Week 4 HW: Protein Design Part 1

South American Rattlesnakes (Crotalus durissus terrificus) with Crotamine protein
Part A: Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- I intake approx. 5 * 1023 Daltons of amino acids when ingesting 500 grams of meat. This is based off results indicating I ingest approx. 1021 Daltons of amino acids when ingesting 1 gram of meat
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Humans eat beef but don’t become cattle, and eat fish but don’t become fish because genetic information from the lifeform being ingested isn’t transferred wholesale. Much of the genetic material being eaten is broken down during digestion and more importantly a human beings’ cells follow instructions derived from their DNA. Human beings’ cells utilize amino acids from the lifeform being ingestion, but perform this utilization according to specific genetic instructions. The lifeform being ingested and its amino acids are the raw materials the cell uses for various means.
- Why are there only 20 natural amino acids?
- There are several broad reasons why there are only 20 standard natural amino acids. The first reason is that early on in the history of evolution, this group of amino acids became more or less ’locked in’, meaning that once the basic relationship between three letter codons and these 20 standard natural amino acids became widely distributed across the kingdom of life, it becamde too risky/dangerous from an evolutionary standpoint to alter this core set. Another reason is that the group of 20 gives enough range in structure and chemistry to build a large chunk of what evolution or directed evolution might desire. The other reasons seem to amount to various types of evolutionary trade-offs. Adding more than 20 amino acids to this standard set would add additional, potentially unwanted complexity, while decreasing the number of amino acids in the set might lead to issues with a lack of uniqueness with amino acids side chain sharing, which would in turn limit the functional flexibility of amino acids to do things like fold precisely.
- Can you make other non-natural amino acids? Design some new amino acids.
- Yes you can. My attempts to design some new amino acids usng SwissSideChain and the Cryo-EM structure of Receptor Tyrosine Kinase ROS1 PDB file in PyMol (open-source) are shown below:
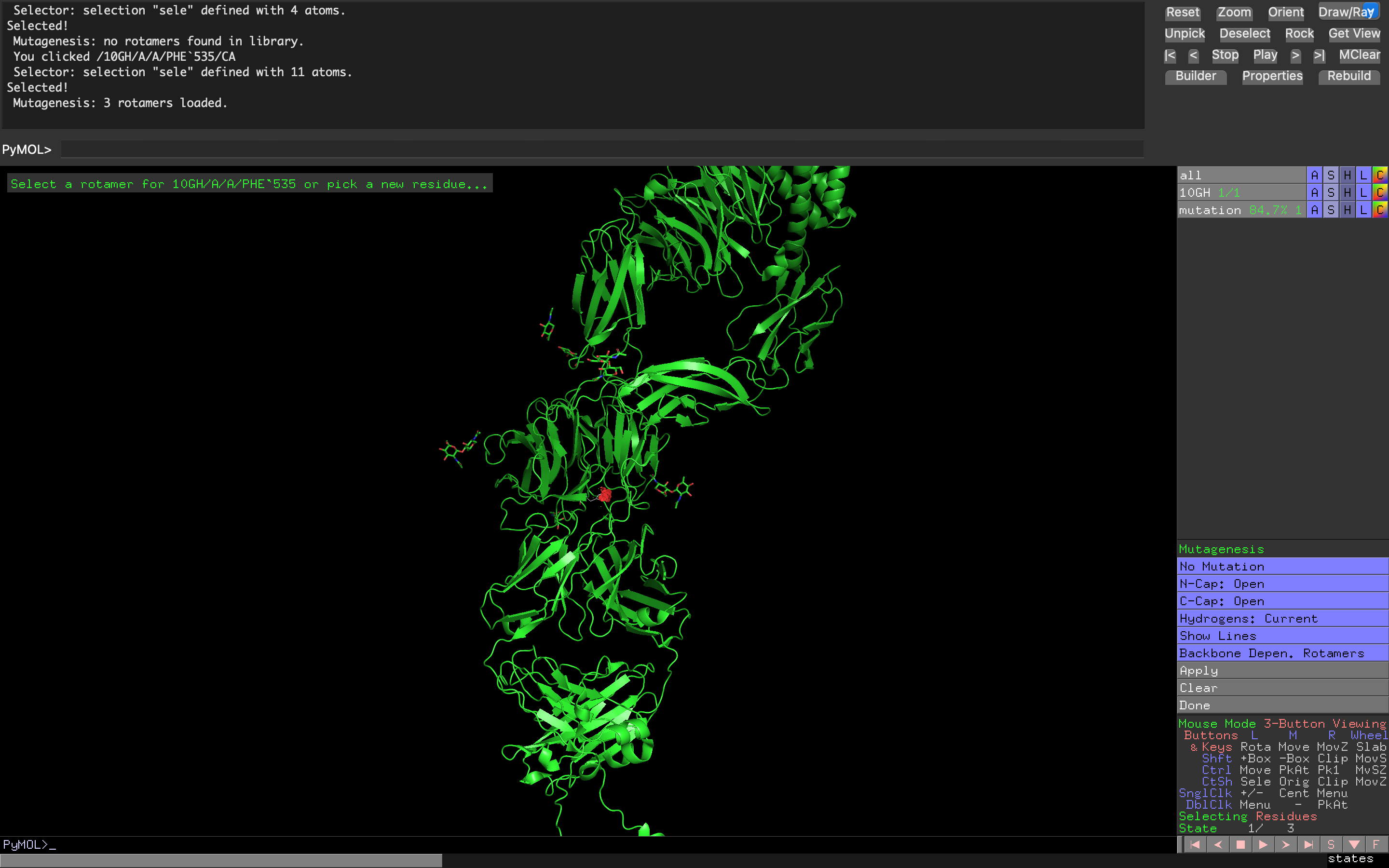 Attempt at creating a non-natural amino acid residue mutation of Tyrosine Kinase ROS1 using cyclohexanecarboxylic acid
Attempt at creating a non-natural amino acid residue mutation of Tyrosine Kinase ROS1 using cyclohexanecarboxylic acid
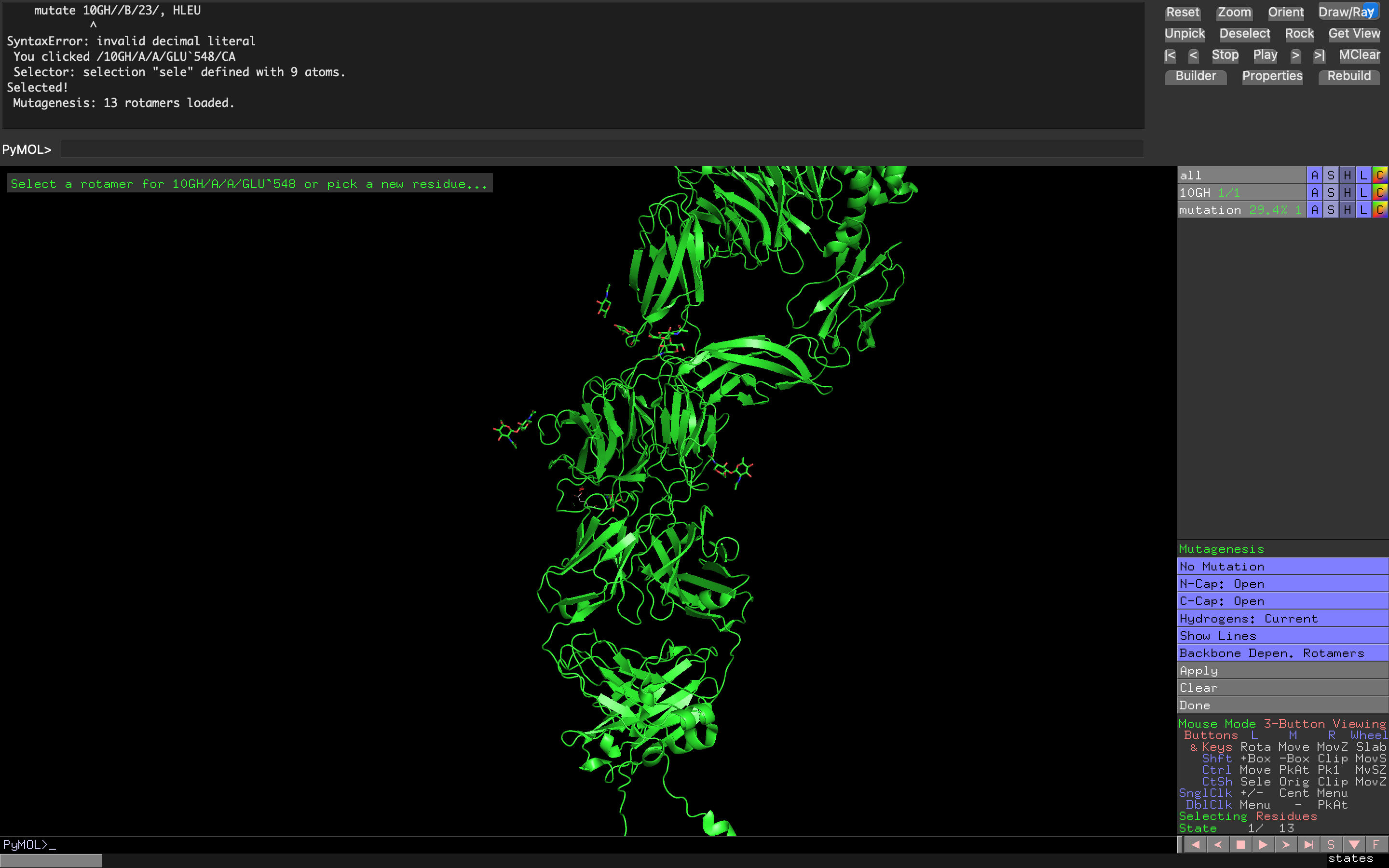 Attempt at creating a non-natural amino acid residue mutation of Tyrosine Kinase ROS1 using cyclopropanecarboxylic acid
Attempt at creating a non-natural amino acid residue mutation of Tyrosine Kinase ROS1 using cyclopropanecarboxylic acid
- Where did amino acids come from before enzymes that make them, and before life started?
- Amino acids come from metabolic molecules within the cell. These metabolic molecules consist of carbon atom chemical backbone, inorganic nitrogen, and enzyme-facilitated chemical reactions. Before life as we know it started, amino acids originated from abiotic (not from living organisms) chemical reactions on Earth before the emergence of life as we know it. The chemical reactions occurred in the atmosphere, hydrothermal and oceanic vents, and via meteorite and comet (i.e., extraterrestrial) delivery
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- If I made an a-helix using D-amino acids, I’d expect left handedness because the majority of alpha helices are built from L-amino acids, which exhibit right handedness.
- Can you discover additional helices in proteins?
- Yes additional helices can be discovered in proteins. These can include non-alpha helices and even π-helices. These can be discovered via x-ray crystallography, structure databases, or with the aid of AI prediction tools.
- Why are most molecular helices right-handed?
- Most molecular helices are right-handed because L-amino acides force the helix backbones to conform to right-handedness. Selection pressures also favors this form of handedness in the helix for hydrogen atom bonding and overall functionality
- Why do β-sheets tend to aggregate?
β-sheets tend to aggregate because the edges of β-sheets have exposed nitrogen and hydrogen (donor) atomic pairs and carbon-oxygen pairs (acceptor) atoms. What happens is that when sheets get in close proximity these atomic pairs snap together, almost like a zipper.
- What is the driving force for β-sheet aggregation?
- The driving force for β-sheet aggregation is the attraction of the donor and acceptor atomic pairs
Supporting prompts for this section listed below:
- What is the driving force for β-sheet aggregation?
| Supporting Prompt | Model |
|---|---|
| What is a Dalton as a unit of measurement? | Perplexity |
| How many molecules of amino acids are located within a single gram of edible meat, on average? Do NOT hallucinate when answering this question | Perplexity |
| What is Avogadro’s number and why is it relevant within the context of the answer to the previous prompt? What does mol mean in the context of g/mol? Do NOT hallucinate when answering these questions | Perplexity |
| 10^{21} * 500 equals how much? | Perplexity |
| From a biological and genetic standpoint why is it that when an organism ingests food, they don’t become the organism they’re ingesting (i.e., if a human being eats salmon, why don’t they become a salmon)? Where do amino acids fit into this explanation? Do NOT hallucinate when answering these questions | Perplexity |
| Why are there only 20 amino acids found in nature? Do NOT hallucinate when answering this question | Perplexity |
| To elaborate on a point made in the 5th part of the answer to the previous prompt: what is a side chain in the context of amino acids? Do NOT hallucinate when answering this question | Perplexity |
| Share me resources for designing non-natural amino acids rather simply/with relatively little friction or extensive technical knowledge | Google AI |
| How can I quickly design a non-natural amino acids? What internet resources exist to help me do this? | Google AI |
| Having issues downloading SwissSidechain into PyMol. I’m on a MacOS. Do I need Python downloaded on my local machine for successful downloading of SwissSidechain? Do NOT hallucinate when answering these questions | ChatGPT |
| It’s telling me it’s installed but that Swiss Sidechain is Not Loaded. I downloaded the SwissSidechain package. Please tell me how to proceed | ChatGPT |
| I’m using ‘3.1.6.1’, 3.0, 3000000, 1749441553, ‘963e47f43382e009c2cd391f0747a8c20ef108e7’, 0’ of PyMol | ChatGPT |
| Getting ModuleNotFoundError: No module named ‘swisssidechain’ | ChatGPT |
| When I go to select the SwissSidechain folder from my Downloads, I only can open the file. I can’t select the entire thing. Is that an issue? | ChatGPT |
| Yeah when I try to just select the entire folder from PyMol I can’t do. How should I proceed with the install? | ChatGPT |
| Ok. Now I have only a .zip and am able to select it. Getting the following error: “Plugin “PySwissSidechain2” has been installed but initialization failed. Tell me how to proceed | ChatGPT |
| It says ‘Unable to find license file’. Not understanding what’s going on here | ChatGPT |
| No I don’t have a PyMol license | ChatGPT |
| How do I install Conda on Mac? Is Miniconda downloadable on the web? | ChatGPT |
| Should I download the graphical or command Miniconda install? | ChatGPT |
| Yes | ChatGPT |
| Getting “zsh: command not found: conda” | ChatGPT |
| For this code, how do I find my username? Users/ | ChatGPT |
| Getting “zsh: no such file or directory: /Users/j_d_r/miniconda3” | ChatGPT |
| Yes | ChatGPT |
| Getting “Do you accept the Terms of Service (ToS) for https://repo.anaconda.com/pkgs/main? [(a)ccept/(r)eject/(v)iew]:” What should I write? | ChatGPT |
| Getting “PackagesNotFoundError: The following packages are not available from current channels: - pymol” | ChatGPT |
| Ok | ChatGPT |
| Ok. I’ve opened up the Open-Source version of PyMol and installed SwissSideChain. Now tell me how to mutate some residues from some Protein Data Bank (PDB) structures based on one of the non-natural L- or D-sidechains of the SwissSidechain database. Basically I want to create some graphical representations of non-natural amino acids using SwissSideChain in the Open-Source version of PyMol and I would like you to provide me a step by step process for how to get these visuals (when the visuals are created I also want to be able to know their names and other relevant information) | ChatGPT |
| What does a PDB structure mean within the context of proteins? | Perplexity |
| What’s a common protein downloaded from RCSB PDB? | Perplexity |
| Not totally understanding how SwissSidechain works. Do I just install random .pdb online, open that in PyMol, and then run SwissSidechain against it? | ChatGPT |
| Ok. At RCSB PDB and am looking to download a PyMol compatible file extension. What file extension should I go for? | ChatGPT |
| What is the name of a .pdb file? PDBx/mmCIF? | ChatGPT |
| Whenever I click ‘PySwissSidechain’ from Plugin –> Legacy Plugins my Open-Source PyMol window fails and I see this in Terminal: “libc++abi: terminating due to uncaught exception of type NSException”. What’s going on? How do I proceed with running the SwissSidechain extension? | ChatGPT |
| Yes do this | ChatGPT |
| Seeing this command on SwissSidechain: “Command line Use the command: Mutate Object//Chain/ResNumber/, Newres For instance, to mutate residue number ‘82’ on chain ‘E’ in object ‘complex’ into Homoleucine (HLEU), write: Mutate complex//E/82/, HLEU” What does each piece of this command mean in biological speak? How do I find residues and non-natural amino acids to choose from? | ChatGPT |
| In SwissSideChain, what is the short code for the non-natural amino acid ‘Cha’ and ‘Ca’? | Perplexity |
| Amino acids are produced by enzymes, correct? DO NOT hallucinate when answering this question | Perplexity |
| So based on the answer to the last prompt, where exactly do amino acids come from? Where exactly do they originate? DO NOT hallucinate when answering this question | Perplexity |
| When we say ’enzyme‑catalyzed pathway’ we mean a chemical reaction that an enzyme speeds up, correct? Do NOT hallucinate when answering this question | Perplexity |
| What is an enzyme pathway in the context converting a precursor into an amino acid? Is it simply a chemical reaction? Something else? Do NOT hallucinate when answering this question | Perplexity |
| In the answer 3 prompts ago, there was mention of “Carbon Skeleton” and “Intermediates of glycolysis, the citric acid (TCA) cycle, and the pentose phosphate pathway” when describing them. What does this mean in simple terms? Are we saying these provide the chemical structure of an amino acid to keep it fundamentally sound? Do NOT hallucinate when answering these questions | Perplexity |
| Ok. Based on available literature and if relevant/necessary, the information shared in response to the previous prompts, how did amino acids originate before the emergence of life as we know it on this planet? Was it via chemical process in cyanobacteria or another form of bacteria or archaea? Do NOT hallucinate when answering these questions | Perplexity |
| An a-helix within the context of genomics and synthetic biology is an alpha helix correct? How do D-amino acids relate to alpha helices? Do NOT hallucinate when answering these questions | Perplexity |
| Not understanding. In the answer to the last prompt you stated, " In genomics and synthetic biology, “α-helix” or “a-helix” refers to the standard protein alpha helix: a right‑handed secondary structure formed by regular backbone hydrogen bonding, almost always built from L‑amino acids in natural proteins.” Then you said, “A chain made entirely of L‑amino acids naturally forms a right‑handed α‑helix” Did hallucination occur here? If so, where and how? | Perplexity |
| If this is the case, how can an α-helix using D-amino acids exhibit right-handedness? Not seeing how this would work? Do NOT hallucinate when answering this question | Perplexity |
| Explain the following: –The difference between how protein is described in a nutritional context and what proteins are/how they are described in a biological or biotechnological context –My hunch is additional helices in naturally-occurring proteins can be found, although I’m not sure how. Confirm or deny this hunch. If my hunch is correct, tell me why additional helices for proteins found in nature can be found and some methods for discovering these additional helices. Do NOT hallucinate when explaining these items. If you there’s risk of exaggeration or outputting something not confirmed or based on sources, don’t output it | Perplexity |
| What are molecular helices in a biological context? How do they differ from other types of helices (if at all)? Do NOT hallucinate when answering these questions | Perplexity |
| What’s the handedness of most molecular helices? What explains their handedness? Do NOT hallucinate when answering these questions | Perplexity |
| What does this mean in the answer to the last prompt? “L-amino acids dictate right-handed protein helices (opposite would clash sterically).” What are L-amino acids again? Does the L stand for lysate? Explain all this to me as if I was a reasonably educated 14 year-old. Do NOT hallucinate when answering these questionss | Perplexity |
| What are β-sheets in a biological context? Do we call them beta-sheets? What does the β stand for? What exactly are they? Do NOT hallucinate when answering these questions | Perplexity |
| What causes beta-sheets to come together as a larger aggregate? Is it molecular forces from difference ionic charges on atoms comprising the beta-sheets? Something else? Do NOT hallucinate when answering these questions | Perplexity |
| From the answer to the previous prompt, “H-bond donors (N-H) and acceptors (C=O)”" means hydrogen bond donors and carbon acceptors, correct? Do NOT hallucinate when answering this question. What does the C stand for? Do NOT hallucinate when answering these questions | Perplexity |
Part B: Protein Analysis and Visualization
I selected the crotamine protein found in the South American rattlesnake, the protein’s abilities to penetrate cells as a toxin allows it to serve as a template for antivenom and potentially targeted therapies.
The amino acid sequence of the crotamine protein protein is below 1
AAF34911.1 crotamine [Crotalus durissus terrificus] MKILYLLFAFLFLAFLSEPGNAYKQCHKKGGHCFPKEKICLPPSSDFGKMDCRWRWKCCKKGSGK
- This protein is 65 amino acids long. Its most frequent amino acid in this protein is lysine, which appears 11 times
- There are 250 protein sequence homologs for the crotamine protein after running it in UniProt BLAST (ID: Q9PWF3)
- Yes, this protein belongs to the crotamine-myotoxin family.
Structural answers to this question are listed below:
- The protein structure was discovered in 2005 2. It appears to be a mixed bag from a structural quality standpoint based on the NMR Structure Validation Report below:
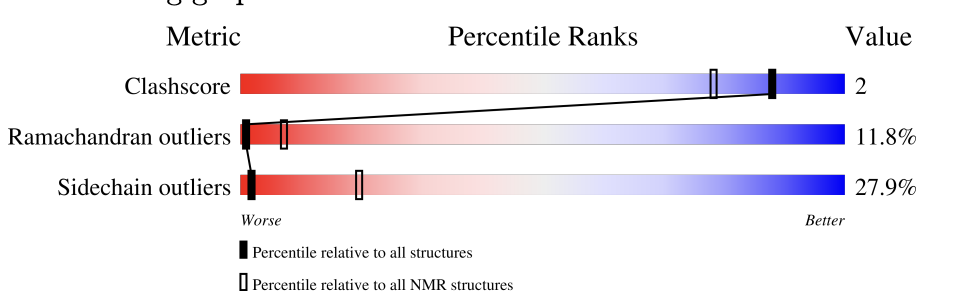 Crotamine protein RCSB NMR Structure Validation Report
Crotamine protein RCSB NMR Structure Validation Report - No, there are no other molecules in the solved structure apart from the protein
- It’s a defensin-like protein of the myotoxin family
- The protein structure was discovered in 2005 2. It appears to be a mixed bag from a structural quality standpoint based on the NMR Structure Validation Report below:
3D visualization software (NGL Outputs)
- “Cartoon”, “Ribbon”, and “Ball and Stick” combo
.png)
- Protein coloring by secondary structure. I think it might have more sheets than helices, although I’m not sure
.png)
- Protein by residue type. It seems like it has a good mix of hydrophobic vs hydrophilic residues
.png)
- Protein surface visualization. It doesn’t seem like it has a lot of holes, although I’m not sure
.png)
- “Cartoon”, “Ribbon”, and “Ball and Stick” combo
All supporting prompts listed below:
| Supporting Prompt | Model |
|---|---|
| I have a FASTA file and a GenBank identifier for a protein I found on NCBI.gov. How do I then find the amino acid sequence for that given protein? What will it look like? Do NOT hallucinate when answering this question | Perplexity |
| Confirming that the beta-keratin 2 [Gekko gecko] protein has a 3D structure, correct? | Perplexity |
| Does the beta-keratin 2 [Gekko gecko] protein have a 3D structure? | Google AI |
| Understood. Are there any slug mucus proteins that have a 3d structure? | Google AI |
| Gotcha. Tell me if there are any serpent-derived proteins that have a 3D structure | Google AI |
| What is the K amino acid? Is it potassium? | Perplexity |
| What are main protein sequence homologs in the context of genomics and biotechnology? What do they look like? | Perplexity |
| Show me the format of protein sequence homologs in UniProt database. Show me what they usually look like. Do NOT hallucinate when producing this output | Google AI |
| Find me resolution info depicting the quality of the structure described in this tab | Gemini |
| So I’ve heard a good quality structure has higher resolution. How can I obtain information on the quality o the structure from this tab? Is it possible? | Gemini |
| Based on the scores in the 2nd page of this tab, what is the quality of the structure? Where exactly does the black dot reside here? | Gemini |
| Based on this tab, tell me if there are any other molecules in the structure of the Crotamine protein beyond the protein itself? If there are, what are they? Do NOT hallucinate when answering this question | Gemini |
| Where can I find the structure classification family for this protein in this tab? Show me where I can find this | Gemini |
| Tell me what I’m looking at and how it helps determine the structure classification family of the crotamine protein | Gemini |
| Tell me if and/or how I can view this protein as a “cartoon” “ribbon” and “ball and stick” using this NGL viewer tool | Gemini |
| See the plus icon but can’t click on it | Gemini |
| I want to color the protein by secondary structure to determine whether or not it has more helices or shets. How can I do this in this viewer? | Gemini |
| Can you see the successful secondary structure output here? If not, say so | Gemini |
| I now want to color the protein by residue type to determine whether or not it has more helices or shets. How can I do this in this viewer? | Gemini |
| I no longer want to see secondary structures or helices or sheets. I want to see the protein’s residue types | Gemini |
| Right now my chain-related Color Scheme options are ChainID, ChainIndex, and ChainName. Which one should I choose? | Gemini |
| Yes it seems to be recognizing the entire structure as Chain A. Does this mean that most of the residue type is of 1 type? And does this mean that the residues are mostly hydrophobic or hydrophilic? | Gemini |
| Ok – now how would I visualize the surface of the protein. How can I see if it has holes or binding pockets? | Gemini |
Part C: Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
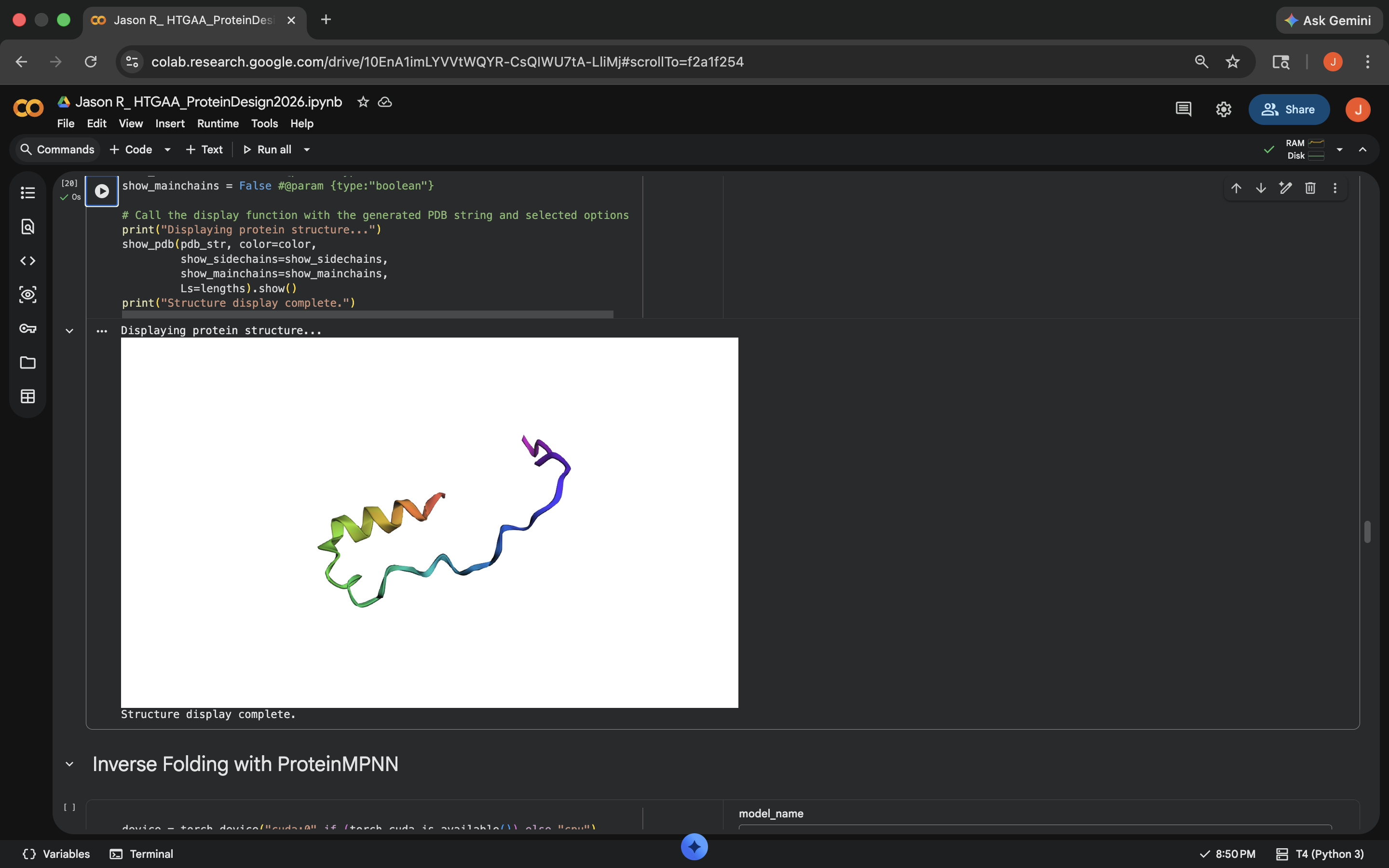
See picture below:
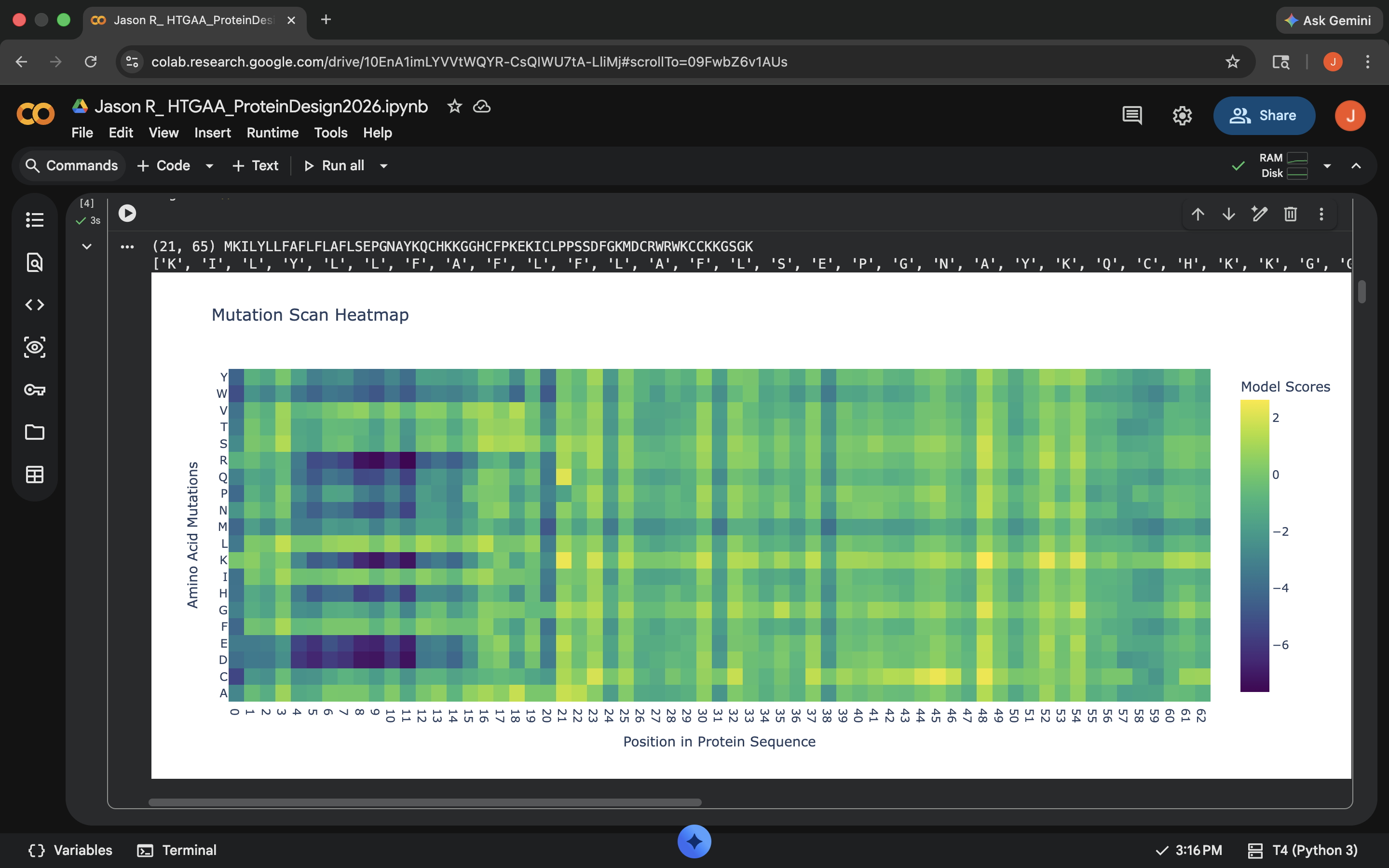 Crotamine Protein Deep Mutational Scan
Crotamine Protein Deep Mutational ScanThere seem to be these really interesting columns that occur as one moves rightward past position 20 in the heatmap. There is a single almost uniform top-down color for these columns, which according to Gemini, indicate a special sensitivity. These are apparently disulfide bridges, which are quite important for holding the protein together (these are referred to as ‘anchors’)
Latent Space Analysis
See picture below
.png) Non-Crotamine tagged point cloud showing embedded proteins
Non-Crotamine tagged point cloud showing embedded proteinsYes they do–there are approximations of similar proteins found throughout the cloud.
.png) Crotamine tagged point cloud showing embedded proteins and Crotamine’s location among embeddings
Crotamine tagged point cloud showing embedded proteins and Crotamine’s location among embeddingsThere are other proteins at the edge of the cloud that belong to either other viruses, or other organisms that might be found in similar locations/habitats to the South American rattlesnake (mice, chickens, humans), or other types of toxins or species carrying toxins. See picture below:
.png) Crotamine tagged point cloud showing embedded proteins and Crotamine’s location relative to its neighborhood
Crotamine tagged point cloud showing embedded proteins and Crotamine’s location relative to its neighborhood
C2. Protein Folding
Approach 1: It doesn’t look like the predicted coordinates match the original structure in the PDB much at all (see pictures below). Think this might be due to the fact that the original inputted structures in the PDB for this protein were apparently a bit of a mixed bag according to its NMR Structure Validation Report results (see previous section).
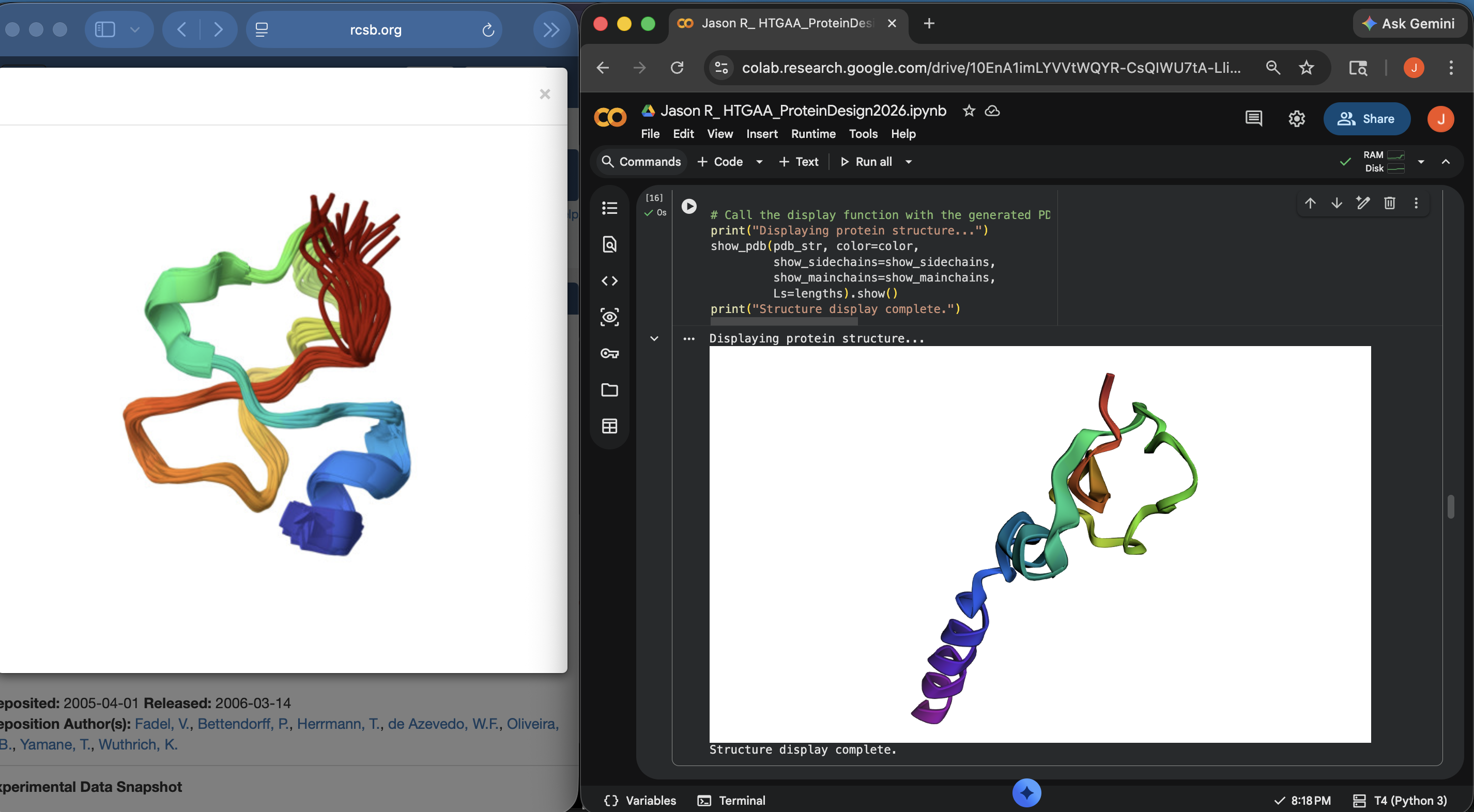 Crotamine Protein PDB < > ESM Side-by-Side Structural Comparison (1)
Crotamine Protein PDB < > ESM Side-by-Side Structural Comparison (1)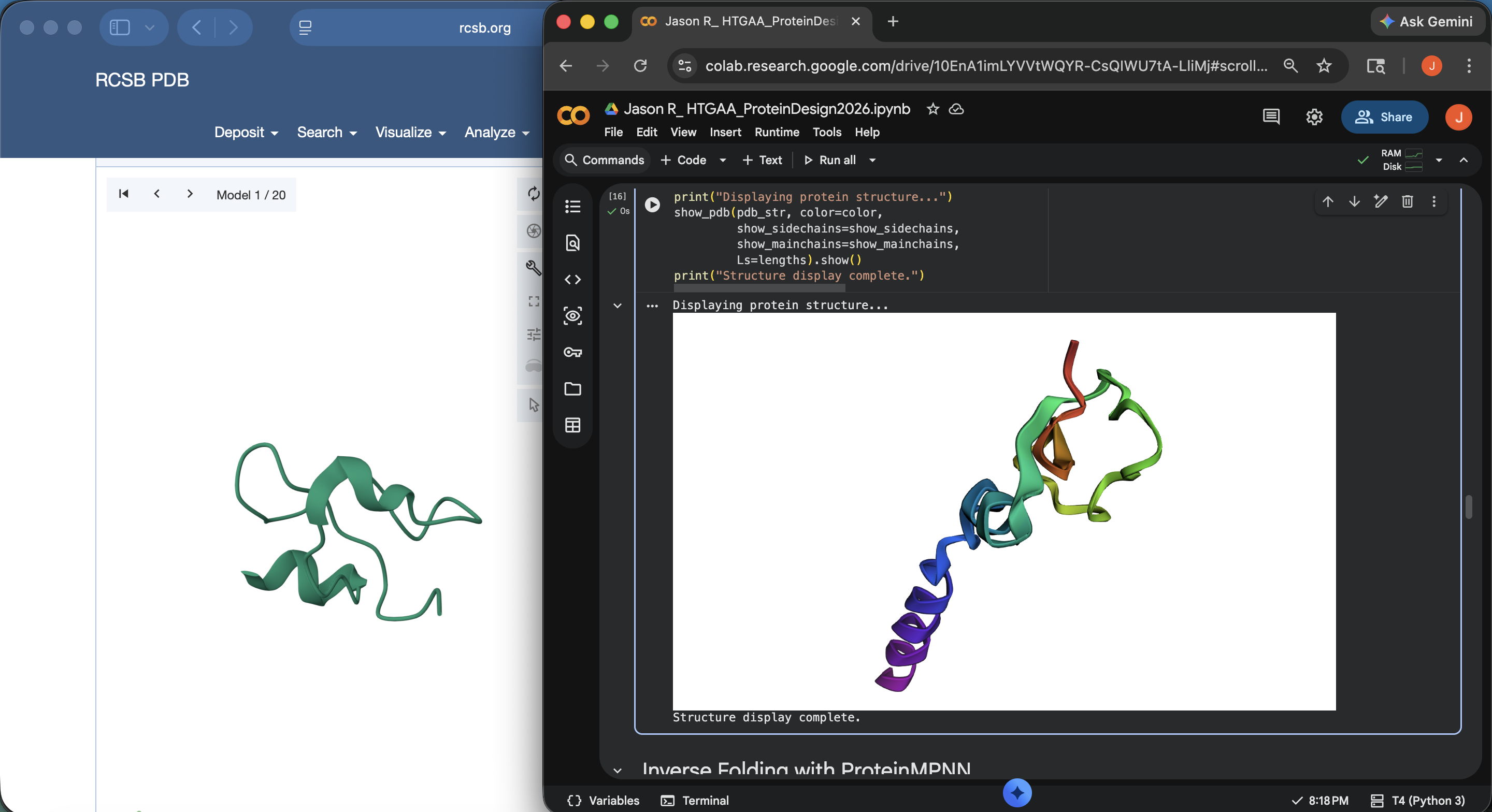 Crotamine Protein PDB < > ESM Side-by-Side Structural Comparison (2)
Crotamine Protein PDB < > ESM Side-by-Side Structural Comparison (2)Approach 2: Asked Gemini about this discrepancy and was recommended to input the mature sequence containing the last 42 residues. The result still visually seems to have some discrepancies compared with the original PDB visual upon first glance, but far less than the outputs under Approach 1 (see photo below)
 Crotamine Protein PDB < > ESM Side-by-Side Structural Comparison (3) – Post-Mature Sequence Input
Crotamine Protein PDB < > ESM Side-by-Side Structural Comparison (3) – Post-Mature Sequence InputI tried the following mutations:
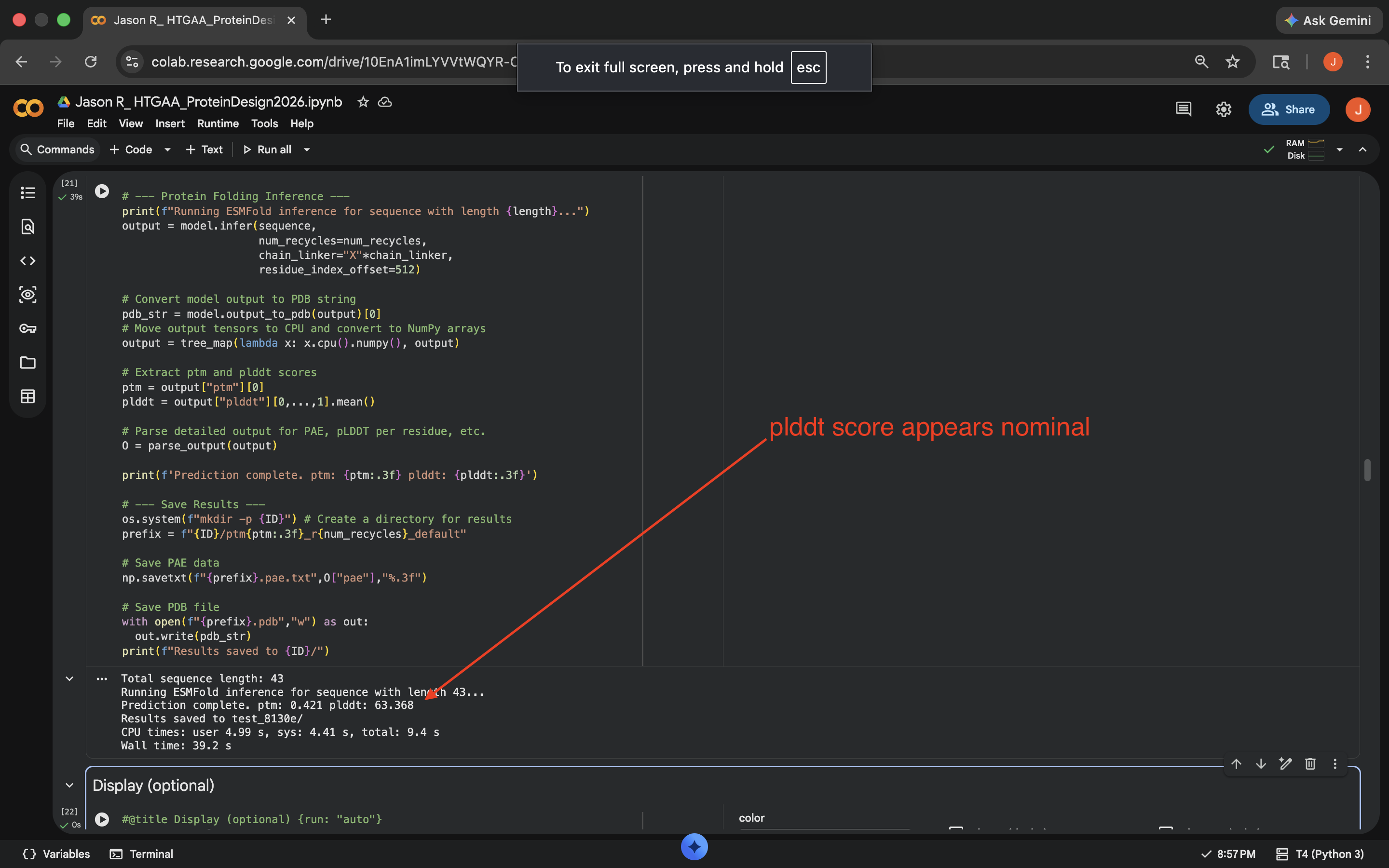
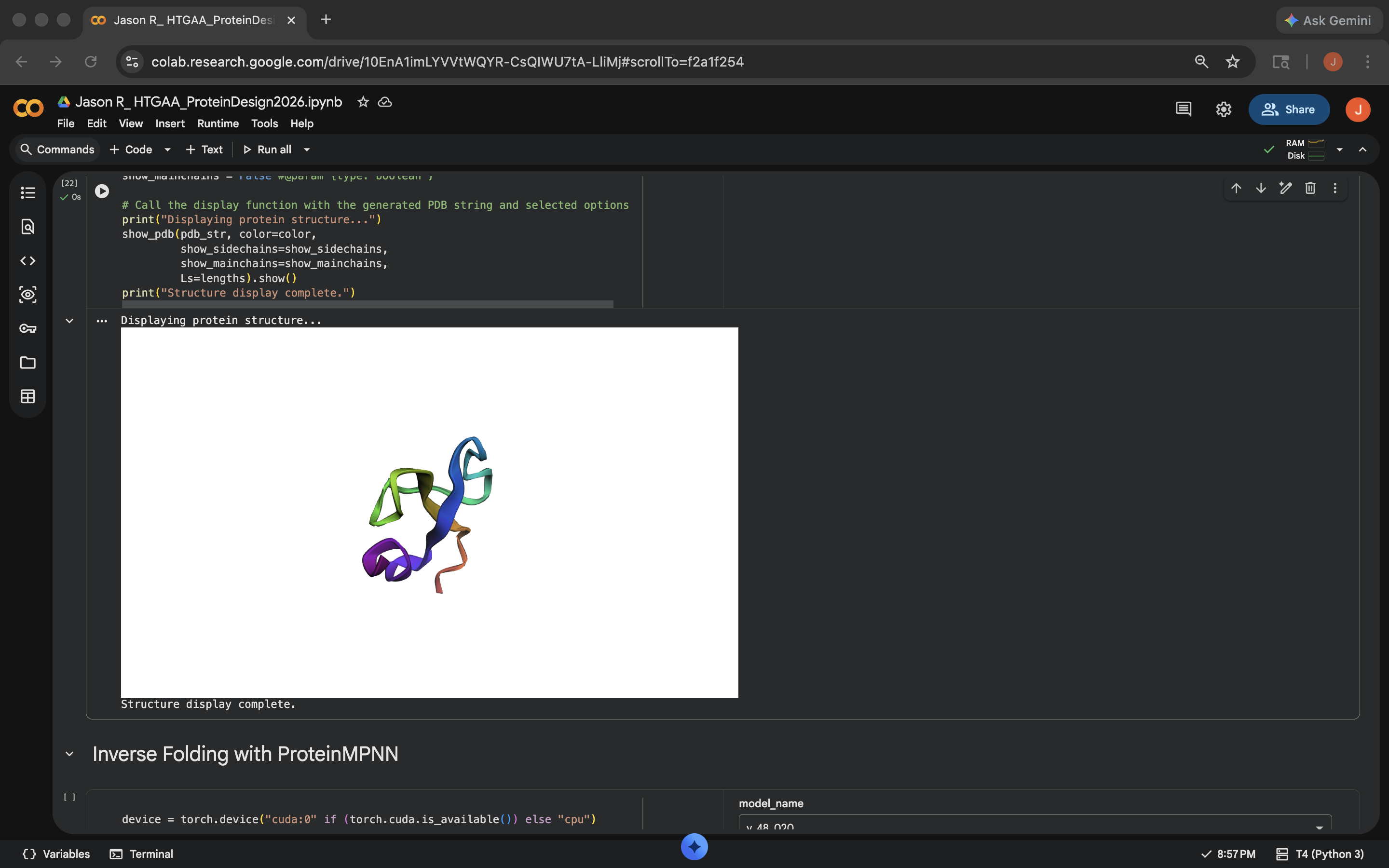
Cytesine Break: Per Gemini suggestion, replaced all the C residues to A (alanine). This notably decreased the predicted Local Difference Test (pLDDT) score of the outputed mutated sequence and the visual also seemed to indicate less structural integrity to the protein, implying less resilience to this mutation (see photo below)
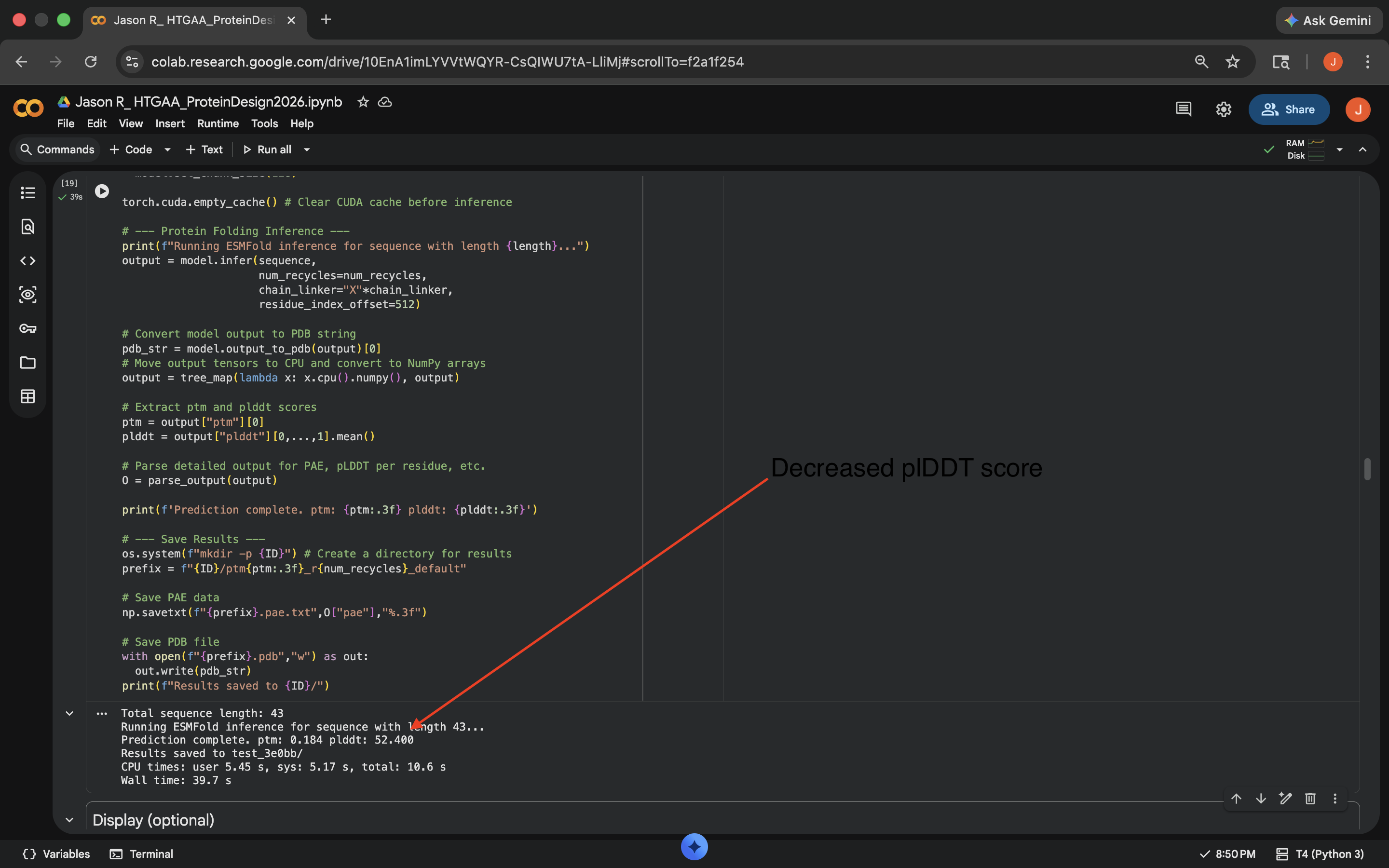
 Crotamine Cytesine Break Mutation Results
Crotamine Cytesine Break Mutation ResultsCharge Swap: Per Gemini suggestion, replaced all the K (Lysine) and R (Arginine) residues to D (Aspartic Acid). This decreased the pLDDT score of the outputed mutated sequence a bit, but less than the Cytesine Break, indicating the protein was more comparably resilient to this mutation (see photo below)
 Crotamine Charge Swap Mutation Results
Crotamine Charge Swap Mutation Results
C3. Protein Generation
- Based on the heatmap below, there seems to be a difference between the predicted sequence probabilities here and the original heatmap generated in the ‘Deep Mutational Scans’ subsection. 48 of the 65 positions were changed and the sequence recovery rate was around 0.26, which didn’t seem all that promising
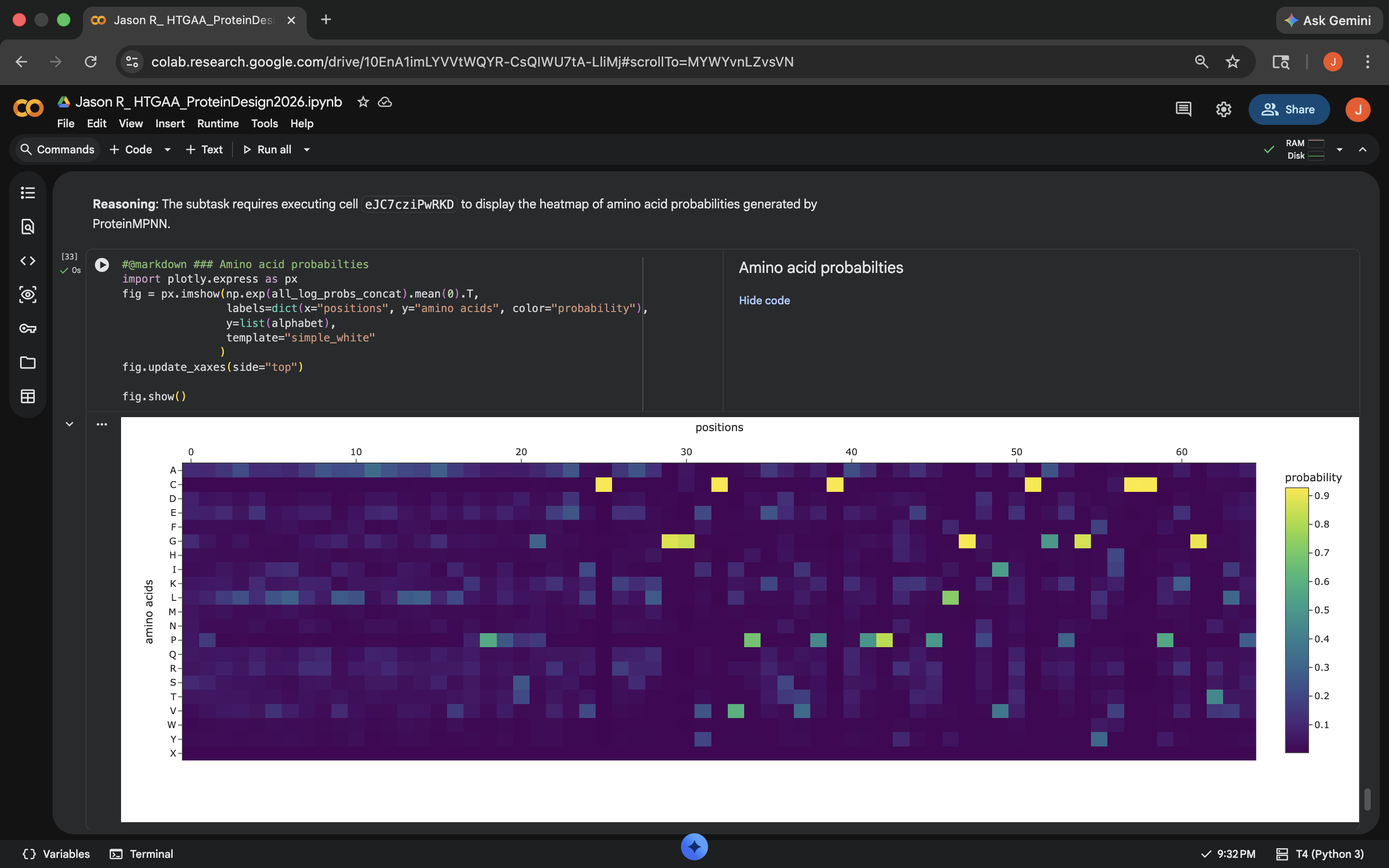
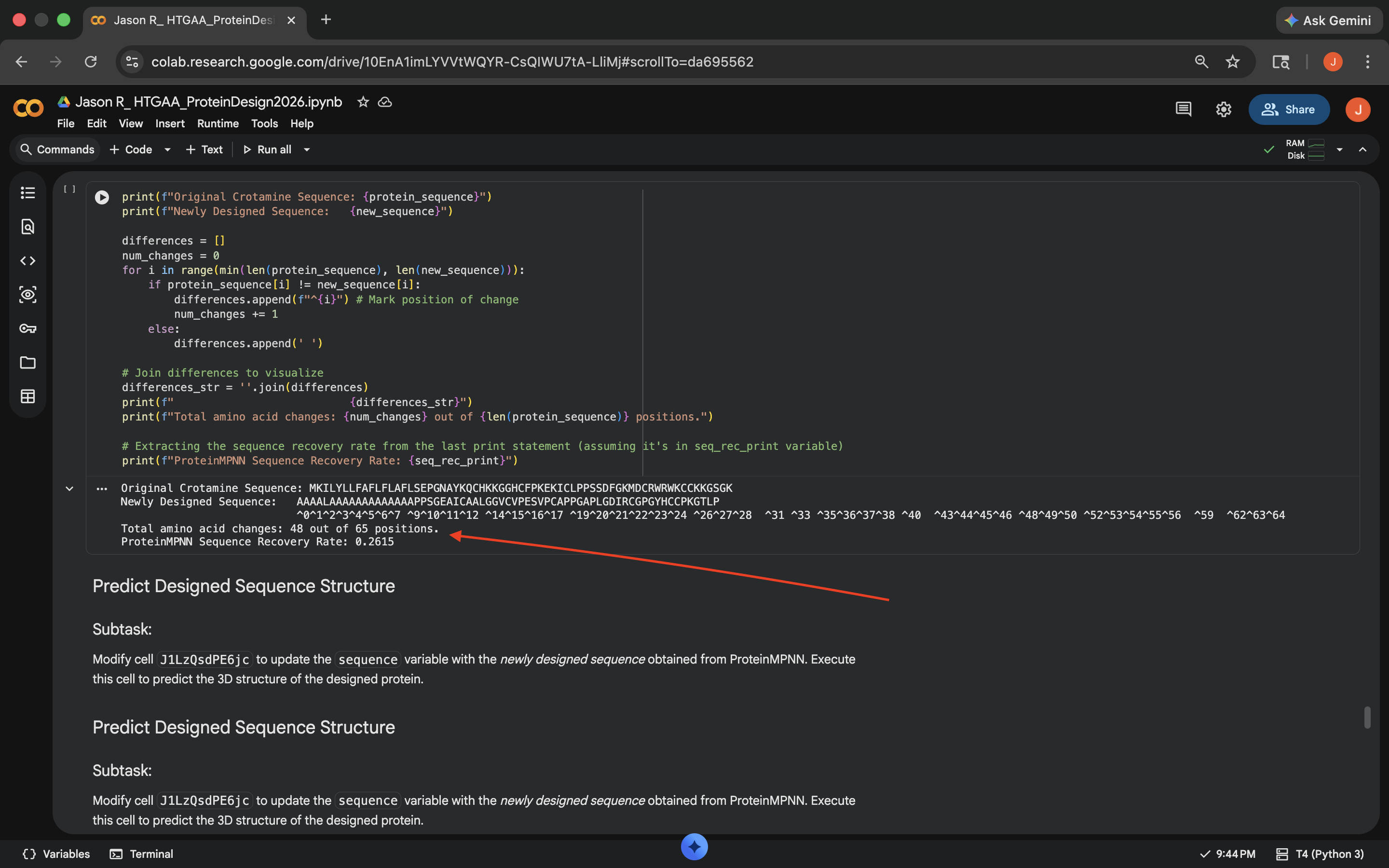 Crotamine ProteinMPNN Results (1)
Crotamine ProteinMPNN Results (1) - Visually, this re-inserted ESMFold output also looks structurally different than the original protein structure (see photo below)
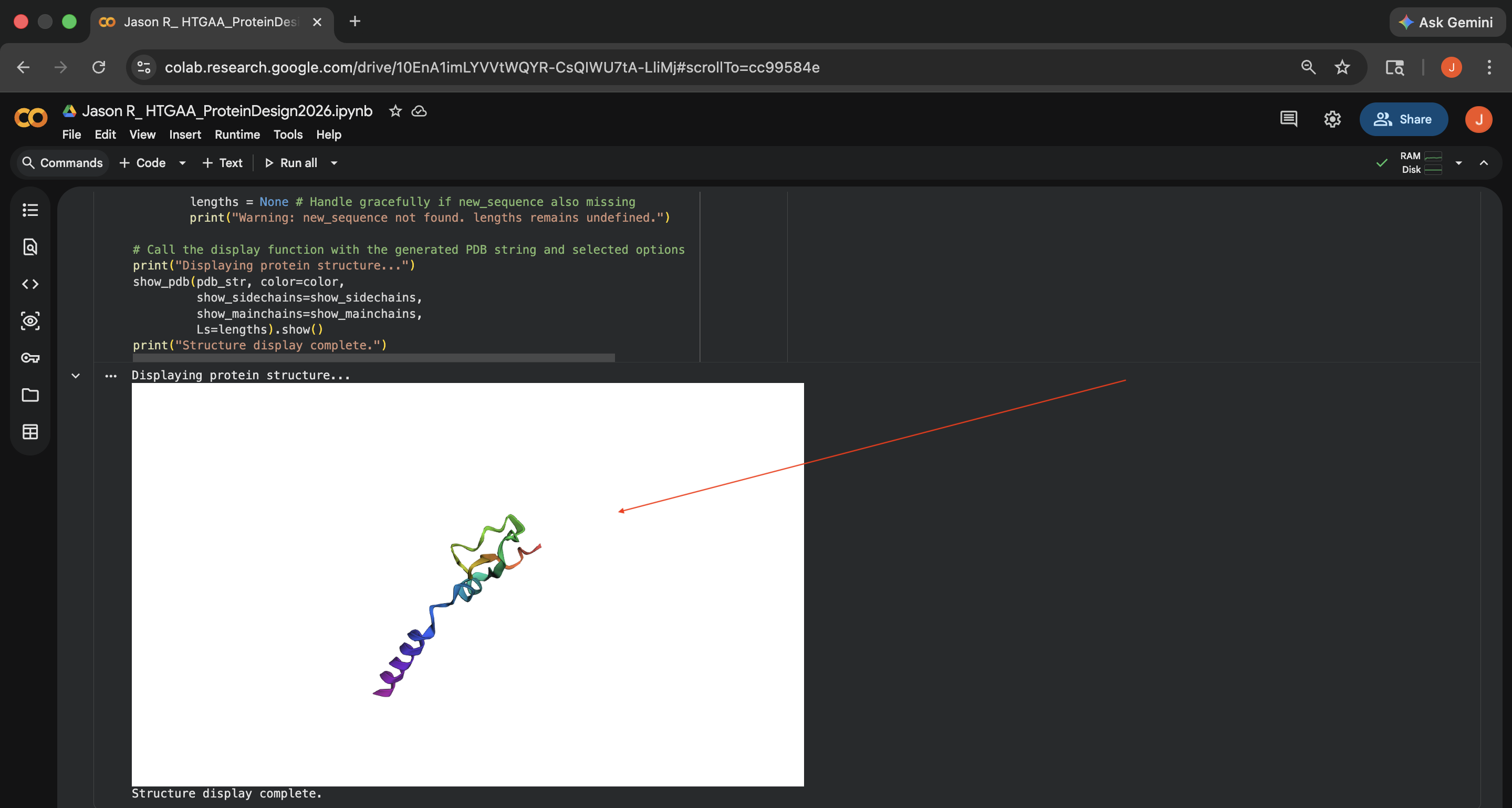 Crotamine ProteinMPNN Results (2)
Crotamine ProteinMPNN Results (2)
All supporting promopts for this section listed below:
| Supporting Prompt | Model |
|---|---|
| Want to choose a GPU to run this. Believe the ability to select GPUs should be in the bottom right but am not seeing this. Direct me to where I should go on this page for GPU selection | Gemini |
| Looking over the heatmap under the ‘Mutation Scans’ section of the code, what stands out regarding the inputted protein sequence (MKILYLLFAFLFLAFLSEPGNAYKQCHKKGGHCFPKEKICLPPSSDFGKMDCRWRWKCCKKGSGK)? Are there any particular mutations or patterns that stand out and why? Do NOT hallucinate when answering this question | Gemini |
| I want to make sure I perform the Latent Space Analysis located under ‘Latent Space Analysis’ correctly, as in I want to make sure I perform Latent Space Analysis on the specific protein sequence I inputted under the previous section ‘Mutation Scans’. Is there anything I need to do or input? Does the code in ‘Latent Space Analysis’ just take the specific protein sequence I inputted under the previous section ‘Mutation Scans’ and run with it? Do NOT hallucinate when answering this question | Gemini |
| I want to make sure I perform the Latent Space Analysis located under ‘Latent Space Analysis’ correctly, as in I want to make sure I perform Latent Space Analysis on the specific protein sequence I inputted under the previous section ‘Mutation Scans’. It looks like I just inputted some incorrect code in the cell under ‘Latent Space Analysis’ that starts with ‘Latent Space Analysis’ Fix the errors in the code and make sure to input the appropriate information so the protein sequence from the previous ‘Mutation Scans’ cell can be analyzed here. Do NOT hallucinate when addressing this query | Gemini |
| Ok, so it looks like there’s output in the ‘3D T-SNE visualization of Protein Sequence Embeddings’ subsection of the ‘Latent Space Analysis’ section. Help me understand where my initially inputted Crotamine protein sequence (MKILYLLFAFLFLAFLSEPGNAYKQCHKKGGHCFPKEKICLPPSSDFGKMDCRWRWKCCKKGSGK) is located within the 3D plot. Do NOT hallucinate when addressing this query | Gemini |
| Ok, so it looks like there’s output in the ‘3D T-SNE visualization of Protein Sequence Embeddings’ subsection of the ‘Latent Space Analysis’ section. Help me understand where my initially inputted Crotamine protein sequence (MKILYLLFAFLFLAFLSEPGNAYKQCHKKGGHCFPKEKICLPPSSDFGKMDCRWRWKCCKKGSGK) is located within the 3D plot. It just seems like there are a lot of protein sequences in this plot and while I can drag over and find things manually, I’m not sure where to start or how to efficiently find the area in this plot where the inputted sequence is located. Any assistance would be useful. Do NOT hallucinate when addressing this query | Gemini |
| Yes let’s do that. Let’s give it a color that stands out. | Gemini |
| Now that the protein visualization has been generated in the ‘Run ESMFold’ section, I’m instructed to do the following: “Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?” There’s a couple things here. The first is that the outputted protein structure in ESMFold doesn’t really seem to match up all that well with the original protein structure in the Protein Data Bank (PDB). This might be because the original protein structure in the PDB was apparently a bit of a mixed bag from a quality standpoint. Not sure how to proceed, and that hesitation is twofold: 1) Is there anything I can do regarding the discrepancy between the outputted protein structure in ESMFold and the original protein structure in the PDB? If this is most likely out of my control/if there’s nothing that can be changed from an input or coding perspective, say so 2) To accomplish the instructions listed above in the quotation marks, should I just go to the subsection that says ‘sequence’ in the first cell under the ‘Run ESMFold’ section and just start randomly changing letters? Does that make sense? Regardless of whether or not this is the best approach for fulfilling the instructions, how would I know if the outputted structure(s) are resilient (or not) to mutations? Would it just be discerned from degree of change in outputted structures (i.e., if the structures change a lot, it likely isn’t resilient and vice versa)? Do NOT hallucinate when addressing these questions | Gemini |
| What does the pLDDT acryonym stand for? | Gemini |
| My instructions for the ‘Inverse Folding with ProteinMPNN’ section are to ‘Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.’ I assume there’s some customization to be done here, either through coding or through inputting sequence information. What would be the most likely sensible plan to proceed? Success seems to be that the compared protein sequence and the original one look visually rather similar. Any next steps on how to begin working this workflow would be appreciated. Do NOT hallucinate when addressing this query | Gemini |
| My instructions for the ‘Inverse Folding with ProteinMPNN’ section are to ‘Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.’ From there I need to put results back into ESMFold to see if the visuals match up. I assume there’s some customization to be done here, either through coding or through inputting sequence information. What would be the most likely sensible plan to proceed? Any next steps on how to begin working this workflow would be appreciated. Do NOT hallucinate when addressing this query | Gemini |
| My instructions for the ‘Inverse Folding with ProteinMPNN’ section are to ‘Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.’ From there I need to put results back into ESMFold to see if the visuals match up. I assume there’s some customization to be done here, either through coding or through inputting sequence information. What would be the most likely sensible plan to proceed? Any next steps on how to begin working this workflow would be appreciated. Do NOT hallucinate when addressing this query | Gemini |
| Do these steps outputted from the answer to the previous prompt take into account earlier sequence information inputted into previous cells outside the ‘Inverse Folding with ProteinMPNN’ section, specifically information on the protein sequence in question we want to visually compare across the ‘Inverse Folding with Protein MPNN’ and ‘Run ESMFold’ sections (YKQCHKKGGHCFPKEKICLPPSSDFGKMDCRWRWKCCKKGSGK)? If not, how can this be addressed? Do NOT hallucinate when addressing these questions | Gemini |
| For the last step, just executed, I want a more 1-to-1 comparison between the results in the diagram under ‘Visualize Amino Acid Probabilities’ an the results located in the cell under ‘Mutation Scans’ earlier in the notebook (https://colab.research.google.com/drive/10EnA1imLYVVtWQYR-CsQIWU7tA-LliMj#scrollTo=09FwbZ6v1AUs&line=2&uniqifier=1) Can we make the X and Y axis of the heatmap just executed match this original heatmap with ‘Position in Protein Sequence’ with numbers left to right in the X axis and ‘Amino Acid Mutations’ in the Y axis? Also want ‘Model Scores’ scored from 2 (more yellow-ish) to -6 (more purple-ish) a-la this original heatmap If there are parts of this instruction you don’t understand or that are not possible to execute, say so. After this is resolved, we’ll return to the original workflow from the answer to the previous prompt | Gemini |
| Address the error found when running the ‘Run ESMFold’ cell | Gemini |
| Everything under ‘Configure ProteinMPNN with Crotamine Structure’ doesn’t appear to be working. What’s going on? | Gemini |
Part D: Group Brainstorm on Bacteriophage Engineering
- My William & Mary Node Bacteriophage Engineering Group Brainstorm Google Doc. can be found here 3.