Phage Lysis Protein Design Challenge L-Protein Engineering (Mutagenesis) Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class. L PROTEIN SEQ:
Overview | Background In this two-day lab, you will design and build your very own IANN using a library of plasmids from the Ron Weiss lab and human embryonic kidney (HEK) 293 cells. IANNs differ from traditional synthetic genetic circuits because IANNs can perform analog computations, rather than being limited to digital computations. IANNs are also universal function approximators–given an adequate number of intracellular artificial neurons, you can use an IANN to achieve any input/output behavior you’d like.
Post Lab Questions | Mandatory for All Students Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively? Lycopene: crtE, crtB , crtI
B-carotene: crtE, crtB, crtI, crtY (converts lycopene to b-carotene)
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene? We need the antibiotic resistance gene because plasmids are not naturally mantained. This gene lets only plasmid containing e.coli survive ensuring the stable expression of the pathway for the pigment. Without the gene cells loose the plasmid, deleting the wanted pathway.
Subsections of Labs
Week 1 Lab: Pipetting
Week 5; Phage Lysis Protein Design Challenge
Phage Lysis Protein Design Challenge
L-Protein Engineering (Mutagenesis)
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
To create the following mutations, I combined the seuqnce conservation analysis with the experimental mutational data. Cluster gave a good signaling to the mutations found in wild types of the protein, giving a map to where to and not to make changes without distroyign completely the whole protein and its domains, which gives the lytic effect to it. I created the changes and then compared with the lab tested L protein mutants to see if this variations have already been investigated.
It is a change in the DNAJ interactive domain. We make a change from Q to a polar uncharged residue to E a negative one. This could disrupt the binding.
We are making a mutaltion in the interactive DNAJ domain. The change is conservative as we keep the characteristics of the aminoacis being positive charged.
We chose this positon to create a mutation in the transmembrane domain. We change L TO A, reducing the side chain slightly and still keeping the hydrophobic property.
Position 65, means we are making another mutation in the transmembrenae domain. We are changing R a charged residue to L, a hydrophobic one. This big change may improve the helix giving more stability to the pore and its lytic effect.
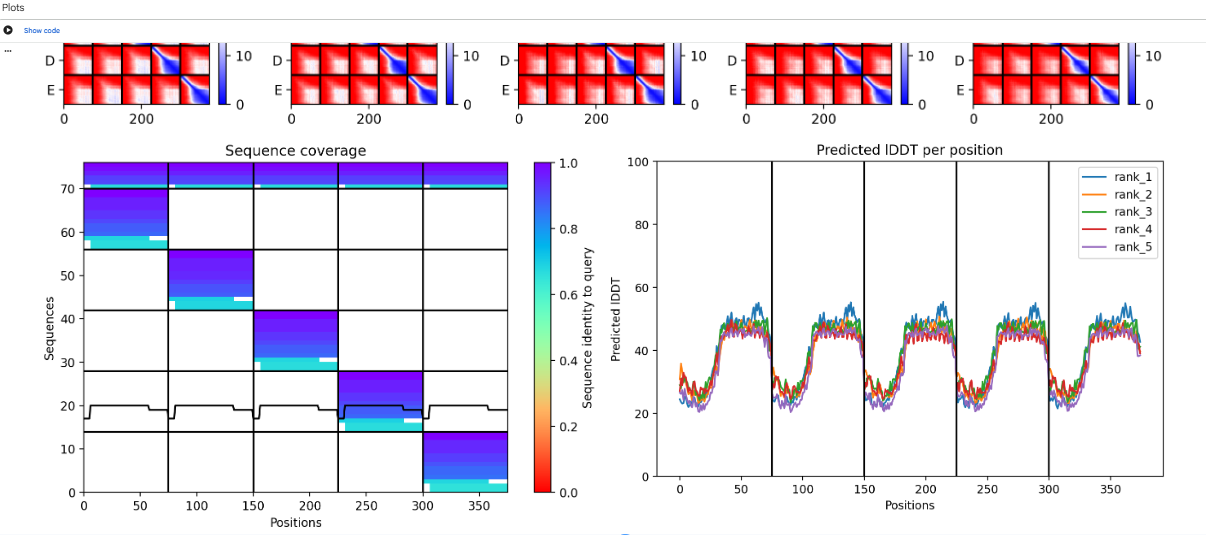
“ptm”:0.23,“iptm”:0.17
Alphafold2Multimer
The computational design results tell us that the L-protein soluble domain (1–40) lacks a folding trigger, as evidenced by pLDDT scores <40. However, the transmembrane region (41–75) successfully forms a multimeric bundle. Future design iterations should focus on mutations that increase the pLDDT of the soluble domain or co-fold the sequence with DnaJ to observe if stability increases at the binding interface.
Week 7: Neuromorphic Circuits
Overview | Background
In this two-day lab, you will design and build your very own IANN using a library of plasmids from the Ron Weiss lab and human embryonic kidney (HEK) 293 cells. IANNs differ from traditional synthetic genetic circuits because IANNs can perform analog computations, rather than being limited to digital computations. IANNs are also universal function approximators–given an adequate number of intracellular artificial neurons, you can use an IANN to achieve any input/output behavior you’d like.
Overview | Concepts Learned & Skills Gained
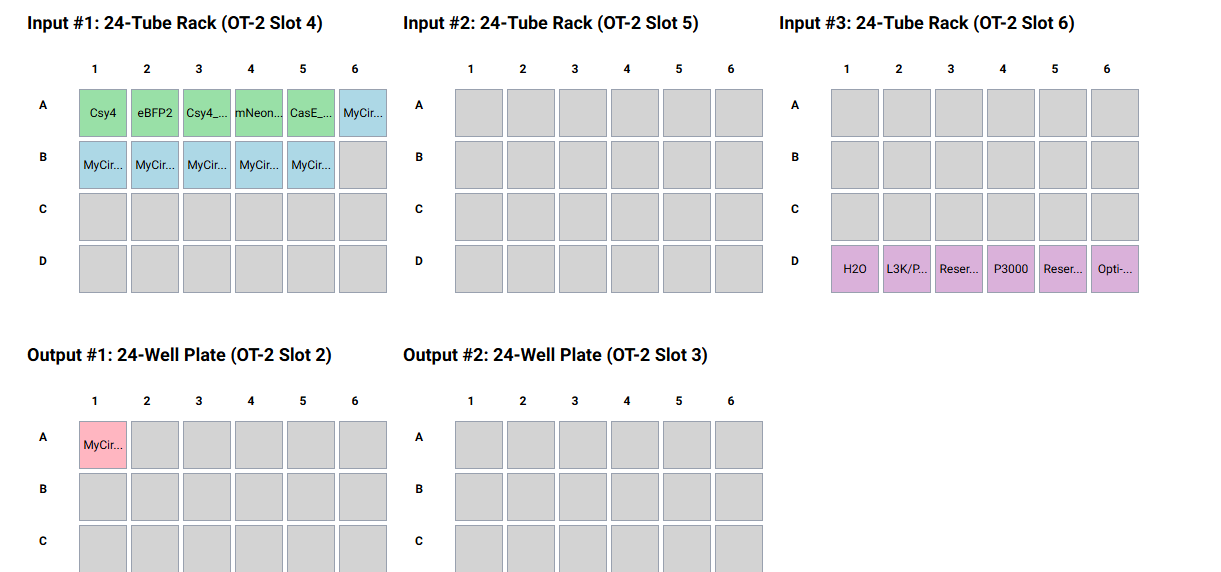
This is a lab with a dry and wet component. In the dry lab component, you will design a neuromorphic circuit in groups of 3. Once your design has been finalized, you will write instructions for an OT-2 to build your circuit for you. In the wet lab component, a TA will upload your OT-2 instructions and you will observe the OT-2 building and transfecting your IANN into HEK293 cells.
Circuit Assembling
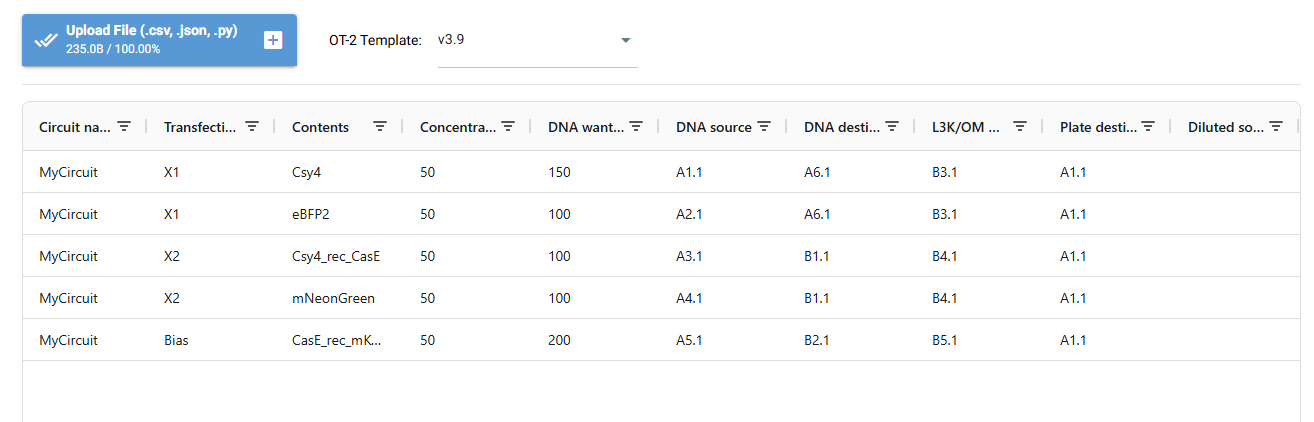
Genetic Circuit Parts Available
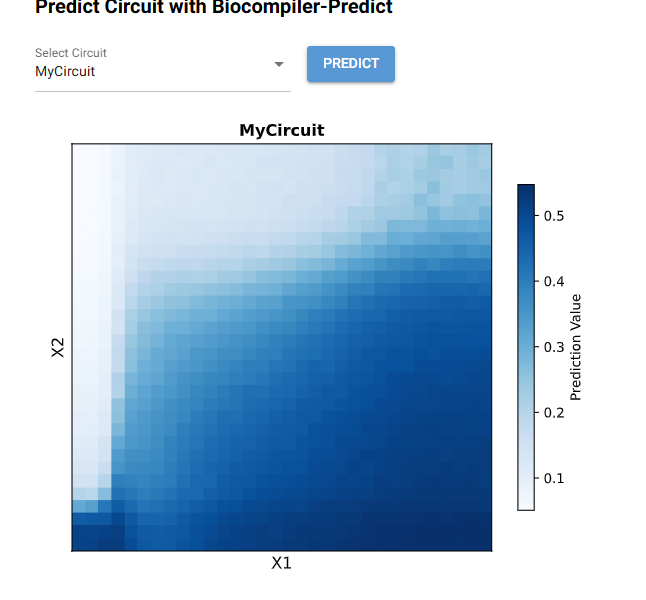
My Circuit
Experiment Layout
Prediction
When looking at the template, the designed I ended up creating has the same 2-layered genetic network with a bias input. THe circuit uses endoribonucleases to process signals inside the cell and flourescent proteins to report activity we are looking for.
Components Chosen:
X1
Csy4: first layer ern enzyme
eBFP2: flourescent marker for X1 (BLUE)
Csy4 acts as a sensor detecting upstream signals and process DNA.
X2
Csy4_rec_CasE: CasE expression is controlled by the sequences recognized by Cys4 from X1.
mNeonGreen:reports activity in X2 (GREEN)
BIAS
CasE_rec_mKO2: the bias helps tune the overall circuit response, giving a baseline signal and it not being stricly ON/OFF.
Week 12: Bioproduction of Beta-Carotene and Lycopene
Post Lab Questions | Mandatory for All Students
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively?
Lycopene: crtE, crtB , crtI
B-carotene: crtE, crtB, crtI, crtY (converts lycopene to b-carotene)
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene?
We need the antibiotic resistance gene because plasmids are not naturally mantained. This gene lets only plasmid containing e.coli survive ensuring the stable expression of the pathway for the pigment. Without the gene cells loose the plasmid, deleting the wanted pathway.
What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures?
Temperature: if we decrease the temperature (30°C) the growth rate slows down but give a higher pigment yield. If we increase to 37°C, we get a faster growth but could ahve less pigment due to misfolding or metabolic burden.
Media:
-2YT: get higher biomass and more precursor a vailability.
-LB: ok production
Fructose: Enhances the carbon flux boositng the isoprenoid pathway which gives more pigment.
Generally describe what “OD600” measures and how it can be interpreted in this experiment.
it measures light scaterring at 600nm. In this experiment is used to normalize pigment production of the cell, we interpret it as a porxy for the cell concentration. If theres a higher OD600 = high scaterring = more cells, lower OD600 = low scaterring = few cells.
What are other experimental setups where we may be able to use acetone to separate cellular matter from a compound we intend to measure?
Acetone can be used to extract lipids from cells, isolate chlorophyll from plants, precipitate proteins from small molecules in solution and extract carotenoids like in this lab. We can use acetone when we are targeting hydrophobic molecules.
Why might we want to engineer E. coli to produce lycopene and beta-carotene pigments when Erwinia herbicola naturally produces them?
Because E.coli grows faster and can be easily geneticallu manipulated, meaning it can be scalable. We trade the natural ability of herbicola for a a host thats efficient and easy to control.
Post Lab Questions | For Committed Listeners Only
Let’s get in touch with our metabolic pathway
What are the enzymes of the carotene pathway?
CrtE: produces GGPP precursor
CrtB: phytoene synthase
CrtI: phytoene desaturase-> carotene
CrtY: lycopene cyclase-> b-carotene
IspA: farnesyl pyrophosphate synthase
Dxs: MEP pathway entry
Within this pathway, which is the rate determining step (the step that takes the longest)? Which enzyme is responsible for this step?
Crtl, the desaturation step, because it is a multi step oxidation. It makes a 4 sequential desaturation reactions on a single substrate making it a bottleneck.
Notes for design of a DNA construct for bioproduction
The first thing to do is to decide what organism you are going to use for this (E. coli or S. cerevisiae) for production. Which would you choose and why (emphases on production differences)?
I would choose E.coli as it has a fast growth, is easy to clone and has a high short-term yield compared to s.cerevisae that takes a longer time and its more complex to manipulate and harder to scale up. The advantage with using yeast is the mevalonate pathway, that produces some isoprenoid precursors.
Now choose one of the enzymes and lets outline the parts of the construct for expression.
Crtl, as it is a limiting factor. the basic structure for the construct for the expression is composed by the promoter, RBS, crtl gene and a terminator.
Promoter: t7 as is extremely strong, and inducible(good for a high level production)
For E. coli lets create a expression vector that works as a plasmid you choose E. coli let’s create a expression vector that works as a plasmids.
Now, for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites, terminators, operators and promoters. The last ones are the most important in terms of enzyme or protein production. Let’s elaborate further on this biopart.
A promoter is a dna regulatory seq. upstream of a gene that binds with RNA polymerase to initiate transcription. Its function to determinate when, where and how much a gene is expressed.
What types of promoters do we have?
Thre are 4 types of promoters:
Constitutive: is always active, no regulation
Inducible: is activated by a signal/molecule
Repressible: Active by default, and its turned off by a repressor or metabolite
Synthetic: is engineered for tunability
If we wanted to turn off the transcription of a gene in response to a metabolite, what type of promoter would be most useful? What if we wanted this to increase in the presence of the metabolite?
If we wanted to turn off the transcription of a gene in response to a metabolite, we would need to use a repressible promoter like trp, which gets turned off with high levels of cellular tryptophan.
If we wanted to increase the transcription with the presence of a metabolite, we would need a inducible promoter. For example t7lac, that can be induced by IPTG.
Now choose one of the genes of the metabolic pathway previously described (Carotene/lycopene )and choose one enzyme to make an expression construct. What promoter could you use for this? Why did you choose it?
CrtI (phytoene desaturase)
Promoter: t7 as is strong and IPTG inducible.
Because it is an inducible promoter and strong is good for the use of producing carotene/lycopene production. We couñd first have a big dense first growth of cells and then induce them into the enzyme production.
It is a specific DNA sequence where replication is initiated. In plasmids it also can determined the copy number and its host compability.
What types of origin of replication do we have?
ORIs are categorized by location and function.
organism type
plasmid function
replication mechanism
eukaryotic timing and activity
Some type in bacteria are ColE1, pMB1 used for high protein expression, p15A that’s compatible with ColE1 plasmids, pSC101 has a low metabolic burden, and RSF1030, pBBR1, who have a broad host range.
(Extra) What are compatibility groups?
Compatibility groups are categories to classify plasmids that could coexist inside a bacterial cell. Plasmids with the same compatibility group cant coexist in the same cell because they have the same machinery for replication, meaning they’ll fight for it and the loosing one would dissapear.
Now for the previously chosen promoter and gene what will be the best origin of replication?
Maybe we could use pMB1 or ColE1, as they have a high copy number which ensures the abundant enzyme prodution.
(Mandatory for Global listeners, Optional MIT/Harvard) Elaborate further on other bioparts like RBS, terminators, operators you would use for a correct design and further bioproduction?
As we are using CrtI, we need a strong RBS LIKE B0034.
RBS: is located 5-10bp upstream of the start codon, its strength control the translation efficiency. After our rbs, we shoudl add a spacer sequences so the SD core is unsobstructed and present.
Terminators, signals RNA polymerase to stop transcription.
As we are using t7 for the promoter we are gonna used the same apporach for terminator to match to the rna polymerase.
Operators are dna sequences where repressor proteins bind to physically block transcription. we used LacO downstream our t7 promoter. LacI repressor protein would bind our lacO operator and block t7rna polymerase from proceeding with translation.
(Hot! Extra points) What are aptamers and riboswitches and how can they be used for metabolic tuning or engineering in prokaryotes?
Aptamers are short, structured RNA/DNA sequences that bind to specific molecules called ligands with high affinity and specificity.
Riboswitches are RNA (usually 5’utr of mrna) that contains an aptamer domain and an expression platform. When target binds the structure changes in the rna either turning on or off translation.
They can be used for tuning or engineering acting like biosensors and dynamically regulating the production. If lycopene precursors accumulate they can downregulate competing pathway enzymes, self balancing the circuit and keeping the production steady.
(Extra points) Now what approach can be used to join all these parts together? Make a quick analysis of their sequence in search of possibilities (search for restriction sites, etc)
there are 3 main strategies that can be used for the joining of the parts
restriction enzyme cloning, were we cut with restriction enzymes and then ligate the compatible ends.
gibson assembly, where we 3 enzymes in one reaction. Exonuclease creates the single stranded overhangs at the 5’ ends, polymerase fills the gaps and ligase joins it all together.
golden gate assembly, that uses type IIS restriction enzymes like BsaI and BsmBI, to cut outside their recognition sequence giving 4bp overhangs, that later are put together.
From our sequence, we looked fofr restriction sites and found where are multiple internal sites inside crtI gene, like NdeI, BamHI, PstI, and XbaI.
Given this and the other site found otuside our construct, it would be better to go ahead with Gibson Assembly because classic enzymes site are inside our gene of interested which blocks classical restriction cloning. XhoI and NcoI, are outside the construct but would still need the adding of restriction site to primers, making it a harder process. With Gibson we dont need any restriction sites, just design the 20-40bp overlappping regions between our parts and our backbone and the reaction would do the assembling seamlessly in one reaction.
(Extra Hot!!! Extra Points) Try to elaborate further on a biosynthetic pathway you would want to engineer in E. coli for production of a metabolite or product. What use could this bio-product have? Imagine dream applications!!!
Sangre de Drago / Dragon’s Blood Bioproduction
Sangre de Drago is a deep red latex-like sap that comes from Croton lechleri tree native to the Amazon. It has been used for ages by native people to aid cuts and reduce inflammation in skin. It has become popular in the skincare industry for antiaging and regeneration because of its potent antioxidant and collagen stimulating properties. This properties come from 2 bioactive compounds that stimulate wound repair mechanism, taspine and dimethylcedrusin.
AS its popularity grows Croton lechleri starts to be overharvest risking its extinction and with it history and tradition. If we engineered E.coli to produce this active compounds it gives us an alternative, letting the forest untouched and this indigenous knowledge honored and preserved. Both active compounds can be produced using phenylpropanoid/lignan biosynthetic pathways, as we start with phenylalanine which E.coli produces naturally. For the design we could use 2 different plasmids which different ORIs so they are compatible to divide the multiple reactions, as we need 9 different plant enzymes for this production.
This process helps with the cutting of a native forest and the preservation of tradition. At the same time, as we dont depend on the harvesting for this bioactive, we have more chances to
use this knowledge and innovate it. Just the overall creation of this procress giving us actives for wounds healing and repairing in a more accesible way, we could create in situ woudn repair bandages. Using this pathway with regulation mechanisms like riboswitches. This can be embedded into biocompatible hydrogel bandages that continuously produce the healing coumpounds in situ as it heals and self regulates thru the sensing of inflammatory markers. After its job is done, it degrades and dies once eveything is healed and close off. This would mean the creationg of a lviing self regulated wound healing bandage, producing its own medicine on demand based of centuries of indigenous pharmacological knowledge from Amazonian communities in Ecuador, Colombia and Peru.
(Extra points) For S. cerevisiae create an integration cassette for homologous recombination.
As well as for prokaryotes, eukaryotic DNA designs need bioparts used for construction of a function design and further expresion. Now search for a biosynthetic pathway if interested and describe one of the genes of the pathway.
Artemisinin production in yeast
In 2006, they created a possible way to mass produce artemisinic acid, a key precursor of the potent antimalaria drug artemisinin, from yeast rather than its harvesting from Artemesia annua, by transplanting the pathway into the S. cerevisiae. The yeast alreayd makes FPP naturally, but it was needed for a higher yield of it, this was done by the replacement of HMG1 for tHMG1, a truncated without feedback inhibition domain. 3 copies of this gene where added into the genome. After this, 4 genes of the plant where added, to recreate the pathway in plant into the yeast that uses FFP and transforms it into artemisinic acid, now being produce in higher levels.
Now, remember that for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites or Kozak sequences, terminators, and promoters. List the ones you could use for DNA design.
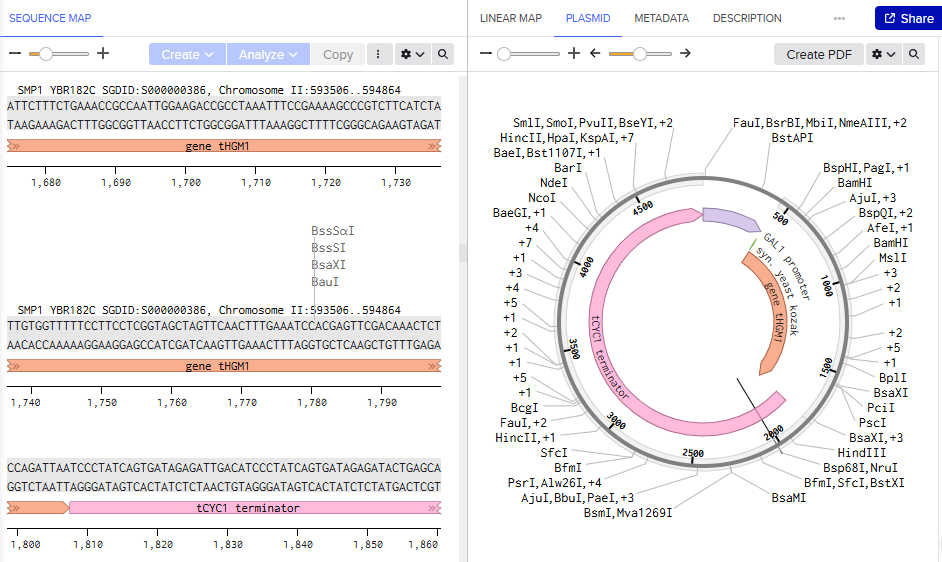
Promoter: the expression system uses the gal1 promoter region as it is inducible and repressible on glucose media. This works for the overexpression of tHGM1, as we need cells to first reach a high density and them start the pathway.
Once mRNA is made, the ribosome needs to find the startign point. On yeast ribosomes scans along it looking for an AUG. The sequence helps ribosome pause rather than skipping it.
As we want to recreate the pathway modification for the accumulation of FPP, we took the truncated HGM1, so it doesnt stop producing and we can have what we need to go ahead and get a higher yield of the pro active compound.
tCYC1, is a usual used terminator for yeast constructs. After the ribosome hits the stop codon in our gene, rna polymerase 2 is still running downstream so we need a sequence to stop. The terminator singals the clevage and polyadenylation of mrna to be done, so the mrna is stabilzief and ready to be exported.
In yeast engineering we use DNA construction designs for making genome integration. What chromosome site could you use for integration of these and why?
Genomic integration site is critical to choose while as it afects the expression stability and the gene effects that could disrupt essential functions. Some sites are HO locus- chromosome IV, auxotrophic loci, delta sites, easy clone sites, and rDNA locus. In actuality people choose to integrate it in easy clone sites thru CRISPR giving a markerless result. In the paper done in 2006(before CRISPR), it was used the auxotrophic loci, where 3 copies of the gene where integrated thru out homologous recombination.
(Hot! Extra points) Following the next chart of how a DNA integration cassette should be designed and with the previously chosen parts elaborate the DNA sequence you could use to synthesize with Twist.