Week 5; Phage Lysis Protein Design Challenge
Phage Lysis Protein Design Challenge
L-Protein Engineering (Mutagenesis)
- Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
L PROTEIN SEQ:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
https://www.uniprot.org/uniprotkb/P03609/entry#sequences
To create the following mutations, I combined the seuqnce conservation analysis with the experimental mutational data. Cluster gave a good signaling to the mutations found in wild types of the protein, giving a map to where to and not to make changes without distroyign completely the whole protein and its domains, which gives the lytic effect to it. I created the changes and then compared with the lab tested L protein mutants to see if this variations have already been investigated.

- Q33E
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQERSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- It is a change in the DNAJ interactive domain. We make a change from Q to a polar uncharged residue to E a negative one. This could disrupt the binding.
ptm":0.28,“iptm”:0.21
- R19K
METRFPQQSQQTPASTNRKRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- We are making a mutaltion in the interactive DNAJ domain. The change is conservative as we keep the characteristics of the aminoacis being positive charged.
“ptm”:0.26,“iptm”:0.19
- S35A
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRASTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- Change near the end of the soluble domain, the change of S TO A, makes a big change form a polar residue to a small non polar one.
“ptm”:0.25,“iptm”:0.17
- L59A
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSALEAVIRTVTTLQQLLT
- We chose this positon to create a mutation in the transmembrane domain. We change L TO A, reducing the side chain slightly and still keeping the hydrophobic property.
“ptm”:0.24,“iptm”:0.17
- R65L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRASTLYVLIFLAIFLSKFTNQLLLSLLEAVILTVTTLQQLLT
- Position 65, means we are making another mutation in the transmembrenae domain. We are changing R a charged residue to L, a hydrophobic one. This big change may improve the helix giving more stability to the pore and its lytic effect.
“ptm”:0.23,“iptm”:0.17
Alphafold2Multimer

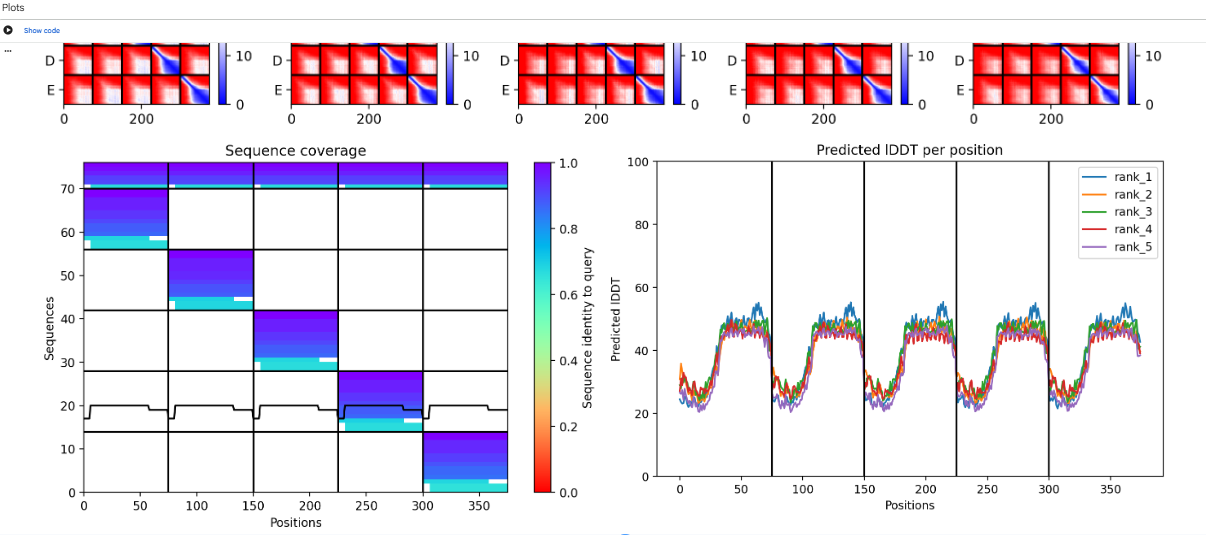
The computational design results tell us that the L-protein soluble domain (1–40) lacks a folding trigger, as evidenced by pLDDT scores <40. However, the transmembrane region (41–75) successfully forms a multimeric bundle. Future design iterations should focus on mutations that increase the pLDDT of the soluble domain or co-fold the sequence with DnaJ to observe if stability increases at the binding interface.