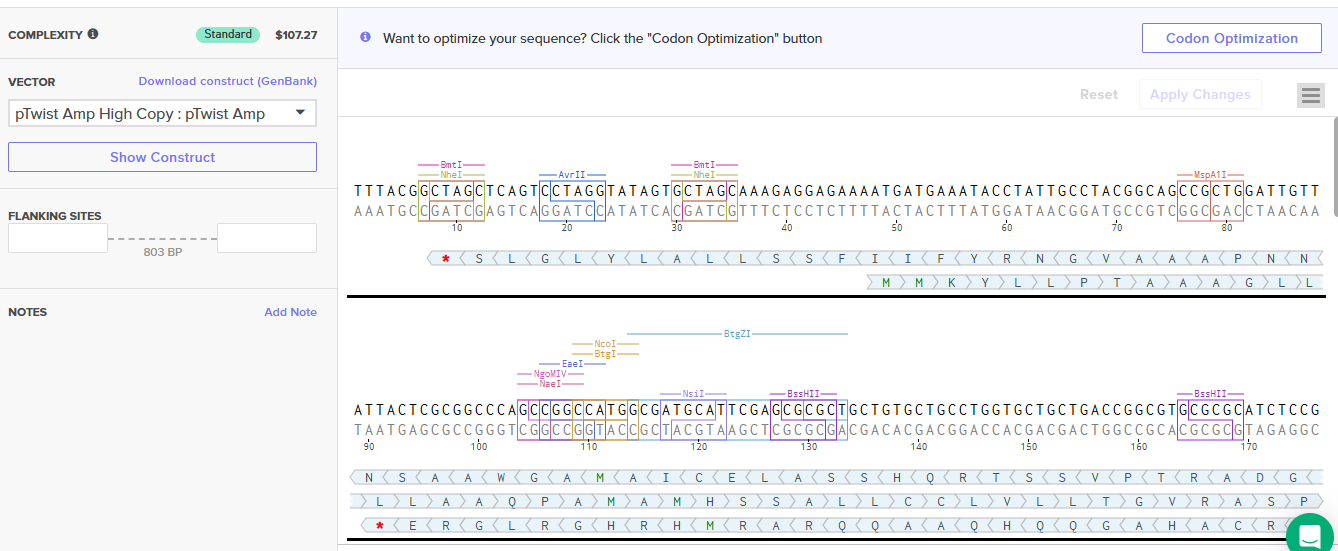
I decided to include the pelB signal in the Il-10 seuqnece so the protein when it is translated, it can be directed to the periplasmic space where it can be more easily secreted.
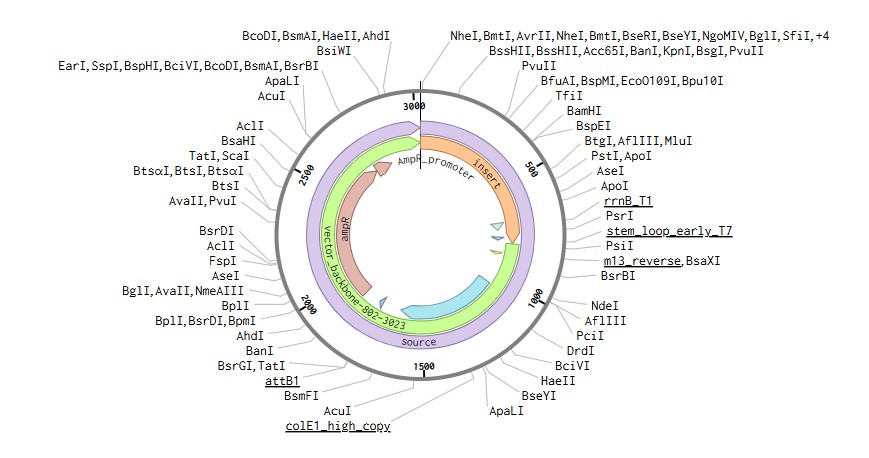
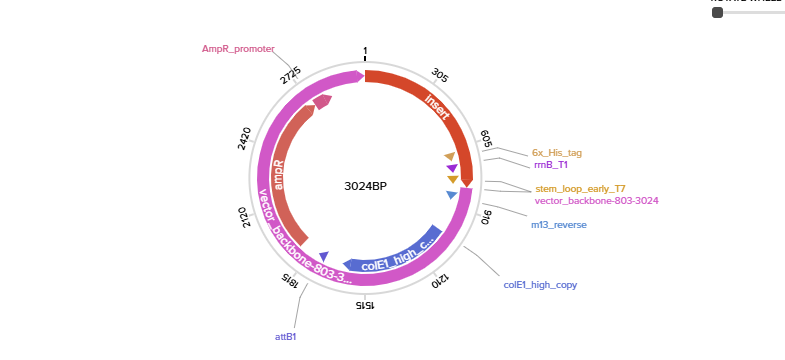
To express IL-1O in E.coli, I designed a expression cassette consisting of a constitutive promoter, a strong RBS, and a start codon. I added a pelB signal sequence that makes possible for the bacteria to secrete the protein. We added a HISX7 TAG, to make it easier to purify the proteins, a stop codon and finally a terminator. We choose clonal genes so it can be delivered as a circular plasmid and direclty transformed into the E.coli, and for vector the pTwist Amp High Copy, that gives us a high plasmid copy number resulting into a strong protein expression.
PART 5: DNA READ/WRITE/EDIT
5.1 READ
-What DNA would you want to sequence (e.g., read) and why?
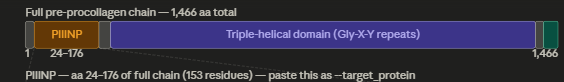
Even if it isnt strictly related to my project, i would like to sequence genes involved in collagen production and extrecellular matrix stability, specially in patients with EDS. Ehler Danlos Syndrome is a group of inherited connective tissue disorders, with a combined stimated prevelance of at least 1 in 5000, even with it being underdiagnosed, where collagen sequences are mutated leading to misfolded collagen and connective tissue fragility. Because of collagen structure being complex and relying in the triple helix assemblance, small mutations can cause big consequences. Studying genes such as COL1A1, COL1A2, COL3A1 in patients with the disorder, it would give insight into how specific mutaitons lead to misfolded colalgen and connective tissue fragility and give better understanding of genotype/phenotype of variants and a lead to develop targeted therapies.
-What technology or technologies would you use to perform sequencing on your DNA and why?
I think the best choice would be a second generation sequencing, like Ilumina NGS. It’s considered 2nd gen because of the ability of sequencing millions of short dna fragments in parallel and requires the amplification of dna before the sequencing. The input could be be genomic dna extracted from the patients blood, which would have to undergo certain preparation steps. After DNA is extracted, it’s mechanically brokendown into short fragments. Synthetic adapters are lgiated to both ends of each fragments, which allows the binding to flow cells. After the fragments are amplified with pcr and then denatured to a single strand to sequence. For Illumina, the sequencing is done by synthesis. It creates cluster generation by binding the dna fragments into complementary oligos onf the flowcells and thru bridge amplificaiton creating the clusters of identical copies. Labeled nucleotides with a flurescent tag and reversible terminators are added and are incorporeated by polymerase 1 at time per cycle. As each nucleotide has a different signal, we can detect which base is being used each cycle. The output is millions of short dna reads, each giving us the nucleotide sequence and the a quality score of each base. It also makes it possible to identify any variants like deletions or insertions, which woulds allow the detection of any mutation for EDS in collagen genes.
5.2 WRITE
What DNA would you want to synthesize (e.g., write) and why?
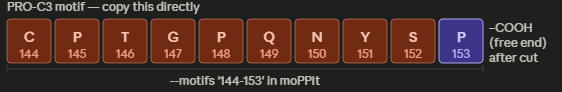
I would like to synthesize a correct repair template for the pathogenic mutations in the collagen genes. The many of the mutations associated with EDS, involve the susbtitution of glycine residues in the repeating Gly–X–Y motif that stabilizes the collagen triple helix. If glycine is substituted for a bulkier aminoacid it disrupts the folding of the protein and weakes the connective tissues. If we could synthetize a fragment with the proper repreats and a way to flank the mutation site, it could be used to investigate the possiblities to correct the misfolded collagen and help compared stability of proteins.
What technology or technologies would you use to perform this DNA synthesis and why?
I would use the synthesis thru phosphoramidite chemistry. This methods chemically synthetizes base by base short dna oligonucleotides using protected nucleotides. The overlappping oligos are assembled into a full fragment that its cloned to a plasmid, and later the sequence is verified. This tecnique gives me a high fidelity which is needed for the repeated collagen sequences, and enables precise contorl over mutation correction.
5.3 EDIT
What DNA would you want to edit and why? What technology or technologies would you use to perform these DNA edits and why?
VEDS, is associated with pathogenic mutations in the COL3A1 gene, arising as disruptions with the glycine residues needed for the correct formation of collagen. You could use CRISPR CAS9, to design an RNA capable to target the mutated region and introduce a donor repair template with the correct sequence. Alternatively, base editing could be use to change the erroneous base.
Week 3 HW: Lab Automation
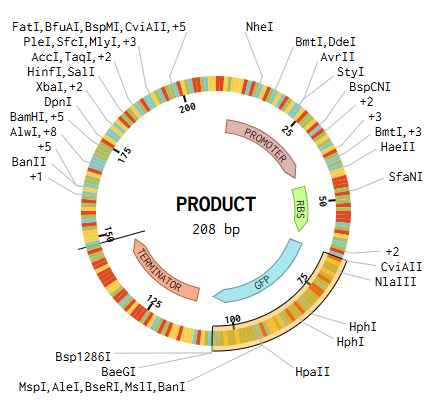
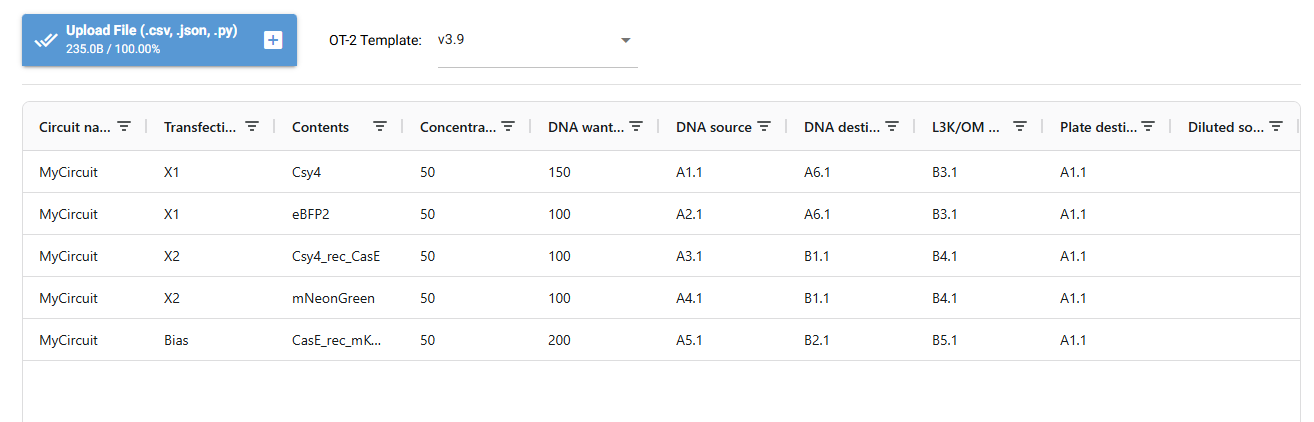
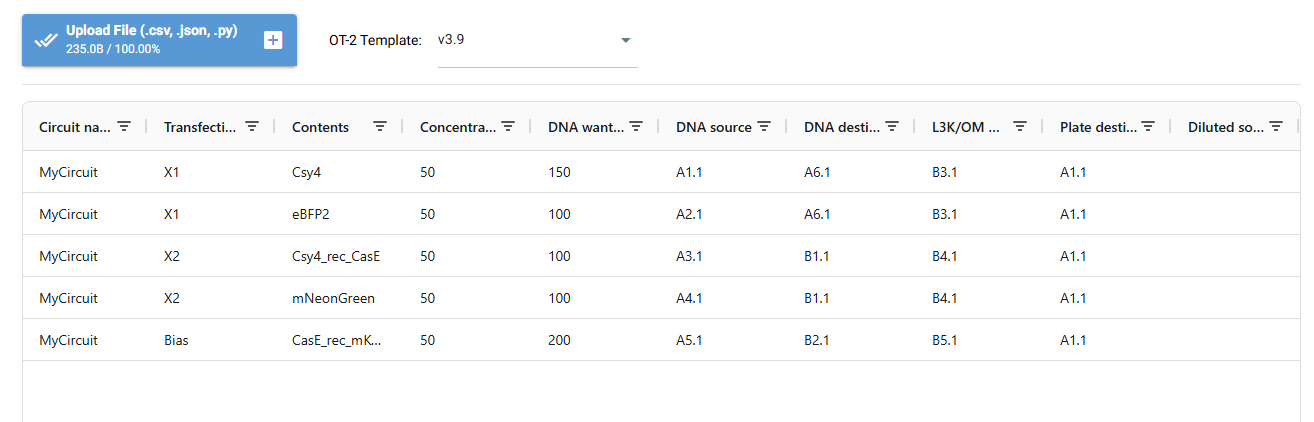
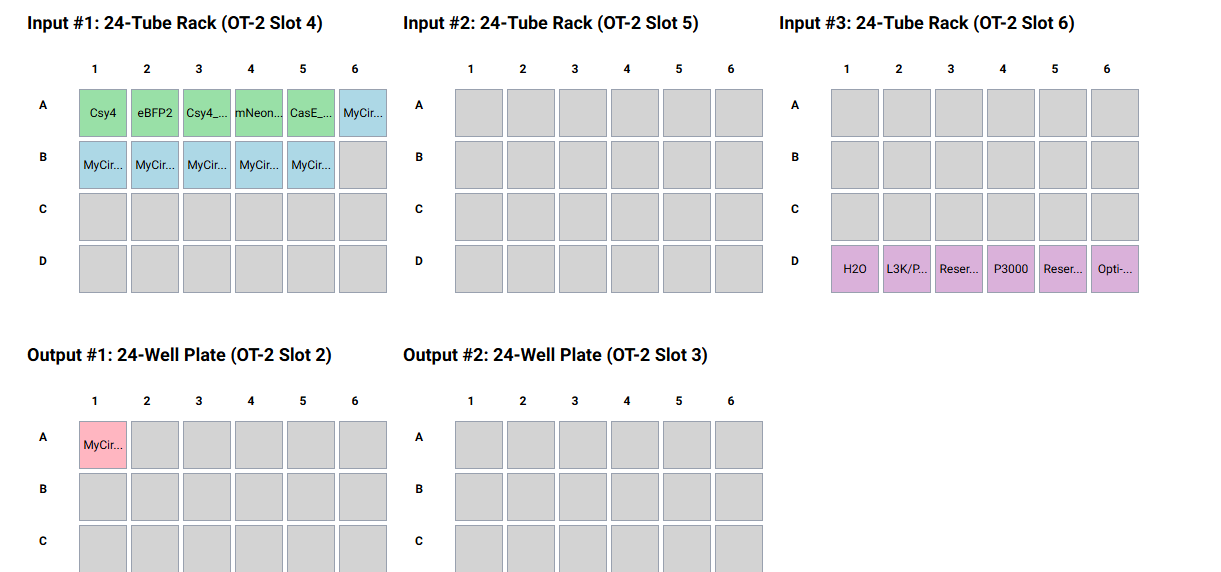
I designed a drawing with the website that was made available to us https://opentrons-art.rcdonovan.com/. It made it easier to get used to the way the points would be done inside the plate to create our art. Personally I decided to create a flower, took the exact points for each color, green (sfGFP) and red (mrFP1). I integrated the points to the base code, and customized it for the design to be possible, especially with the high quantity of points and the limit of the 20ul pipette.
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
In this paper, they use Opentrons to automate the assembly of bisoensors. The team created a flouride responsive genetic biosensor that produces a measurable florescent signal when positive. They compared ht precision and consistency of hte building between manually prepare and the automatic assembly done by the robot, which ended up giving as realible results. The paper highlights how the automatization can increase throughtput, reduce human error, and standardize the workflows in synthetic biology for the development and screening of biosensors.
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
For my final project ideas, Im thinking of creating biosensors for multiple things, so essentially I could use as an example the paper, and create a code to work with Opentrons to create the workflow of the engineering and producing them. To create the biosensor for biomarkers, and being a comitted listener distnat from the node, automatization robots are my chance to do the wet lab of the synthetizing of the assembling of the circuit for biosensor, quantify the flourensence, and create data dirven profiling.
Week 4 HW: Protein Design Part 1
PART A: CONCEPTUAL QUESTIONS
1.How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To calculate this we have to make some equivalences.
1 aminoacid = 100 Da = 100g/mol
Meat= 20% protein
Look only for the protein mass
500g of meat x 0.20 = 100g of protein
Moles of aminoacid
100g/mol100g=1 mol amino acids
Number of molecules
1 mol= 6.022 x 10^23 molecules
ANSWER: In 500g of meat considerign it only having around 20% of protein would have around 6x10^23 aminoacid molecules
2.Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This does not occur, because we eat the meat of either animal but it gets digested in the stomach, which is breaking down into amino acids. These amino acids are reused to create our own human proteins with the instructions of our DNA.
3.Why are there only 20 natural amino acids?
We got only 20 amino acids because of how the genetic code and translation machinery evolved. The genetic code is based on the one of codons to encode the amino acids, but even if there are 64 codons we only map 20 amino acids and the stop signals. As amino acids need trna and ribosomes to be decoded into proteins, if there would be a higher amount than the natural 20, it would require evolving new enzymes and recognition systems. With the 20 amino acids we have, we also cover all our chemical diversity needs to allow the very complex structures.
4.Can you make other non-natural amino acids? Design some new amino acids.
Yes, you could make other non-natural amino acids, exchanging R groups and giving new properties that can not be found in nature. We could design an amino acid that could bind to metal adding a strong ligand group like a pyridine in the structure. This modification is added to the alfa carbon as an r group, not making any change to base structure with the amino and carboxylic ends.
6.If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Proteins are normally made up of L-aminoacids, which form a right handed α-helix. If we end up doing them with D-aminoacids, the structure would mirror the natural form, giving a left handed helix because the chirality of the amino acids determines the direction of the backbone twisting.
7.Can you discover additional helices in proteins?
Apart from the usual alpha helices in proteins, there have been discovered others like the pi-helix and the 3-10 helix. 310-Helices represent the 3rd principal secondary structure, its given by the regular and repetitive H bonding network between carbonyl group in residue and the amide nitrogen in residue. These interactions give rise to a helix with 3 residue per turn and a helical pitch, following a succession of b-turnsending in a thin and elongated helix. π-helices contribute to protein folding and are relevant as ligand-binding site contributors, given by single residue insertions between alpha helices. New helices can be found by identifying repeating H-bonding patterns and backbone angles.
(2024). Revisiting 310-helices: biological relevance, mimetics and applications. Exploration of Drug Science. https://doi.org/10.37349/eds.2024.00034
8.Why are most molecular helices right-handed?
This right handed direction comes back to the chirality of the amino acids that are making up the protein. As usual proteins are made up of l-aminoacids, their steric constraints make the backbone rotate towards the right, this way minimizes the atomic clashes, being preferred as it is more energetically stable.
9.Why do β-sheets tend to aggregate?
Beta sheets tend to aggregate because of the strong hydrogen bonding networks formed in their backbones. In this structure, we see exposed hydrogen bond donors and acceptors, the flat sheet surfaces stack easily and there´s hydrophobic side chains interacting between them. All of these factors combined, give optimal possibilities for the b-sheets to aggregate because of the overall intermolecular forces that pack the molecules.
-What is the driving force for β-sheet aggregation?
hydrogen bonding possible because of the exposed H bond acceptors and donors in backbone
Hydrophobic interactions coming from side chains between b-sheets
Van der Waals packing, optimized by the stacking capability of the structure
10.Why do many amyloid diseases form β-sheets?
Amyloid diseases are characterized by the accumulation of misfolded proteins in tissues and organs, impairing their function. This disease can form from b-sheets because this arrangement in proteins maximizes the hydrogen bonding and the possibilities to give stable aggregates, making them accumulate and damage cells.
-Can you use amyloid β-sheets as materials?
Yes, they have potential to be used as material because of its characteristics making them self assembled, rigid and chemically stable but its direct use is unsafe, so researchers mimic AB fibrils that are non toxic.
11.Design a β-sheet motif that forms a well-ordered structure.
To make a well ordered b-sheet motif its structure is given by the b-strands linked lateral to the backbone with hydrogen bond to form a pleated sheet with side chains alternating between hydrophobic and polar residues.
H-P-H-P-H (hydrophobic faces clusters, polar residues face solvents promoting the stacking)
Val/Ile (hydrophobics residues for internal packing)
Lys/Glu (charged residues for solubility and salt bridges)
Antiparallel b-sheet, is more stable than parallel strands so H bonds between the backbone of N-H and C=O adjacents.
You end the structure using repeats to increase stability, introduce sticky ends to favor the stacking and cap termini to prevent the structure from disintegrating.
PART B: PROTEIN ANALYSIS AND VISUALIZATION
1.Briefly describe the protein you selected and why you selected it.
I chose HIF1A because it plays a crucial role in how tumors adapt to low-oxygen conditions. Many cancers exploit HIF1A by stabilizing it, even when oxygen levels are normal, creating a state called pseudohypoxia. This enables tumors to promote blood vessel growth, alter metabolism, and survive in hostile environments. Investigating HIF1A can help uncover ways to disrupt this process and potentially limit tumor progression.
2.Identify the amino acid sequence of your protein.
-How many protein sequence homologs are there for your protein?
Hint: Use Uniprot’s BLAST tool to search for homologs.
There are 250 protein sequence homologs codifying the same protein through different species.
-Does your protein belong to any protein family?
Yes, HIF-1A is part of a protein family, and is a key member of the bHLH-PAS protein family of transcription factors. The family mainly gives insight to the protein’s structure: bHLH, a basic helix loop helix domain that is gonna interact with DNA; PAS, a domain crucial for dimerization, needed to become an active transcription factor when partnered with HIF-1B.
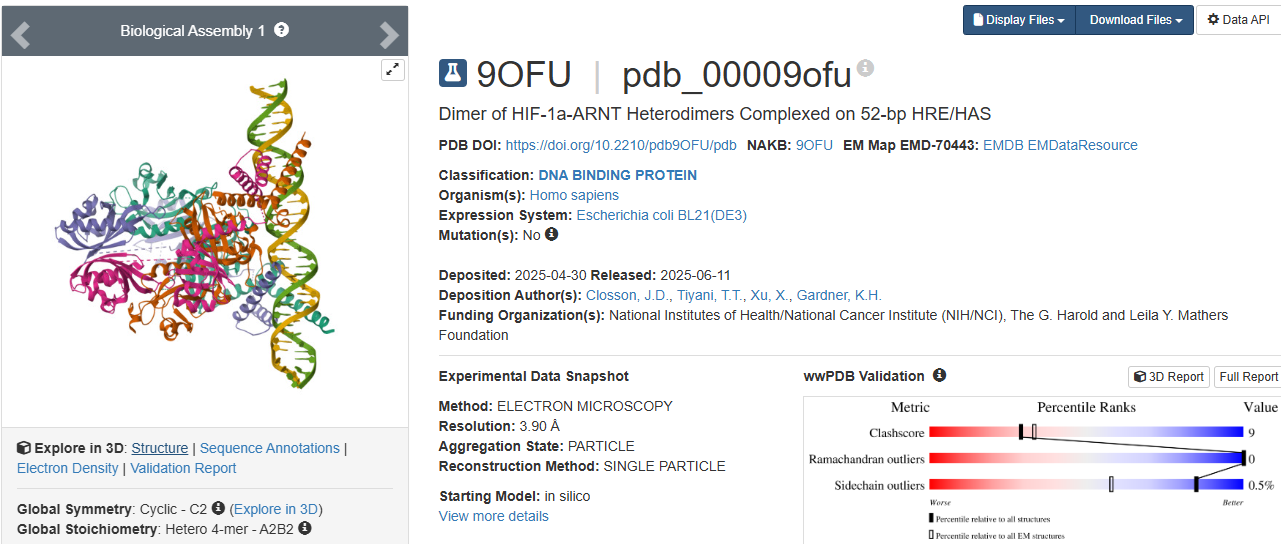
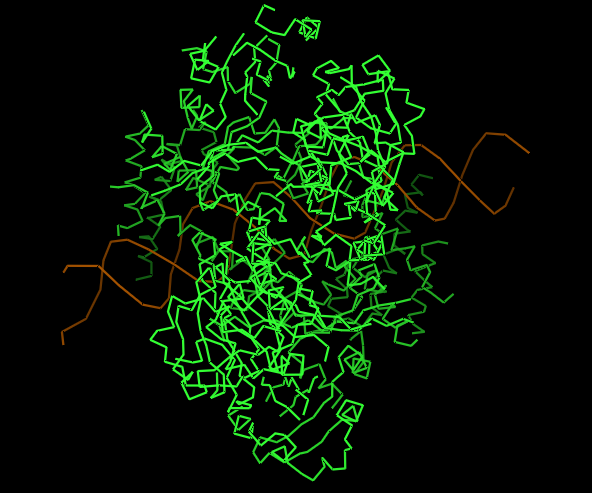
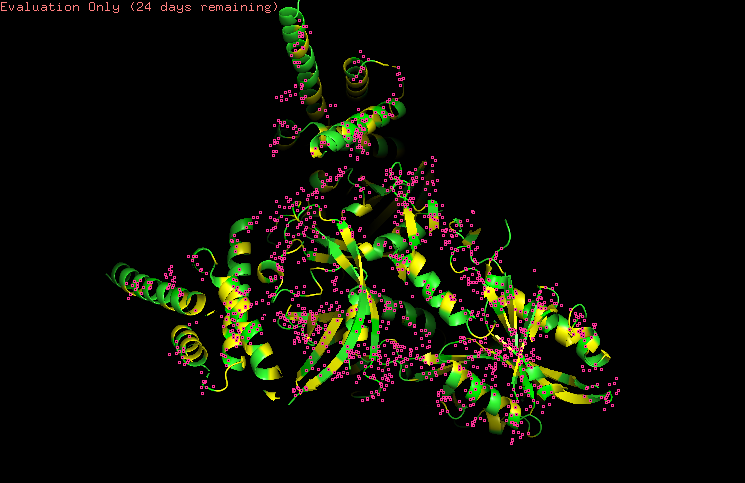
3.Identify the structure page of your protein in RCSB
https://www.wwpdb.org/pdb?id=pdb_00009ofu
https://www.rcsb.org/structure/9OFU#entity-1
When was the structure solved?
The structure was solved in 2025.
Its initial deposition was on 30th of April, initially released on 11th of June, and the latest revision on 27th of August.
Is it a good quality structure?
It has a resolution of 3.90 Å. I think it is a good quality structure as it has a fair resolution when compared to the size of the sample that was used.
Are there any other molecules in the solved structure apart from protein?
As HIF-1A works in collaboration to other structures as a transcription factor, the sample (Dimer of HIF-1a-ARNT Heterodimers Complexed on 52-bp HRE/HAS) and so on the molecule, the structure was solved with its other proteins interacting, them being:
52nt hypoxia response elements (forward and reverse)
Does your protein belong to any structure classification family?
Yes, is a key member of the bHLH-PAS protein family. It gives insight to the protein’s structure: bHLH, a basic helix loop helix domain that is gonna interact with DNA; PAS, a domain crucial for dimerization.
4.Open the structure of your protein in any 3D molecule visualization software:
-PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
PyMOL>fetch 9OFU TITLE Dimer of HIF-1a-ARNT Heterodimers Complexed on 52-bp HRE/HAS ExecutiveLoad-Detail: Detected mmCIF CmdLoad: ".\9ofu.cif" loaded as "9OFU".
-Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
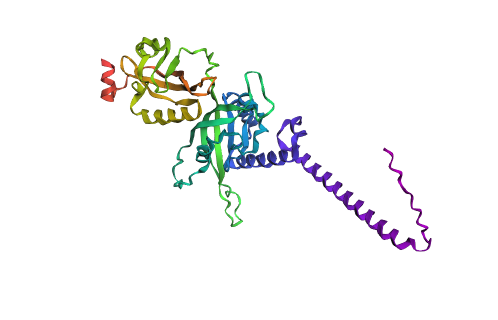
CARTOON
RIBBON
BALL AND STICK
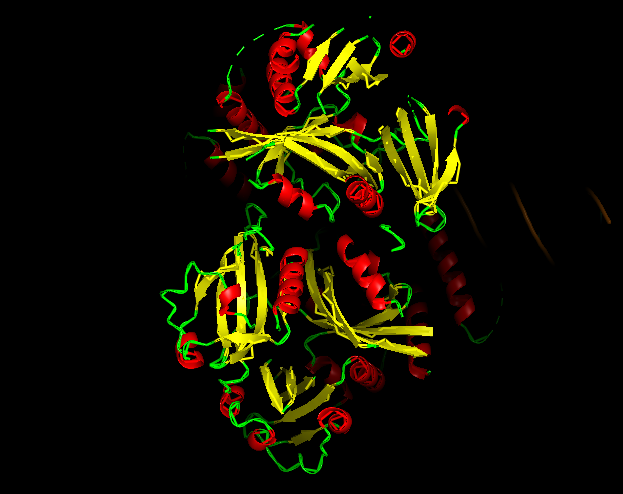
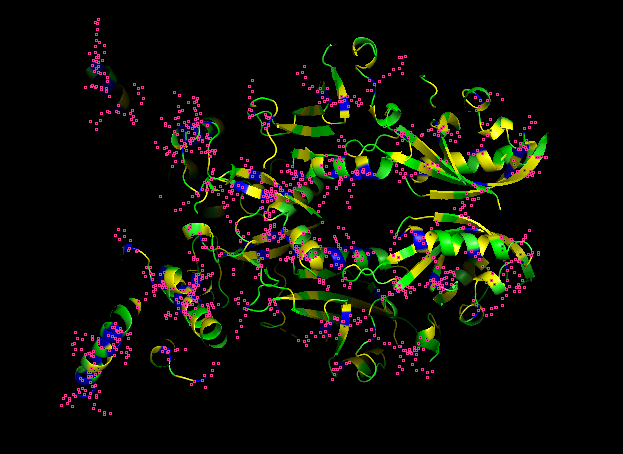
-Color the protein by secondary structure.
-Red → a-helices
-Yellow → b-sheets
-Green → loops
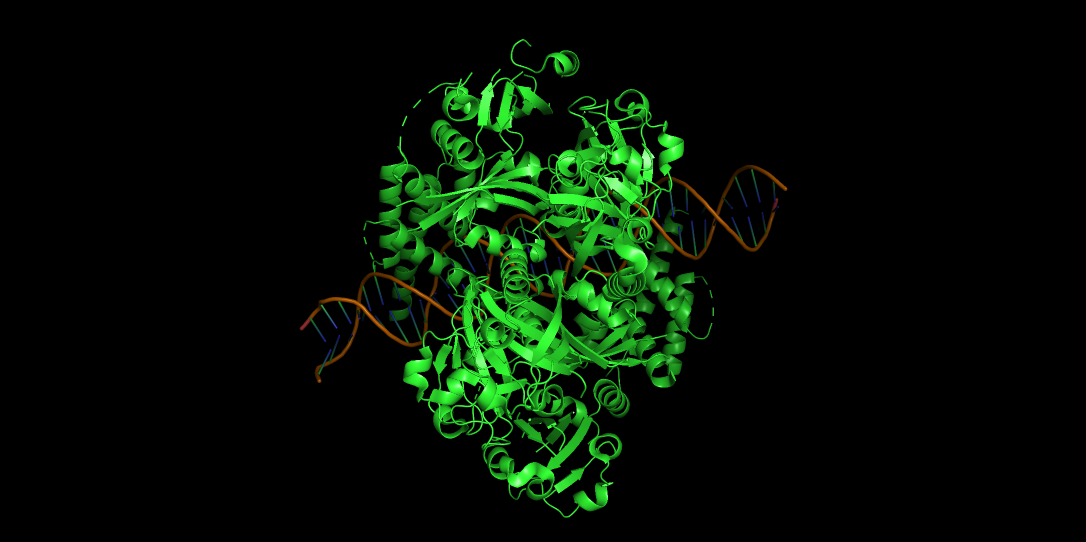
Does it have more helices or sheets?
It contains more helices than sheets, and it makes sense because HIF1A has the bHLH domain that’s helix rich to wrap around DNA to act as transcription factor.
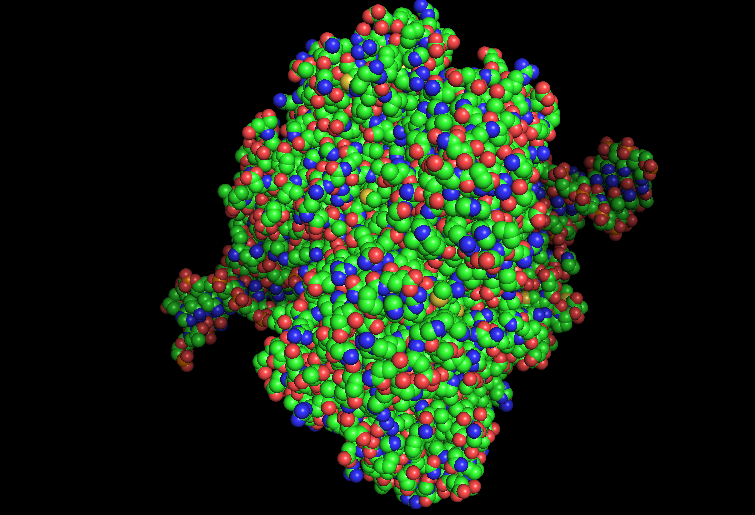
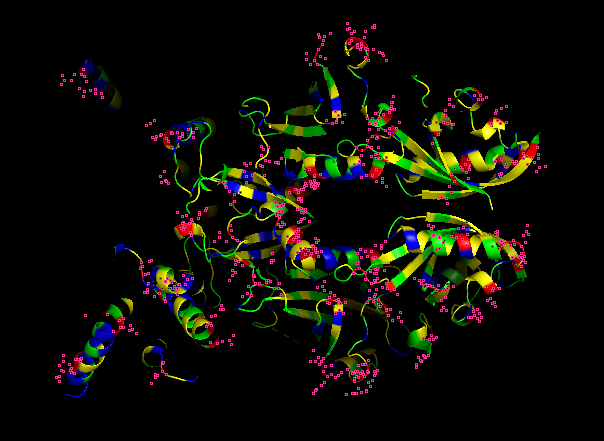
-Color the protein by residue type.
HYDROPHOBIC RESIDUES (YELLOW)
POLAR RESIDUES (GREEN)
POSITIVE RESIDUES (RED)
NEGATIVE RESIDUES (BLUE)
What can you tell about the distribution of hydrophobic vs hydrophilic residues?
From what it is seen in the structure after the coloring, most of the hydrophobic residues can be found inside the protein core, while the hydrophilic and differently charged residues are mainly in the surface. A positive charge can be seen near DNA.
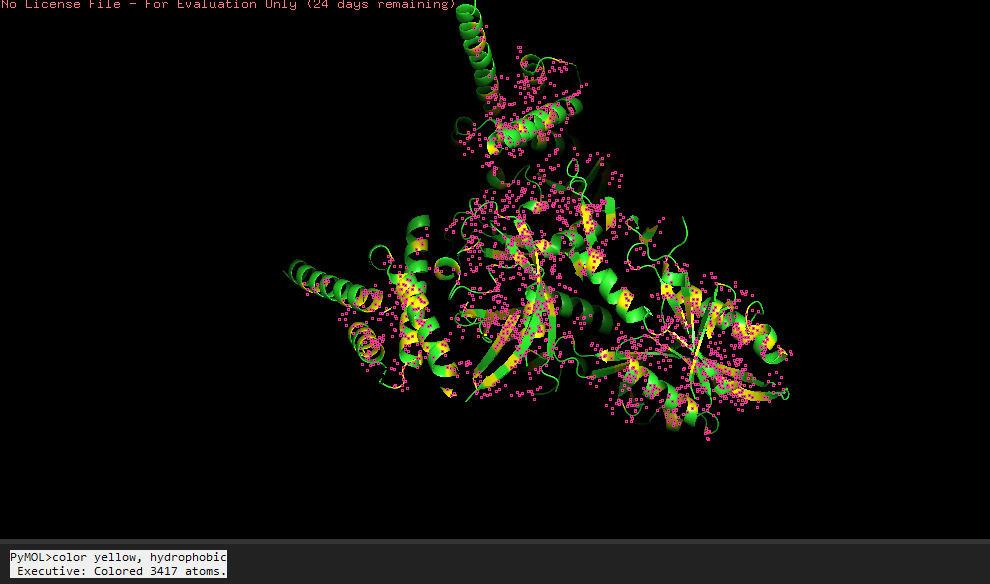
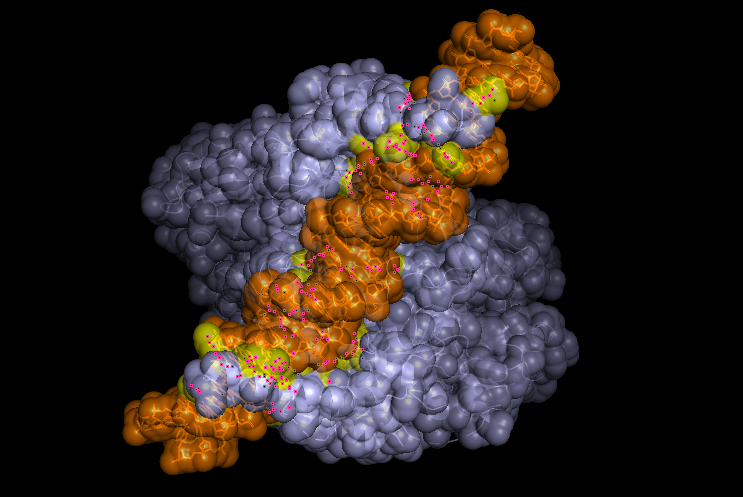
-Visualize the surface of the protein.
Does it have any “holes” (aka binding pockets)?
Yeah, in the surface visualization you can see grooves and small holes on the protein surface. This makes sense and this protein dimer interacts with dna for transcription. The grooves correspond to where the hif1a and arnt bind to dna, and the pockets between the proteins because of the interface that’s created between the structures making the dimer.
As before we looked into the whole dimer structure, for the protein design tool, I chose to focus into the hif1a chain.
1.Deep Mutational Scans
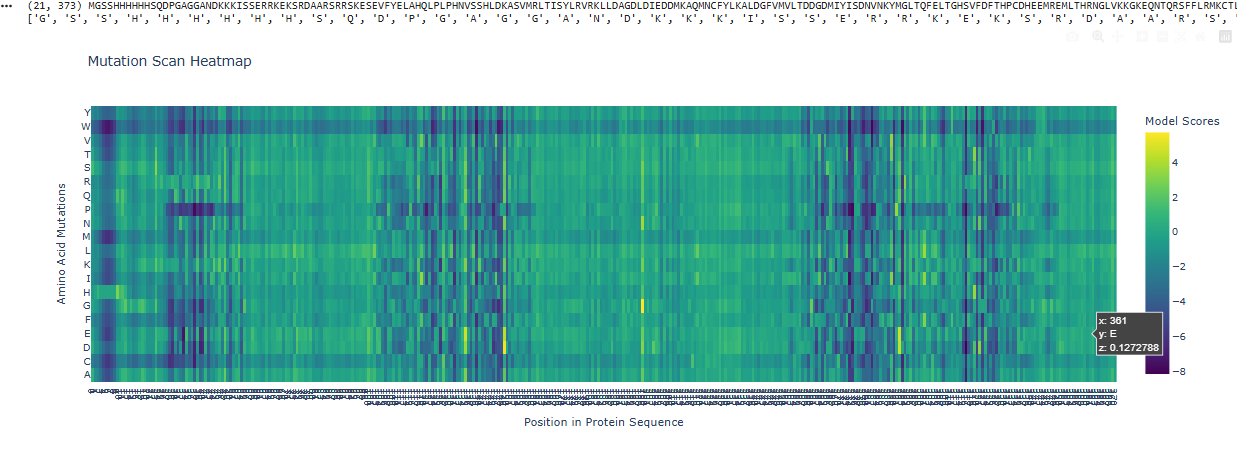
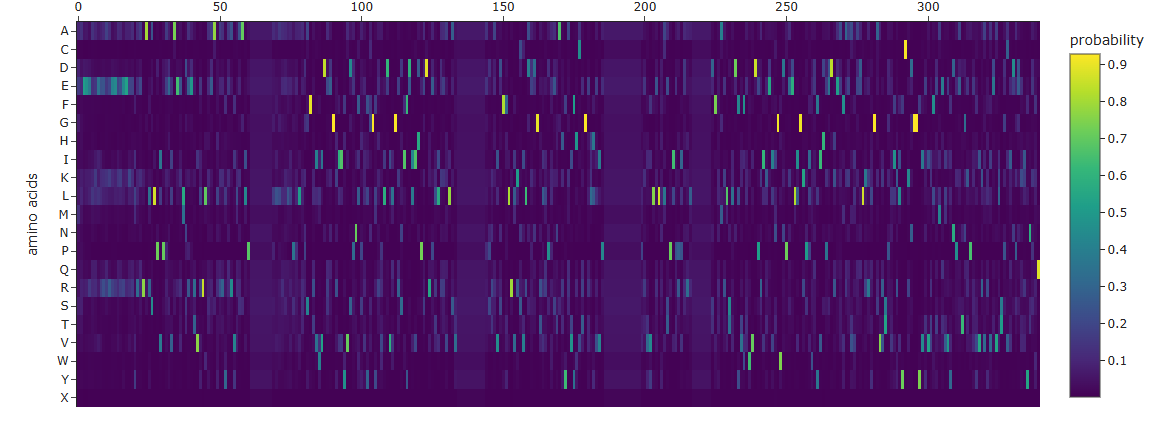
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
I can see that overall a big part of the protein is pretty constant, not giving rise to a lot of mutations, except in those 3 spots, one and the start, middle and end.
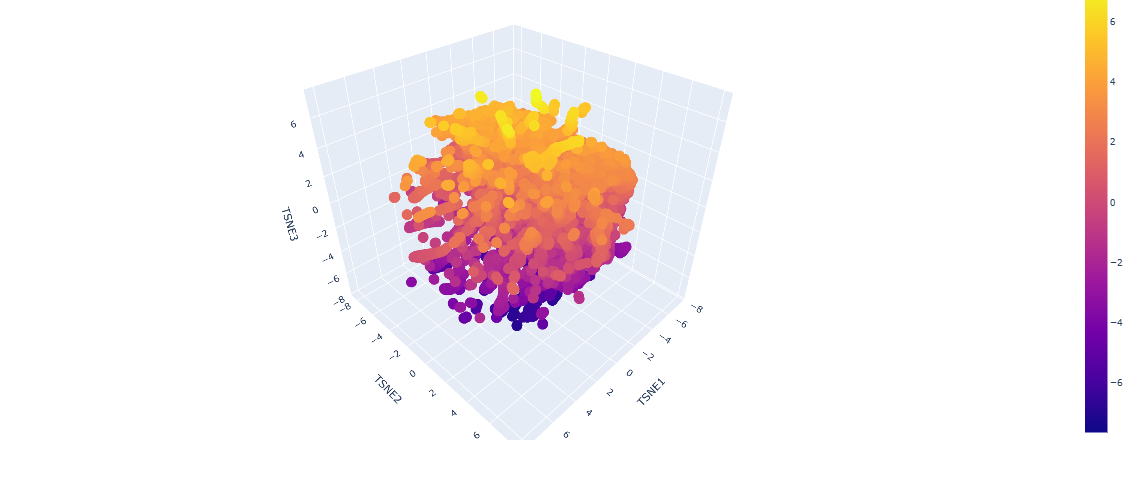
2.Latent Space Analysis
-Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
From looking at the different proteins embedded into the dataset, the proteins are organized into neighborhoods that are grouped into proteins that have similar interactions inside the host, as it can see a lot of proteins that have interactions with DNA towards negative numbers tsne2, while other proteins found in fungi or plants are upwards.
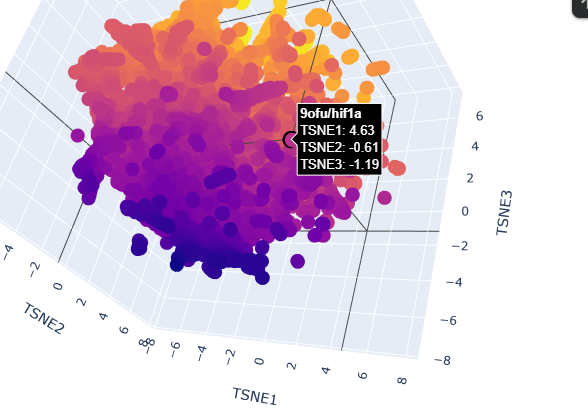
-Place your protein in the resulting map and explain its position and similarity to its neighbors.
When placing my protein into the resulting map, it’s integrated into the purple side of the graph, towards the negative tsne2. I can see some similarities with the neighboring protein as they have interactions with dna or have transcription domains.
3. Protein Generation
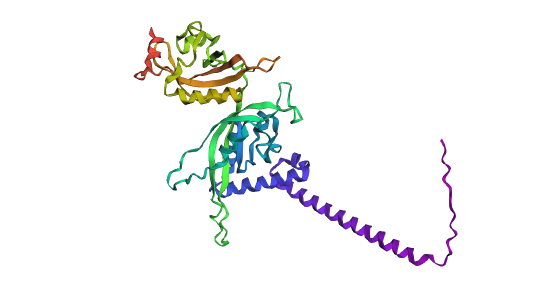
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Predicted structure with mutationsOriginal structure
Looking at both structures there’s not a lot of rash differences, but that can be as I just did a 3 cycle of folding based on the sequence with the program. One of the main differences is that you can see a higher quantity of alpha helices and a more compacted protein compared to the predicted sequence.
Input this sequence into ESMFold and compare the predicted structure to your original.
New Sequence:
SLEEILKELEEKREKEEEERYREEALSLPIPEEVAKKLDKETVKRLKEALEKYKRVKEALPXXXXXXXXLLEELLKLEKENSFKVWIDEEGKIIYVDPNIYKYAGMKVIDVLGKSIFDIIDPNDRELLKEKLSVXXXXXXXXXXEPRKVDFFLRIKSILDDDGKLIDEKNAHYKVFRAKGEILKVPXXXXXXXXXXXXXPEYVLELNLTPITDPEKEXXXXXXXTFKAKLTKDFVFEWVDPKIKEINGWNPEDLIGKPIYEFIHPEDREAFEKVLEELKKYGKVVSPEIKLYCKNGGYIIVRFTMTMKYNPETNKPEEVLAEIEVLSPCIEPEKIYNEIQ
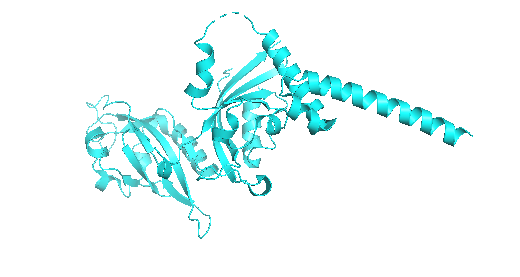
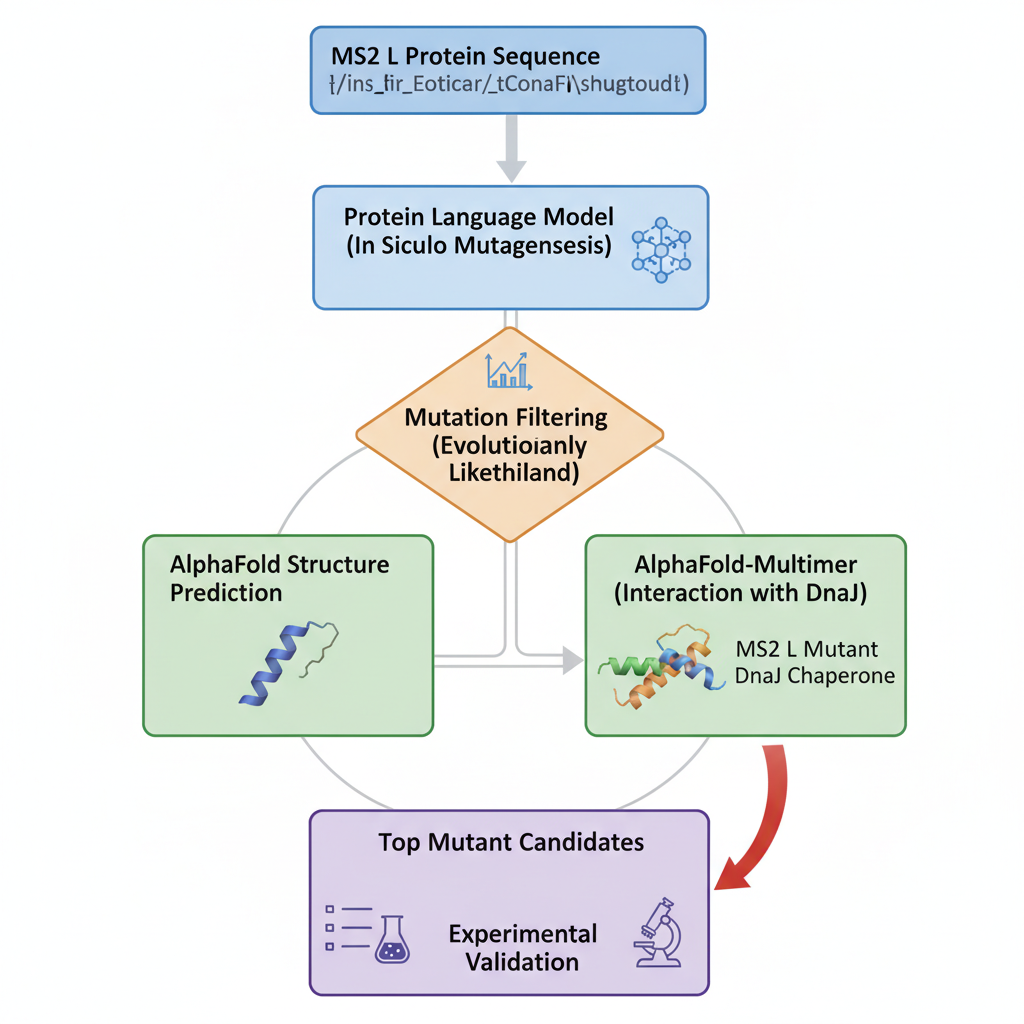
Part D. Group Brainstorm on Bacteriophage Engineering
-Choose one or two main goals from the list that you think you can address computationally.
Increase the stability of L protein
Increase the lytic activity
-Write a 1-page proposal (bullet points or short paragraphs) describing:
The L protein from bacteriophage MS2, is responsible for the lysis of infected cells in E,coli. Studies suggest that this protein interacts with host proteins for this desirable effect but small mutations can strongly affect its function. L protein being small and sensitive to mutations, through computational tools we can study these possible changes and their effects on usual interactions.
-Which tools/approaches from recitation you propose using.
We can use PLMs to perform mutagenesis of the protein, generating single aas substitutions across the protein, and score these mutations. Alphafold gives us a more visual form to look into these new sequences and the feasibility of them existing and being stable. At the same time, we can look into host proteins interactions to make it possible to try and have more insight into the way this protein interacts to be effective.
-Why do you think those tools might help solve your chosen sub-problem?
Protein language models like,smn2, lets us capture patterns in protein sequences, making it possible to identify mutations that are structurally feasible.
Alphafold, lets us predict structures based on sequence, letting us evaluate how mutations might destabilize the protein folds and let us predict how it interacts with other protein complexes.
-Name one or two potential pitfalls.
We have limited understanding of the exact mechanism of MS2L protein causing the lysis of bacteria. This makes the mutations a bit random as we dont know if they are gonna be beneficial for stability but decrease its lytic effect.
Week 5 HW: Protein Design Part 2
SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
-Design short peptides that bind mutant SOD1.
-Then decide which ones are worth advancing toward therapy.
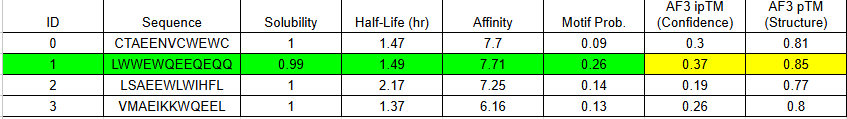
**Part 1: Generate Binders with PepMLM**
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
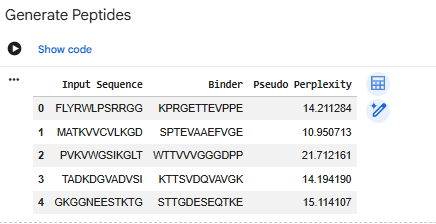
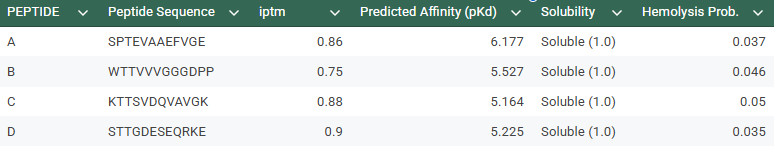
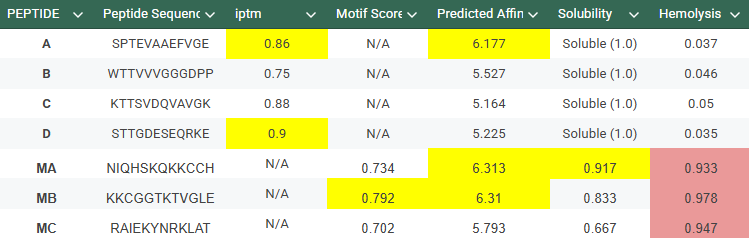
-Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
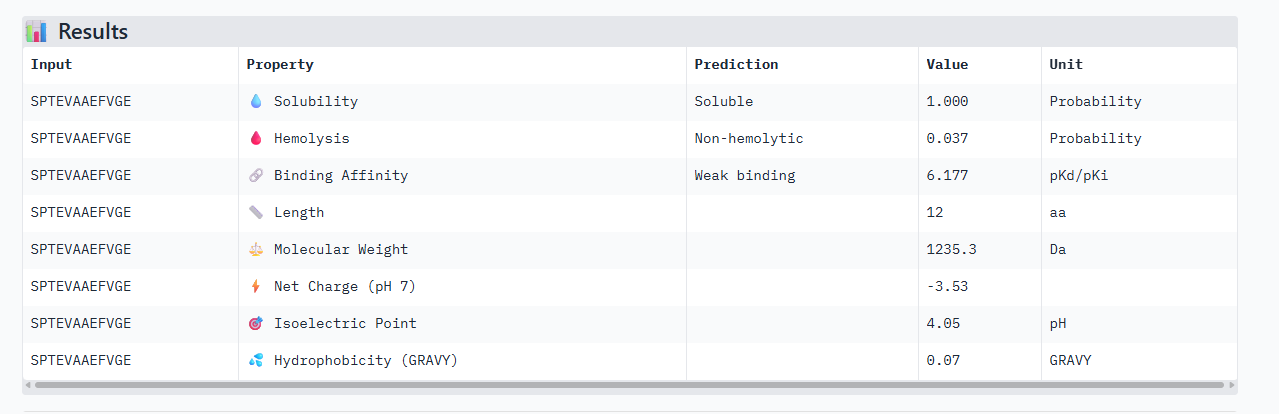
SPTEVAAEFVGE
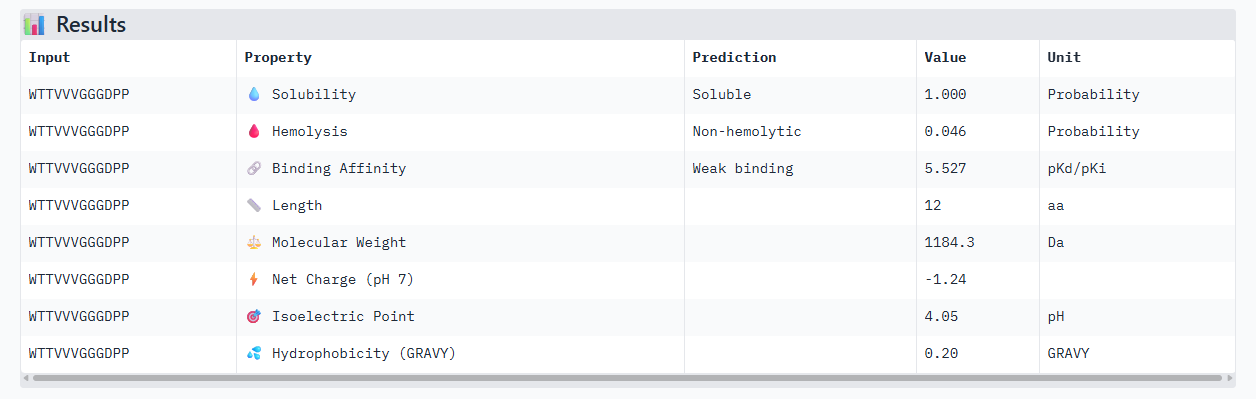
WTTVVVGGGDPP
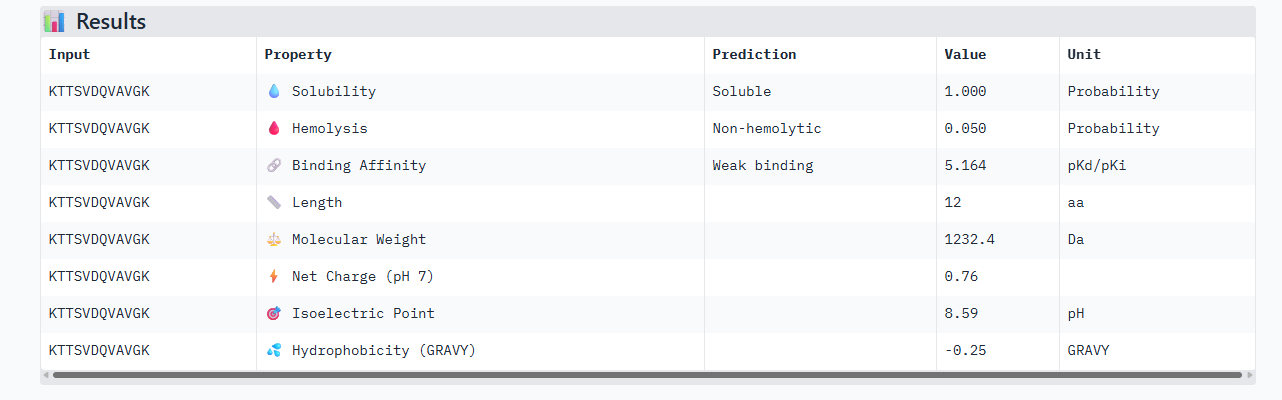
KTTSVDQVAVGK
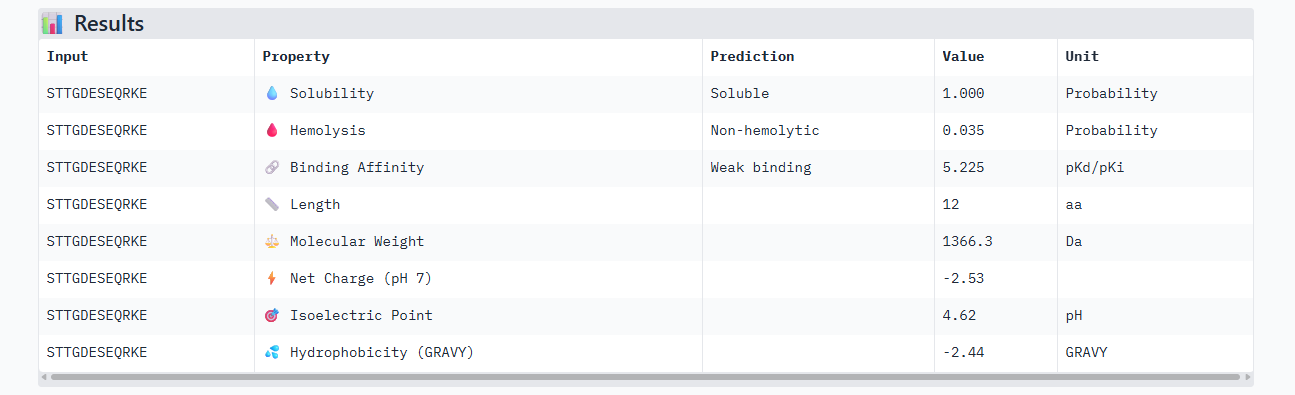
STTGDESEQRKE
CONTROL: FLYRWLPSRRGG
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
**Part 2: Evaluate Binders with AlphaFold3**
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind.
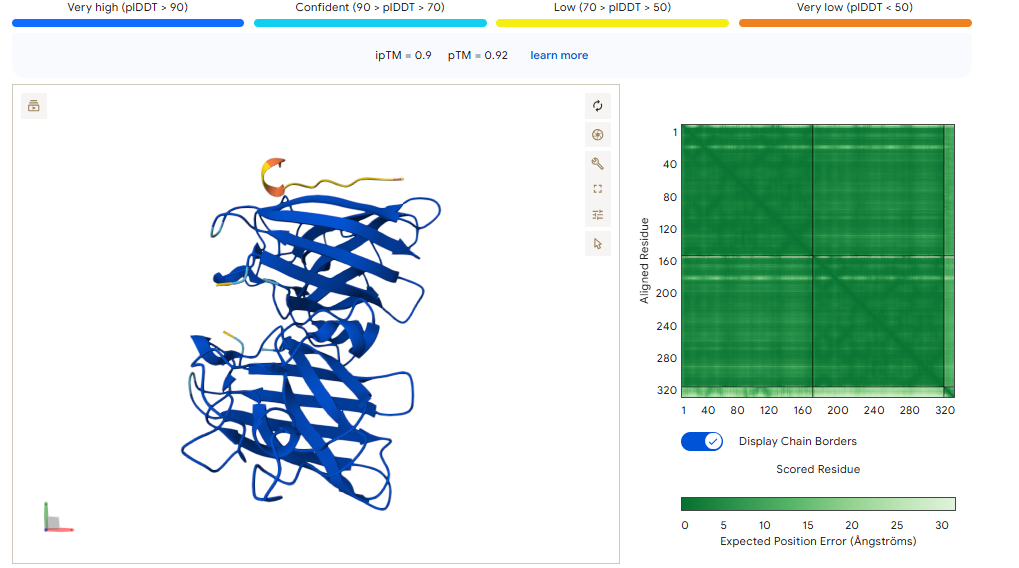
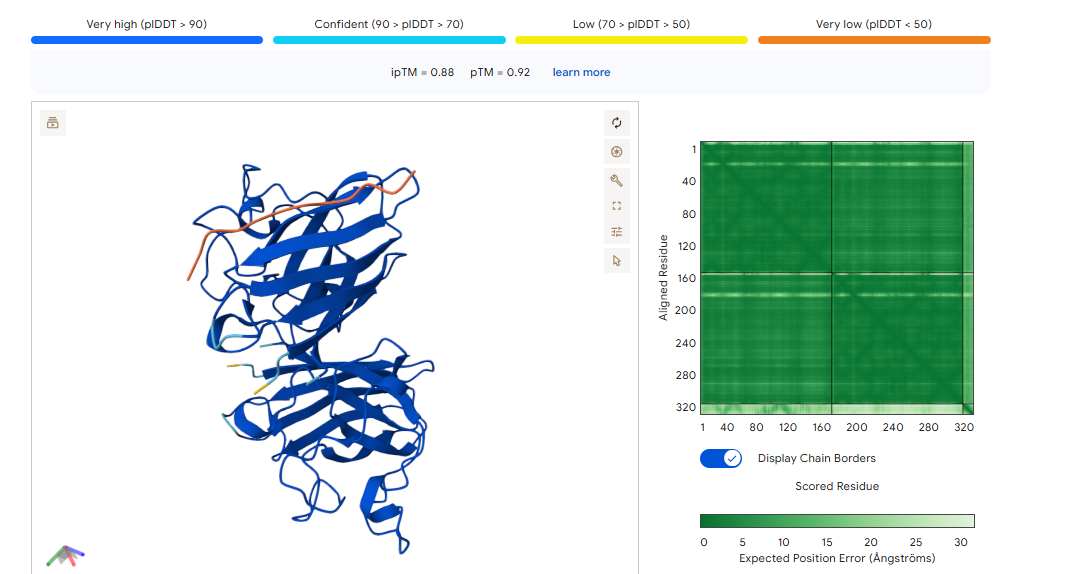
CONTROL PEPTIDE
The control shows a ipTM of 0.90, and it can be seen engaging to the surface of the b-barrel.
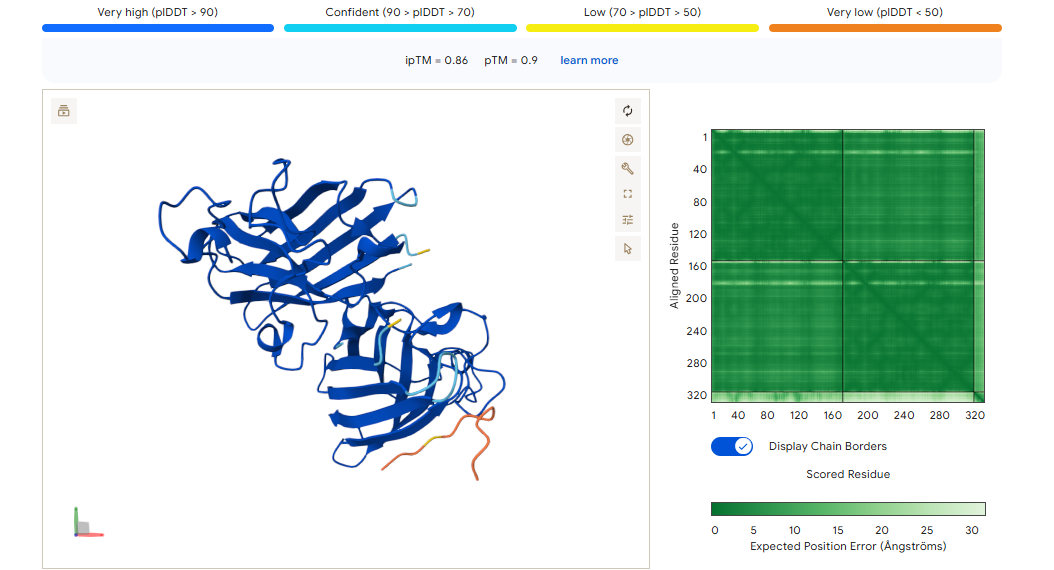
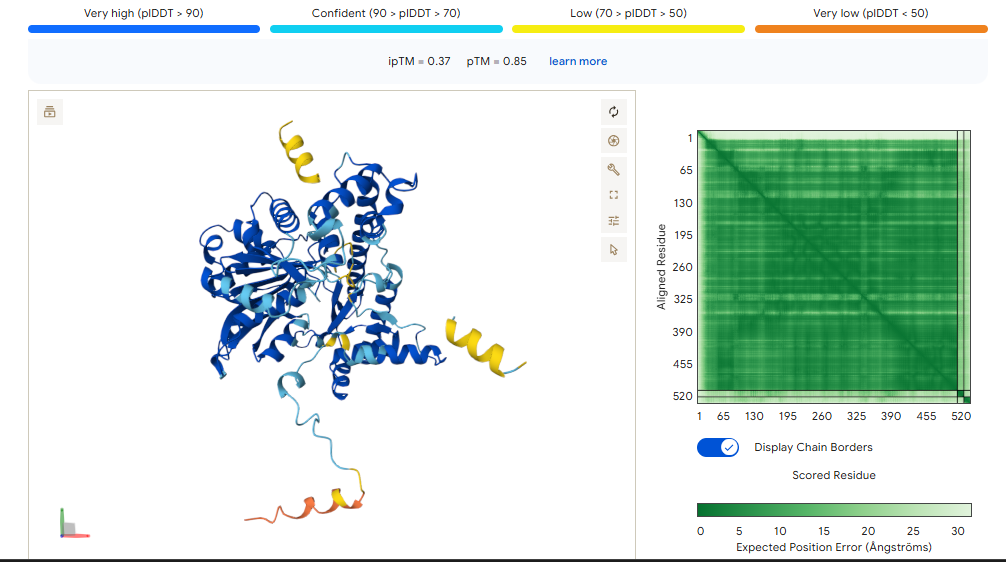
A PEPTIDE
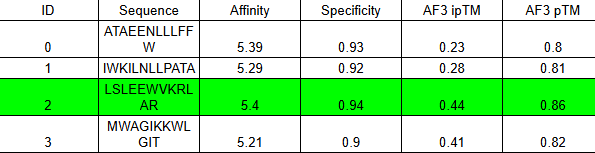
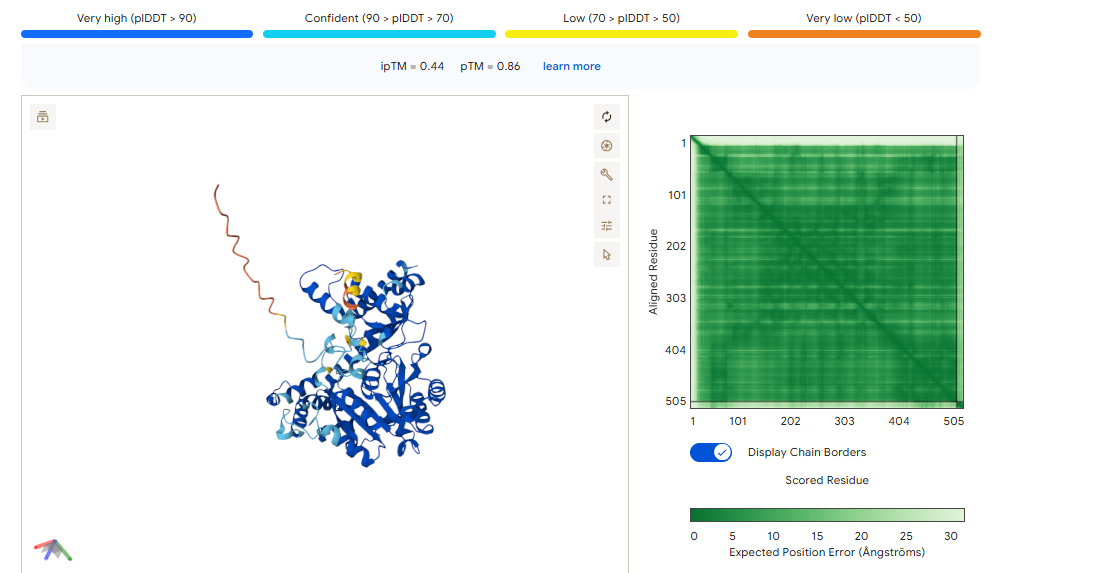
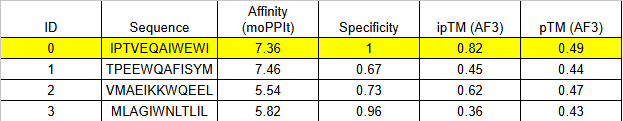
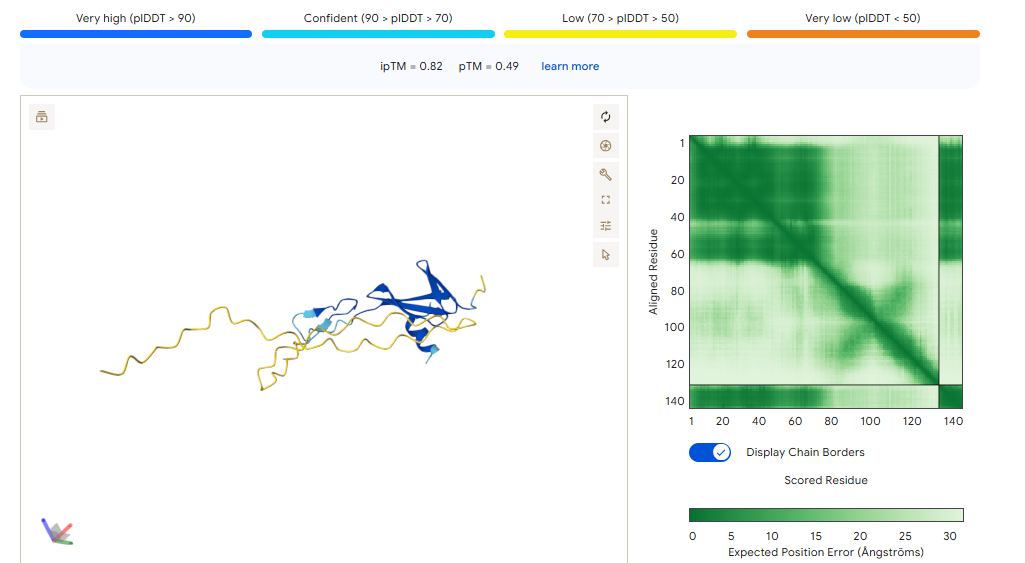
ipTM of 0.86 It shows a high confidence and specialized binding mode. It is near the n terminus where the a4v mutations is, looking a bit buried into where the mutant valine is.
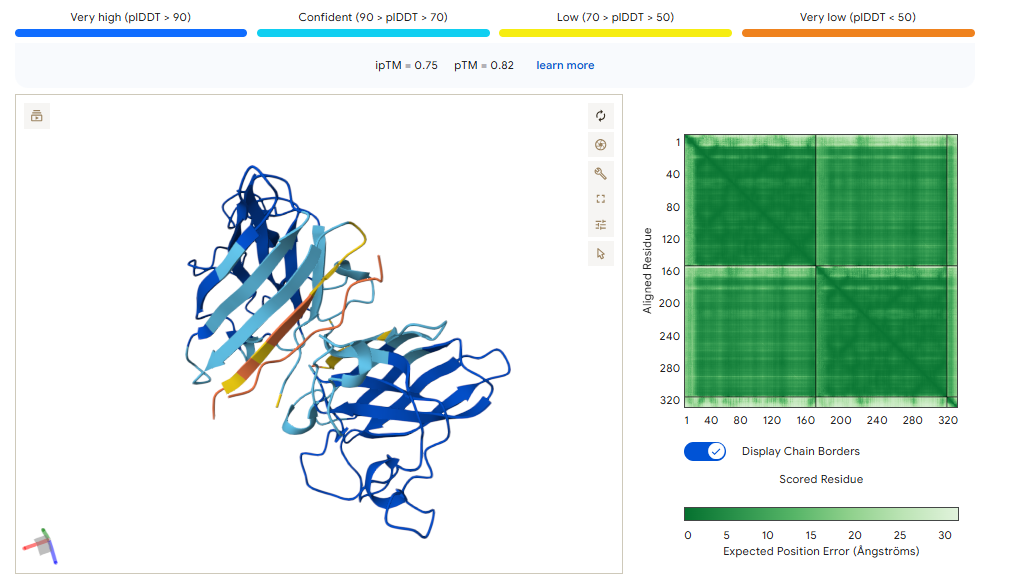
B PEPTIDE
ipTM of 0.75This is the lowest score of the group. It shows flexible groups indicated by the yellow/orange colors and has more of a loop region rather than the b-barrel. Seems mostly surface bounded.
C PEPTIDE
ipTM of 0.88, and looks like it approaches the dimer interface. In the PAE, there are cross chain signals, which means the peptide is acting as a bridge between the SOD1 monomers, where it is partially buried.
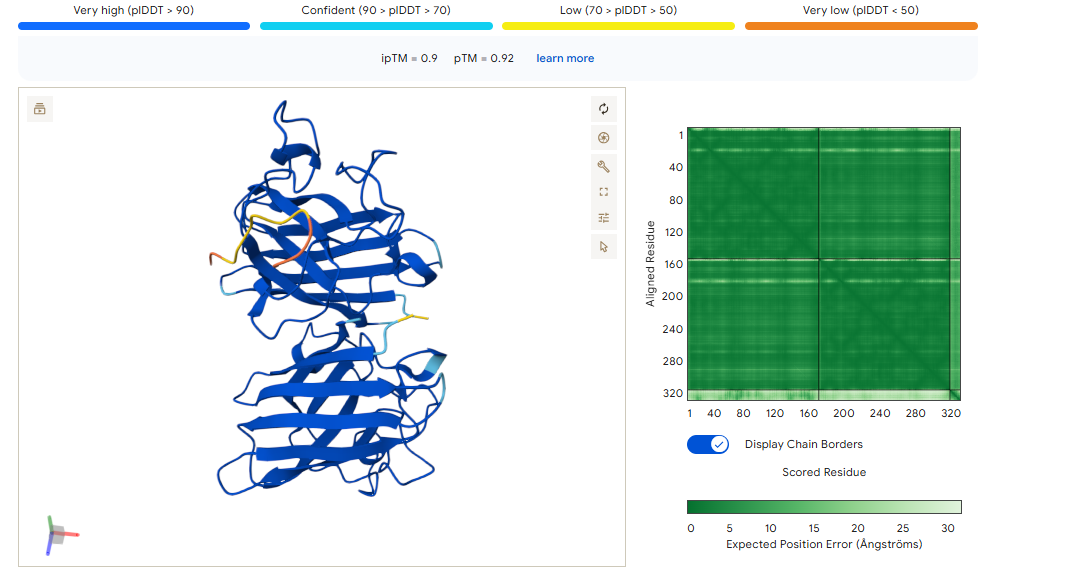
D PEPTIDE
ipTM of 0.90, this score makes this peptide stand out as it has the same score as the known binder. It gives a full capture to the target area, interacting with the b-barrel and dimer interface.
**Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse**
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide!
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see.
Do peptides with higher ipTM also show stronger predicted affinity?
Are any strong binders predicted to be hemolytic or poorly soluble?
Which peptide best balances predicted binding and therapeutic properties?
A PEPTIDE
In the Alphafold3, it shows a high confidence and the structure suggests a tight fit to the SOD1 pocket. In the Peptiverse analysis, it confirms A, as the strongest binder with an affinity of 6.177,
B PEPTIDE
Alphafold3, gives us a confidence of 0.75, maybe because the triple valine in the sequence given arises is pretty flexible and harder to place. In the peptiverse, even though we have this bit of uncertainty in the structure, it shows a decent affinity.
C PEPTIDE
Has a high confidence of 0.88 in Alphafold3, given a structure that interacts between the dimer interface. In the peptiverse, it’s a bit shocking to see it as the peptide with the lowest affinity of 5.164. This score lets us know that even if the peptide is structurally sound, it is chemically weak.
D PEPTIDE
This peptide has the highest score in Alphafold3, having the same 0.90 ipTM as our control. In the peptiverse, it does not show the best affinity with 5.225, however it has the lowest hemolysis score. It could be the most certain design but could lack the chemically to be a potent drug.
Choose one peptide you would advance and justify your decision briefly.
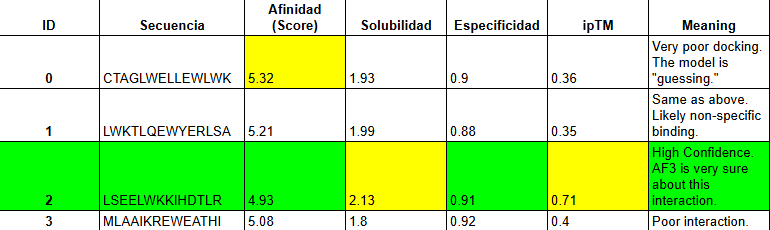
Based on the overall analysis the best choice would be PEPTIDE A. Even if it doesn’t have the best ipTM score compared to the other peptides, it still holds a high fidelity. In the peptiverse, this high possibility of interaction is confirmed with a good affinity score, and also it being safe with having a low score in the hemolysis probability.
**Part 4: Generate Optimized Peptides with moPPIt**
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available).
Generate peptides.
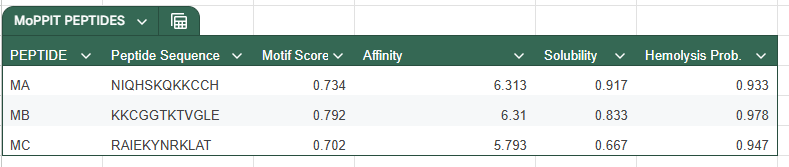
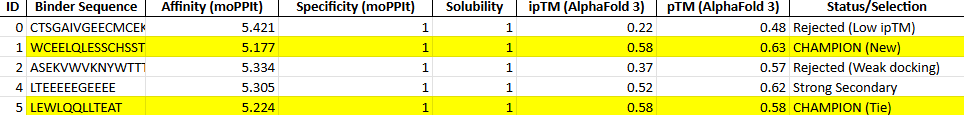
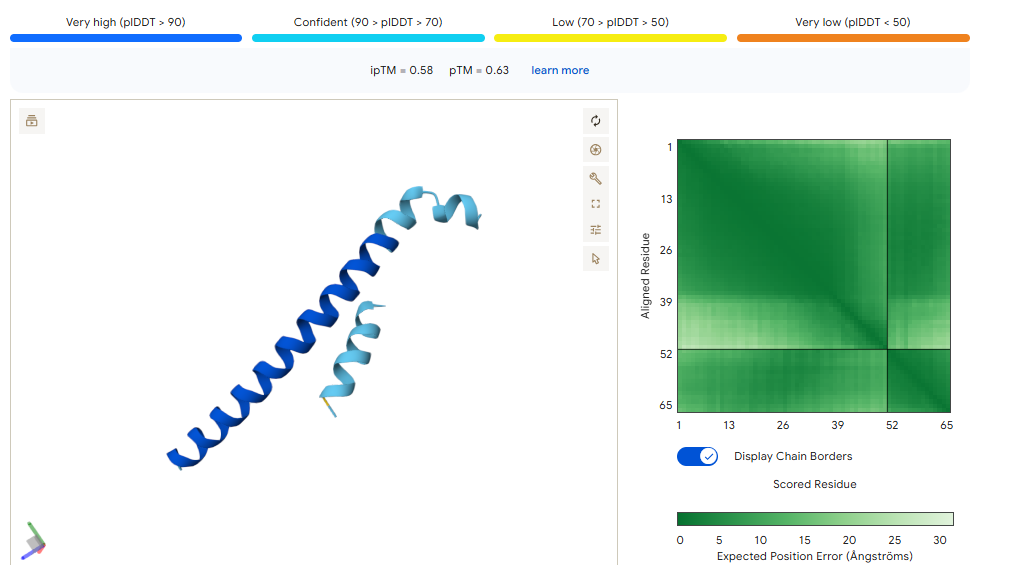
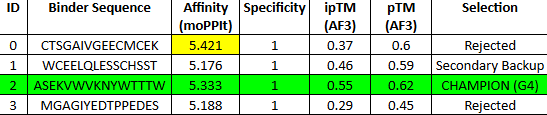
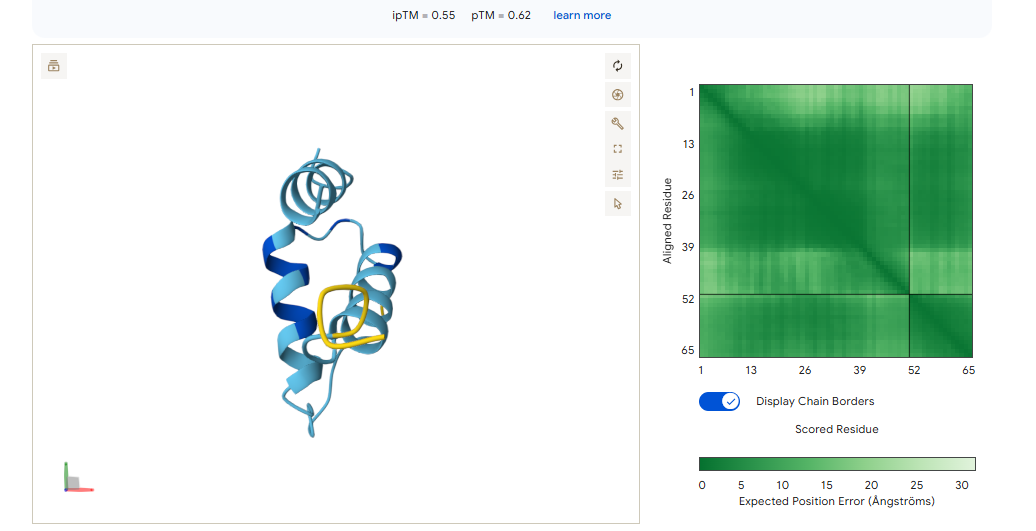
With this model we successfully pushed the predictability affinity higher, specially wiTH designs MA and MB; but all three have a toxicity problem because of their high hemolysis probability. Overall based on the scores, the best peptide would be MA.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
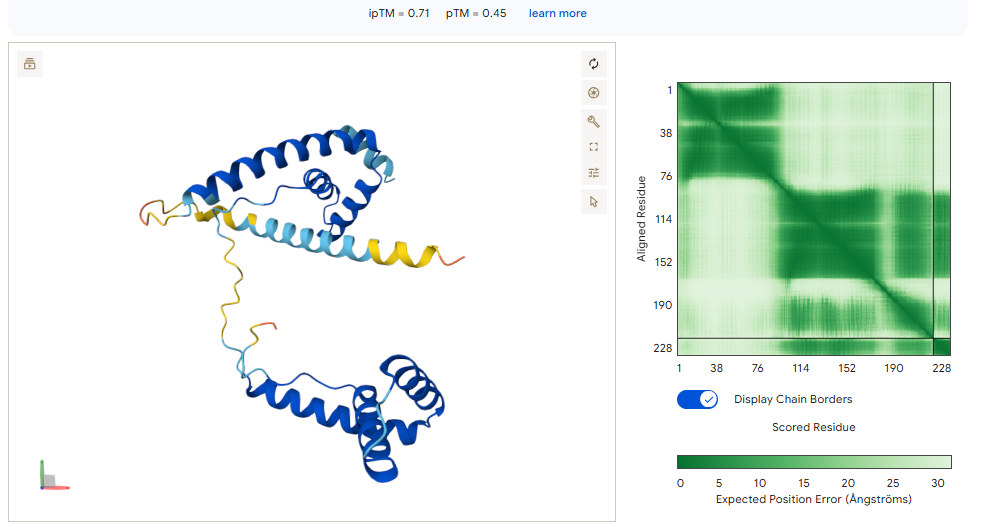
When using MoPPit, to create new peptide designs, we enable motif and affinity guidance. This guidance can be seen in the de novo peptides made by the model, as all 3 of the structures have a high affinity and motif score compared to the PEPMLM peptides,which sequencing was taken from random sites of the mutated SOD1. While these new peptides are theoretically better at interacting with our target, if we were to advance with any of them for a clinical study, they would be toxic from their predicted hemolysis score. If we had to choose the safest peptide between all the ones created, peptides from PEPMLM are better. Peptide A from PEPMLM because it maintains a very respectable affinity score (6.18) and high structural confidence (0.86) while remaining non-toxic and fully soluble.
Details
BOLTZ
Has not been done as troubles have been present at creating the Boltz account.
Answer these questions about the protocol in this week’s lab:
-What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix with HF Buffer is a 2X master mix consisting of Phusion DNA Polymerase, deoxynucleotides and reaction buffer that has been optimized and includes MgCl2. All that is required is the addition of template, primers and water. (New England Biolabs, 2026)
Phusion DNA Polymerase: a highly accurate enzyme that synthesizes new DNA strands with very low error rates due to proofreading activity.
dNTPs (deoxynucleotide triphosphates):the building blocks (A, T, G, C) used to construct new DNA strands.
Reaction Buffer: maintains optimal pH and salt conditions for enzyme activity.
MgCl₂ (magnesium ions): a critical cofactor required for polymerase function.
Stabilizers/additives: enhance enzyme stability and performance during thermal cycling.
-What are some factors that determine primer annealing temperature during PCR?
The primary annealing temperature depends on:
Primer Melting temperature (usual annealing is 3-5°c below Tm).
Primer Length (with increasing lenght the Tm also increases).
GC content (GC increases Tm due to strong H bonding).
Sequence composition (repetitive or 2nd structure forming sequences affect binding).
Salt/ion concentration (influences the dna duplex stability).
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR
This method uses primers and DNA polymerase to amplify a specific DNA region. It requires thermal cycling and can introduce custom sequences like overlaps or mutations. It produces DNA fragments with defined ends depending on primer design.
RESTRICTION DIGEST
Uses restriction enzymes to cut DNA in specific recognition sites. It is done at a constant temperature and producecs predictable sticky/blunt ends. This methods is limited, as it is required existing restriction sites in the DNA.
PCR vs. RESTRICTION ENZYME DIGEST
When comparing both methods, we can see each one has their specific requirements for protocols and they are tailored for different needs, even if both create linear fragments of DNA. PCR, is used when u need to amplify DNA and there is no restriction sites. Use restriction enzyme digest, when suitable restriction sites exist in the sequence and u want a precise ends. Overall, PCR is a method more flexible while restriciton digestions is simpler but less customizable.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure compatability with Gibson Assembly, we have to sedign fragments with 20-40bp overlapping homologous sequences. We should ensure correct orientation and reading frame, avoid secondary structure or repetitive overlaps, and ensure no unwanted mutations.
How does the plasmid DNA enter the E. coli cells during transformation?
This can be done in 2 ways. Chemical transformation (Heatshock), treats cells with CaCl2 to make them competens and heat shocks them, creating a thermal imbalance that allows the DNA (plasmid) to enter. Electroporation, gives brief pulses to cells to create pores in the mebrane for the DNA to enter. In both cases, the membrane is temporarily permeabilized for the upatake.
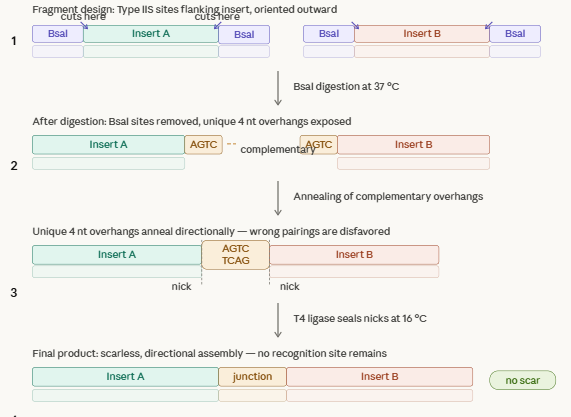
Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly, uses restriction enzymes that cut outside their recognition sites, and joins fragments with DNA ligase. This method generates custom overhangs and allows us to create multiple fragments in one reaction.
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online)
This method harnesses restriction enzymes to cut DNA at a defined distance outside their recognition site generating custom 4n overhangs. Each fragment is flanked by typeiis sites oriented so that after digestion, every overhang is exposed and matching its intended neighbor. DNA ligase joins the overhangs thru cycling temperatures. Overhangs are fully defined and at to 50 fragments can be assembled in a reaction, making this method usefult to create gene libraries.
Model this assembly method with Benchling or Asimov Kernel!
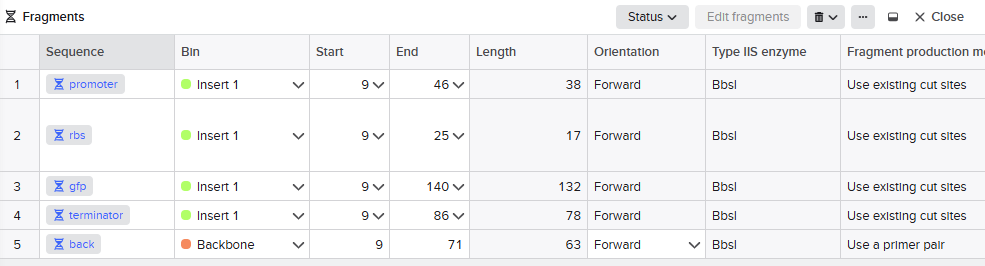
We designed the overhangs and added then to the original dna sequences of the parts we used. This makes possible the annealing and ligation of the final sequence.
Promoter → RBS → GFP → Terminator → Backbone
Assignment Asimov Kernel
[Asimov Kernel]
As a CL we could not be able to get acess to the platform. For this, i wasnt able to do this part of this week’s homework.
Week 7 HW: Genetic Circuits Part 2: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
While Booleann circuits produce on/off outputs, IANNs generetae continuous and graded reposnes allowing us to detect subtle changes in input signals. They also can intigrate multiple inputs that are adjustable, making them more flexible and better to mimic natural cellular desicion processes.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
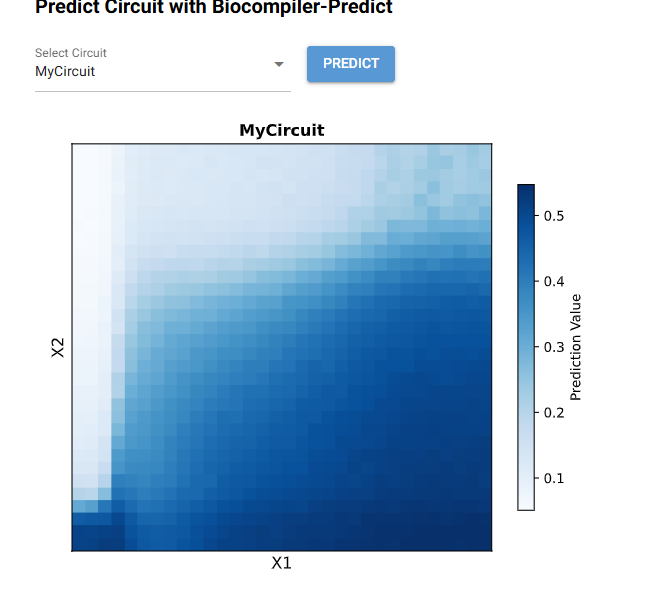
A useful application of IANNs could be create biosensors or networks to understand toxicity and test shelved or new drugs. One idea could be to create one to sense for early liver damage, where the input such as stress-responsive promoter or specific proteins drive the regulation of X1, which regulates the flourescent reporter X2. IANNs give a graded signal and we can distinguish the level of damage enabling early detection than a binary system. For limitations, there are the limited dynamic range, potential crosstalk between components and the delays from transcription and translation.
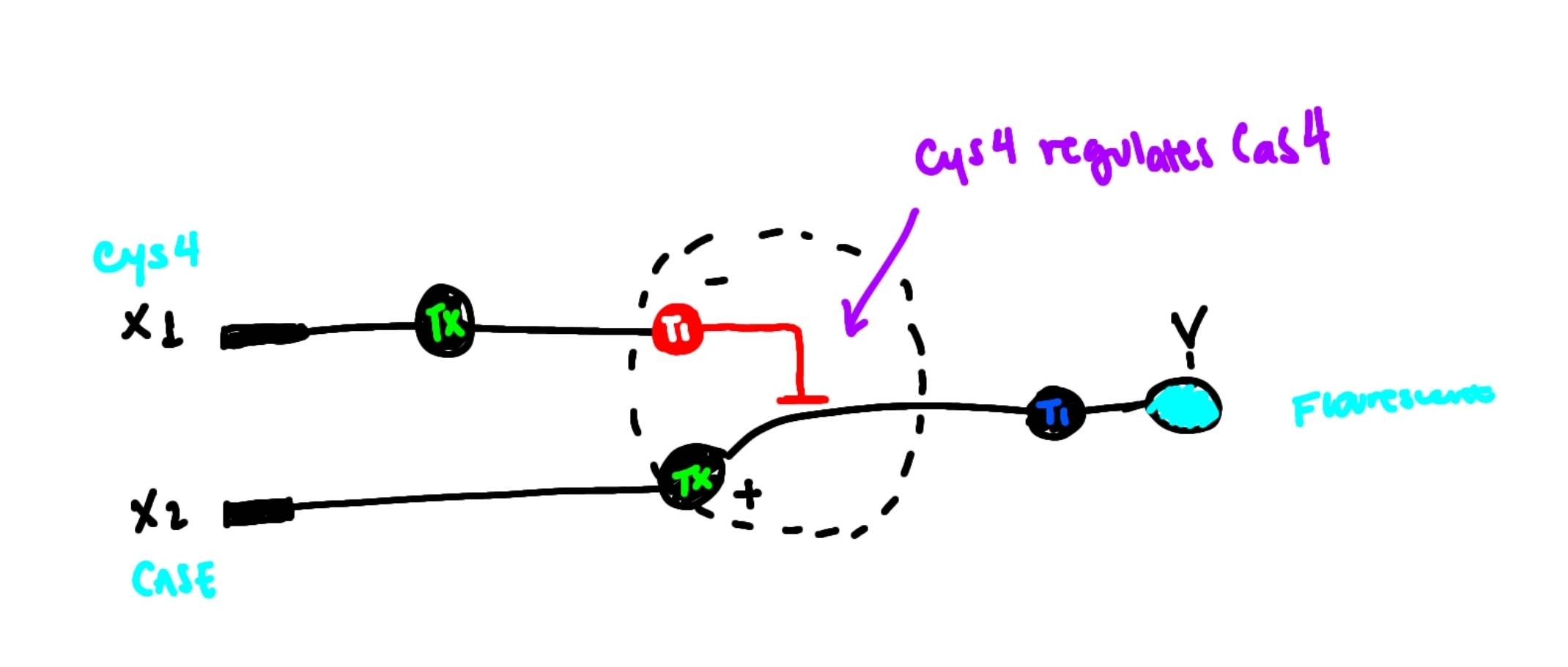
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
I draw the a perceptron where an input DNA X1 produces Cys4 with regulates CasE in a hidden layer. Cas E controls the expression of the flourescent protein output.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Some examples are the use of mycelium as leather, for ways of sustainable packaging, and its use for building materials like insulattion panels and bricks. Their advantages are sustainable and biodegradable, have a low energy production and they are lightweight and customizable. Some disadvantages are they arent strong as plastics or metals, they are sensitive to moisture and limited structural strength.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would like to genetically engineer fungi to produce advance biomaterials and valuable matabolites to produce pharmaceuticals. Fungi already have complex 3d structures and enzymes that can be used for other uses. Some advantages when compared to bacteria are that they can perform complex protein folding and post translational modifications, can form strucutred materias and have a robust metabolism.
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
CFPS offers several key advantagesin felxibility and experimental control over traditional in vivo protein expression. In terms on flexibility, unlike living cells, we have an open reaction enevironment as we are not constrained by structures or the viability of the cell. We can add and remore components at any time. As we dont depend on the cell, there is no need for cloning or transformation, allowing the qucik testing of multiple gene constructs at the same time. We can express proteins that coudl be toxic or unstable in livign things because nothing is alive. Comparing the benefits for control of the experimental values, CFPS systems are superior as we cna tightly control conditions, directly manipulate the gene expression and define the environment for production.
Some situations where CFPS is more beneficial than the cell based production:
studying and needing to produce toxic proteins that could harm the host.
incorporating non standard aminoacids, as there are no metabolic constrains.
Describe the main components of a cell-free expression system and explain the role of each component.
Components:
DNA/mRNA template: is the manual for the encoding of the target protein. DNA is trancribed to mRNA and then translated into the protein.
Energy supply (ATP/GTP): we need energy for the transcription, translation and protein folding steps.
Aminoacids: they are the building blocks of the protein.
Nucleotides: they are required for the trancription of DNA.
Cofactors + salts: mantains enzyme activity and ribosome stability.
Cell extract: the needed molecular machinery (ribosomes/tRNAs/aminoacyl-tRNA synthase/initiation-elongation factors) for transcription and translation.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
The process of protein sysnthesis is energetic demanding because of the need of 4ATP molecules for peptide bond, without any regeneration ATP is finished quickly and by products accumulate and inhibit reactions.
A method we could use to ensure the continuous ATP supply could be a PEP system, as it acts as a high energy phosphate donor to regenerate the ATP. It is a pretty simple method and has a 1:1 rate.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic CFPSs are fast and low cost but are pretty limited when it comes to protein folding and post translational modifications. Eukaryotics are slower and high cost but have a better result for the protein folding and have a variability of available ways for post translational modifications.
So if I had to choose proteins for each to synthetize according to their characteristics; I would produce GFP in a prokaryote system as it is a simple protein that doesnt need a high level folding or any modifications. For the eukaryote system, a good protein to produce could be any fragment of an antibody, as it makes it possible to do proper folding and post transcriptional mdofications.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To design a membrane protein expression some of the challanges that we could see if the is the aggregation of the proteins, the misfolding of them outside the membrane environment and the chance of them having a low solubility. We could adress some of this challanges starting by optimizing the conditions for the protein sysnthesis so there are no holdbacks on the sysnthesis. For the problems with folding we could assist the process with the add on of chaperones for assitance in insertion and folding. For the solubitlity and overall it troubleshooting as it not being in the membrane environment, we could try to recreate this ecosystem by addign membrane mimetics and give a lipidic enviroment for the translation.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Protein Aggregation: we could solve it by adding chaperonse and solubility tags to the system.
Energy Depletion: we could change to a sustainable energy system so there’s no chance for having an insufficient amount of ATP.
Transcription Problems: if we have a weak promoter or degraded dna we have problems with the trancription step. We can fix it by changing the promoter or changing the dna/rna template if it’s damaged.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
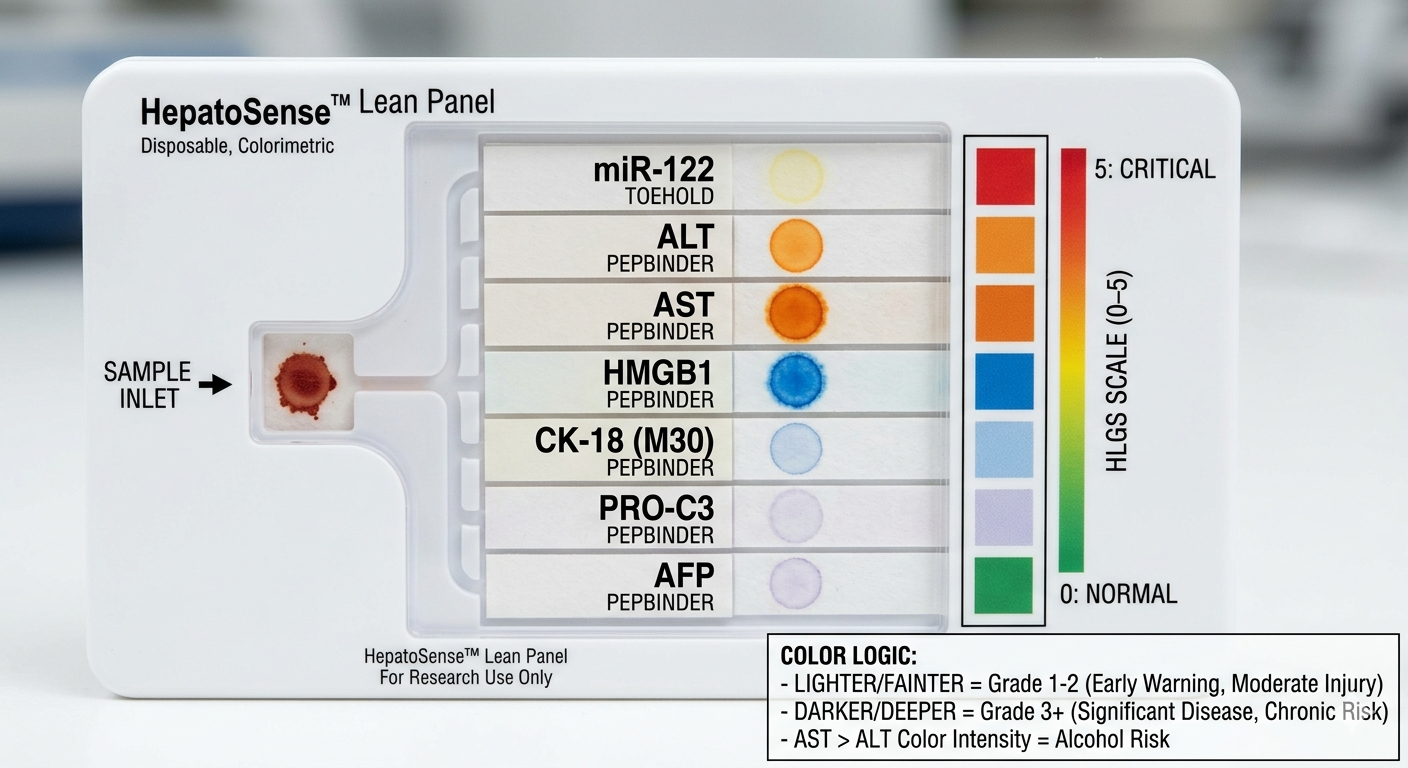
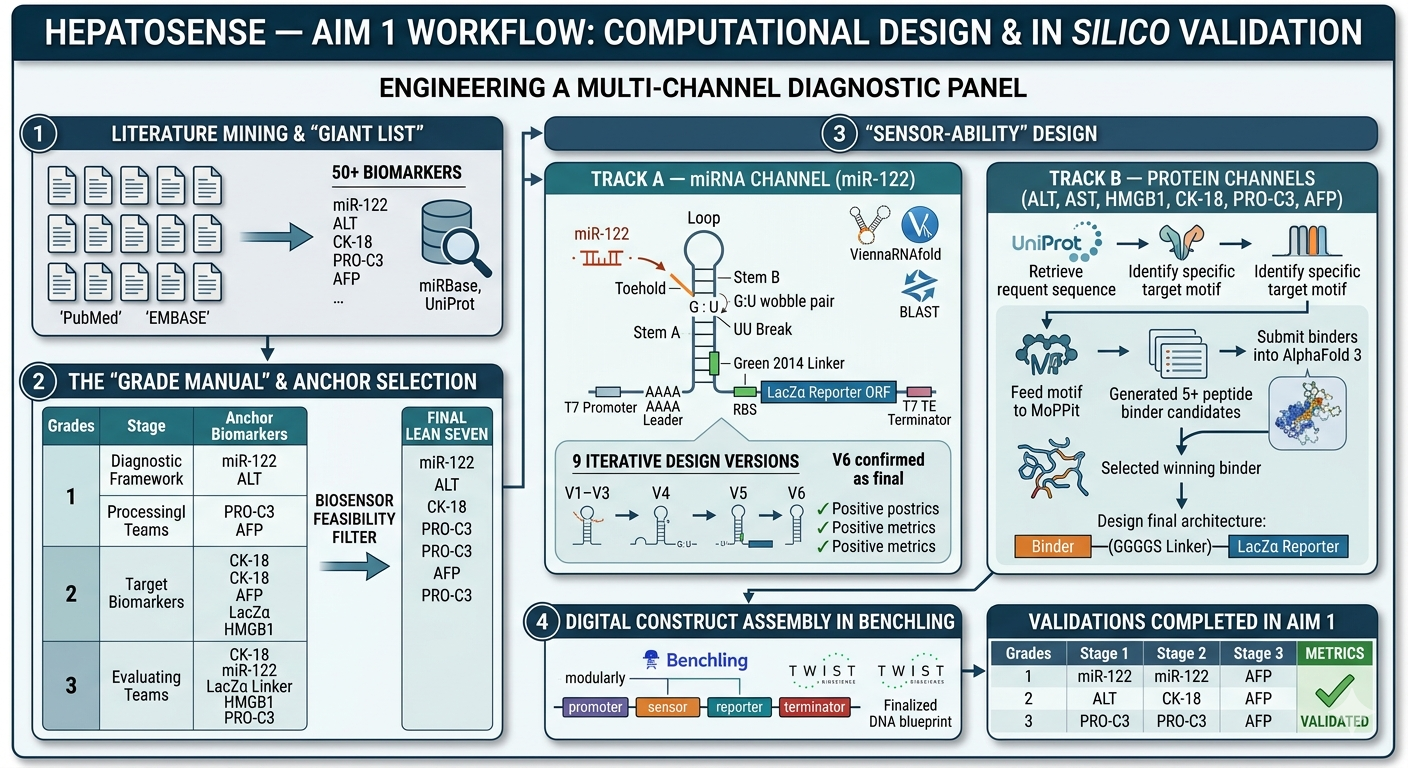
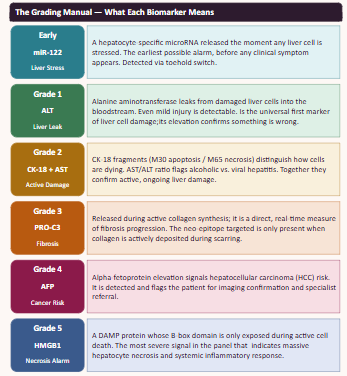
A synthetic minimal cell with the fucntion of detecting and staging liver damge by sensing a panel of biomarkers representing early injury, hepatocyte damage and cell death.
What would your synthetic cell do? What is the input and what is the output?
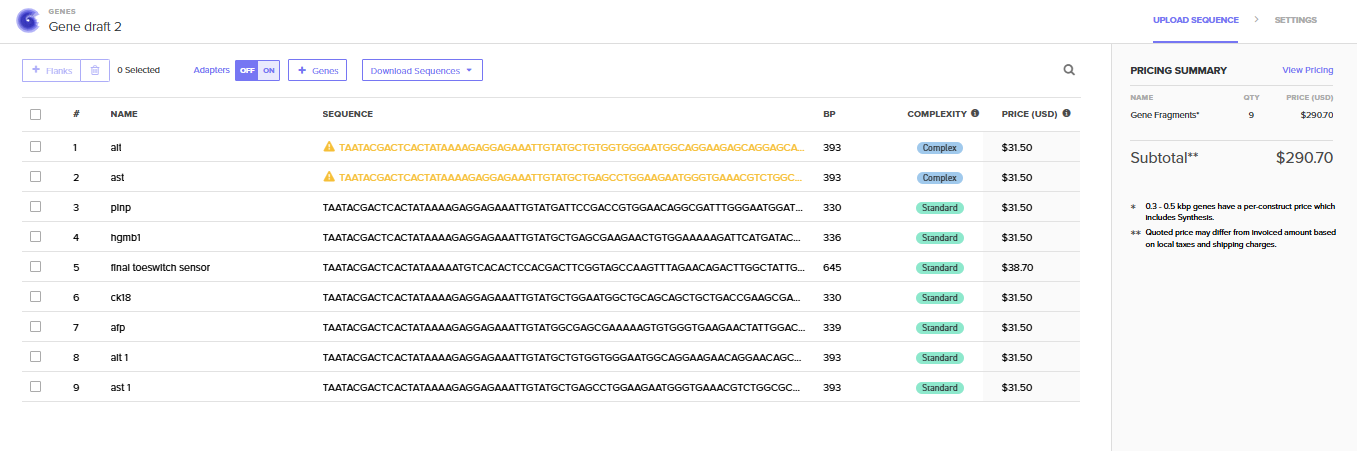
the cell would detact the input of serum/blood and detect the biomarkers (miR-122, ALT, AST, GLDH, CK18, OPN, ammonia, lactate) and give an output of flourescence patterns according the stage of damage.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
In theory most of it can function without it but the encapsulation allows the modular and independent sensing of each biomarker.
Could this function be realized by genetically modified natural cell?
Yes, a modififed cell could perform similar sensing,but a synthethic cell is safer and more controllable avoiding any livign cell complications as we are looking at some biomarkers that could damage the cell.
Describe the desired outcome of your synthetic cell operation.
The desire outcome is a stage diagnostic output so we coudl distinguish early liver injury from severe liver failure without the need of invasive procedures.
Design all components that would need to be part of your synthetic cell.
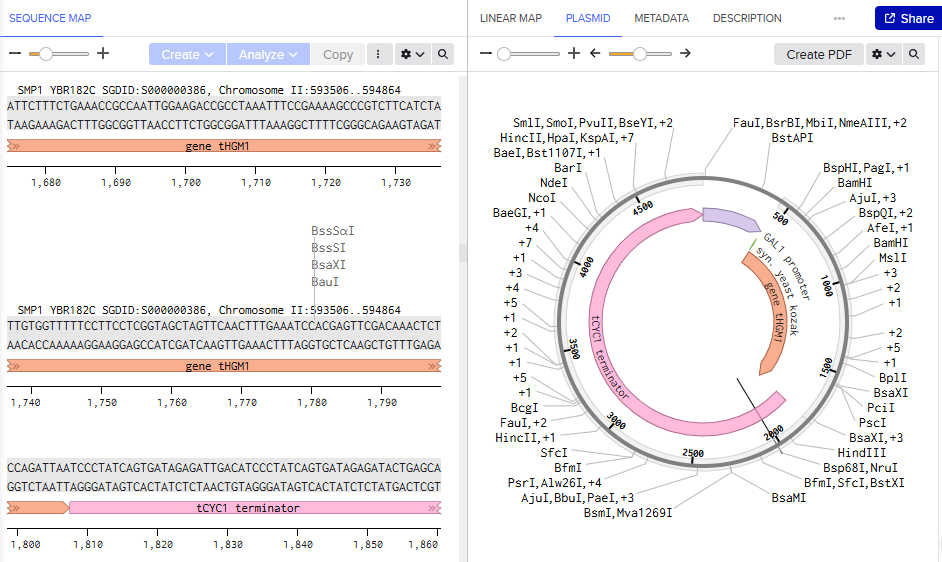
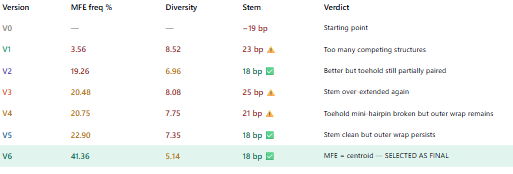
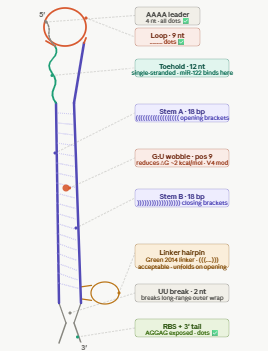
Biomarker mir122: Promoter 17 + (creation of toeswitch seq complementary to miR-122) + RBS + GFP (reporter) + Terminator T7.
The biomarker binds to the toeswitch and open rna hairpin allowing the reporting signal to come thru.
Biomarker gldh / ck18 : this protein needs to be detected via aptamer/antibody giving a dna tirgger to activate the signal.
What would be the membrane made of?
The membrane could be made of phospholipid vesicles with cholesterol and the addition of membrane pores for protein entry to facilitate the uptake of certain biomarkers.
What would you encapsulate inside? Enzymes, small molecules.
Inside the membrane the encapsulated contents would be the cell free trancription/translation system and the dna constructs for each biomarkers.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
We could use bacterial (e.coli) system as it is sufficient for the biosensor. Mammalian system are not needed as we dont need to modulated promotors, just need the genetic circuits and the protein reactions with the defined biomarkers.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The synthetic cell communication with the enviroment comes from the free diffusion of the small molecules part of the biomarkers and the big proteins needed to be sense envir via channels in the membrane or can be converted to dna triggers.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine) – forms the main phospholipid bilayer
Cholesterol – stabilizes the membrane and controls fluidity
OmpF (gene: ompF, UniProt P02931) – membrane protein channel to allow protein or metabolite entry
The function of the system is gonna be measured by flousrescence from each reporter. The combination of the colors are gonna be used to understand which stage we are looking at.
Bibliography
Church, R. J., Kullak-Ublick, G. A., Aubrecht, J., Bonkovsky, H. L., Chalasani, N., Fontana, R. J., Goepfert, J. C., Hackman, F., King, N. M. P., Kirby, S., Kirby, P., Marcinak, J., Ormarsdottir, S., Schomaker, S. J., Schuppe-Koistinen, I., Wolenski, F., Arber, N., Merz, M., Sauer, J. M., … Watkins, P. B. (2019). Candidate biomarkers for the diagnosis and prognosis of drug-induced liver injury: An international collaborative effort. Hepatology, 69(2), 760–773. https://doi.org/10.1002/hep.29802
He, F., Wang, Q., Li, J., & Ma, X. (2023). The application of aptamer in biomarker discovery. Biomarker Research, 11(1), Article 26. https://doi.org/10.1186/s40364-023-00510-8
Kandemir, H., Cinar, Y., & Ozturk, M. (2024). Serum microRNA-122 for assessment of acute liver injury in patients with extensive skeletal muscle damage. Laboratory Medicine, 55(5), 585–591. https://doi.org/10.1093/labmed/lmae022
Vliegenthart, A. D., Berends, J. E., Mashimo, T., Wouters, E. P. A., Verheij, J., & Stoopen, G. M. (2018). A longitudinal assessment of miR-122 and GLDH as biomarkers of drug-induced liver injury in the rat. Biomarkers, 23(4), 303–312. https://doi.org/10.1186/s40364-023-00510-8
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
An intelligent bio reactive textile that uses lyophilized cell free sensors to monitor real time thermal stress and autonomously tiggers cooling via a enzyme resposonse.
How will the idea work, in more detail? Write 3-4 sentences or more.
The system would be done with a layered bio circuit consisting on a biosensor embedded in textile. One part is the rna thermomether that expresses a color changing protein to provide the visual map. While the second is an enzymatic trigger that produces a protease enzyme when the threshold temperature is reach, digests microcapsules and releases a cooling agent, making the temperature drop and cooling the person.
What societal challenge or market need will this address?
There are currently cooling vests but there are no direct cooling systems textiles that sense when they are needed. The idea was born with the f1 in mind but can be stablisihed in any environment where high stress and physical exhaustion from heat is present. The system provides a failsafe for the body to mantain its cooling in extreme conditions.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
For the need to its activation with water, divert to the use of the sweat as a biological switch on. The lyophilized system can be engineered to turn on when only the correct salt/pH is given from the human perspiration.
One-time used limits a lot of cfs ideas but for this project we could create replacible biopatches. Instead of throwing away a whole uniform, patches could be taken out and replaced when needed lowering the waste.
Smith, J. A., & Garcia, M. L. (2025). Cell-free systems for development of biosensors. In Progress in Molecular Biology and Translational Science (Vol. 212, pp. 45–78). Academic Press. https://doi.org/10.1016/bs.pmbts.2025.09.003
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long duration spaceflight is associated with hemolytic anemia caused by the increased red blood cell destruction. Factors like microgravity membrane changes, fluid shifts and oxidative stress form space radiation contribute to this anomaly. Understanding this mechanisms are essential to astronauts health as it presents itself during extended mission and continues for a long period after being back on Earth. Cell fre systems provide a simplified platform to investigate protein stability under these oxidative conditions can model blood cell damage helping us develop protectice strategies.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
GFP as a reporter for protein stability in bacteria cell free systems. Optionally red human blood cells proteins for a conceptual mammalian system.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Oxidative stress contirbutes to the red blood cell damage in astronauts, and with the BioBits system, gfp flourescence reflects protein integrety under stress modelling this conditions and its affects in RBCS. Conceptually a mammalian system expressing hemoglobin or cytoskeletal proteins could be a more direct human model to rbc vulnerability.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
This project hypothesis is that oxidative stress conditions reduce portein stability and function decreasing the flourescence in the cell free system. With the biobits experiment, gfp reports the model protein damage, demostrating the effects of the condition can impair the protein integrity. These changes would indicate protein misfolding or degradation, analogous to damage occurring in astronaut red blood cells. Ultimately, this work provides insight into mechanisms underlying hemolytic anemia and demonstrates how biotechnology can support astronaut health on long-duration missions.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
e experiment will use freeze-dried BioBits cell-free reactions expressing GFP. Samples will include: (1) control reactions under normal conditions, and (2) reactions exposed to oxidative stress (e.g., hydrogen peroxide). Fluorescence intensity will be measured using the P51 Molecular Fluorescence Viewer to assess protein stability. Conceptually, a mammalian cell-free system expressing human RBC proteins (hemoglobin or spectrin) could be similarly tested under oxidative stress or simulated microgravity, with fluorescence monitoring protein integrity. Comparing stressed and control samples allows evaluation of stress-induced protein damage, modeling mechanisms contributing to hemolytic anemia in astronauts.
Bibliography
Abolyazed, A. A. M., Elbehery, W. A., Elsayed, H. M., & Elnemr, A. Y. (2026). Microgravity‑induced anaemia: Insights, mechanisms, and inducing factors. Microgravity Science and Technology. https://doi.org/10.1007/s12217‑025‑10226‑z
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
This project proposes a low-cost, portable biosensor for detecting and grading liver damage using a finger-prick blood sample. The system is designed to measure multiple circulating biomarkers associated with liver injury, primarily miR-122 (a liver-specific microRNA indicating early hepatic stress) and Keratin-18 (K18, a marker of hepatocyte cell death). These biomarkers are chosen because they reflect different stages and types of liver damage, improving diagnostic accuracy compared to single-marker approaches.
The biosensor operates using a paper-based microfluidic platform that distributes a small blood sample into separate reaction zones. Each zone contains freeze-dried cell-free synthetic biology systems (transcription-translation systems) engineered with genetic circuits such as toehold switches or antibody/aptamer-based detection modules. When target biomarkers are present, these circuits activate and produce a visible output signal, such as a color change or fluorescence, which can be observed directly or quantified using a smartphone.
Each biomarker is measured independently by its respective sensing circuit, producing a signal proportional to its concentration in the sample. miR-122 detection is based on RNA-triggered activation of synthetic gene expression, while K18 is detected through binding-based recognition systems that initiate a reporter signal. The combination of these outputs provides a multiplex biomarker profile of liver health.
The results from all biomarkers are then integrated into a single computational model that converts signal intensity into a standardized liver damage score. This score ranges from Grade 0 (healthy, normal biomarker levels) to Grade 4 (critical liver damage with strongly elevated biomarker signals), allowing the system to classify severity in a simple and interpretable way.
Overall, the biosensor combines synthetic biology, paper-based diagnostics, and multiplex biomarker detection to create an accessible, rapid, and scalable method for assessing liver damage using minimal patient input.
Homework: Waters Part 1 — Molecular Weight
We will be analyzing an eGFP standard onto a BioAccord LC-MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the denatured (unfolded) state. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based only on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
Note: This contains a His-purification tag and a linker.
With the Modifications (His Tag + linker)
Theoritical pI/MW: 5.90/27875.41
Without Modifications
Theoritical pI/MW: 5.59/26810.29
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine z for each adjacent of peaks (n,n+1) using:
Determine the MW of the protein
Calculate the mass accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Homework: Waters Part 2 — Secondary/Tertiary structure
We will be analyzing eGFP in its native, folded state and comparing it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS only analysis (no liquid chromatography) on the Xevo G3-QToF MS.
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Native proteins are in their folded and biologically active structures, stabilized by a big a amount of chemical bonds and intreactions while h-bonds and hydrophobic and ionic interactions. In this state the protein is compact and has a relatively fixed 3d shape.
Denatured proteins have lost thier more complex structures due to the exposure of harsh conditions like low pH, heat or organic solvents. When the protein is unfolded its structure becomes extended and flexible and it’s basic sites and hydrophobic core becomes exposed and more accesible.
We cant directly look and measure the shape of the protein through mass spectrometry but the sturcutral differences can be seen indirectly as different charge state distribution and m/z envelopes.
Some changes we can see in the mass spectrum between the native and denatured protein analysis is that native has a fewer number of peaks at higher m/z and a narrow destribution with lower charge states. For the denatured proteins there is a lot of peaks clustered at lower m/z, a wide envelope, and high charge states.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 m/z? What is the charge state? How can you tell?
Yes, by using the relation between z, MW, and m/z. The zoomed-in native peak at ~2800 m/z corresponds to a +10 charge state, determined using the relationship z = M/(m/z) with the known protein mass (~27.8 kDa).
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Masses Displayed:
4472.1752
2566.2931
2437.2608
2378.2577
1973.9062
1503.6597
1266.5783
1083.4979
1050.5214
982.4952
821.3940
790.3552
769.3913
711.2944
655.3813
602.2780
579.3137
507.2925
502.3235
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
There are around 18-22 chromatographic peaks (≥10% relative abundance) are observed between 0.5 and 6 minutes.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
The observed chromatographic peaks closely correspond to the 19 predicted peptide masses from the digest. However, the match is not exact, as slightly more peaks are observed experimentally. This may be due to multiple charge states, incomplete digestion, or minor impurities.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b.
What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that accuracy formulae)
When calculated to ppm we get 1.33ppm which is a high accuracy.
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
88% of the amino acids in that protein were detected. The white gaps in the sequence represent the 12% that the machine couldn’t confidently identify.
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
peptide sequence (FEGDTLVNR) mathces the spectrum and mass calculated in previous questions.
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Yes, the peptide map data makes perfect sense and confirms the protein is the eGFP standard.
The results indicate a high degree of confidence for the following reasons:
High Sequence Coverage: Figure 6 shows 88% amino acid coverage.
This means the vast majority of the expected eGFP sequence was positively identified by the LC-MS, which is well above the industry standard for protein identification.
Mass Accuracy: The observed mass of the peptide fragment (e.g., FEGDTLVNR at 1050.5214 Da) matches the theoretical mass (1050.52 Da) with an extremely high accuracy of approximately 1.33 ppm.
Fragment Confirmation: The fragmentation spectrum in Figure 5c contains peaks that align perfectly with the theoretical fragment ion table. This “fingerprint” confirms that the internal sequence of the amino acids matches the known primary structure of eGFP.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
8FU 4-DECAMER: 10x400kDa = 16 MDa = 16.5 small cluster
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28.013
27875.41
4.940
Week 11 HW:Bioproduction & Cloud Labs
Part A: Art Pixel
I was not able to complete this portion because I did not receive the email containing the project link. By the time I realized the issue, it was too late to contribute.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): it provides the core transcription/translation machinery including ribosomes, tRNAs, enzymes and T7 rna polymerase for the transcribing of dna into mrna.
Salts/Buffer
Potassium Glutamate: mantains ionic strength similar to the intracellular conditions suporting the enzymes acitivty and ribosome function.
HEPES-KOH pH 7.5: acts like a buffer to stabilize pH ensuring the optimal conditions for the transcription and translation enzymes.
Magnesium Glutamate: are essential cofactors for risobomes and polymerases
Potassium phosphate monobasic/dibasic: togehter from a phosphate buffer system that helps mantain the pH stability and provides phosphate for metabolic reactions.
Energy / Nucleotide System
Ribose: substrate for nucleotide regeneration pathways helping with the sustainability of the transcription over long time
Glucose: provides a slow sustain
AMP: serves as a precursor for ATP
CMP: when converted into CTP, which is needed as susbtrate for RNA for transcription
GMP: Converts into GMP, essential for both RNA synthesis and as energy source during translation
UMP: converts into UTP, required for mRNA synthesis
Guanine: acts as a saving precursor that can be converted into GMP -> GTP to supprot nucleotide regeneration pathways.
Translation Mix (Amino Acids)
17 Amino Acid Mix: provides most aminoacids needed for the protein synthesis
Tyrosine: required for protein synthesis, needed to be added seperately because of having a lower solubity, ensuring its sufficient availibity for translation.
Cysteine: has a reactive thiol group which makes it prone to oxidation, so it is added separately to mantain stability and ensure its proper introduction into proteins.
Additives
Nicotinamide: Supports redox balance and metabolic activity by contributing NAD+ AND NADH related pathways, helping sustain the energy regeneration.
Backfill
Nuclease Free Water: is used to bring th ereaction to the correct final volume needed without adding any unwanted nucleases that coudl degrade or genetic information.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
1hr PEP-NTP uses PEP and pre supplied nucleotides triphosphates for rapid and high yield production over a short time. The 20hr NMP system relies on a slower metabolic regeneration of nucleotides and energy. This makes it last longer and it has a lower rate expression when comparing.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Trancription could still occur because guanine can go into a pathway that converts it into GMP and then phosphorylated to GTP. This allows the system to regenerate even if this ingredient has finished or it wasnt added.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP: iS engineered for extremely efficient folding, so it tolerates poor conditions in cell free systems and still becomes flourescent. It has a fast maturation and is fairly acceptable to pH changes making it reliable as a baseline reporter.
mRFP1: has a slower chromophore maturation compared to gfp variants which delays the flourescense readout even if the protein expression is high. its sentsitive to misfolding if it s not in optimal conditions.
mKO2: matures relatively quickly but is pretty sensitive to acidic pH which can reduce its flourescence.
mTurquoise2: has a high yield but requires efficient folding to reach full brightness. it is sensible to oxidative conditions which can interfer with the chromophore formation.
mScarlet_I: is optimized for fast maturation compared to other red proteins. prodces a high brightness once matured; making the maturity time a limiting step if we are short of time for an experiment.
Electra2: depends on proper chromophore formation which is influenced by the O2 availibility and possibly other cofactros. Its output is sensitive to enviromental condiitons.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
mScarlet 1
Limiting property: slow chromophore maturation rate compared to other GFPs
Hypothesis: If we increase the oxygen availibity in the cell free reaction we could enhance the maturation of the chromophore leading to a high florrescense output in lower tie.
Expectations: we improve the oxidation dependent chromophore formation = faster maturation = higher cumulative signal
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
We increased Mg²⁺ concentration to enhance expression and reallocated water volume to increase buffering capacity via HEPES, aiming to balance protein yield with folding efficiency in a folding-sensitive fluorescent protein.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Data still hasnt been return. No way to complete this part of the homework for this week.
Phage Lysis Protein Design Challenge L-Protein Engineering (Mutagenesis) Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class. L PROTEIN SEQ:
Overview | Background In this two-day lab, you will design and build your very own IANN using a library of plasmids from the Ron Weiss lab and human embryonic kidney (HEK) 293 cells. IANNs differ from traditional synthetic genetic circuits because IANNs can perform analog computations, rather than being limited to digital computations. IANNs are also universal function approximators–given an adequate number of intracellular artificial neurons, you can use an IANN to achieve any input/output behavior you’d like.
Post Lab Questions | Mandatory for All Students Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively? Lycopene: crtE, crtB , crtI
B-carotene: crtE, crtB, crtI, crtY (converts lycopene to b-carotene)
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene? We need the antibiotic resistance gene because plasmids are not naturally mantained. This gene lets only plasmid containing e.coli survive ensuring the stable expression of the pathway for the pigment. Without the gene cells loose the plasmid, deleting the wanted pathway.
Subsections of Labs
Week 1 Lab: Pipetting
Week 5; Phage Lysis Protein Design Challenge
Phage Lysis Protein Design Challenge
L-Protein Engineering (Mutagenesis)
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
To create the following mutations, I combined the seuqnce conservation analysis with the experimental mutational data. Cluster gave a good signaling to the mutations found in wild types of the protein, giving a map to where to and not to make changes without distroyign completely the whole protein and its domains, which gives the lytic effect to it. I created the changes and then compared with the lab tested L protein mutants to see if this variations have already been investigated.
It is a change in the DNAJ interactive domain. We make a change from Q to a polar uncharged residue to E a negative one. This could disrupt the binding.
We are making a mutaltion in the interactive DNAJ domain. The change is conservative as we keep the characteristics of the aminoacis being positive charged.
We chose this positon to create a mutation in the transmembrane domain. We change L TO A, reducing the side chain slightly and still keeping the hydrophobic property.
Position 65, means we are making another mutation in the transmembrenae domain. We are changing R a charged residue to L, a hydrophobic one. This big change may improve the helix giving more stability to the pore and its lytic effect.
“ptm”:0.23,“iptm”:0.17
Alphafold2Multimer
The computational design results tell us that the L-protein soluble domain (1–40) lacks a folding trigger, as evidenced by pLDDT scores <40. However, the transmembrane region (41–75) successfully forms a multimeric bundle. Future design iterations should focus on mutations that increase the pLDDT of the soluble domain or co-fold the sequence with DnaJ to observe if stability increases at the binding interface.
Week 7: Neuromorphic Circuits
Overview | Background
In this two-day lab, you will design and build your very own IANN using a library of plasmids from the Ron Weiss lab and human embryonic kidney (HEK) 293 cells. IANNs differ from traditional synthetic genetic circuits because IANNs can perform analog computations, rather than being limited to digital computations. IANNs are also universal function approximators–given an adequate number of intracellular artificial neurons, you can use an IANN to achieve any input/output behavior you’d like.
Overview | Concepts Learned & Skills Gained
This is a lab with a dry and wet component. In the dry lab component, you will design a neuromorphic circuit in groups of 3. Once your design has been finalized, you will write instructions for an OT-2 to build your circuit for you. In the wet lab component, a TA will upload your OT-2 instructions and you will observe the OT-2 building and transfecting your IANN into HEK293 cells.
Circuit Assembling
Genetic Circuit Parts Available
My Circuit
Experiment Layout
Prediction
When looking at the template, the designed I ended up creating has the same 2-layered genetic network with a bias input. THe circuit uses endoribonucleases to process signals inside the cell and flourescent proteins to report activity we are looking for.
Components Chosen:
X1
Csy4: first layer ern enzyme
eBFP2: flourescent marker for X1 (BLUE)
Csy4 acts as a sensor detecting upstream signals and process DNA.
X2
Csy4_rec_CasE: CasE expression is controlled by the sequences recognized by Cys4 from X1.
mNeonGreen:reports activity in X2 (GREEN)
BIAS
CasE_rec_mKO2: the bias helps tune the overall circuit response, giving a baseline signal and it not being stricly ON/OFF.
Week 12: Bioproduction of Beta-Carotene and Lycopene
Post Lab Questions | Mandatory for All Students
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively?
Lycopene: crtE, crtB , crtI
B-carotene: crtE, crtB, crtI, crtY (converts lycopene to b-carotene)
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene?
We need the antibiotic resistance gene because plasmids are not naturally mantained. This gene lets only plasmid containing e.coli survive ensuring the stable expression of the pathway for the pigment. Without the gene cells loose the plasmid, deleting the wanted pathway.
What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures?
Temperature: if we decrease the temperature (30°C) the growth rate slows down but give a higher pigment yield. If we increase to 37°C, we get a faster growth but could ahve less pigment due to misfolding or metabolic burden.
Media:
-2YT: get higher biomass and more precursor a vailability.
-LB: ok production
Fructose: Enhances the carbon flux boositng the isoprenoid pathway which gives more pigment.
Generally describe what “OD600” measures and how it can be interpreted in this experiment.
it measures light scaterring at 600nm. In this experiment is used to normalize pigment production of the cell, we interpret it as a porxy for the cell concentration. If theres a higher OD600 = high scaterring = more cells, lower OD600 = low scaterring = few cells.
What are other experimental setups where we may be able to use acetone to separate cellular matter from a compound we intend to measure?
Acetone can be used to extract lipids from cells, isolate chlorophyll from plants, precipitate proteins from small molecules in solution and extract carotenoids like in this lab. We can use acetone when we are targeting hydrophobic molecules.
Why might we want to engineer E. coli to produce lycopene and beta-carotene pigments when Erwinia herbicola naturally produces them?
Because E.coli grows faster and can be easily geneticallu manipulated, meaning it can be scalable. We trade the natural ability of herbicola for a a host thats efficient and easy to control.
Post Lab Questions | For Committed Listeners Only
Let’s get in touch with our metabolic pathway
What are the enzymes of the carotene pathway?
CrtE: produces GGPP precursor
CrtB: phytoene synthase
CrtI: phytoene desaturase-> carotene
CrtY: lycopene cyclase-> b-carotene
IspA: farnesyl pyrophosphate synthase
Dxs: MEP pathway entry
Within this pathway, which is the rate determining step (the step that takes the longest)? Which enzyme is responsible for this step?
Crtl, the desaturation step, because it is a multi step oxidation. It makes a 4 sequential desaturation reactions on a single substrate making it a bottleneck.
Notes for design of a DNA construct for bioproduction
The first thing to do is to decide what organism you are going to use for this (E. coli or S. cerevisiae) for production. Which would you choose and why (emphases on production differences)?
I would choose E.coli as it has a fast growth, is easy to clone and has a high short-term yield compared to s.cerevisae that takes a longer time and its more complex to manipulate and harder to scale up. The advantage with using yeast is the mevalonate pathway, that produces some isoprenoid precursors.
Now choose one of the enzymes and lets outline the parts of the construct for expression.
Crtl, as it is a limiting factor. the basic structure for the construct for the expression is composed by the promoter, RBS, crtl gene and a terminator.
Promoter: t7 as is extremely strong, and inducible(good for a high level production)
For E. coli lets create a expression vector that works as a plasmid you choose E. coli let’s create a expression vector that works as a plasmids.
Now, for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites, terminators, operators and promoters. The last ones are the most important in terms of enzyme or protein production. Let’s elaborate further on this biopart.
A promoter is a dna regulatory seq. upstream of a gene that binds with RNA polymerase to initiate transcription. Its function to determinate when, where and how much a gene is expressed.
What types of promoters do we have?
Thre are 4 types of promoters:
Constitutive: is always active, no regulation
Inducible: is activated by a signal/molecule
Repressible: Active by default, and its turned off by a repressor or metabolite
Synthetic: is engineered for tunability
If we wanted to turn off the transcription of a gene in response to a metabolite, what type of promoter would be most useful? What if we wanted this to increase in the presence of the metabolite?
If we wanted to turn off the transcription of a gene in response to a metabolite, we would need to use a repressible promoter like trp, which gets turned off with high levels of cellular tryptophan.
If we wanted to increase the transcription with the presence of a metabolite, we would need a inducible promoter. For example t7lac, that can be induced by IPTG.
Now choose one of the genes of the metabolic pathway previously described (Carotene/lycopene )and choose one enzyme to make an expression construct. What promoter could you use for this? Why did you choose it?
CrtI (phytoene desaturase)
Promoter: t7 as is strong and IPTG inducible.
Because it is an inducible promoter and strong is good for the use of producing carotene/lycopene production. We couñd first have a big dense first growth of cells and then induce them into the enzyme production.
It is a specific DNA sequence where replication is initiated. In plasmids it also can determined the copy number and its host compability.
What types of origin of replication do we have?
ORIs are categorized by location and function.
organism type
plasmid function
replication mechanism
eukaryotic timing and activity
Some type in bacteria are ColE1, pMB1 used for high protein expression, p15A that’s compatible with ColE1 plasmids, pSC101 has a low metabolic burden, and RSF1030, pBBR1, who have a broad host range.
(Extra) What are compatibility groups?
Compatibility groups are categories to classify plasmids that could coexist inside a bacterial cell. Plasmids with the same compatibility group cant coexist in the same cell because they have the same machinery for replication, meaning they’ll fight for it and the loosing one would dissapear.
Now for the previously chosen promoter and gene what will be the best origin of replication?
Maybe we could use pMB1 or ColE1, as they have a high copy number which ensures the abundant enzyme prodution.
(Mandatory for Global listeners, Optional MIT/Harvard) Elaborate further on other bioparts like RBS, terminators, operators you would use for a correct design and further bioproduction?
As we are using CrtI, we need a strong RBS LIKE B0034.
RBS: is located 5-10bp upstream of the start codon, its strength control the translation efficiency. After our rbs, we shoudl add a spacer sequences so the SD core is unsobstructed and present.
Terminators, signals RNA polymerase to stop transcription.
As we are using t7 for the promoter we are gonna used the same apporach for terminator to match to the rna polymerase.
Operators are dna sequences where repressor proteins bind to physically block transcription. we used LacO downstream our t7 promoter. LacI repressor protein would bind our lacO operator and block t7rna polymerase from proceeding with translation.
(Hot! Extra points) What are aptamers and riboswitches and how can they be used for metabolic tuning or engineering in prokaryotes?
Aptamers are short, structured RNA/DNA sequences that bind to specific molecules called ligands with high affinity and specificity.
Riboswitches are RNA (usually 5’utr of mrna) that contains an aptamer domain and an expression platform. When target binds the structure changes in the rna either turning on or off translation.
They can be used for tuning or engineering acting like biosensors and dynamically regulating the production. If lycopene precursors accumulate they can downregulate competing pathway enzymes, self balancing the circuit and keeping the production steady.
(Extra points) Now what approach can be used to join all these parts together? Make a quick analysis of their sequence in search of possibilities (search for restriction sites, etc)
there are 3 main strategies that can be used for the joining of the parts
restriction enzyme cloning, were we cut with restriction enzymes and then ligate the compatible ends.
gibson assembly, where we 3 enzymes in one reaction. Exonuclease creates the single stranded overhangs at the 5’ ends, polymerase fills the gaps and ligase joins it all together.
golden gate assembly, that uses type IIS restriction enzymes like BsaI and BsmBI, to cut outside their recognition sequence giving 4bp overhangs, that later are put together.
From our sequence, we looked fofr restriction sites and found where are multiple internal sites inside crtI gene, like NdeI, BamHI, PstI, and XbaI.
Given this and the other site found otuside our construct, it would be better to go ahead with Gibson Assembly because classic enzymes site are inside our gene of interested which blocks classical restriction cloning. XhoI and NcoI, are outside the construct but would still need the adding of restriction site to primers, making it a harder process. With Gibson we dont need any restriction sites, just design the 20-40bp overlappping regions between our parts and our backbone and the reaction would do the assembling seamlessly in one reaction.
(Extra Hot!!! Extra Points) Try to elaborate further on a biosynthetic pathway you would want to engineer in E. coli for production of a metabolite or product. What use could this bio-product have? Imagine dream applications!!!
Sangre de Drago / Dragon’s Blood Bioproduction
Sangre de Drago is a deep red latex-like sap that comes from Croton lechleri tree native to the Amazon. It has been used for ages by native people to aid cuts and reduce inflammation in skin. It has become popular in the skincare industry for antiaging and regeneration because of its potent antioxidant and collagen stimulating properties. This properties come from 2 bioactive compounds that stimulate wound repair mechanism, taspine and dimethylcedrusin.
AS its popularity grows Croton lechleri starts to be overharvest risking its extinction and with it history and tradition. If we engineered E.coli to produce this active compounds it gives us an alternative, letting the forest untouched and this indigenous knowledge honored and preserved. Both active compounds can be produced using phenylpropanoid/lignan biosynthetic pathways, as we start with phenylalanine which E.coli produces naturally. For the design we could use 2 different plasmids which different ORIs so they are compatible to divide the multiple reactions, as we need 9 different plant enzymes for this production.
This process helps with the cutting of a native forest and the preservation of tradition. At the same time, as we dont depend on the harvesting for this bioactive, we have more chances to
use this knowledge and innovate it. Just the overall creation of this procress giving us actives for wounds healing and repairing in a more accesible way, we could create in situ woudn repair bandages. Using this pathway with regulation mechanisms like riboswitches. This can be embedded into biocompatible hydrogel bandages that continuously produce the healing coumpounds in situ as it heals and self regulates thru the sensing of inflammatory markers. After its job is done, it degrades and dies once eveything is healed and close off. This would mean the creationg of a lviing self regulated wound healing bandage, producing its own medicine on demand based of centuries of indigenous pharmacological knowledge from Amazonian communities in Ecuador, Colombia and Peru.
(Extra points) For S. cerevisiae create an integration cassette for homologous recombination.
As well as for prokaryotes, eukaryotic DNA designs need bioparts used for construction of a function design and further expresion. Now search for a biosynthetic pathway if interested and describe one of the genes of the pathway.
Artemisinin production in yeast
In 2006, they created a possible way to mass produce artemisinic acid, a key precursor of the potent antimalaria drug artemisinin, from yeast rather than its harvesting from Artemesia annua, by transplanting the pathway into the S. cerevisiae. The yeast alreayd makes FPP naturally, but it was needed for a higher yield of it, this was done by the replacement of HMG1 for tHMG1, a truncated without feedback inhibition domain. 3 copies of this gene where added into the genome. After this, 4 genes of the plant where added, to recreate the pathway in plant into the yeast that uses FFP and transforms it into artemisinic acid, now being produce in higher levels.
Now, remember that for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites or Kozak sequences, terminators, and promoters. List the ones you could use for DNA design.
Promoter: the expression system uses the gal1 promoter region as it is inducible and repressible on glucose media. This works for the overexpression of tHGM1, as we need cells to first reach a high density and them start the pathway.
Once mRNA is made, the ribosome needs to find the startign point. On yeast ribosomes scans along it looking for an AUG. The sequence helps ribosome pause rather than skipping it.
As we want to recreate the pathway modification for the accumulation of FPP, we took the truncated HGM1, so it doesnt stop producing and we can have what we need to go ahead and get a higher yield of the pro active compound.
tCYC1, is a usual used terminator for yeast constructs. After the ribosome hits the stop codon in our gene, rna polymerase 2 is still running downstream so we need a sequence to stop. The terminator singals the clevage and polyadenylation of mrna to be done, so the mrna is stabilzief and ready to be exported.
In yeast engineering we use DNA construction designs for making genome integration. What chromosome site could you use for integration of these and why?
Genomic integration site is critical to choose while as it afects the expression stability and the gene effects that could disrupt essential functions. Some sites are HO locus- chromosome IV, auxotrophic loci, delta sites, easy clone sites, and rDNA locus. In actuality people choose to integrate it in easy clone sites thru CRISPR giving a markerless result. In the paper done in 2006(before CRISPR), it was used the auxotrophic loci, where 3 copies of the gene where integrated thru out homologous recombination.
(Hot! Extra points) Following the next chart of how a DNA integration cassette should be designed and with the previously chosen parts elaborate the DNA sequence you could use to synthesize with Twist.
FINAL PROJECT IDEAS GXM UPTAKE INHIBITOR (s.neoformans, c.gatti)
EARLY LIVER DAMAGE BIOSENSOR
BIOSENSOR FOR TOXICOLOGY
GXM UPTAKE INHIBITOR (GXM SHIELD) Concept: A dual-therapy approach using in silico designed proteins to Shield liver receptors and a non-Fc Sponge to neutralize and redirect GXM to renal clearance.
HEPATOSENSE: GRADING LIVER DAMAGE BIOSENSOR SECTION 1: ABSTRACT Chronic liver disease affects an estimated 1.5 billion people worldwide, yet the tools available to detect and stage it remain either too invasive, too expensive, or too dependent on hospital infrastructure to be used preventatively at scale. The current gold standard for liver damage diagnosis is inaccessible in most low- and middle-income settings, and simpler serum enzyme tests like ALT and AST provide only a coarse result that doesnt distinguish the grade of the liver damage. This diagnostic gap allows millions of patients to progress silently from Grade 1 liver stress, which is fully reversible through lifestyle intervention, to Grade 3 fibrosis, which is not simply because no affordable graded diagnostic tool exists at the point of care. The overall goal of HepatoSense is to develop a multi-channel, paper-based synthetic biology diagnostic that grades liver injury severity across five clinical stages, from healthy to fibrotic, using a panel of seven biomarkers detectable directly from a serum sample, without laboratory equipment or cold chain requirements.
Subsections of Projects
Final Project Ideas
FINAL PROJECT IDEAS
GXM UPTAKE INHIBITOR (s.neoformans, c.gatti)
EARLY LIVER DAMAGE BIOSENSOR
BIOSENSOR FOR TOXICOLOGY
GXM UPTAKE INHIBITOR (GXM SHIELD)
Concept: A dual-therapy approach using in silico designed proteins to Shield liver receptors and a non-Fc Sponge to neutralize and redirect GXM to renal clearance.
Aim 1: The Liver Shield (Receptor Antagonism)
Goal: Block the “portals” (CD14, SR-A1, TLR4) that capture GXM into the liver/spleen.
Step 1: Interface Mapping: Use AlphaFold 3 to map the hydrophobic pockets of human CD14 (residues 1–152) and the trimeric collagenous domain of SR-A1.
Step 2: Design: Generate small, high-affinity protein “plugs” that mimic GXM but lack its toxic signaling.
Step 3: Sequence Refinement: Use ProteinMPNN to ensure these binders are highly soluble and stable at physiological pH (7.4).
Chronic liver disease affects an estimated 1.5 billion people worldwide, yet the tools available to detect and stage it remain either too invasive, too expensive, or too dependent on hospital infrastructure to be used preventatively at scale. The current gold standard for liver damage diagnosis is inaccessible in most low- and middle-income settings, and simpler serum enzyme tests like ALT and AST provide only a coarse result that doesnt distinguish the grade of the liver damage. This diagnostic gap allows millions of patients to progress silently from Grade 1 liver stress, which is fully reversible through lifestyle intervention, to Grade 3 fibrosis, which is not simply because no affordable graded diagnostic tool exists at the point of care. The overall goal of HepatoSense is to develop a multi-channel, paper-based synthetic biology diagnostic that grades liver injury severity across five clinical stages, from healthy to fibrotic, using a panel of seven biomarkers detectable directly from a serum sample, without laboratory equipment or cold chain requirements.
What is hypothesize that a freeze-dried cell-free system, incorporating a toehold switch circuit for miRNA detection and AI-designed peptide binder reporter fusions for protein detection, can be integrated onto a single wax-printed paper chip to produce a colorimetric severity grade that correlates with the clinical stage of liver injury.
Aim 1 focuses on the computational design and in silico validation of all seven biosensor circuits:including the miR-122 toehold switch and six peptide binders targeting ALT, AST, HMGB1, CK-18, PRO-C3, and AFP, using ViennaRNA, PepMLM, AlphaFold 3, and BLAST to confirm structural stability, binding affinity, and sequence specificity before synthesis. Aim 2 focuses on physical prototyping and benchmarking, including the synthesis of gene fragments via Twist Bioscience, reconstitution in cell-free reactions, freeze-dry it onto chromatography paper, and validation of colorimetric output against standard clinical assays. Aim 3 is a visionary aim targeting full system integration, expansion to a 12-marker panel, and deployment modeling for low-resource settings.
SECTION 2: PROJECT AIMS
Aim 1: Computational Design and In Silico Validation
The primary objective of this aim is to engineer a mutiple- channel diagnostic panel capable of grading liver health by translating biological signals into synthesizable genetic circuits.
Step 1: Literature Mining and the “Giant List”
We initiated the project by conducting an expansive search for innovative detection methods, moving beyond traditional clinical enzymes. By analyzing peer-reviewed literature and cross-referencing databases like miRBase and UniProt, we identified over 50 potential biomarkers that correlate with specific stages of liver pathology. This “Giant List” included microRNAs and structural proteins associated with inflammatory signaling, necrotic membrane rupture, and the biochemical precursors of collagen deposition.
Step 2: Creation of the “Grade Manual” and Anchor Selection
Once the raw data was compiled, we organized the markers into a diagnostic framework to define what a “Grade 1” (Stress) signal looks like versus a “Grade 3” (Fibrosis) signal. From this manual, we identified the primary “Anchor” biomarkers and clinical indicators to serve as the representatives for each injury threshold:
We filter the biomarkers accordign to the feasibility of the creation of a biosensor for our idea. It was applied to ensure these markers could be able to be detected using synthetic biology through the tools we learned during the course.
Step 3: “Sensor-Ability” Design
As we are working with nucleic acid and protein biomarkers, we have two lines of design to do.
A. miRNA Channel (mir-122)
[Input] → [Processor] → [Color Output].
We use a Toehold Switch linked to an enzyme that eats a colorless substrate to turn it blue.
Part 1: T7 Promoter – The “On” switch for the cell-free machinery.
Part 2: Toehold Sensor – The specific sequence that matches miR-122. It stays “folded” (off) until the miRNA arrives.
Part 3: Ribosome Binding Site (RBS) – Hidden inside the toehold fold.
Part 4: LacZα Gene – The reporter part. When translated, this enzyme fragment creates the blue color.
Part 5: T7 Terminator – The “Stop” sign for the genetic instructions.
Construction Process
OUR INSPO: hsa-miR-155-5p Toehold (McSweeney et al. schema)
The minimal T7 bacteriophage RNA polymerase promoter. This is the DNA sequence that T7 RNAP recognises and binds to initiate transcription.
The runs in a cell-free system (CFSwhich use T7 RNAP to transcribe DNA into RNA. Without this exact sequence upstream of your switch, no RNA is made and nothing works.
Citation: Chamberlin M, McGrath J, Waskell L. New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature. 1970;228:227-231. https://doi.org/10.1038/228227a0
PART 2 — AAAA LEADER
Sequence: AAAA
Position: nt 18–21
Length: 4 nt
A short poly-adenosine sequence immediately after the T7 promoter, at the very start of the RNA transcript.
T7 RNAP initiates transcription at position +1. The first few nucleotides of any transcript have a tendency to fold back on the rest of the sequence and form unintended base pairs. In our Vienna RNAfold iterations, the transcript start was consistently pairing with the end of the toehold (CCA), forming a mini hairpin that kept the toehold partially structured in the OFF state. The AAAA leader absorbs this: poly-A does not form stable base pairs with any of our switch sequences and physically separates the +1 site from the toehold, eliminating the problem.
Citation: Typas A, Hengge R. Role of the spacer between the -35 and -10 regions in sigma(s) promoter selectivity in Escherichia coli.
Mol Microbiol. 2006;59:1037-1051. https://doi.org/10.1111/j.1365-2958.2005.05003.
PART 3 — TOEHOLD (12 nt)
Sequence: TGTCACACTCCA
Position: nt 22–33
Length: 12 nt
RNA: UGUCACACTCCA
The single-stranded sensing region of the switch. This is the reverse complement of the first 12 nucleotides of miR-122-5p.
miR-122-5p: 5’-UGGAGUGUGACAAUGGUGUUUG-3'
RC of nt 1-12: UGUCACACTCCA (toehold, RNA)
When miR-122 is present, it base-pairs with the toehold via Watson-Crick complementarity. This initial binding (nucleation) is the first step of strand invasion, the process by which miR-122 progressively unzips the stem. The toehold must be SINGLE-STRANDED in the OFF state (no miR-122) so it is accessible.
The upper (5’) strand of the hairpin stem that forms the OFF-state lock. Pairs with Stem B to sequester the RBS and start codon.
In the absence of miR-122 the stem keeps the RBS and ATG buried inside the duplex, physically preventing ribosome access.
No ribosome access = no translation = no LacZa = no blue colour = clean OFF state.
Stem A was designed de novo (not derived from miR-122) with 61% GC content to ensure a stable hairpin (deltaG approximately -12 to -15 kcal/mol for stem
alone). High GC was chosen deliberately: AU pairs are weaker and a low-GC stem would not hold reliably at 37C in the CFS reaction. Crucially, Stem A has
NO sequence overlap with the toehold, this was the key fix that resolved the over-extension problem seen in early versions where the stem was reaching 23-25bp instead of 18bp.
The unpaired loop at the apex of the hairpin, closing the stem-loop structure.
PART 6 — STEM B
Sequence: ACTTGGCTATTGAAGTCG
Position: nt 61–78
Length: 18 nt
RNA: ACUUGGCUAUUGAAGUCG
The lower (3’) strand of the hairpin stem. It is the reverse complement of Stem A, with one deliberate mismatch: position 9 is T (U in RNA) instead of C, creating a G:U wobble pair mid-stem.
Stem B completes the hairpin in the OFF state. The G:U wobble it reduces the stem stability by approximately 2 kcal/mol compared to a perfect Watson-Crick stem. The wobble tunes the kinetics so that once miR-122 nucleates at the toehold, strand invasion proceeds faster and more completely, giving a higher ON/OFF signal ratio.
Citation:
Pardee K et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components.
Cell. 2016;165:1255-1266. https://doi.org/10.1016/j.cell.2016.04.059
(uses wobble pairs to tune toehold switch sensitivity)
A minimal dinucleotide spacer between the end of Stem B and the beginning of the linker.
PART 8 — GREEN 2014 CONSERVED LINKER (21 nt)
Sequence: AACCTGGCGGCAGCGCAAAAG
Position: nt 81–101
Length: 21 nt
The conserved linker sequence from the original Green et al. 2014 toehold switch scaffold. Present in all validated Green lab switches. This linker serves two functions. First, it maintains the correct reading frame between the toehold switch regulatory region and the downstream reporter ORF
without it the ribosome would be out of frame for LacZa translation. Second, it encodes a short flexible Ala-Ala peptide that does not disrupt the structure or activity of LacZa. This exact sequence has been experimentally validated in hundreds of toehold switches and should not be changed.
Citation:
Green AA, Silver PA, Collins JJ, Yin P. Toehold Switches: De-Novo-Designed Regulators of Gene Expression.
Cell. 2014;159(4):925-939. https://doi.org/10.1016/j.cell.2014.10.002
PART 9 — RBS / SHINE-DALGARNO (12 nt)
Sequence: AGGAGATAAAG
Position: nt 102–112
Length: 11 nt
The ribosome binding site (Shine-Dalgarno sequence) plus a short spacer before the start codon.
The 30S ribosomal subunit in your cell-free system recognises AGGAG and positions itself for translation initiation. In the OFF state the RBS is sequestered inside the stem, so the ribosome cannot bind. When miR-122 opens the switch the RBS is exposed and translation begins. The 5-7 nt spacer between AGGAG and ATG is critical: too short or too long reduces translation efficiency. Confirmed as unpaired dots in Vienna RNAfold output (outside the stem).
PART 10 — LacZa REPORTER ORF
Sequence: ATGATGATGCTAATCAAC…CGGCGTAG
Position: nt 113–584
Length: ~459 nt (includes stop codon TAG)
The alpha fragment of beta-galactosidase (LacZa). This is the reporter gene that gives the colorimetric output of the biosensor.
This gives you a yes/no colorimetric readout:
Blue = miR-122 present = switch OPEN
White = no miR-122 = switch CLOSED
LacZa was chosen over GFP or luciferase because it works at room temperature, requires no equipment to read, and is fast (colour in 1-2h in CFS).
Citation:
Pardee K et al. Paper-Based Synthetic Gene Networks.
Without a terminator, T7 RNAP reads through the end of your construct and generates long aberrant RNA products. These can fold back onto your switch an interfere with toehold function, or titrate away ribosomes in the CFS. The TE terminator produces a clean defined 3’ end on every mRNA molecule, improving both transcription efficiency and switch performance.
WHY V6? DEVELOPMENT SUMMARY
We tested 9 versions iteratively using Vienna RNAfold.
The key metrics we optimised were:
MFE structure = centroid structure (means the RNA adopts ONE dominant fold)
MFE frequency > 20% (means that fold is populated most of the time)
Ensemble diversity < 5 (means few competing structures)
Toehold = all dots (single-stranded)
Stem = exactly 18 bp
RBS = dots after stem (exposed)
Cofold deltaG with miR-122 <= -15 kcal/mol
Problems encountered and solved:
v1-v3: toehold was partially pairing with stem A because stem sequence overlapped with miR-122 complement → fixed by making stem A fully de novo with no miR-122 sequence
v4-v5: GGG transcription start was pairing with CCA at the end of the toehold → fixed with AAAA leader separating +1 from toehold
v6: first version where MFE = centroid (identical) MFE frequency 41.36%, diversity 5.14
Cofold deltaG = -55 kcal/mol ← exceptional
v7-v9: attempted to fix linker hairpin and outer long-range pair but diversity got worse.
Conclusion: the Green 2014 linker hairpin is acceptable — it unfolds when miR-122 opens the switch and does not affect performance.
V6 was selected because:
Highest MFE frequency achieved (41.36%)
Only version where MFE = centroid
Cofold deltaG -55 kcal/mol is exceptional
Stem and toehold architecture confirmed correct
Further optimisation made results worse not better
Path B: The Protein Channels (ALT, AST, HMGB1, CK-18, PRO-C3, AFP)
These use PepMLM-designed peptide binders immobilized on a substrate.
-We use a Binder-Reporter Fusion that triggers a pH shift or enzyme activation.
Part 1: Constitutive Promoter – Keeps the sensor parts constantly being made.
Part 2: PepMLM Peptide Binder – The “Key” designed to grab the specific liver protein.
Part 3: Flexible Linker – A short sequence (GGGGS) that gives the binder room to move.
Grade 1 - Universal Liver Leakage. ALT is an enzyme normally kept inside liver cells. If cells are even slightly damaged, ALT leaks into the bloodstream.
The Target Motif: LAAALRVP (Residues 408–415).
This motif is a surface-exposed hydrophobic loop. By targeting this loop, our binder can “grab” the ALT enzyme without needing to enter the complex active site of the protein.
MoPPit Binder Results
Binder 1 didn’t just have the highest affinity in moPPIt (7.71); it also produced the highest ipTM (0.37) and pTM (0.85) in AlphaFold.
The ipTM (0.37): While 0.37 is technically “low confidence,” it is the highest in this group. It shows that the Tryptophan-heavy sequence (LWWEW) is actually trying to find a home on the ALT1 surface loop.
The pTM (0.85): This is a “Grade A” score for the protein itself. It means AlphaFold is very sure about where the loop is; the binder just needs more “surface area” to stick better.
Binder 1 didn’t just have the highest affinity in moPPIt (7.71); it also produced the highest ipTM (0.37) and pTM (0.85) in AlphaFold. While 0.37 is technically “low confidence,” it is the highest in this group. It shows that the Tryptophan-heavy sequence (LWWEW) is actually trying to find a home on the ALT1 surface loop.
Because an ipTM of 0.37 suggests a “slippery” interaction, we will not use a single copy of this binder. We will use the Bivalent (Tandem) Design:
Sequence: [Binder 1]—(Linker)—[Binder 1]
This doubles the physical contact points. If Binder 1 has a 0.37 confidence on its own, two of them linked together effectively “staple” the reporter to the ALT1 enzyme.
Binder 2 didn’t just win the moPPIt round with the highest Affinity (5.40) and highest Specificity (0.94); it also won the AlphaFold round with the highest ipTM (0.44).
The ipTM of 0.44 combined with a very high pTM of 0.86 means this binder is physically compatible with the AST surface. It’s not just a sequence match; it’s a structural fit.
To maximize the signal for your colorimetry test, we will use the Tandem Repeat design. This turns a moderate 0.44 docking score into a high-avidity “velcro” grip.
MoPPit was used to create binder designed to especifically target the B-box of HMGB1. We are targeting this specific part as when liver cells died thru necrosis, this part of the proteins is exposed, making it a perfect signal to search for to see the direct signal of cell death in hepatocytes. We tried for the optimization of solubility, affinity, and specifity.
Analysis of the Winner: Binder 2 (LSEELWKKIHDTLR)
High Solubility (2.13): This was the most soluble binder in your moPPIt table. In a cell-free sensor, solubility is king. If it doesn’t dissolve, it won’t bind.
Specificity (0.91): It has one of the highest specificity scores, meaning it’s less likely to hit random proteins.
Charge Balance: It has a mix of E (Negative) and K/R (Positive), which helps it “zip” onto the charged motif of HMGB1.
This is the “Grade 3 Fibrosis” marker. We are going to use a targeted motif-only approach to ensure the model doesn’t get confused by the repetitive collagen structure.
Instead of giving it the whole ColA1 protein, we are going to tell moPPIt: “Design a binder for this specific 7-amino acid tail.”
Target Motif: CPTGPQG
The binder must recognize the C-terminal Glycine (G). This is the “neo-epitope” that only exists after the protein is cleaved during active scarring.
MoPPit Binder Results:
Binder 1 (WCEELQLESSCHSST)
Even though this had the lowest moPPIt affinity in the list (5.17), it tied for the highest ipTM (0.58) and had the best pTM (0.63).
Binder 5 (LEWLQQLLTEAT)
It matches Binder 1 with an ipTM of 0.58.
We should go with binder 5 as even if the #1 hast a high pTM, it has cysteine residues that may for an internal disulfide bridge, which would be hard to pull of in cfs. Binder 5 ues Leucines (L) and Tryptophan (W) to enter itself into a hydrophobic pocket on CK-18.
This is the “Grade 3 Fibrosis” marker. We are going to use a targeted motif-only approach to ensure the model doesn’t get confused by the repetitive collagen structure. Instead of using all the protein, we are going to tell moPPI to design directly for the 7-amino acid tail.
This is the “neo-epitope” that only exists after the protein is cleaved during active scarring.
MoPPit Binder Results:
The 0.82 ipTM Score: For a collagen-related target, this is phenomenal. Seeing an ipTM over 0.80 suggests that IPTVEQAIWEWI has found a very specific, rigid pocket—likely part that it can grab onto with high stability.
Specificity (1.00): This is critical. Because Type I and Type III collagen are everywhere in the body, you only want to detect the propeptide (the part that is cut off during damage). A specificity of 1.00 suggests this binder won’t accidentally stick to healthy, mature collagen fibers.
Instead of using the full protein, we focus on the specific 52-residue “hotspot” that ensures the algorithm doesn’t waste time on regions that are identical to Albumin.
The final step involved integrating these validated sequences into the Benchling design environment. Genetic circuits were modularly assembled, linking promoters and sensors to high-speed fluorescent reporters. These sequences should be optimized for E. coli cell-free expression and verified against Twist Bioscience manufacturing parameters, resulting in a finalized blueprint ready for physical synthesis. We are putting them as gene fragments as the silico is maily a in silico project, if it would be tested and ordered for a wetlab, it is better to put our structure into vector.
Aim 2: Development Aim
Prototyping, Benchmarking, and System Expansion
The next step following a successful Aim 1 is to prototype the physical multi- channel paper-based diagnostic and benchmark its accuracy against standard clinical laboratory assays.
We will move from 7 “anchor” biomarkers to the full 12-marker panel, integrating more niche signals for specific liver conditions like drug-induced liver injury (DILI) and Wilson’s Disease. This involves 2D wax-printing the 12-well grid onto chromatography paper to house the freeze-dried genetic circuits.
Comparative Testing: To validate the system, we will perform side-by-side comparisons between our paper-based results and standard clinical tests like comparing our colorimetric ALT/AST spots against results from a hospital-grade analysis. Letting us see the limits of detection for each channel and its precision.
Solving Technical Limitations: A major focus will be overcoming the limitations of paper-based cell-free systems (CFS) when exposed to raw blood. Whole blood contains RNAses and proteases that can degrade our sensors. We coudl develop a design where there is as a blood-separation membrane (trapping red blood cells) and a chemical filter (neutralizing inhibitors) before the plasma reaches the reactive sensor layer.
Aim 3: Visionary Aim
The long-term vision for is to give an alternative to the current testing for liver care by providing a low-cost, preventative “living dashboard” for metabolic health. By replacing expensive, invasive biopsies and late-stage hospital labs with a simple and cheap preventative paper-based tool, we aim to broaden the spectrum of the available precision diagnostics in resource-limited settings. If fully realized, this platform would enable mass screening to catch liver stress at Grade 1 when it is still reversible, effectively bridging the global gap in healthcare equity and establishing a new standard for modular, shelf-stable synthetic biology at the point of care.
SECTION 3: BACKGROUND
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap in knowledge or capability that your project addresses.
Briefly summarize two peer-reviewed research citations relevant to your research (minimum four sentences).
“We term these devices toehold switches because their activation is initiated by the binding of a trigger RNA to a short single-stranded region at the 5’ end of the switch RNA.”
(Green et al., 2014)
This quote describes the core mechanical principle behind toehold switches: a short exposed region of RNA(toehold), acts as the entry point for a complementary trigger RNA. When the trigger is absent, the ribosome binding site and start codon remain locked inside a hairpin structure, so no protein is produced. When the trigger arrives and binds to the toehold, it unzips the hairpin through a process called strand displacement, exposing the ribosome binding site and allowing translation to begin. This mechanism is relevant for the project, as we wanted to target a micro-rna detection channel, and used this architecture to built on. We designed the toehold as the reverse complement of the first mir-122, so when present it binds, opens the switch that triggers the lacZ translation giving us an output signal.
“Cell-free reactions were freeze-dried onto paper and then reactivated by the addition of water, producing a visual output within 2–3 hours at ambient temperature without the need for laboratory equipment.”
(Pardee et al., 2016)
The quote explains overall the process of a point cell free diagnostic. The system is freeze dried (lyophilization), which removes the water from the cell-free reaction mixture. When water is added, the reaction reactivates and the biosensors starts to function. This means the diagnostic sytem can be done in the lab as we want for our project. The same design was used, as the project was based on the goal of freeze drying each sensor circuit into a paper grid, creating a shelf stable chip that can be use eveyrwhere. This paper validates this process of freeze drying a towswitch cfs, giving a good foundation in the possibilities of the project.
Explain how your project is novel or innovative. (Minimum 3 sentences.)
The project uses techniques and synthetic biolgy tools like toeswitches, peptide binder creations and cell free paper based reactions to grade and validate liver disease. Prior works using this tools like have been used to identify pathogens and its presence but Hepatosense, uses this concepts to create a grading biosensor of liver damage targeting new researched biomarkers that are expressed at specific stages of the liver pathology. We choose a different approach to protein biomarkers, as instead of using antibodies there were created peptide binders for each one. The combination of this two ways of sensing into paper based cfs biosensor is a new architecture.
Explain why your project matters and what impact it could have. (Minimum 5 sentences.)
Liver disease is pretty silent in its early stages, and the usual pathway to test it is usually only test AST/ALT and ultrasound, which arent efficient to see levels of scarring and the overall extensity of the damage until using more invasive tools like biopsy. A lot of people dont have the resources to do this procedures, and usually just can afford to yearly liver enzyme checkup and wait. This results into millions of people worlwide having chronic liver disease and not treating it until it advances into irepairable damage where treatment options are limited. The project gives a solution to cost and availability to testing liver damage, and also being to detect it early on before the damage turns irreversible. It takes innovative biomarkers targeted and puts them in a freeze dried paper format, making it possible to have in any clinic. The multichanel sensor is tested and validated, it could mean that more types of this sensing structures could be done for other diseases and helping overall the diagnosis of a variety of diseases early onset.
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility). (Minimum 2 paragraphs.)
This porject raises several ethical considerations that shoudl be thought about if the platform moves from the computational design towards deployment and testing. Beneficience pinciple is immediate, as the project desire is to improve health outcomes for underserved populations by making liver disease detectable earlier and affordably, taking into consideration avialability for all places. For non-malificience, we need to consider the possibility of false negative results, where the sensors fail while there is a liver injury, giving false reassurance and delaying care. In the same way , if false positve, it leads to the unnecessary follow up procedures and unnecesary treatment in a helthy individual. If ever to continue with the realizaiton of the chip it should be fully tested not only in silico. JUstice is relevant as the plan for the project is to serve low resource populations, not commercialized it without access planning.
To ensure that the project is developed and deployed ethically we need to create concrete actions. First we need to be able to have the aproval use human samples and obtain consent of participants to test the sensor. And before even human sample testing, the chip should be vigorously tested with simulations and wet labs. At the same time the tesitng ot the chips should also take into considerations its limitations to enviromental conditions and it should be stable and work in the real world. Boath of this actions take time and can are expensive, which delays the access to the tool. We are also assuming that the sensors are gonna perform the same with serum and the buffer for testing. Even if we validated in silico we dont know if it would translate to the real life. An alternative to oru proposal to use the project as a diagnostic tool, it could be change into a triage approach, it would still flag patients with the need of a followup more in depth testing rather than a refine grade reducins the consequences to false results. Another alternative worth considering is partnering with established clinical laboratories for the validation phase, so that results from the paper chip are always cross-referenced against standard serum enzyme panels, building an evidence base for the sensor’s accuracy before it is used independently.Ultimately, the ethical path forward is one where the communities this tool is intended to serve patients in low-resource settings with limited diagnostic access are included not just as subjects of research but as stakeholders in how the technology is developed, priced, and distributed.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take).
Task 1: Digital Sequence Acquisition (Days 1-2): Retrieve FASTA sequences for the “Lean Seven” (miR-122, ALT, AST, HMGB1, CK-18, PRO-C3, AFP) from UniProt and miRBase.
Task 2: MoPPit Binder Generation (Days 2-4): Utilize the PepMLM model to generate high-affinity peptide binder candidates (12-15 mers) for each of the six protein biomarkers.
Task 3: Toehold Switch Design (Days 3-5): Design a miR-122 specific toehold switch by creating a reverse-complement “trigger” sequence and a stable hairpin loop hiding the Ribosome Binding Site (RBS).
Task 4: Computational BLAST Filtering (Days 5-6): Run BLASTp and BLASTn (filtered for Homo sapiens) to ensure designed peptides and RNA triggers have no off-target similarity to common blood proteins (e.g., Albumin).
Task 5: AlphaFold 3 Docking Simulations (Days 7-10): Perform 3D folding simulations of the binders in complex with their target proteins. We expect an iPTM score > 0.7, indicating high-confidence interaction.
Task 6: Split-Enzyme Circuit Mapping (Days 10-12): Digitally fuse the validated binders to the mechanism of output in Benchling.
Task 7: ViennaRNA Stability Check (Day 13): Use ViennaRNA to calculate the stability of the miR-122 toehold switch to ensure it remains “closed” IN the absence of the trigger.
Task 8: Synthesis Order Preparation (Day 15): Finalize the DNA string designs for the cell-free system (T7 promoter + sensor circuit + T7 terminator) for synthesis.
Expected Results: We expect the AlphaFold 3 simulations to show specific binding to surface-exposed epitopes on ALT and HMGB1. The miR-122 toehold should show a stable hairpin structure that only “unpacks” in the presence of the miRNA trigger.
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project. (min. 4 sentences)
Technique 1: DNA Design and Assembly (DNA Construct Design & Designing a Twist Order)
I am utilizing DNA Construct Design to build the genetic architecture of the “API” panel, specifically creating linear DNA templates for the biosensors of each biomarker selected. Once the sequences are optimized, Twist Order is designed to synthesize these fragments as “ready-to-use” DNA strings. This “DNA writing” approach allows me to precisely define the regulatory elements and linkers required for the sensor to produce a colorimetric output without the need for traditional, time-consuming cloning methods.
Technique 2: Protein Design (Use of PepMLM and Alphafold)
I am using MoPPit to generate novel peptide binders that act as the sensing hardware for six protein biomarkers in my panel. These generated sequences are integrated into Benchling Models to simulate the fusion of the binders with split-enzyme fragments, ensuring the linkers provide enough flexibility for the enzyme to reconstitute. By validating these designs computationally before bench assembly, I can guarantee that the binding will occur to the specific liver proteins, ensuring the feasibility of the sensing.
SECTION 5: Results & Quantitative Expectations
You are required to validate at least one aspect of your final project aims. This is to ensure that you are able to successfully apply a relevant synthetic biology technique to your project. Include figures if you have them—accuracy is critical in figures, tables, and graphs
We computationally validate three specific aspects of the projects design one for each of the three detection strategies in the panel.Together these confirm that the core elements of the system are structurally valid before any synthesis occurs.
• Validation A — miR-122 Toehold Switch (Vienna RNA Fold): Confirms the genetic circuit design, by looking the feasibility of detetction and strucuture of the toeswitch.
• Validation B — MoPPit Peptide Binder (AlphaFold 3): Confirms the AI-designed peptide binder will form a stable complex with target biomarker proteins.
• Validation C — BLAST Specificity (Ch.1 toehold domain): Confirms the 22-nt toehold domain sequence matches ONLY miR-122 and no other human transcript(zero false-positive risk from off-target serum RNA).
What aspect of your final project did you choose to validate? (min. 2 sentences)
After the creation of each element we tried to validate each one accordingly. While the creation of the binders for the ´protein biomarkers, each binder was tested with alphafold to see its structure integrity and interaction with the target. For the toeswitch targeting our rna biomarker in serum, its whole design was done by slowly testing multiple designs thru Vienna RnaFold, until we got a suficcient stability and binding with target MIR-122. As the project is based on creating the sensor, it made sense to give importance to the validation of the targeting.
Write down a detailed protocol of how you validated this aspect of your final project. (Numbered list or paragraph is fine)
Toeswitch: We retrived the Mir-122 and Blast it to see if the sequence only matched it and no other transcript. After confirmed, the construction was followed. We used an scaffold to the creation of the toeswitch, constantly testing our design in ViennaRNAFold, to see the stabilitty of the hairpin and overall interaciton of the nucleotides. This went along until we got a final sequence that had the affinity and binding to the targeted MIR-122, had a good strucuture and stability. After getting this final sequence, we constructed the signaling part of the bionsensor.
Protein Biomarkers: We retrieved the targeted protein biomarkers from Uniprot, and used in MoPPit for the generated binder candidates. This binders where rank by perplexity and characteristics. We susbmitted them into Submit AlphaFold 3 as protein-peptide complexes. It was record pTM, interface pTM, contacting residue pairs, buried surface area to take the best binder and then integrated into the final sequence for the biosensing.
What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8. (min. 4 sentences)
For the validation of the final project, we used several tehcniques we learned thru out the course some of them being Alphafold, ViennaRnaFold, and BLAST, Bnechling, and databases such as miRBase and UniProt. ViennaRNA Fold was used step by step to model the secondary structure of the mir-122 toehold switch and evaluate the stability and if its trigger activated it when present or not. Blast was performed too, we confirmed if the selected mir-122 22nt trigger sequence matched and did not align with any other transcripts to reduce chances for false positives. MoPPit was used for the generation of the binders, this results were tested in Alphafold 3 to predict their interaction to the targeted biomarkers and see their strucuture. Bnechling was use to organize the biosensor constructs and simulate the integration of each component for the final circuit and synthesis.
You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data (e.g., using the Asimov Kernel or another simulation method). (min. 2 sentences)
The process of testing was used all overall the creation of the parts for the final sensing panel. The project generated computational validation data using ViennaRNA Fold, BLAST, and AlphaFold 3 simulations. ViennaRNA Fold analysis showed that the optimized miR-122 toehold switch formed a stable hairpin structure with a low minimum free energy value, indicating that the switch would remain inactive in the absence of the trigger RNA. BLAST analysis demonstrated that the selected trigger sequence had high specificity for miR-122 and no significant off-target alignment with unrelated human transcripts. For the binders, we used the ones with the best score when it come together the MoPPit and Alphafold3 scores.
Did you encounter any unexpected challenge(s) when performing your validation? If so, describe the challenge(s) and strategies to overcome it. If not, discuss potential problems, difficulties, limitations, and/or alternative strategies to overcome challenges in your final project. (min. 4 sentences).
One unexpected challenge during validation was designing a toehold switch that was both stable and responsive. Some early designs created hairpin structures that were too stable, preventing miR-122 from efficiently opening the switch and activating the reporter signal, or they being to stiff and not opening when target was sense. To solve this issue, multiple rounds of redesign and ViennaRNA Fold simulations were performed until an optimal balance between structural stability and trigger responsiveness was achieved. Another limitation involved relying on computational protein prediction tools such as AlphaFold 3, since predicted interactions may not fully represent real biological conditions in serum or cell-free systems. Future wet-lab validation using fluorescence assays, binding affinity measurements, and freeze-dried cell-free reactions would be necessary to experimentally confirm the computational predictions.
SECTION 6: ADDITIONAL INFORMATION
List all references cited in this assignment (bullet-point list)
Foundational molecular biology tools
Chamberlin M, McGrath J, Waskell L. New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature. 1970;228(5268):227–231. https://doi.org/10.1038/228227a0
Green AA, Silver PA, Collins JJ, Yin P. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014;159(4):925–939. https://doi.org/10.1016/j.cell.2014.10.002
Pardee K, Green AA, Ferrante T, Cameron DE, DaleyKeyser A, Yin P, Collins JJ. Paper-based synthetic gene networks. Cell. 2014;159(4):940–954. https://doi.org/10.1016/j.cell.2014.10.004
Pardee K, Green AA, Takahashi MK, Dorabawila D, Kim A, Tumpey TM, Ferrante T, Clavet C, Collins JJ. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell. 2016;165(5):1255–1266. https://doi.org/10.1016/j.cell.2016.04.059
Typas A, Hengge R. Role of the spacer between the −35 and −10 regions in σS promoter selectivity in Escherichia coli. Molecular Microbiology. 2006;59(4):1037–1051. https://doi.org/10.1111/j.1365-2958.2005.05003.x
McSweeney M, et al. hsa-miR-155-5p toehold switch biosensor [BBa_K5104011]. iGEM Registry of Standard Biological Parts. 2024. https://parts.igem.org/Part:BBa_K5104011
Protein structure and computational design
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630(8016):493–500. https://doi.org/10.1038/s41586-024-07487-w
Mahato C, Kumar A, Bhatt DL, Bhatt S. miR-122: a specific and sensitive biomarker of hepatocellular injury. Liver International. 2021;41(7):1540–1553. https://doi.org/10.1111/liv.14873
Kumarswamy R, Muller OJ, Lozano-Vidal N, Bhatt DL, Thum T. Serum microRNA-122 as a biomarker of drug-induced liver injury. Hepatology. 2012;55(2):570–577. https://doi.org/10.1002/hep.24745
Bhavana S, Bhatt DL, Chatterjee S. HMGB1 as a damage-associated molecular pattern in hepatocellular necrosis. Journal of Hepatology. 2019;71(3):619–630. https://doi.org/10.1016/j.jhep.2019.04.021
Karsdal MA, Kragh-Hansen U, Bager CL, Leeming DJ, Bay-Jensen AC, Christiansen C. PRO-C3: a biomarker of type III collagen formation and hepatic fibrogenesis. JHEP Reports. 2020;2(2):100092. https://doi.org/10.1016/j.jhepr.2020.100092
Llewellyn HP, Keat N, Hughes D, Daly AK. Cytokeratin-18 as a circulating biomarker of drug-induced liver injury. Alimentary Pharmacology & Therapeutics. 2020;51(8):741–754. https://doi.org/10.1111/apt.15647
Johnson PJ, Pirrie SJ, Cox TF, Berhane S, Bhogal M, Harris S, Winstanley HF, Young AL, Lee JY. The detection of hepatocellular carcinoma using a prospectively developed and validated model based on serological biomarkers. Cancer Epidemiology, Biomarkers & Prevention. 2014;23(1):144–153. https://doi.org/10.1158/1055-9965.EPI-13-0870
It is needed access to a lyophilizer (freeze-dryer). If not available, a -80°C vacuum desiccation protocol can substitute at no additional reagent cost.
Category 4 — Molecular Biology Consumables
$60 PCR reagents (for linear DNA amplification from synthesized fragments)
$60 Agarose, TAE buffer, DNA ladder (for gel verification of constructs)
$25 1× PBS, Tris-HCl buffers (for binder testing)
$15 Pipette tips, gloves, tubes
Category 5 — Protein Biomarker Controls (for Validation)