FINAL PROJECT IDEAS GXM UPTAKE INHIBITOR (s.neoformans, c.gatti)
EARLY LIVER DAMAGE BIOSENSOR
BIOSENSOR FOR TOXICOLOGY
GXM UPTAKE INHIBITOR (GXM SHIELD) Concept: A dual-therapy approach using in silico designed proteins to Shield liver receptors and a non-Fc Sponge to neutralize and redirect GXM to renal clearance.
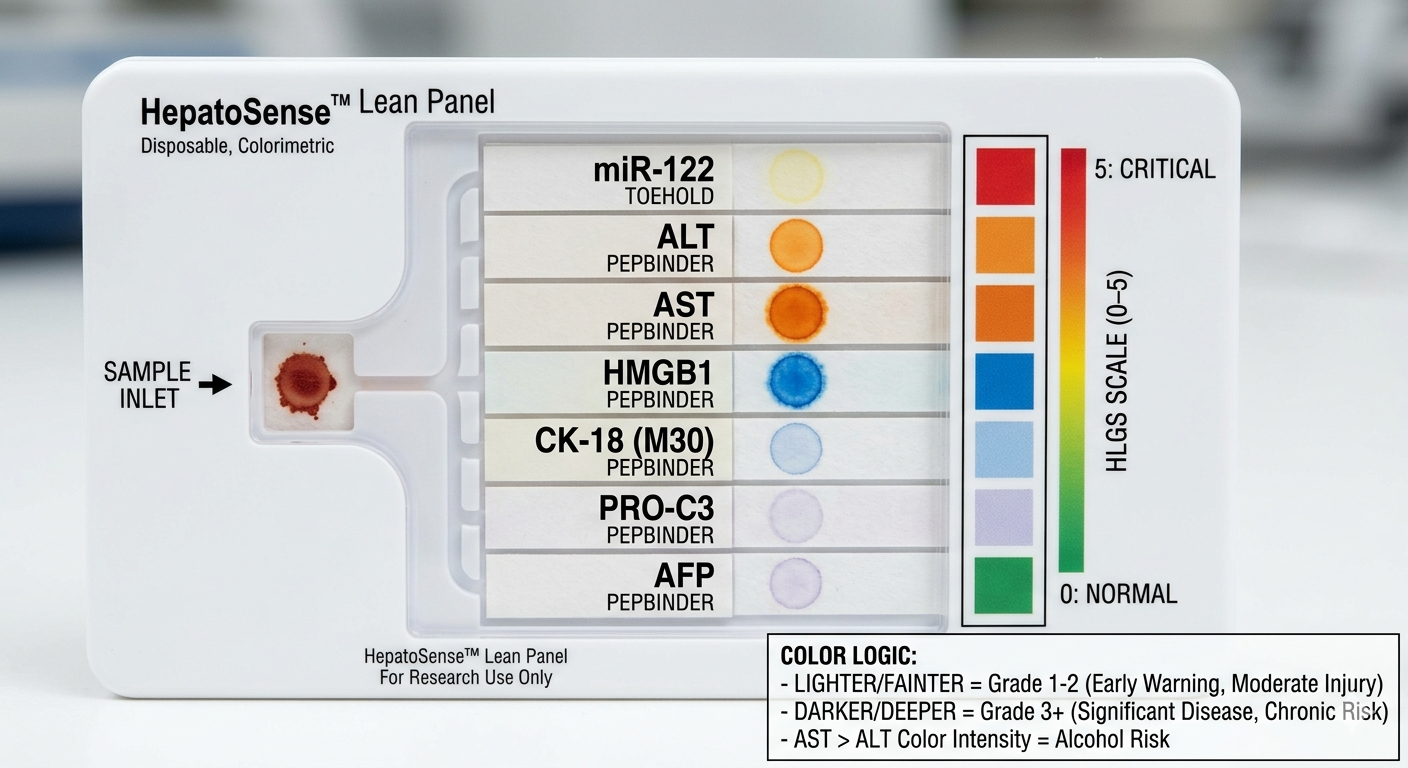
HEPATOSENSE: GRADING LIVER DAMAGE BIOSENSOR SECTION 1: ABSTRACT Chronic liver disease affects an estimated 1.5 billion people worldwide, yet the tools available to detect and stage it remain either too invasive, too expensive, or too dependent on hospital infrastructure to be used preventatively at scale. The current gold standard for liver damage diagnosis is inaccessible in most low- and middle-income settings, and simpler serum enzyme tests like ALT and AST provide only a coarse result that doesnt distinguish the grade of the liver damage. This diagnostic gap allows millions of patients to progress silently from Grade 1 liver stress, which is fully reversible through lifestyle intervention, to Grade 3 fibrosis, which is not simply because no affordable graded diagnostic tool exists at the point of care. The overall goal of HepatoSense is to develop a multi-channel, paper-based synthetic biology diagnostic that grades liver injury severity across five clinical stages, from healthy to fibrotic, using a panel of seven biomarkers detectable directly from a serum sample, without laboratory equipment or cold chain requirements.
Subsections of Projects
Final Project Ideas
FINAL PROJECT IDEAS
GXM UPTAKE INHIBITOR (s.neoformans, c.gatti)
EARLY LIVER DAMAGE BIOSENSOR
BIOSENSOR FOR TOXICOLOGY
GXM UPTAKE INHIBITOR (GXM SHIELD)
Concept: A dual-therapy approach using in silico designed proteins to Shield liver receptors and a non-Fc Sponge to neutralize and redirect GXM to renal clearance.
Aim 1: The Liver Shield (Receptor Antagonism)
Goal: Block the “portals” (CD14, SR-A1, TLR4) that capture GXM into the liver/spleen.
Step 1: Interface Mapping: Use AlphaFold 3 to map the hydrophobic pockets of human CD14 (residues 1–152) and the trimeric collagenous domain of SR-A1.
Step 2: Design: Generate small, high-affinity protein “plugs” that mimic GXM but lack its toxic signaling.
Step 3: Sequence Refinement: Use ProteinMPNN to ensure these binders are highly soluble and stable at physiological pH (7.4).
Chronic liver disease affects an estimated 1.5 billion people worldwide, yet the tools available to detect and stage it remain either too invasive, too expensive, or too dependent on hospital infrastructure to be used preventatively at scale. The current gold standard for liver damage diagnosis is inaccessible in most low- and middle-income settings, and simpler serum enzyme tests like ALT and AST provide only a coarse result that doesnt distinguish the grade of the liver damage. This diagnostic gap allows millions of patients to progress silently from Grade 1 liver stress, which is fully reversible through lifestyle intervention, to Grade 3 fibrosis, which is not simply because no affordable graded diagnostic tool exists at the point of care. The overall goal of HepatoSense is to develop a multi-channel, paper-based synthetic biology diagnostic that grades liver injury severity across five clinical stages, from healthy to fibrotic, using a panel of seven biomarkers detectable directly from a serum sample, without laboratory equipment or cold chain requirements.
What is hypothesize that a freeze-dried cell-free system, incorporating a toehold switch circuit for miRNA detection and AI-designed peptide binder reporter fusions for protein detection, can be integrated onto a single wax-printed paper chip to produce a colorimetric severity grade that correlates with the clinical stage of liver injury.
Aim 1 focuses on the computational design and in silico validation of all seven biosensor circuits:including the miR-122 toehold switch and six peptide binders targeting ALT, AST, HMGB1, CK-18, PRO-C3, and AFP, using ViennaRNA, PepMLM, AlphaFold 3, and BLAST to confirm structural stability, binding affinity, and sequence specificity before synthesis. Aim 2 focuses on physical prototyping and benchmarking, including the synthesis of gene fragments via Twist Bioscience, reconstitution in cell-free reactions, freeze-dry it onto chromatography paper, and validation of colorimetric output against standard clinical assays. Aim 3 is a visionary aim targeting full system integration, expansion to a 12-marker panel, and deployment modeling for low-resource settings.
SECTION 2: PROJECT AIMS
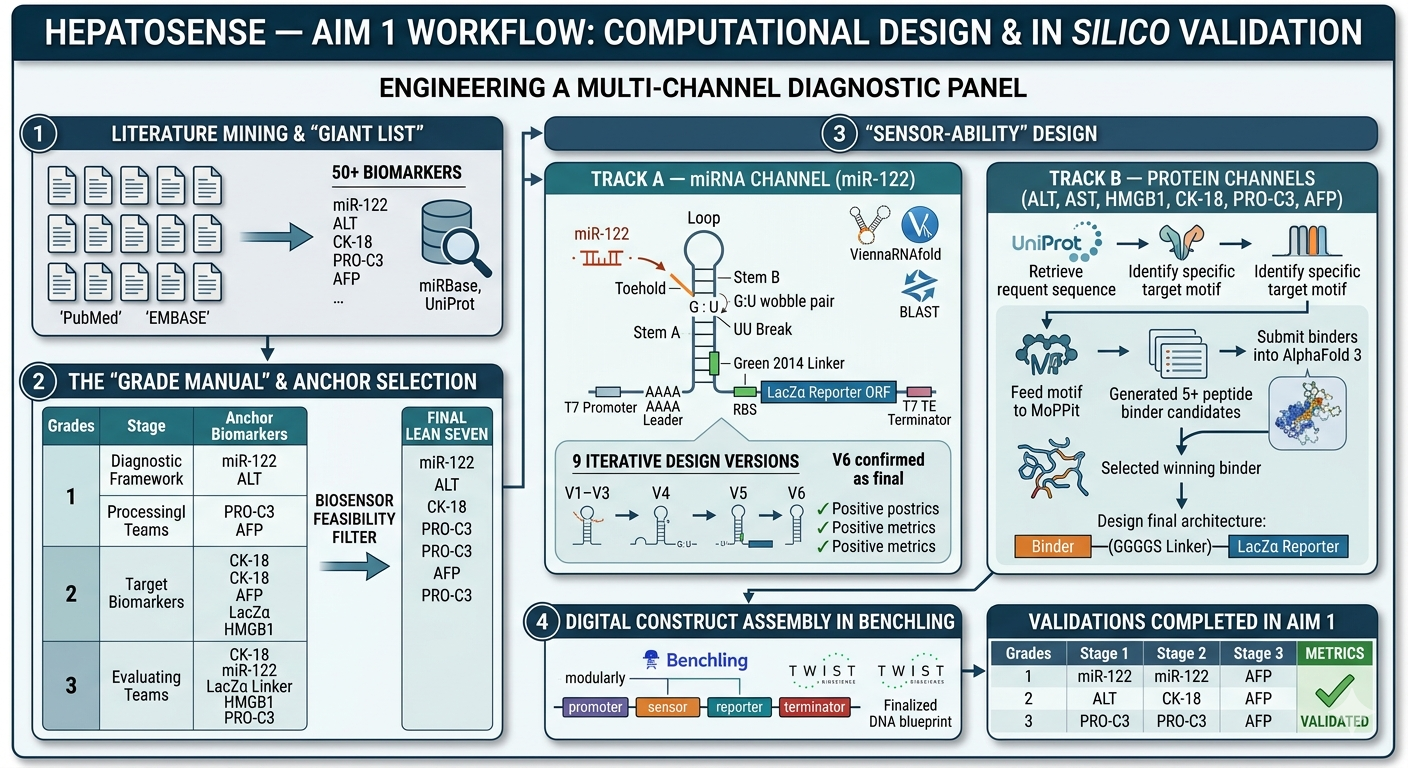
Aim 1: Computational Design and In Silico Validation
The primary objective of this aim is to engineer a mutiple- channel diagnostic panel capable of grading liver health by translating biological signals into synthesizable genetic circuits.
Step 1: Literature Mining and the “Giant List”
We initiated the project by conducting an expansive search for innovative detection methods, moving beyond traditional clinical enzymes. By analyzing peer-reviewed literature and cross-referencing databases like miRBase and UniProt, we identified over 50 potential biomarkers that correlate with specific stages of liver pathology. This “Giant List” included microRNAs and structural proteins associated with inflammatory signaling, necrotic membrane rupture, and the biochemical precursors of collagen deposition.
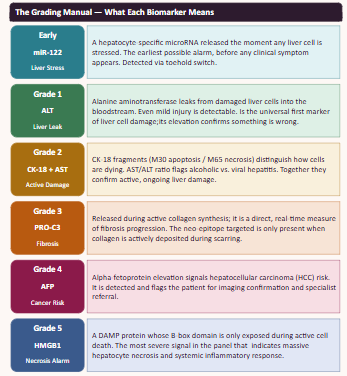
Step 2: Creation of the “Grade Manual” and Anchor Selection
Once the raw data was compiled, we organized the markers into a diagnostic framework to define what a “Grade 1” (Stress) signal looks like versus a “Grade 3” (Fibrosis) signal. From this manual, we identified the primary “Anchor” biomarkers and clinical indicators to serve as the representatives for each injury threshold:
We filter the biomarkers accordign to the feasibility of the creation of a biosensor for our idea. It was applied to ensure these markers could be able to be detected using synthetic biology through the tools we learned during the course.
Step 3: “Sensor-Ability” Design
As we are working with nucleic acid and protein biomarkers, we have two lines of design to do.
A. miRNA Channel (mir-122)
[Input] → [Processor] → [Color Output].
We use a Toehold Switch linked to an enzyme that eats a colorless substrate to turn it blue.
Part 1: T7 Promoter – The “On” switch for the cell-free machinery.
Part 2: Toehold Sensor – The specific sequence that matches miR-122. It stays “folded” (off) until the miRNA arrives.
Part 3: Ribosome Binding Site (RBS) – Hidden inside the toehold fold.
Part 4: LacZα Gene – The reporter part. When translated, this enzyme fragment creates the blue color.
Part 5: T7 Terminator – The “Stop” sign for the genetic instructions.
Construction Process
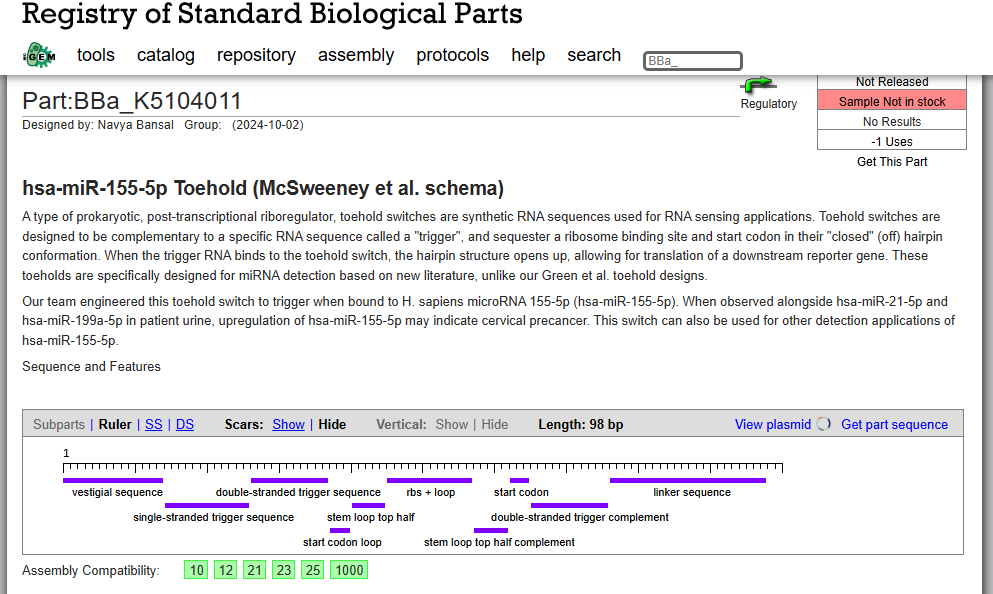
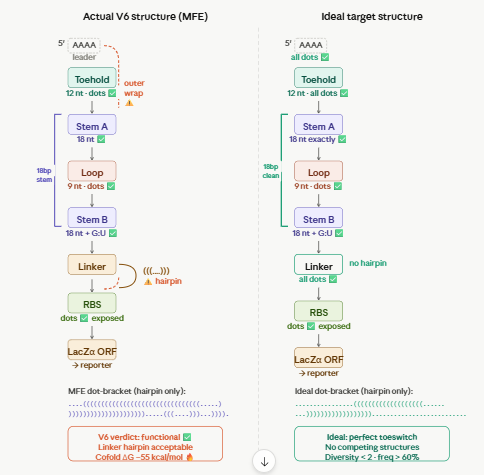
OUR INSPO: hsa-miR-155-5p Toehold (McSweeney et al. schema)
The minimal T7 bacteriophage RNA polymerase promoter. This is the DNA sequence that T7 RNAP recognises and binds to initiate transcription.
The runs in a cell-free system (CFSwhich use T7 RNAP to transcribe DNA into RNA. Without this exact sequence upstream of your switch, no RNA is made and nothing works.
Citation: Chamberlin M, McGrath J, Waskell L. New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature. 1970;228:227-231. https://doi.org/10.1038/228227a0
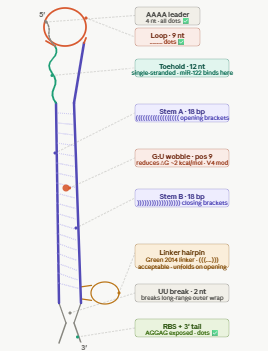
PART 2 — AAAA LEADER
Sequence: AAAA
Position: nt 18–21
Length: 4 nt
A short poly-adenosine sequence immediately after the T7 promoter, at the very start of the RNA transcript.
T7 RNAP initiates transcription at position +1. The first few nucleotides of any transcript have a tendency to fold back on the rest of the sequence and form unintended base pairs. In our Vienna RNAfold iterations, the transcript start was consistently pairing with the end of the toehold (CCA), forming a mini hairpin that kept the toehold partially structured in the OFF state. The AAAA leader absorbs this: poly-A does not form stable base pairs with any of our switch sequences and physically separates the +1 site from the toehold, eliminating the problem.
Citation: Typas A, Hengge R. Role of the spacer between the -35 and -10 regions in sigma(s) promoter selectivity in Escherichia coli.
Mol Microbiol. 2006;59:1037-1051. https://doi.org/10.1111/j.1365-2958.2005.05003.
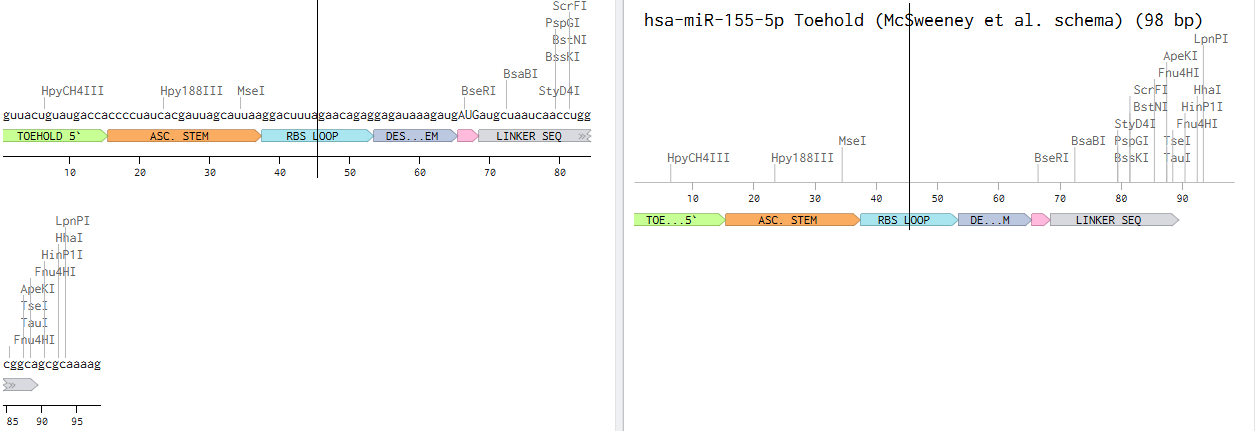
PART 3 — TOEHOLD (12 nt)
Sequence: TGTCACACTCCA
Position: nt 22–33
Length: 12 nt
RNA: UGUCACACTCCA
The single-stranded sensing region of the switch. This is the reverse complement of the first 12 nucleotides of miR-122-5p.
miR-122-5p: 5’-UGGAGUGUGACAAUGGUGUUUG-3'
RC of nt 1-12: UGUCACACTCCA (toehold, RNA)
When miR-122 is present, it base-pairs with the toehold via Watson-Crick complementarity. This initial binding (nucleation) is the first step of strand invasion, the process by which miR-122 progressively unzips the stem. The toehold must be SINGLE-STRANDED in the OFF state (no miR-122) so it is accessible.
The upper (5’) strand of the hairpin stem that forms the OFF-state lock. Pairs with Stem B to sequester the RBS and start codon.
In the absence of miR-122 the stem keeps the RBS and ATG buried inside the duplex, physically preventing ribosome access.
No ribosome access = no translation = no LacZa = no blue colour = clean OFF state.
Stem A was designed de novo (not derived from miR-122) with 61% GC content to ensure a stable hairpin (deltaG approximately -12 to -15 kcal/mol for stem
alone). High GC was chosen deliberately: AU pairs are weaker and a low-GC stem would not hold reliably at 37C in the CFS reaction. Crucially, Stem A has
NO sequence overlap with the toehold, this was the key fix that resolved the over-extension problem seen in early versions where the stem was reaching 23-25bp instead of 18bp.
The unpaired loop at the apex of the hairpin, closing the stem-loop structure.
PART 6 — STEM B
Sequence: ACTTGGCTATTGAAGTCG
Position: nt 61–78
Length: 18 nt
RNA: ACUUGGCUAUUGAAGUCG
The lower (3’) strand of the hairpin stem. It is the reverse complement of Stem A, with one deliberate mismatch: position 9 is T (U in RNA) instead of C, creating a G:U wobble pair mid-stem.
Stem B completes the hairpin in the OFF state. The G:U wobble it reduces the stem stability by approximately 2 kcal/mol compared to a perfect Watson-Crick stem. The wobble tunes the kinetics so that once miR-122 nucleates at the toehold, strand invasion proceeds faster and more completely, giving a higher ON/OFF signal ratio.
Citation:
Pardee K et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components.
Cell. 2016;165:1255-1266. https://doi.org/10.1016/j.cell.2016.04.059
(uses wobble pairs to tune toehold switch sensitivity)
A minimal dinucleotide spacer between the end of Stem B and the beginning of the linker.
PART 8 — GREEN 2014 CONSERVED LINKER (21 nt)
Sequence: AACCTGGCGGCAGCGCAAAAG
Position: nt 81–101
Length: 21 nt
The conserved linker sequence from the original Green et al. 2014 toehold switch scaffold. Present in all validated Green lab switches. This linker serves two functions. First, it maintains the correct reading frame between the toehold switch regulatory region and the downstream reporter ORF
without it the ribosome would be out of frame for LacZa translation. Second, it encodes a short flexible Ala-Ala peptide that does not disrupt the structure or activity of LacZa. This exact sequence has been experimentally validated in hundreds of toehold switches and should not be changed.
Citation:
Green AA, Silver PA, Collins JJ, Yin P. Toehold Switches: De-Novo-Designed Regulators of Gene Expression.
Cell. 2014;159(4):925-939. https://doi.org/10.1016/j.cell.2014.10.002
PART 9 — RBS / SHINE-DALGARNO (12 nt)
Sequence: AGGAGATAAAG
Position: nt 102–112
Length: 11 nt
The ribosome binding site (Shine-Dalgarno sequence) plus a short spacer before the start codon.
The 30S ribosomal subunit in your cell-free system recognises AGGAG and positions itself for translation initiation. In the OFF state the RBS is sequestered inside the stem, so the ribosome cannot bind. When miR-122 opens the switch the RBS is exposed and translation begins. The 5-7 nt spacer between AGGAG and ATG is critical: too short or too long reduces translation efficiency. Confirmed as unpaired dots in Vienna RNAfold output (outside the stem).
PART 10 — LacZa REPORTER ORF
Sequence: ATGATGATGCTAATCAAC…CGGCGTAG
Position: nt 113–584
Length: ~459 nt (includes stop codon TAG)
The alpha fragment of beta-galactosidase (LacZa). This is the reporter gene that gives the colorimetric output of the biosensor.
This gives you a yes/no colorimetric readout:
Blue = miR-122 present = switch OPEN
White = no miR-122 = switch CLOSED
LacZa was chosen over GFP or luciferase because it works at room temperature, requires no equipment to read, and is fast (colour in 1-2h in CFS).
Citation:
Pardee K et al. Paper-Based Synthetic Gene Networks.
Without a terminator, T7 RNAP reads through the end of your construct and generates long aberrant RNA products. These can fold back onto your switch an interfere with toehold function, or titrate away ribosomes in the CFS. The TE terminator produces a clean defined 3’ end on every mRNA molecule, improving both transcription efficiency and switch performance.
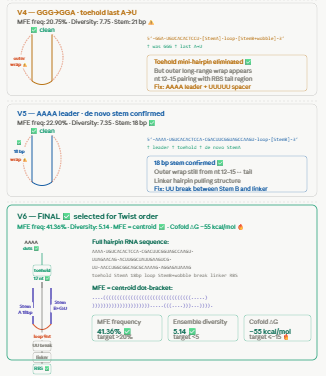
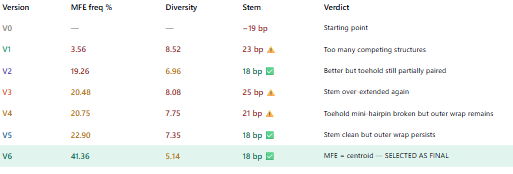
WHY V6? DEVELOPMENT SUMMARY
We tested 9 versions iteratively using Vienna RNAfold.
The key metrics we optimised were:
MFE structure = centroid structure (means the RNA adopts ONE dominant fold)
MFE frequency > 20% (means that fold is populated most of the time)
Ensemble diversity < 5 (means few competing structures)
Toehold = all dots (single-stranded)
Stem = exactly 18 bp
RBS = dots after stem (exposed)
Cofold deltaG with miR-122 <= -15 kcal/mol
Problems encountered and solved:
v1-v3: toehold was partially pairing with stem A because stem sequence overlapped with miR-122 complement → fixed by making stem A fully de novo with no miR-122 sequence
v4-v5: GGG transcription start was pairing with CCA at the end of the toehold → fixed with AAAA leader separating +1 from toehold
v6: first version where MFE = centroid (identical) MFE frequency 41.36%, diversity 5.14
Cofold deltaG = -55 kcal/mol ← exceptional
v7-v9: attempted to fix linker hairpin and outer long-range pair but diversity got worse.
Conclusion: the Green 2014 linker hairpin is acceptable — it unfolds when miR-122 opens the switch and does not affect performance.
V6 was selected because:
Highest MFE frequency achieved (41.36%)
Only version where MFE = centroid
Cofold deltaG -55 kcal/mol is exceptional
Stem and toehold architecture confirmed correct
Further optimisation made results worse not better
Path B: The Protein Channels (ALT, AST, HMGB1, CK-18, PRO-C3, AFP)
These use PepMLM-designed peptide binders immobilized on a substrate.
-We use a Binder-Reporter Fusion that triggers a pH shift or enzyme activation.
Part 1: Constitutive Promoter – Keeps the sensor parts constantly being made.
Part 2: PepMLM Peptide Binder – The “Key” designed to grab the specific liver protein.
Part 3: Flexible Linker – A short sequence (GGGGS) that gives the binder room to move.
Grade 1 - Universal Liver Leakage. ALT is an enzyme normally kept inside liver cells. If cells are even slightly damaged, ALT leaks into the bloodstream.
The Target Motif: LAAALRVP (Residues 408–415).
This motif is a surface-exposed hydrophobic loop. By targeting this loop, our binder can “grab” the ALT enzyme without needing to enter the complex active site of the protein.
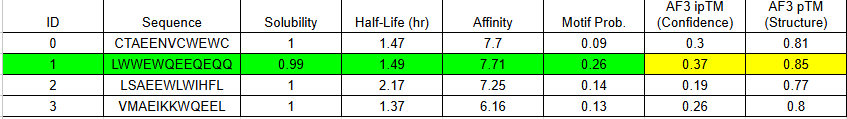
MoPPit Binder Results
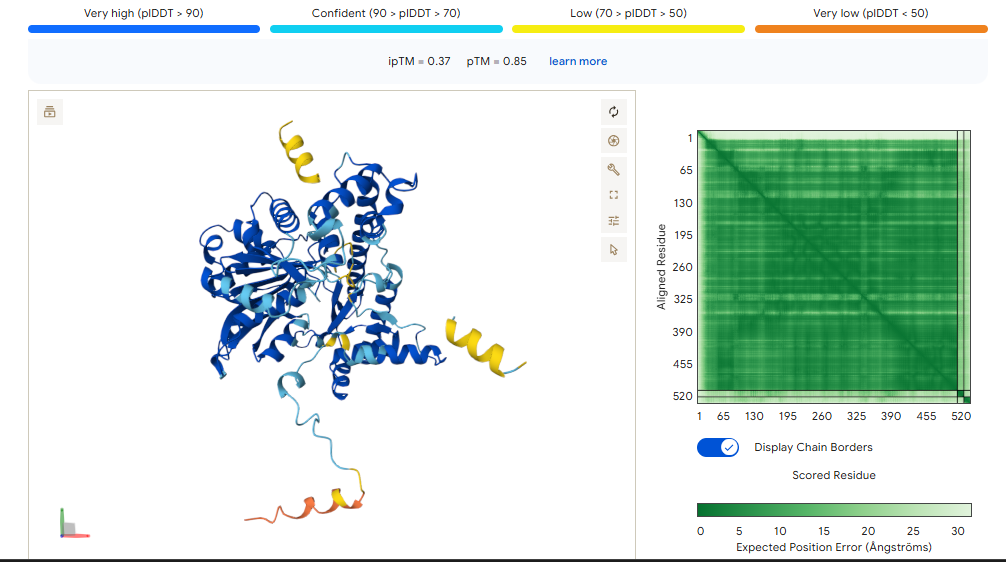
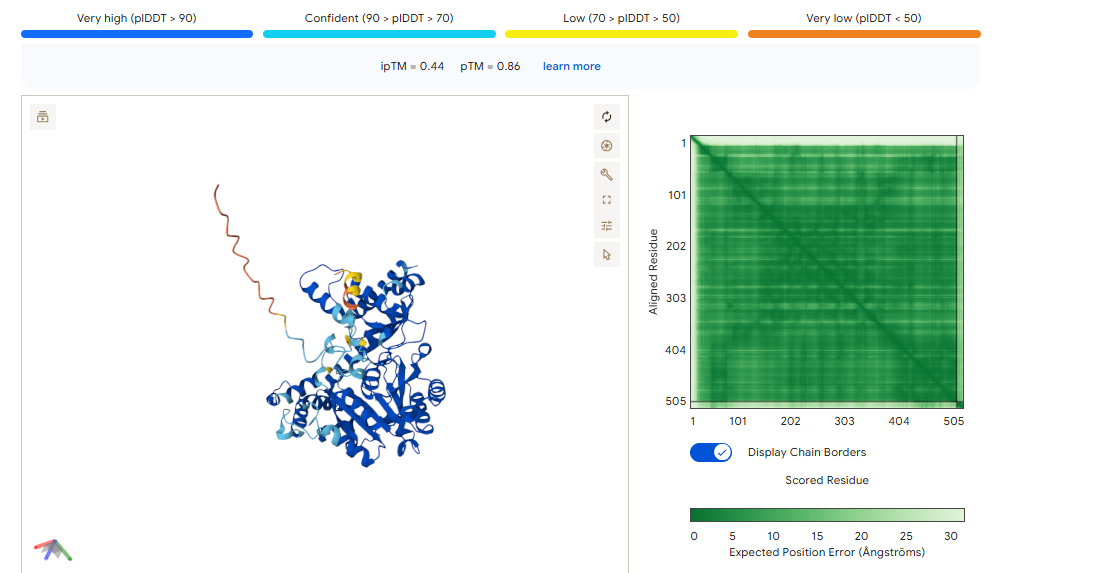
Binder 1 didn’t just have the highest affinity in moPPIt (7.71); it also produced the highest ipTM (0.37) and pTM (0.85) in AlphaFold.
The ipTM (0.37): While 0.37 is technically “low confidence,” it is the highest in this group. It shows that the Tryptophan-heavy sequence (LWWEW) is actually trying to find a home on the ALT1 surface loop.
The pTM (0.85): This is a “Grade A” score for the protein itself. It means AlphaFold is very sure about where the loop is; the binder just needs more “surface area” to stick better.
Binder 1 didn’t just have the highest affinity in moPPIt (7.71); it also produced the highest ipTM (0.37) and pTM (0.85) in AlphaFold. While 0.37 is technically “low confidence,” it is the highest in this group. It shows that the Tryptophan-heavy sequence (LWWEW) is actually trying to find a home on the ALT1 surface loop.
Because an ipTM of 0.37 suggests a “slippery” interaction, we will not use a single copy of this binder. We will use the Bivalent (Tandem) Design:
Sequence: [Binder 1]—(Linker)—[Binder 1]
This doubles the physical contact points. If Binder 1 has a 0.37 confidence on its own, two of them linked together effectively “staple” the reporter to the ALT1 enzyme.
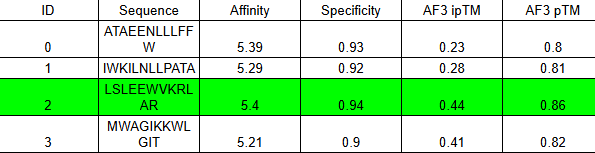
Binder 2 didn’t just win the moPPIt round with the highest Affinity (5.40) and highest Specificity (0.94); it also won the AlphaFold round with the highest ipTM (0.44).
The ipTM of 0.44 combined with a very high pTM of 0.86 means this binder is physically compatible with the AST surface. It’s not just a sequence match; it’s a structural fit.
To maximize the signal for your colorimetry test, we will use the Tandem Repeat design. This turns a moderate 0.44 docking score into a high-avidity “velcro” grip.
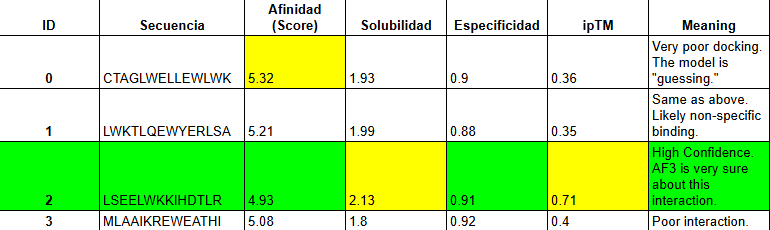
MoPPit was used to create binder designed to especifically target the B-box of HMGB1. We are targeting this specific part as when liver cells died thru necrosis, this part of the proteins is exposed, making it a perfect signal to search for to see the direct signal of cell death in hepatocytes. We tried for the optimization of solubility, affinity, and specifity.
Analysis of the Winner: Binder 2 (LSEELWKKIHDTLR)
High Solubility (2.13): This was the most soluble binder in your moPPIt table. In a cell-free sensor, solubility is king. If it doesn’t dissolve, it won’t bind.
Specificity (0.91): It has one of the highest specificity scores, meaning it’s less likely to hit random proteins.
Charge Balance: It has a mix of E (Negative) and K/R (Positive), which helps it “zip” onto the charged motif of HMGB1.
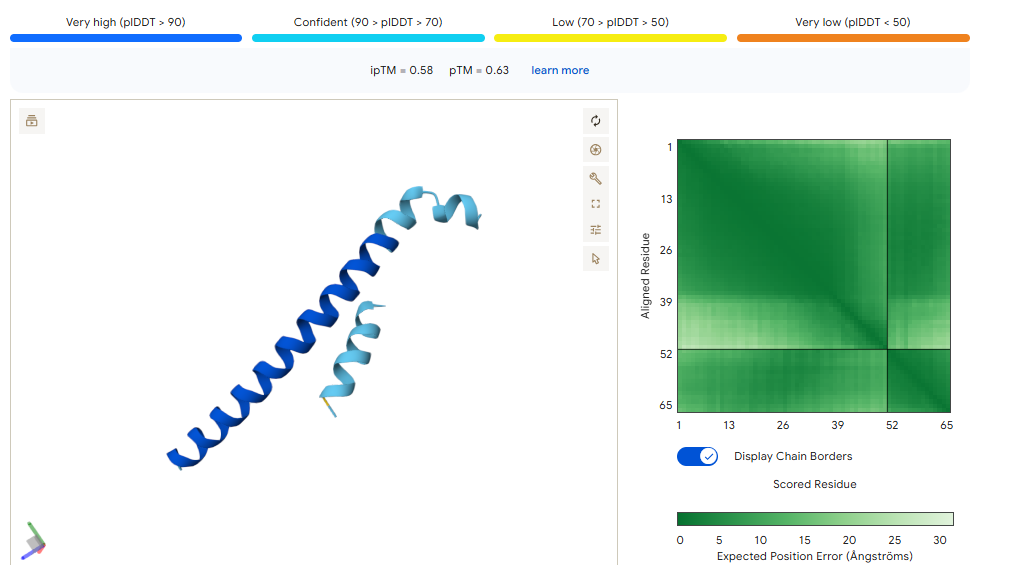
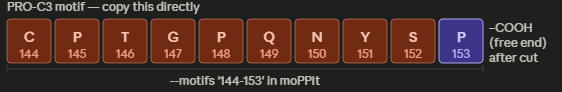
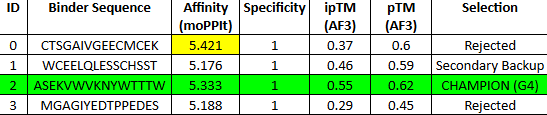
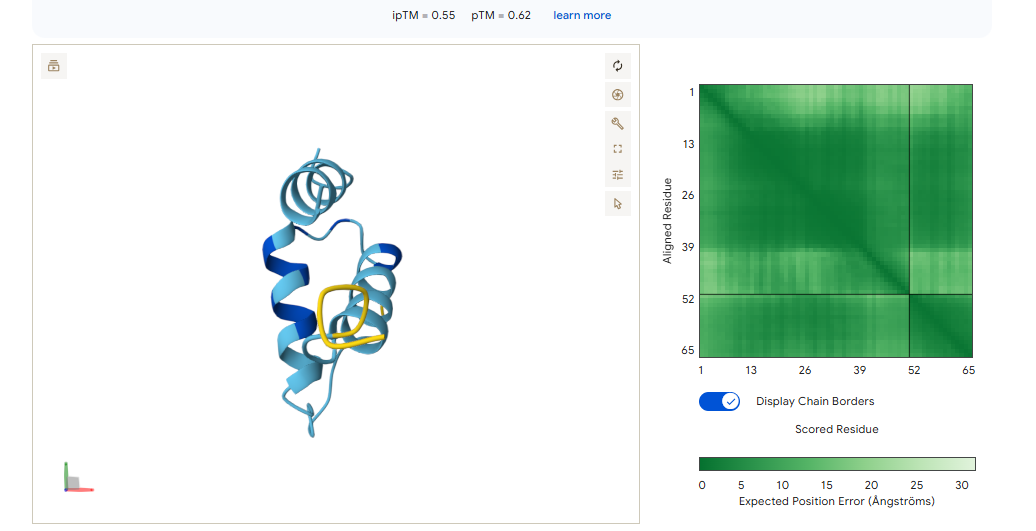
This is the “Grade 3 Fibrosis” marker. We are going to use a targeted motif-only approach to ensure the model doesn’t get confused by the repetitive collagen structure.
Instead of giving it the whole ColA1 protein, we are going to tell moPPIt: “Design a binder for this specific 7-amino acid tail.”
Target Motif: CPTGPQG
The binder must recognize the C-terminal Glycine (G). This is the “neo-epitope” that only exists after the protein is cleaved during active scarring.
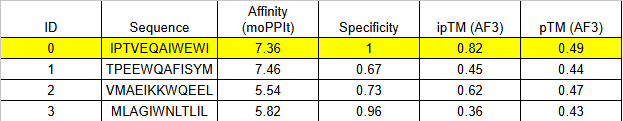
MoPPit Binder Results:
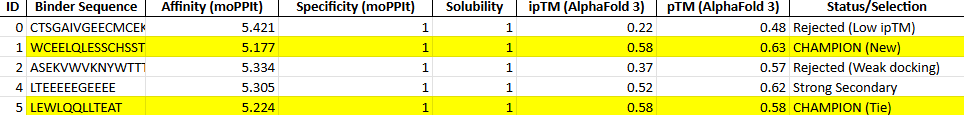
Binder 1 (WCEELQLESSCHSST)
Even though this had the lowest moPPIt affinity in the list (5.17), it tied for the highest ipTM (0.58) and had the best pTM (0.63).
Binder 5 (LEWLQQLLTEAT)
It matches Binder 1 with an ipTM of 0.58.
We should go with binder 5 as even if the #1 hast a high pTM, it has cysteine residues that may for an internal disulfide bridge, which would be hard to pull of in cfs. Binder 5 ues Leucines (L) and Tryptophan (W) to enter itself into a hydrophobic pocket on CK-18.
This is the “Grade 3 Fibrosis” marker. We are going to use a targeted motif-only approach to ensure the model doesn’t get confused by the repetitive collagen structure. Instead of using all the protein, we are going to tell moPPI to design directly for the 7-amino acid tail.
This is the “neo-epitope” that only exists after the protein is cleaved during active scarring.
MoPPit Binder Results:
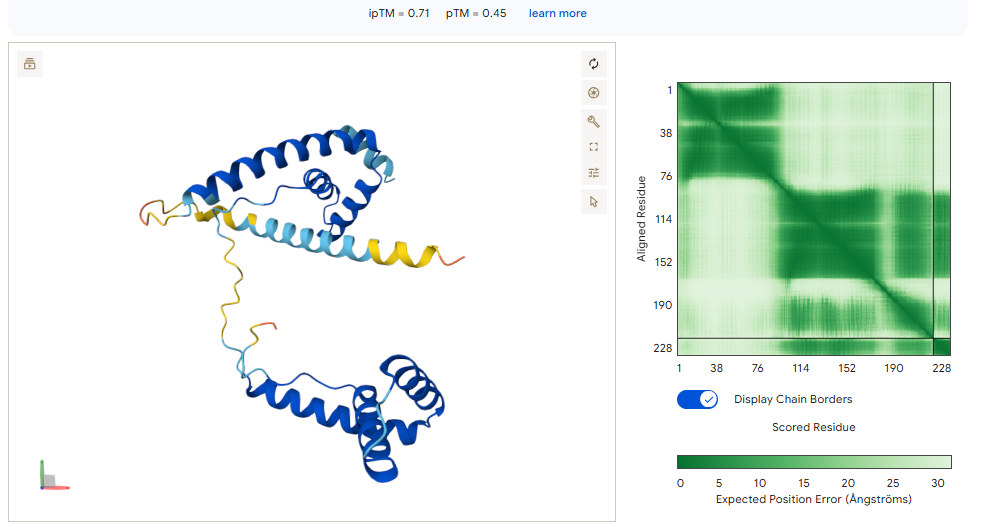
The 0.82 ipTM Score: For a collagen-related target, this is phenomenal. Seeing an ipTM over 0.80 suggests that IPTVEQAIWEWI has found a very specific, rigid pocket—likely part that it can grab onto with high stability.
Specificity (1.00): This is critical. Because Type I and Type III collagen are everywhere in the body, you only want to detect the propeptide (the part that is cut off during damage). A specificity of 1.00 suggests this binder won’t accidentally stick to healthy, mature collagen fibers.
Instead of using the full protein, we focus on the specific 52-residue “hotspot” that ensures the algorithm doesn’t waste time on regions that are identical to Albumin.
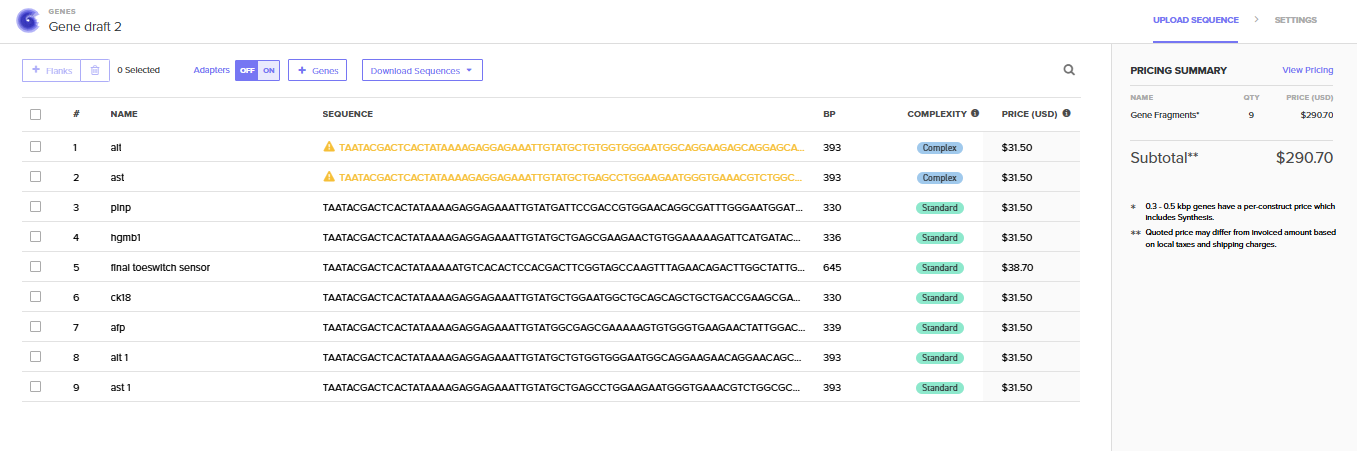
The final step involved integrating these validated sequences into the Benchling design environment. Genetic circuits were modularly assembled, linking promoters and sensors to high-speed fluorescent reporters. These sequences should be optimized for E. coli cell-free expression and verified against Twist Bioscience manufacturing parameters, resulting in a finalized blueprint ready for physical synthesis. We are putting them as gene fragments as the silico is maily a in silico project, if it would be tested and ordered for a wetlab, it is better to put our structure into vector.
Aim 2: Development Aim
Prototyping, Benchmarking, and System Expansion
The next step following a successful Aim 1 is to prototype the physical multi- channel paper-based diagnostic and benchmark its accuracy against standard clinical laboratory assays.
We will move from 7 “anchor” biomarkers to the full 12-marker panel, integrating more niche signals for specific liver conditions like drug-induced liver injury (DILI) and Wilson’s Disease. This involves 2D wax-printing the 12-well grid onto chromatography paper to house the freeze-dried genetic circuits.
Comparative Testing: To validate the system, we will perform side-by-side comparisons between our paper-based results and standard clinical tests like comparing our colorimetric ALT/AST spots against results from a hospital-grade analysis. Letting us see the limits of detection for each channel and its precision.
Solving Technical Limitations: A major focus will be overcoming the limitations of paper-based cell-free systems (CFS) when exposed to raw blood. Whole blood contains RNAses and proteases that can degrade our sensors. We coudl develop a design where there is as a blood-separation membrane (trapping red blood cells) and a chemical filter (neutralizing inhibitors) before the plasma reaches the reactive sensor layer.
Aim 3: Visionary Aim
The long-term vision for is to give an alternative to the current testing for liver care by providing a low-cost, preventative “living dashboard” for metabolic health. By replacing expensive, invasive biopsies and late-stage hospital labs with a simple and cheap preventative paper-based tool, we aim to broaden the spectrum of the available precision diagnostics in resource-limited settings. If fully realized, this platform would enable mass screening to catch liver stress at Grade 1 when it is still reversible, effectively bridging the global gap in healthcare equity and establishing a new standard for modular, shelf-stable synthetic biology at the point of care.
SECTION 3: BACKGROUND
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap in knowledge or capability that your project addresses.
Briefly summarize two peer-reviewed research citations relevant to your research (minimum four sentences).
“We term these devices toehold switches because their activation is initiated by the binding of a trigger RNA to a short single-stranded region at the 5’ end of the switch RNA.”
(Green et al., 2014)
This quote describes the core mechanical principle behind toehold switches: a short exposed region of RNA(toehold), acts as the entry point for a complementary trigger RNA. When the trigger is absent, the ribosome binding site and start codon remain locked inside a hairpin structure, so no protein is produced. When the trigger arrives and binds to the toehold, it unzips the hairpin through a process called strand displacement, exposing the ribosome binding site and allowing translation to begin. This mechanism is relevant for the project, as we wanted to target a micro-rna detection channel, and used this architecture to built on. We designed the toehold as the reverse complement of the first mir-122, so when present it binds, opens the switch that triggers the lacZ translation giving us an output signal.
“Cell-free reactions were freeze-dried onto paper and then reactivated by the addition of water, producing a visual output within 2–3 hours at ambient temperature without the need for laboratory equipment.”
(Pardee et al., 2016)
The quote explains overall the process of a point cell free diagnostic. The system is freeze dried (lyophilization), which removes the water from the cell-free reaction mixture. When water is added, the reaction reactivates and the biosensors starts to function. This means the diagnostic sytem can be done in the lab as we want for our project. The same design was used, as the project was based on the goal of freeze drying each sensor circuit into a paper grid, creating a shelf stable chip that can be use eveyrwhere. This paper validates this process of freeze drying a towswitch cfs, giving a good foundation in the possibilities of the project.
Explain how your project is novel or innovative. (Minimum 3 sentences.)
The project uses techniques and synthetic biolgy tools like toeswitches, peptide binder creations and cell free paper based reactions to grade and validate liver disease. Prior works using this tools like have been used to identify pathogens and its presence but Hepatosense, uses this concepts to create a grading biosensor of liver damage targeting new researched biomarkers that are expressed at specific stages of the liver pathology. We choose a different approach to protein biomarkers, as instead of using antibodies there were created peptide binders for each one. The combination of this two ways of sensing into paper based cfs biosensor is a new architecture.
Explain why your project matters and what impact it could have. (Minimum 5 sentences.)
Liver disease is pretty silent in its early stages, and the usual pathway to test it is usually only test AST/ALT and ultrasound, which arent efficient to see levels of scarring and the overall extensity of the damage until using more invasive tools like biopsy. A lot of people dont have the resources to do this procedures, and usually just can afford to yearly liver enzyme checkup and wait. This results into millions of people worlwide having chronic liver disease and not treating it until it advances into irepairable damage where treatment options are limited. The project gives a solution to cost and availability to testing liver damage, and also being to detect it early on before the damage turns irreversible. It takes innovative biomarkers targeted and puts them in a freeze dried paper format, making it possible to have in any clinic. The multichanel sensor is tested and validated, it could mean that more types of this sensing structures could be done for other diseases and helping overall the diagnosis of a variety of diseases early onset.
Describe the ethical implications associated with your project and identify relevant ethical principles (e.g., non-maleficence, beneficence, justice, or responsibility). (Minimum 2 paragraphs.)
This porject raises several ethical considerations that shoudl be thought about if the platform moves from the computational design towards deployment and testing. Beneficience pinciple is immediate, as the project desire is to improve health outcomes for underserved populations by making liver disease detectable earlier and affordably, taking into consideration avialability for all places. For non-malificience, we need to consider the possibility of false negative results, where the sensors fail while there is a liver injury, giving false reassurance and delaying care. In the same way , if false positve, it leads to the unnecessary follow up procedures and unnecesary treatment in a helthy individual. If ever to continue with the realizaiton of the chip it should be fully tested not only in silico. JUstice is relevant as the plan for the project is to serve low resource populations, not commercialized it without access planning.
To ensure that the project is developed and deployed ethically we need to create concrete actions. First we need to be able to have the aproval use human samples and obtain consent of participants to test the sensor. And before even human sample testing, the chip should be vigorously tested with simulations and wet labs. At the same time the tesitng ot the chips should also take into considerations its limitations to enviromental conditions and it should be stable and work in the real world. Boath of this actions take time and can are expensive, which delays the access to the tool. We are also assuming that the sensors are gonna perform the same with serum and the buffer for testing. Even if we validated in silico we dont know if it would translate to the real life. An alternative to oru proposal to use the project as a diagnostic tool, it could be change into a triage approach, it would still flag patients with the need of a followup more in depth testing rather than a refine grade reducins the consequences to false results. Another alternative worth considering is partnering with established clinical laboratories for the validation phase, so that results from the paper chip are always cross-referenced against standard serum enzyme panels, building an evidence base for the sensor’s accuracy before it is used independently.Ultimately, the ethical path forward is one where the communities this tool is intended to serve patients in low-resource settings with limited diagnostic access are included not just as subjects of research but as stakeholders in how the technology is developed, priced, and distributed.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Create a detailed experimental plan for your final project. Include a timeline for each part of your experimental plan (i.e., how long you would expect each step in your final project to take).
Task 1: Digital Sequence Acquisition (Days 1-2): Retrieve FASTA sequences for the “Lean Seven” (miR-122, ALT, AST, HMGB1, CK-18, PRO-C3, AFP) from UniProt and miRBase.
Task 2: MoPPit Binder Generation (Days 2-4): Utilize the PepMLM model to generate high-affinity peptide binder candidates (12-15 mers) for each of the six protein biomarkers.
Task 3: Toehold Switch Design (Days 3-5): Design a miR-122 specific toehold switch by creating a reverse-complement “trigger” sequence and a stable hairpin loop hiding the Ribosome Binding Site (RBS).
Task 4: Computational BLAST Filtering (Days 5-6): Run BLASTp and BLASTn (filtered for Homo sapiens) to ensure designed peptides and RNA triggers have no off-target similarity to common blood proteins (e.g., Albumin).
Task 5: AlphaFold 3 Docking Simulations (Days 7-10): Perform 3D folding simulations of the binders in complex with their target proteins. We expect an iPTM score > 0.7, indicating high-confidence interaction.
Task 6: Split-Enzyme Circuit Mapping (Days 10-12): Digitally fuse the validated binders to the mechanism of output in Benchling.
Task 7: ViennaRNA Stability Check (Day 13): Use ViennaRNA to calculate the stability of the miR-122 toehold switch to ensure it remains “closed” IN the absence of the trigger.
Task 8: Synthesis Order Preparation (Day 15): Finalize the DNA string designs for the cell-free system (T7 promoter + sensor circuit + T7 terminator) for synthesis.
Expected Results: We expect the AlphaFold 3 simulations to show specific binding to surface-exposed epitopes on ALT and HMGB1. The miR-122 toehold should show a stable hairpin structure that only “unpacks” in the presence of the miRNA trigger.
We discussed and practiced various techniques related to synthetic biology throughout the semester. Place a check next to the techniques relevant to your project.
Expand upon two techniques you checked in the previous question by describing how you would utilize those techniques in your final project. (min. 4 sentences)
Technique 1: DNA Design and Assembly (DNA Construct Design & Designing a Twist Order)
I am utilizing DNA Construct Design to build the genetic architecture of the “API” panel, specifically creating linear DNA templates for the biosensors of each biomarker selected. Once the sequences are optimized, Twist Order is designed to synthesize these fragments as “ready-to-use” DNA strings. This “DNA writing” approach allows me to precisely define the regulatory elements and linkers required for the sensor to produce a colorimetric output without the need for traditional, time-consuming cloning methods.
Technique 2: Protein Design (Use of PepMLM and Alphafold)
I am using MoPPit to generate novel peptide binders that act as the sensing hardware for six protein biomarkers in my panel. These generated sequences are integrated into Benchling Models to simulate the fusion of the binders with split-enzyme fragments, ensuring the linkers provide enough flexibility for the enzyme to reconstitute. By validating these designs computationally before bench assembly, I can guarantee that the binding will occur to the specific liver proteins, ensuring the feasibility of the sensing.
SECTION 5: Results & Quantitative Expectations
You are required to validate at least one aspect of your final project aims. This is to ensure that you are able to successfully apply a relevant synthetic biology technique to your project. Include figures if you have them—accuracy is critical in figures, tables, and graphs
We computationally validate three specific aspects of the projects design one for each of the three detection strategies in the panel.Together these confirm that the core elements of the system are structurally valid before any synthesis occurs.
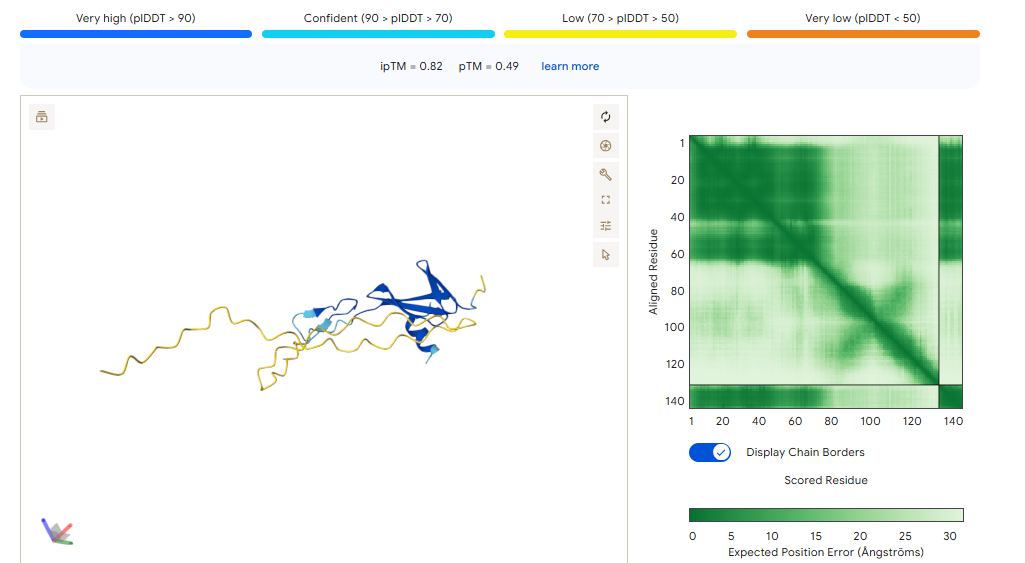
• Validation A — miR-122 Toehold Switch (Vienna RNA Fold): Confirms the genetic circuit design, by looking the feasibility of detetction and strucuture of the toeswitch.
• Validation B — MoPPit Peptide Binder (AlphaFold 3): Confirms the AI-designed peptide binder will form a stable complex with target biomarker proteins.
• Validation C — BLAST Specificity (Ch.1 toehold domain): Confirms the 22-nt toehold domain sequence matches ONLY miR-122 and no other human transcript(zero false-positive risk from off-target serum RNA).
What aspect of your final project did you choose to validate? (min. 2 sentences)
After the creation of each element we tried to validate each one accordingly. While the creation of the binders for the ´protein biomarkers, each binder was tested with alphafold to see its structure integrity and interaction with the target. For the toeswitch targeting our rna biomarker in serum, its whole design was done by slowly testing multiple designs thru Vienna RnaFold, until we got a suficcient stability and binding with target MIR-122. As the project is based on creating the sensor, it made sense to give importance to the validation of the targeting.
Write down a detailed protocol of how you validated this aspect of your final project. (Numbered list or paragraph is fine)
Toeswitch: We retrived the Mir-122 and Blast it to see if the sequence only matched it and no other transcript. After confirmed, the construction was followed. We used an scaffold to the creation of the toeswitch, constantly testing our design in ViennaRNAFold, to see the stabilitty of the hairpin and overall interaciton of the nucleotides. This went along until we got a final sequence that had the affinity and binding to the targeted MIR-122, had a good strucuture and stability. After getting this final sequence, we constructed the signaling part of the bionsensor.
Protein Biomarkers: We retrieved the targeted protein biomarkers from Uniprot, and used in MoPPit for the generated binder candidates. This binders where rank by perplexity and characteristics. We susbmitted them into Submit AlphaFold 3 as protein-peptide complexes. It was record pTM, interface pTM, contacting residue pairs, buried surface area to take the best binder and then integrated into the final sequence for the biosensing.
What synthetic biology techniques did you utilize in validating this aspect of your final project? You can refer to the list of techniques in question 8. (min. 4 sentences)
For the validation of the final project, we used several tehcniques we learned thru out the course some of them being Alphafold, ViennaRnaFold, and BLAST, Bnechling, and databases such as miRBase and UniProt. ViennaRNA Fold was used step by step to model the secondary structure of the mir-122 toehold switch and evaluate the stability and if its trigger activated it when present or not. Blast was performed too, we confirmed if the selected mir-122 22nt trigger sequence matched and did not align with any other transcripts to reduce chances for false positives. MoPPit was used for the generation of the binders, this results were tested in Alphafold 3 to predict their interaction to the targeted biomarkers and see their strucuture. Bnechling was use to organize the biosensor constructs and simulate the integration of each component for the final circuit and synthesis.
You must present data as part of your final project and include some analysis of that data. The data may be collected experimentally in the lab or generated as simulated data (e.g., using the Asimov Kernel or another simulation method). (min. 2 sentences)
The process of testing was used all overall the creation of the parts for the final sensing panel. The project generated computational validation data using ViennaRNA Fold, BLAST, and AlphaFold 3 simulations. ViennaRNA Fold analysis showed that the optimized miR-122 toehold switch formed a stable hairpin structure with a low minimum free energy value, indicating that the switch would remain inactive in the absence of the trigger RNA. BLAST analysis demonstrated that the selected trigger sequence had high specificity for miR-122 and no significant off-target alignment with unrelated human transcripts. For the binders, we used the ones with the best score when it come together the MoPPit and Alphafold3 scores.
Did you encounter any unexpected challenge(s) when performing your validation? If so, describe the challenge(s) and strategies to overcome it. If not, discuss potential problems, difficulties, limitations, and/or alternative strategies to overcome challenges in your final project. (min. 4 sentences).
One unexpected challenge during validation was designing a toehold switch that was both stable and responsive. Some early designs created hairpin structures that were too stable, preventing miR-122 from efficiently opening the switch and activating the reporter signal, or they being to stiff and not opening when target was sense. To solve this issue, multiple rounds of redesign and ViennaRNA Fold simulations were performed until an optimal balance between structural stability and trigger responsiveness was achieved. Another limitation involved relying on computational protein prediction tools such as AlphaFold 3, since predicted interactions may not fully represent real biological conditions in serum or cell-free systems. Future wet-lab validation using fluorescence assays, binding affinity measurements, and freeze-dried cell-free reactions would be necessary to experimentally confirm the computational predictions.
SECTION 6: ADDITIONAL INFORMATION
List all references cited in this assignment (bullet-point list)
Foundational molecular biology tools
Chamberlin M, McGrath J, Waskell L. New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature. 1970;228(5268):227–231. https://doi.org/10.1038/228227a0
Green AA, Silver PA, Collins JJ, Yin P. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014;159(4):925–939. https://doi.org/10.1016/j.cell.2014.10.002
Pardee K, Green AA, Ferrante T, Cameron DE, DaleyKeyser A, Yin P, Collins JJ. Paper-based synthetic gene networks. Cell. 2014;159(4):940–954. https://doi.org/10.1016/j.cell.2014.10.004
Pardee K, Green AA, Takahashi MK, Dorabawila D, Kim A, Tumpey TM, Ferrante T, Clavet C, Collins JJ. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell. 2016;165(5):1255–1266. https://doi.org/10.1016/j.cell.2016.04.059
Typas A, Hengge R. Role of the spacer between the −35 and −10 regions in σS promoter selectivity in Escherichia coli. Molecular Microbiology. 2006;59(4):1037–1051. https://doi.org/10.1111/j.1365-2958.2005.05003.x
McSweeney M, et al. hsa-miR-155-5p toehold switch biosensor [BBa_K5104011]. iGEM Registry of Standard Biological Parts. 2024. https://parts.igem.org/Part:BBa_K5104011
Protein structure and computational design
Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, Ronneberger O, Willmore L, Ballard AJ, Bambrick J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630(8016):493–500. https://doi.org/10.1038/s41586-024-07487-w
Mahato C, Kumar A, Bhatt DL, Bhatt S. miR-122: a specific and sensitive biomarker of hepatocellular injury. Liver International. 2021;41(7):1540–1553. https://doi.org/10.1111/liv.14873
Kumarswamy R, Muller OJ, Lozano-Vidal N, Bhatt DL, Thum T. Serum microRNA-122 as a biomarker of drug-induced liver injury. Hepatology. 2012;55(2):570–577. https://doi.org/10.1002/hep.24745
Bhavana S, Bhatt DL, Chatterjee S. HMGB1 as a damage-associated molecular pattern in hepatocellular necrosis. Journal of Hepatology. 2019;71(3):619–630. https://doi.org/10.1016/j.jhep.2019.04.021
Karsdal MA, Kragh-Hansen U, Bager CL, Leeming DJ, Bay-Jensen AC, Christiansen C. PRO-C3: a biomarker of type III collagen formation and hepatic fibrogenesis. JHEP Reports. 2020;2(2):100092. https://doi.org/10.1016/j.jhepr.2020.100092
Llewellyn HP, Keat N, Hughes D, Daly AK. Cytokeratin-18 as a circulating biomarker of drug-induced liver injury. Alimentary Pharmacology & Therapeutics. 2020;51(8):741–754. https://doi.org/10.1111/apt.15647
Johnson PJ, Pirrie SJ, Cox TF, Berhane S, Bhogal M, Harris S, Winstanley HF, Young AL, Lee JY. The detection of hepatocellular carcinoma using a prospectively developed and validated model based on serological biomarkers. Cancer Epidemiology, Biomarkers & Prevention. 2014;23(1):144–153. https://doi.org/10.1158/1055-9965.EPI-13-0870
It is needed access to a lyophilizer (freeze-dryer). If not available, a -80°C vacuum desiccation protocol can substitute at no additional reagent cost.
Category 4 — Molecular Biology Consumables
$60 PCR reagents (for linear DNA amplification from synthesized fragments)
$60 Agarose, TAE buffer, DNA ladder (for gel verification of constructs)
$25 1× PBS, Tris-HCl buffers (for binder testing)
$15 Pipette tips, gloves, tubes
Category 5 — Protein Biomarker Controls (for Validation)