Week 5 HW: Protein Design Part 2
 Using AlphaFold for Protein Optimization
Using AlphaFold for Protein Optimization
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
- Retrieved human SOD1 sequence via UniProt (see photo below). Introduced A4V mutation via Gemini prompt (see sequence below).
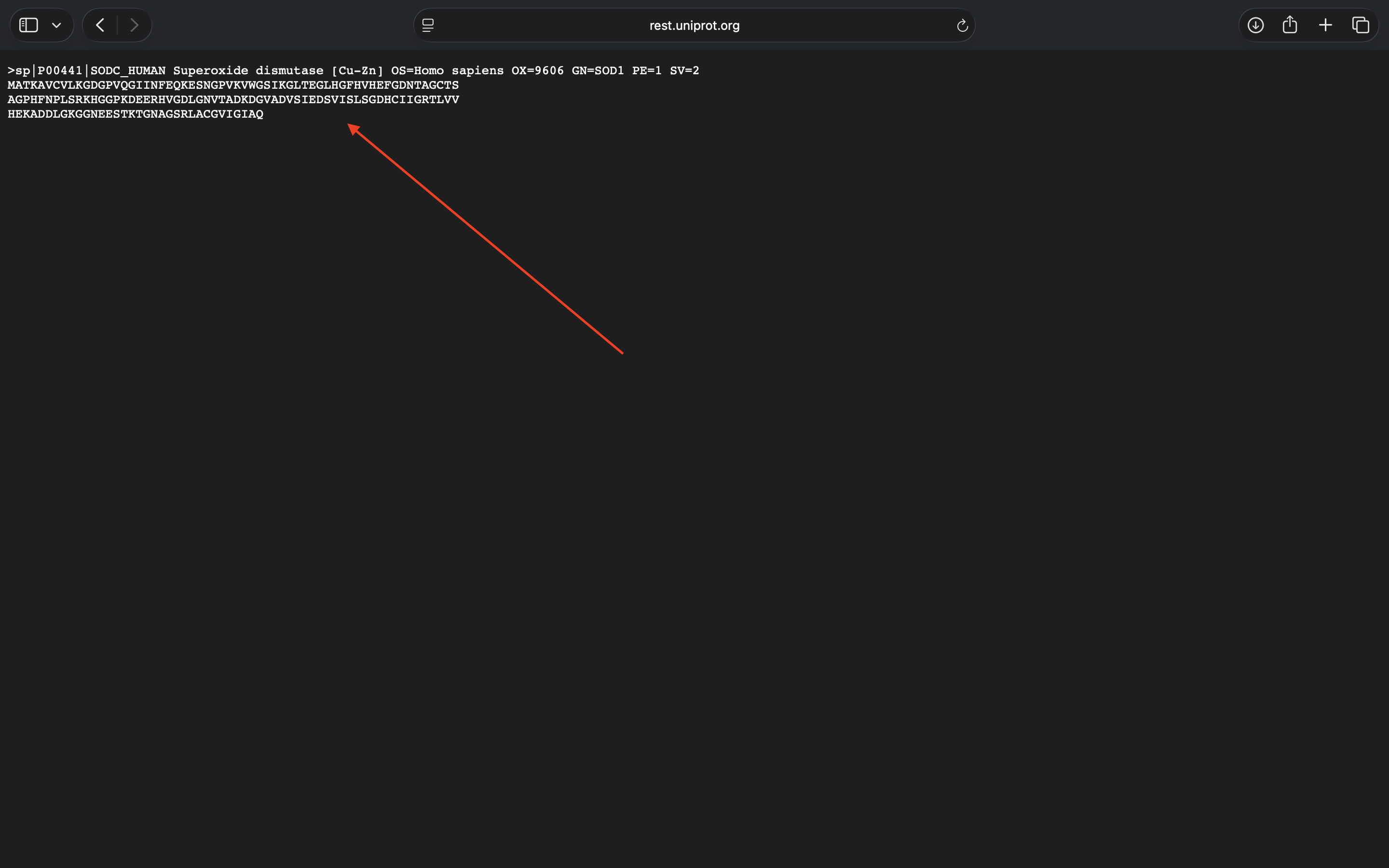 Human SOD1 sequence (A4V mutation not added)
Human SOD1 sequence (A4V mutation not added)
Human SOD1 sequence (A4V mutation added)
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
See results in later questions (answers and photos below)
See photos below
.png)
.png) Generated four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence
Generated four peptides of length 12 amino acids conditioned on the mutant SOD1 sequenceSee photo below
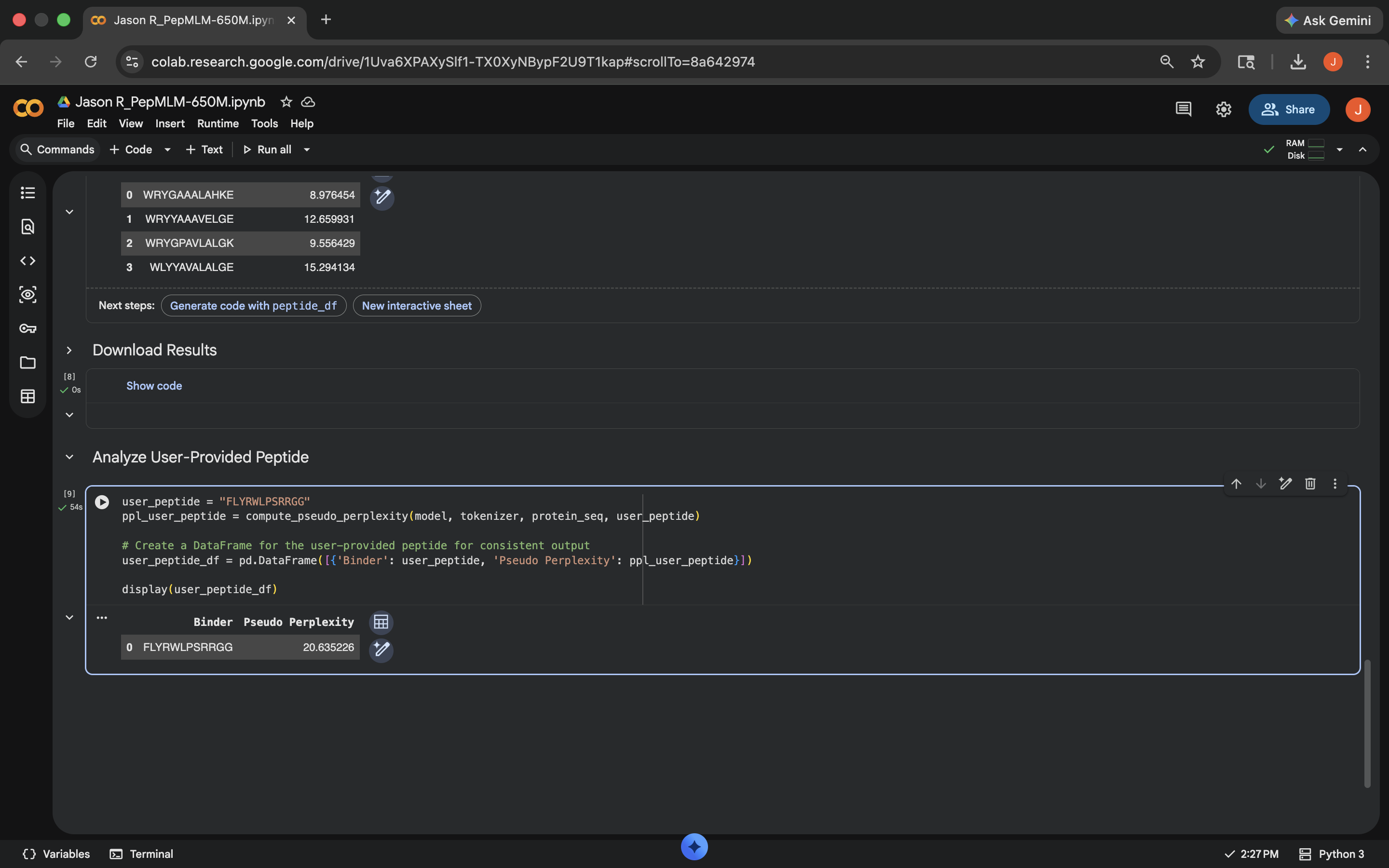 Added known SOD1-binding peptide FLYRWLPSRRGG for comparison
Added known SOD1-binding peptide FLYRWLPSRRGG for comparisonPerplexity scores listed below
| Binder | Perplexity |
|---|---|
| WRYGAAALAHKE | 8.976454 |
| WRYYAAAVELGE | 12.659931 |
| WRYGPAVLALGK | 9.556429 |
| WLYYAVALALGE | 15.294134 |
| FLYRWLPSRRGG | 20.635226 |
NOTE: PepMLM Colab used to generate results above can be found here 1
Supporting prompts for this section listed below:
| Supporting Prompt | Model |
|---|---|
| Why is protein design and models like AlphaFold important in the context of drug discovery and improvements in human health? If I were to describe its importance to a reasonably educated person on the street who doesn’t know much about the subject what would I say? Would something like “Since most diseases are caused by protein-related issues, and because proteins comprise an essential role in human health and physiology, knowing how proteins function and fold can help us design therapeutics with greater precision and efficacy”? What am I missing there and where am I off? Do NOT hallucinate when answering this question | Perplexity |
| In the context of proteins and/or the Superoxide dismutase (SOD1) protein found in Homo sapiens, what is the A4V mutation? What does it entail? What does A4V stand for? Do NOT hallucinate when answering this prompt | Perplexity |
| I have the following Superoxide dismutase (SOD1) protein sequence found in Homo sapiens: MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ. I want to introduce the A4V mutation in this sequence so I can eventually generate relevant peptides to bind to the mutated sequence. How does the sequence need to change to accurately represent this mutation? Do NOT hallucinate when addressing this prompt | Gemini |
| Based on the contents in the ‘Inputs and Parameters’ cell, if I want to generate 4 peptides that each have a 12 amino acid length, other than the ‘Peptide Length’ variable, the other variable I should alter is the ‘Number of Binders’ variable, correct? Or is it the ‘Top K Value’ variable? Not sure which variable I need to alter. Do NOT change any cell content as part of addressing this prompt and do NOT hallucinate | Gemini |
| Now that I’ve generated 4 peptides, each of which are 12 amino acids long, I now want to add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Without changing any code in any of the cells in this workbook, how can I go about doing this? Do NOT hallucinate when addressing this query | Gemini |
| Question regarding the third bullet point under 3. for Method 1. How would the benchmark (FLYRWLPSRRGG)’s properties be known for comparison by performing the actions listed under Method 1 in response to the last prompt? Do NOT hallucinate when answering this question | Gemini |
| Let’s create a new cell under the ‘Download Results’ tab where the following Superoxide dismutase (SOD1)-binding peptide FLYRWLPSRRGG will be analyzed in the exact same way the peptides generated from the ‘Inputs and Parameters’ cell were analyzed in the ‘Generate Peptides’ cell. Do not alter any of the underlying fundamental logic from code in prior cells. Just extend it so the FLYRWLPSRRGG can be analyzed with a Perplexity score in the same way the results from the ‘Inputs and Parameters’ cell were analyzed in the ‘Generate Peptides’ cell. Do NOT hallucinate when performing this task | Gemini |
Part 2: Evaluate Binders with AlphaFold3
- Navigated to AlphaFold Server (see below):
- See peptide results (ipTM scores and binding information) below in 3.
- See ipTM and binding information results below:
WRYGAAALAHKE Peptide:
- ipTM: 0.31; peptide appears to bind near the dimer interface, and appears surface-bound, although it should be noted that the level of confidence indicated by the ipTM score is notably low, which can color the perception of these results. See photo below:
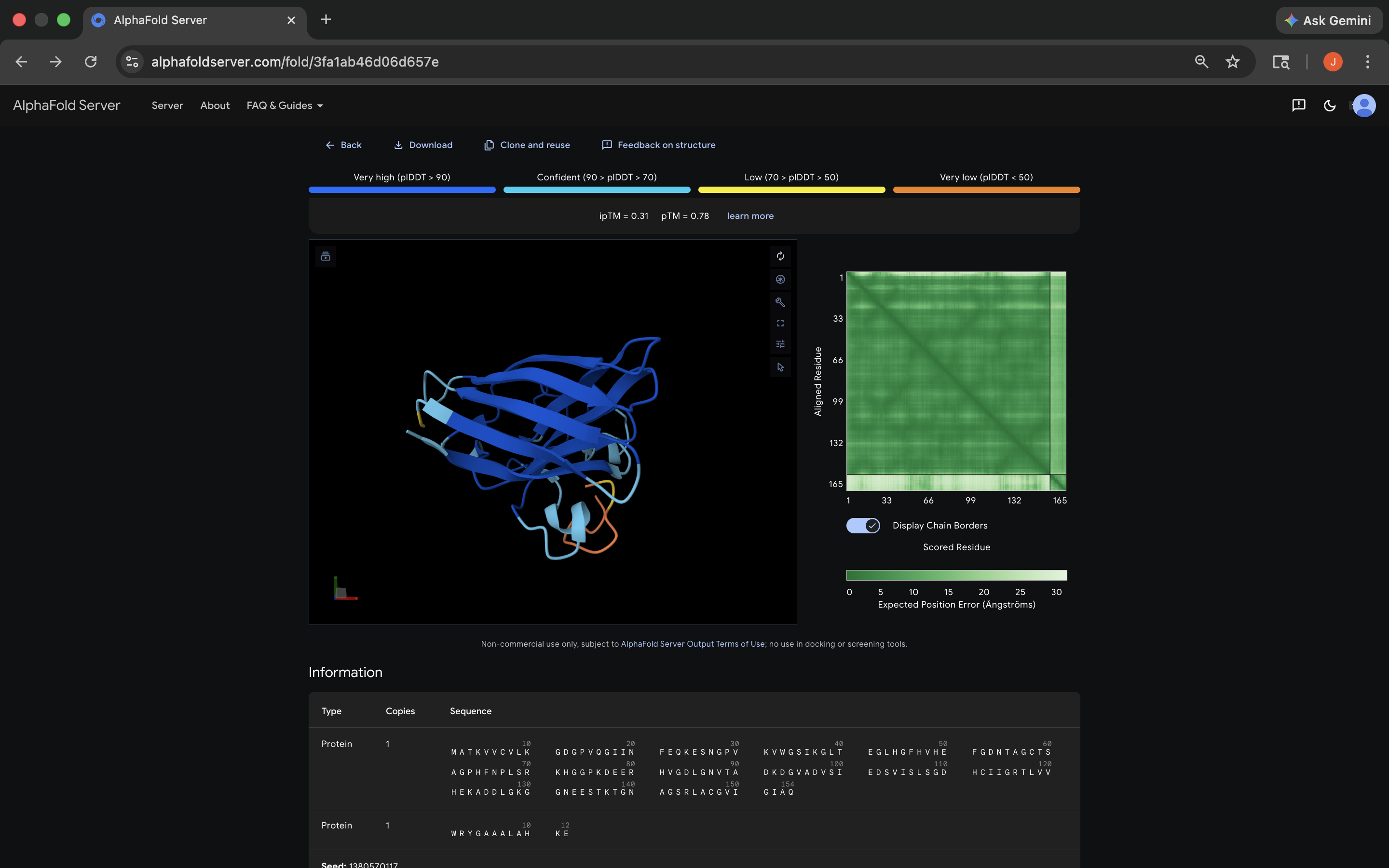 WRYGAAALAHKE peptide AlphaFold Visualization Results
WRYGAAALAHKE peptide AlphaFold Visualization Results
- ipTM: 0.31; peptide appears to bind near the dimer interface, and appears surface-bound, although it should be noted that the level of confidence indicated by the ipTM score is notably low, which can color the perception of these results. See photo below:
WRYYAAAVELGE Peptide:
- ipTM: 0.24; again peptide appears to bind near the dimer interface, and appears surface-bound, although it should be noted again that the level of confidence indicated by the ipTM score is again notably low, which can color the perception of these results. See photo below:
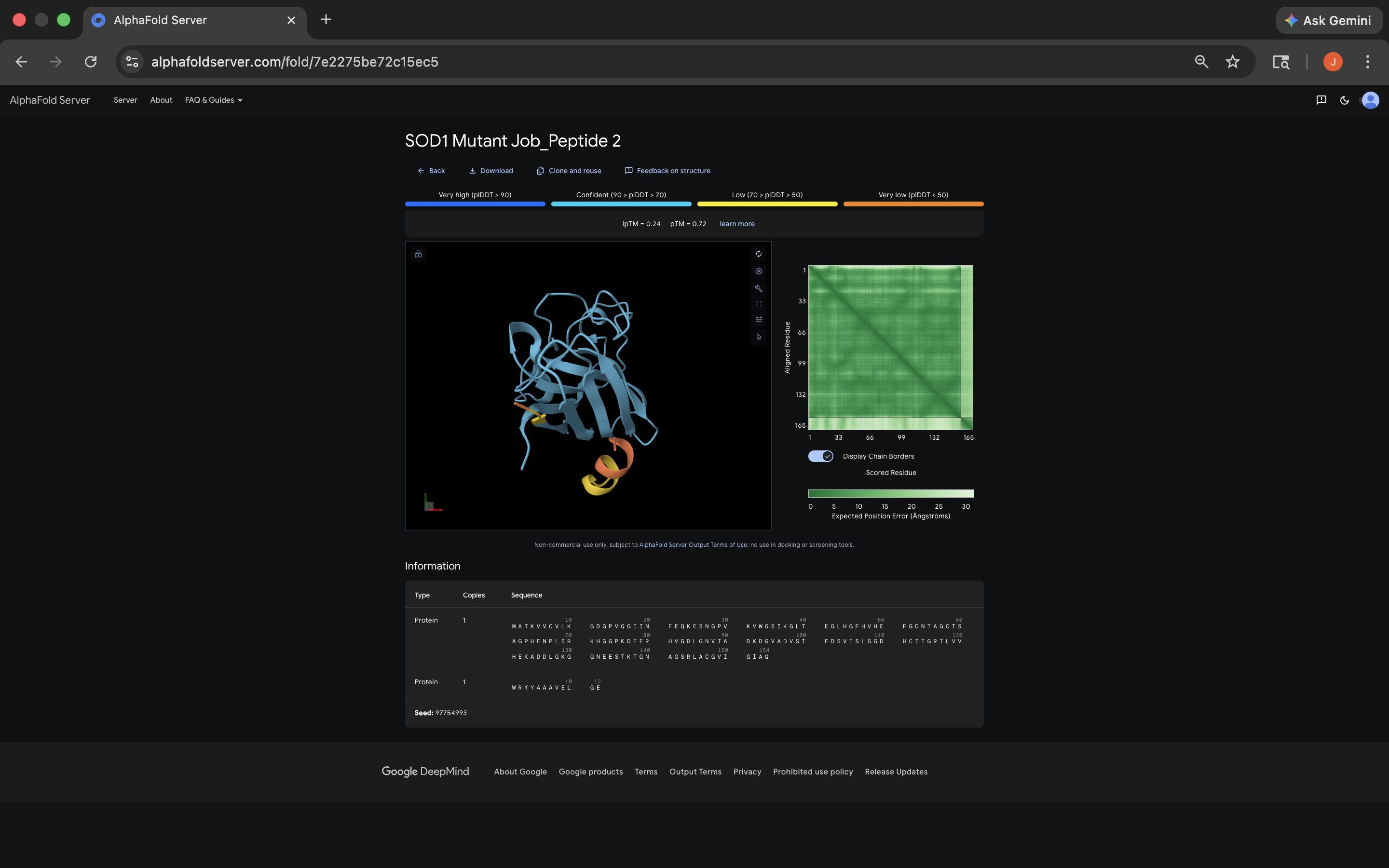 WRYYAAAVELGE peptide AlphaFold Visualization Results
WRYYAAAVELGE peptide AlphaFold Visualization Results
- ipTM: 0.24; again peptide appears to bind near the dimer interface, and appears surface-bound, although it should be noted again that the level of confidence indicated by the ipTM score is again notably low, which can color the perception of these results. See photo below:
WRYGPAVLALGK Peptide:
- ipTM: 0.33; again peptide appears to bind near the dimer interface, and appears surface-bound, although once again the confidence of this assessment is not high based on the ipTM. See photo below:
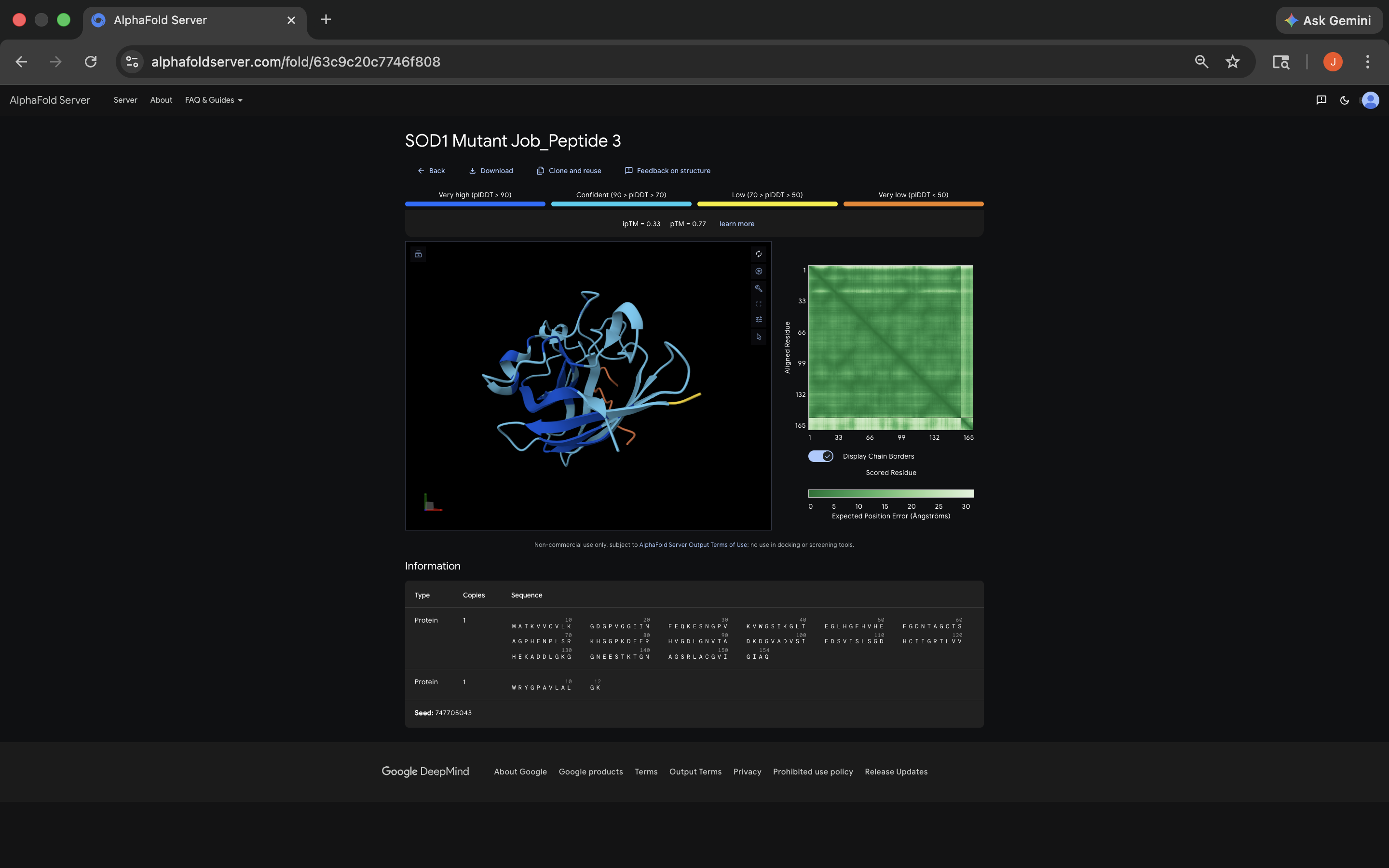 WRYGPAVLALGK peptide AlphaFold Visualization Results
WRYGPAVLALGK peptide AlphaFold Visualization Results
- ipTM: 0.33; again peptide appears to bind near the dimer interface, and appears surface-bound, although once again the confidence of this assessment is not high based on the ipTM. See photo below:
WLYYAVALALGE Peptide:
- ipTM: 0.40; again peptide appears to bind near the dimer interface, and appears surface-bound, although once again the confidence of this assessment is not high based on the ipTM. See photo below:
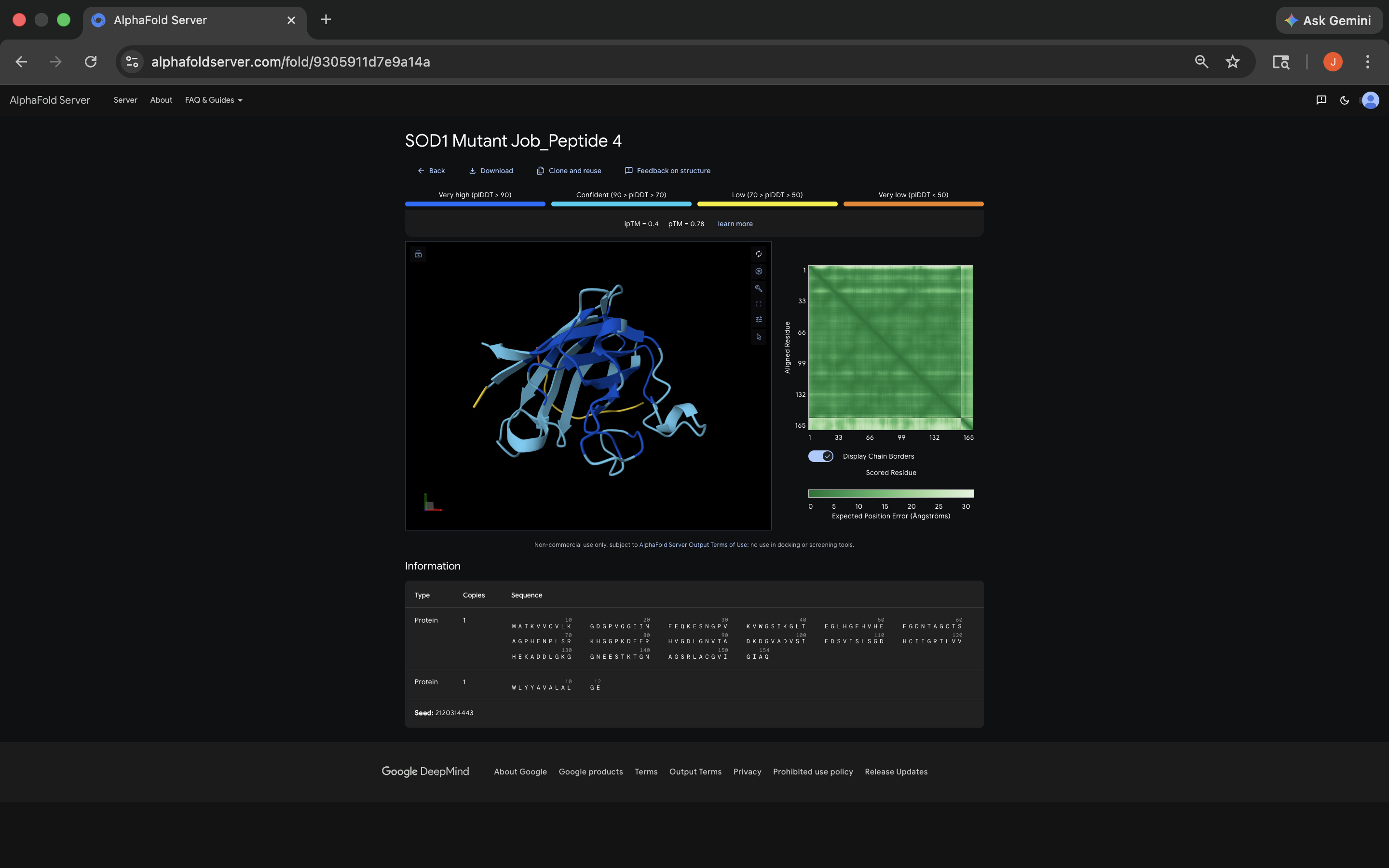 WLYYAVALALGE peptide AlphaFold Visualization Results
WLYYAVALALGE peptide AlphaFold Visualization Results
- ipTM: 0.40; again peptide appears to bind near the dimer interface, and appears surface-bound, although once again the confidence of this assessment is not high based on the ipTM. See photo below:
FLYRWLPSRRGG Peptide:
- ipTM: 0.29; peptide appears to engage with the β-barrel region somewhat and appears surface-bound–again the confidence of this assessment is not high based on the ipTM. See photo below:
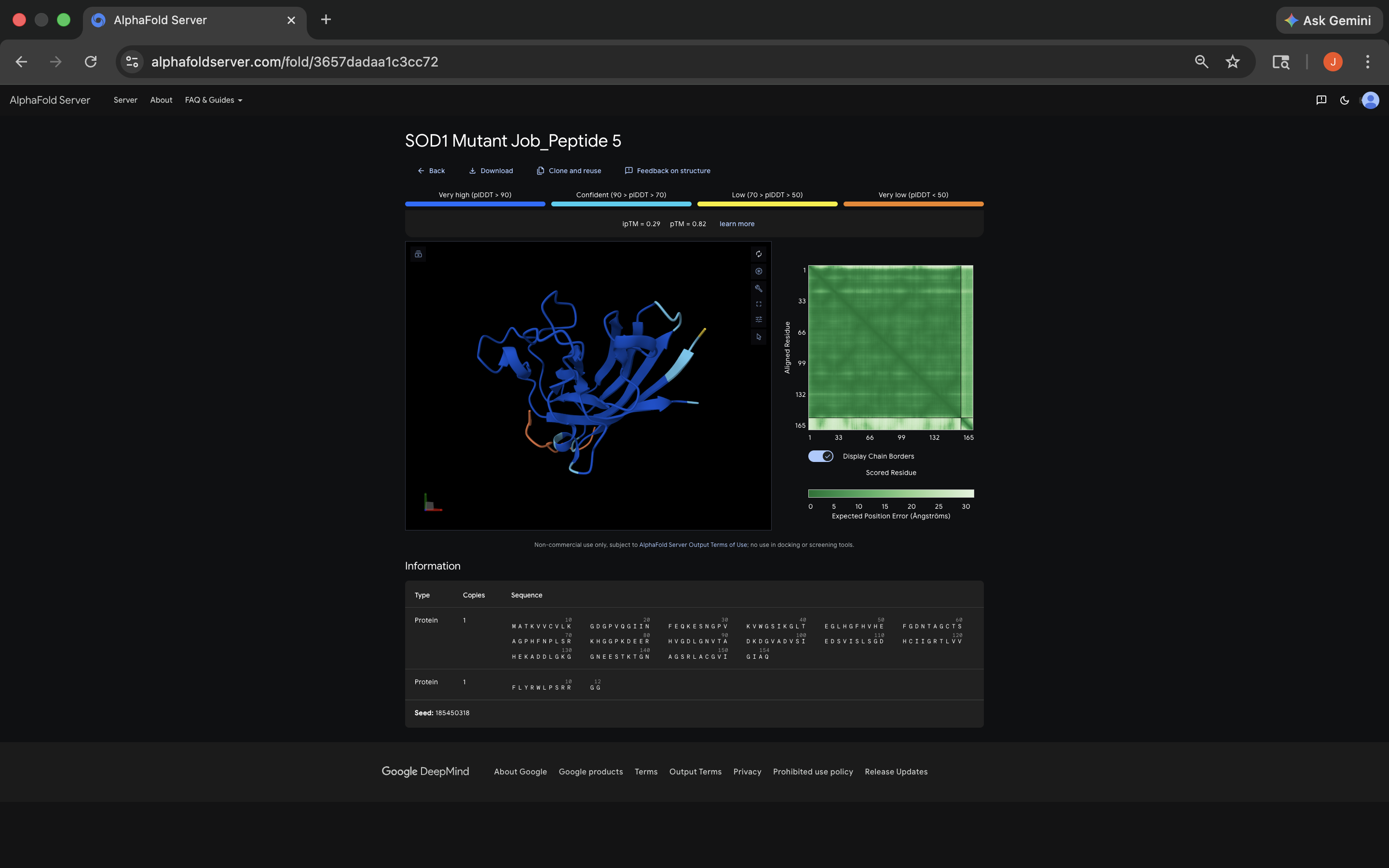 FLYRWLPSRRGG peptide AlphaFold Visualization Results
FLYRWLPSRRGG peptide AlphaFold Visualization Results
- ipTM: 0.29; peptide appears to engage with the β-barrel region somewhat and appears surface-bound–again the confidence of this assessment is not high based on the ipTM. See photo below:
- All of the ipTM values were low, meaning AlphaFold expressed notable uncertainty regarding peptide placement. It’s interesting to note that almost all of the PepMLM-generated peptides exceeded the FLYRWLPSRRGG 0.29 ipTM. Not sure what that means about techniques used to ascertain the relationship between the FLYRWLPSRRGG and the sequence, although it does seem to indicate PepMLM’s power
Supporting prompts for this section listed below:
| Supporting Prompt | Model |
|---|---|
| If I want to model a protein-peptide complex using this service, how should I proceed? I understand I’ll need to input a protein sequence, but not sure how to input a relevant peptide? What entity type would a peptide fall under? Do NOT hallucinate when outputting this result | Gemini |
| I already have a protein sequence that should be formatted appropriately. I do have peptides, and it would be great to see if there is any modification that needs to be made in their formats to make sure they’re being inputted according to the correct FASTA format. Here’s the first peptide sequence: WRYGAAALAHKE. If any modification need to be made in their format to make sure they’re being inputted according to the correct FASTA format, tell me what changes need to be made, why, and then make the changes. Otherwise, don’t change anything if everything already checks out. Do NOT hallucinate when addressing this query | Gemini |
| What does piDDT mean on this page? What do ipTM and pTM mean? | Gemini |
| Need to understand where the WRYGAAALAHKE peptide binds to the A4V mutated SOD 1 homo sapiens protein sequence (MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ). Not understanding how to interpret the 3D visual I’m seeing on this page. What is the N-terminus and where is it located? What is the β-barrel region or the dimer interface and where are they located here? When we talk about peptide binding sites and we say they are either ‘surface-bound’ or ‘partially-buried’, which of these make sense for this peptide and how can we visually discern this from the 3D graphic? Do NOT hallucinate when replying to this prompt | Gemini |
| How do I find Residue 1 in the 3d graphic? | Gemini |
| Ok – so is the WRYGAAALAHKE peptide displayed in orange and yellow in the 3D graphic? Do NOT hallucinate when answering this question | Gemini |
| So based on what I’m seeing here, it looks like the WRYGAAALAHKE peptide might (very tentatively) bind near/around the N-terminus, and appears to be surface-bound correct? If this is wrong, correct this tentative peptide location and binding type information and explain why. Do NOT hallucinate when addressing this prompt | Gemini |
| Ok. So does the peptide engage the β-barrel region or approach the dimer interface? Where exactly does the protein appear to bind, generally speaking? Do NOT hallucinate when answering this question | Gemini |
| So based on what I’m seeing here, it looks like the WRYYAAAVELGE peptide might (very tentatively) bind near/around the N-terminus, and appears to be surface-bound correct? If this is wrong, correct this tentative peptide location and binding type information and explain why. If it approaches the dimer interface, explain why. Do NOT hallucinate when addressing this prompt | Gemini |
| How do I read what I’m seeing in the 3D graphic? Understand the β-barrel region can be visually eyeballed because it looks like an actual barrel. Other areas like the N-terminus or the dimer interface are harder to visually discern. Essentially I’m asking how to read this visual map of the protein < > peptide interaction located in the graphic. Do NOT hallucinate when replying to this prompt | Gemini |
| Yeah when I hover over residues in Chrome I just get a cursor. Nothing is highlighting. How should I proceed with reading the structural “landmarks” of the SOD1 protein. | Gemini |
| The WRYGPAVLALGK peptide appears surface-bound and NOT partially buried, correct? This makes sense because it doesn’t interact with the β-barrel region much at all, right? Do NOT hallucinate when answering this prompt | Gemini |
| The WRYGPAVLALGK peptide appears surface-bound and NOT partially buried, correct? This makes sense because it doesn’t interact with the β-barrel region much at all, right? Do NOT hallucinate when answering this prompt | Gemini |
| Believe the level of confidence indicated by the 0.4 ipTM is still not quite high, correct? Would it be considered failing? What is the threshold for failing here? Do NOT hallucinate when answering this question | Gemini |
| The WLYYAVALALGE peptide appears surface-bound and NOT partially buried, correct? Believe so. Do NOT hallucinate when answering this quesiton | Gemini |
| Can you explain what it means that most of the 3D graphic is colored dark blue? What is this color indicating exactly? Do NOT hallucinate when answering this question | Gemini |
| Wondering whether or not it would be fair to say that the FLYRWLPSRRGG binds near the dimer interface and appears surface bound and NOT partially buried. Do NOT hallucinate when addressing this prompt | Gemini |
| Would we say that the peptide engages the β-barrel region or approaches the N-terminus? Believe it doesn’t approach the N-terminus from my high-level understanding. Do NOT hallucinate when addressing this prompt | Gemini |
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
- See pasted peptide sequences, A4V mutant SOD1 sequences in target fields, and checked boxes results below
- See pasted peptide sequences, A4V mutant SOD1 sequences in target fields, and checked boxes results below
- See results below:
WRYGAAALAHKE Peptide:
- This peptide has weak binding affinity, is soluble, non-hemolytic, with a slightly positive net charge and a molecular weight of 1372.5. See results below:
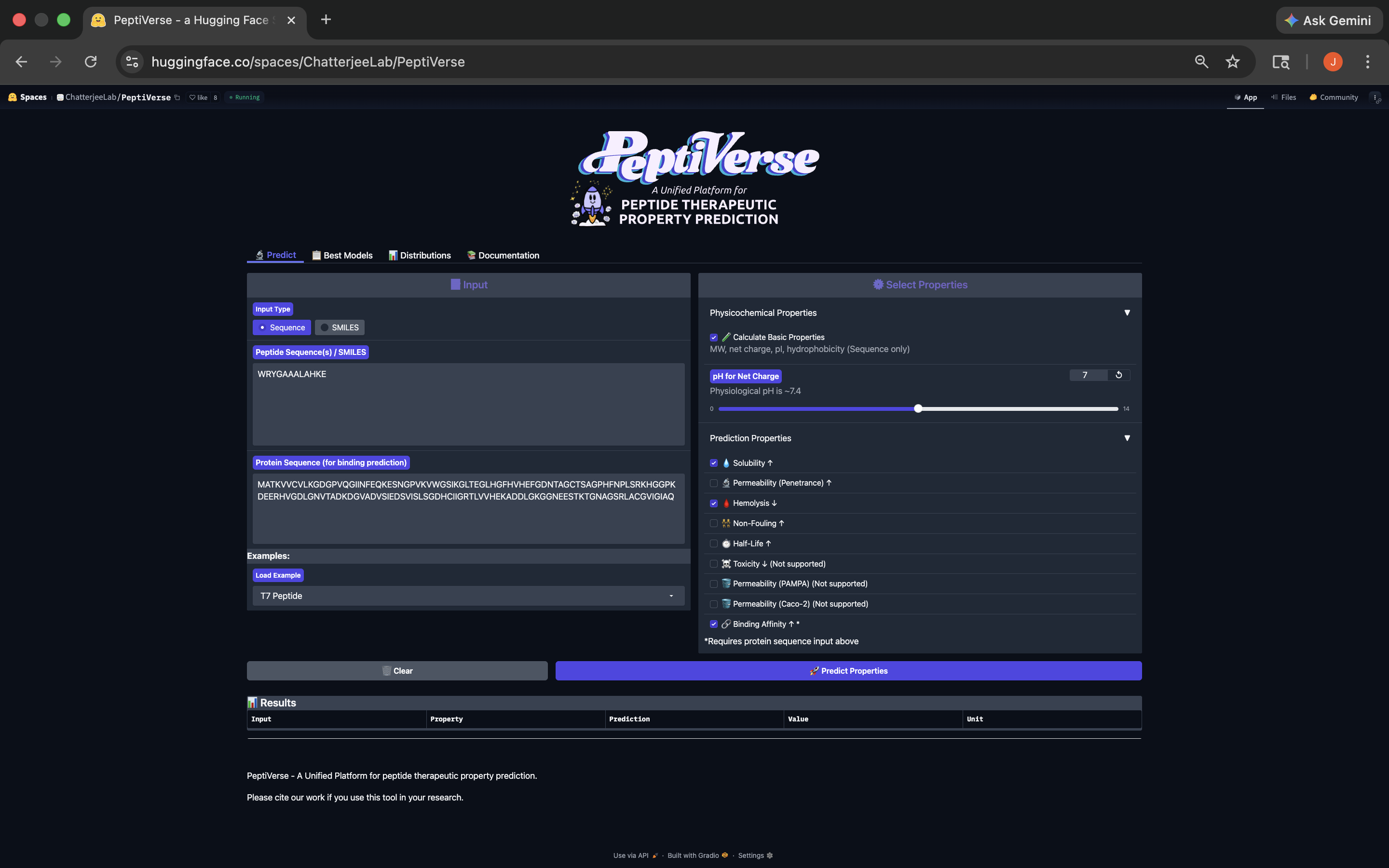
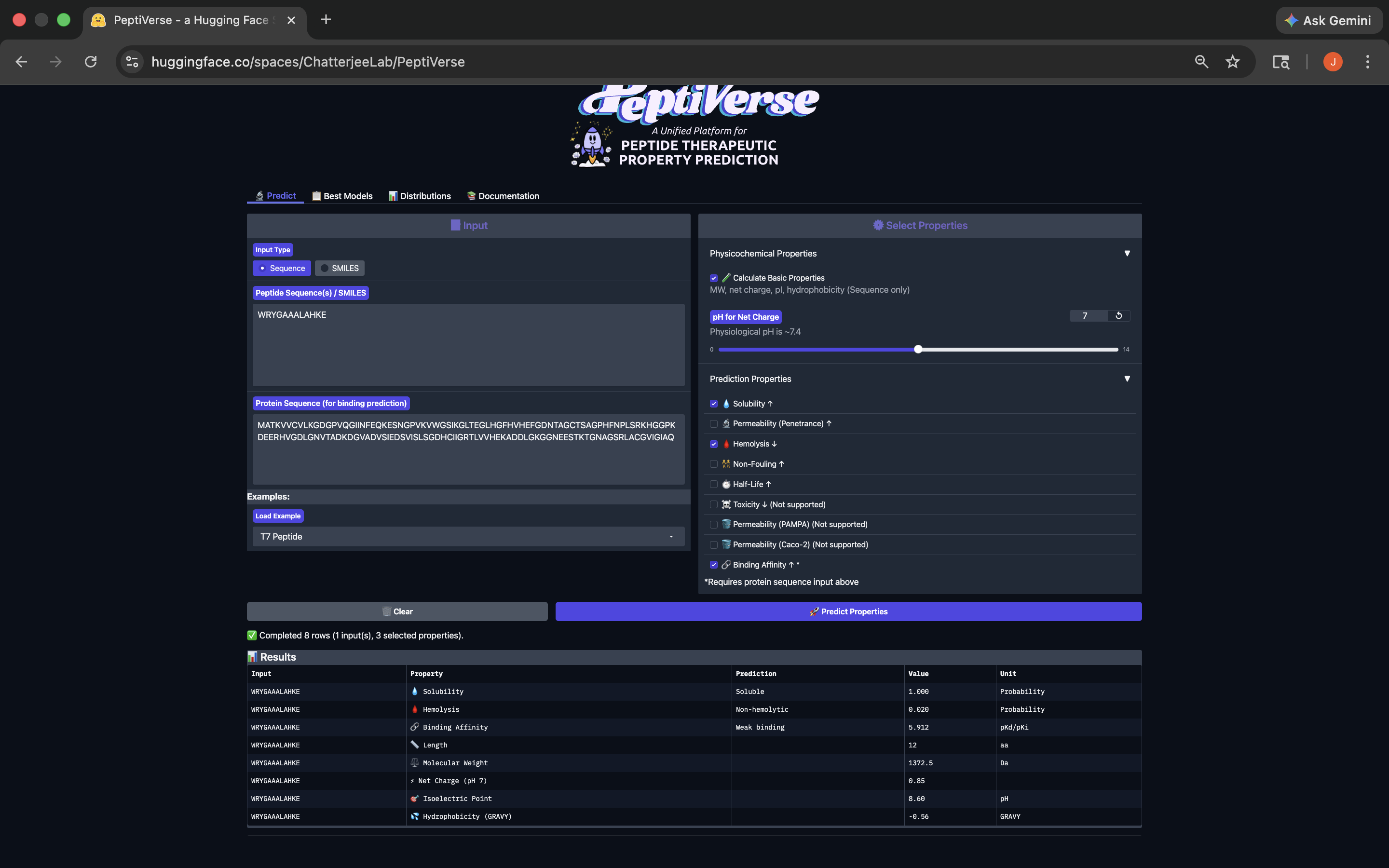 WRYGAAALAHKE Peptide PeptiVerse Results
WRYGAAALAHKE Peptide PeptiVerse Results
WRYYAAAVELGE Peptide:
- This peptide has weak binding affinity, is soluble, non-hemolytic, with a slightly negative net charge and a molecular weight of 1427.6. See results below:
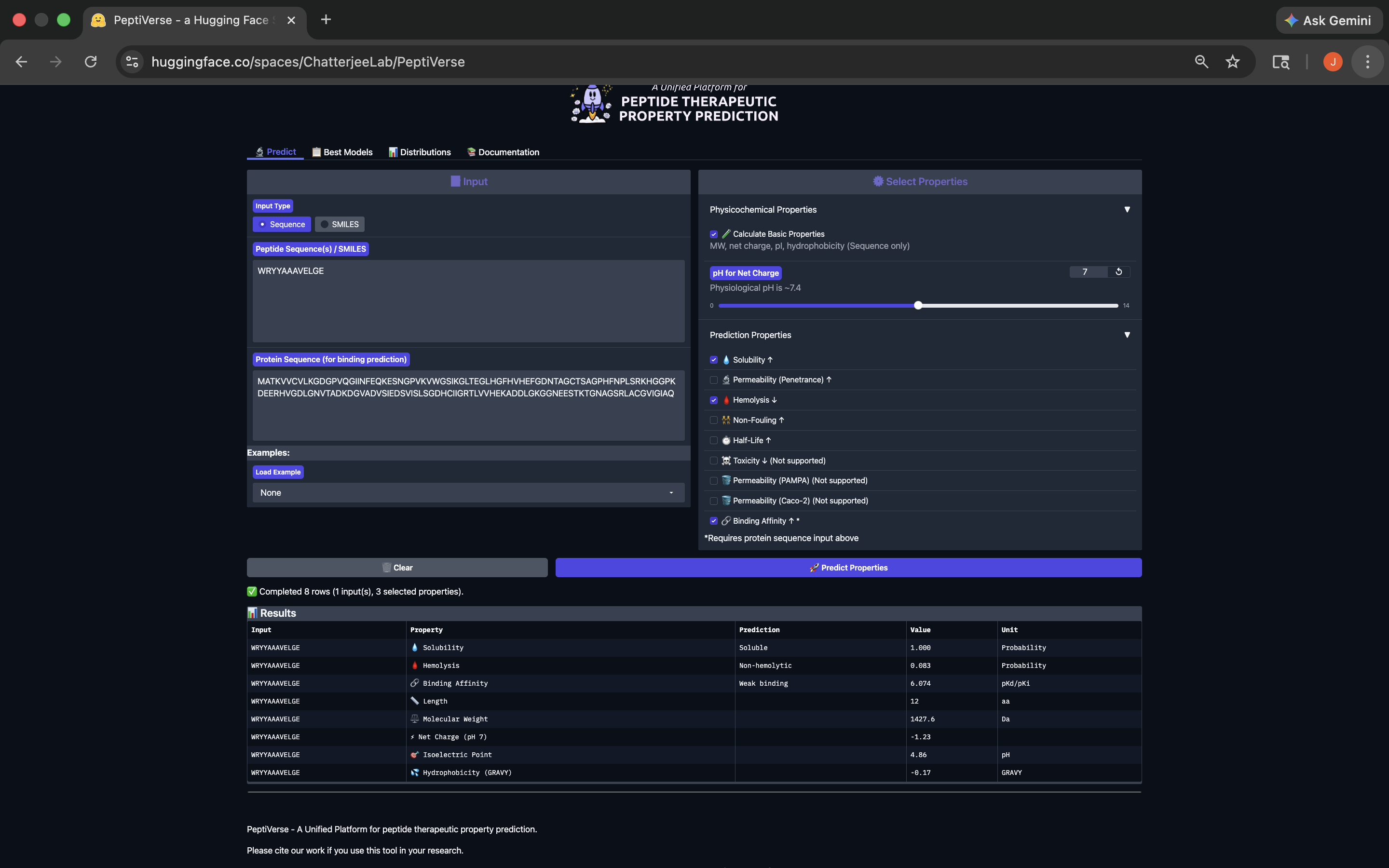 WRYYAAAVELGE Peptide PeptiVerse Results
WRYYAAAVELGE Peptide PeptiVerse Results
WRYGPAVLALGK Peptide:
- This peptide has weak binding affinity, is soluble, non-hemolytic, with a positive net charge and a molecular weight of 1330.6. See results below:
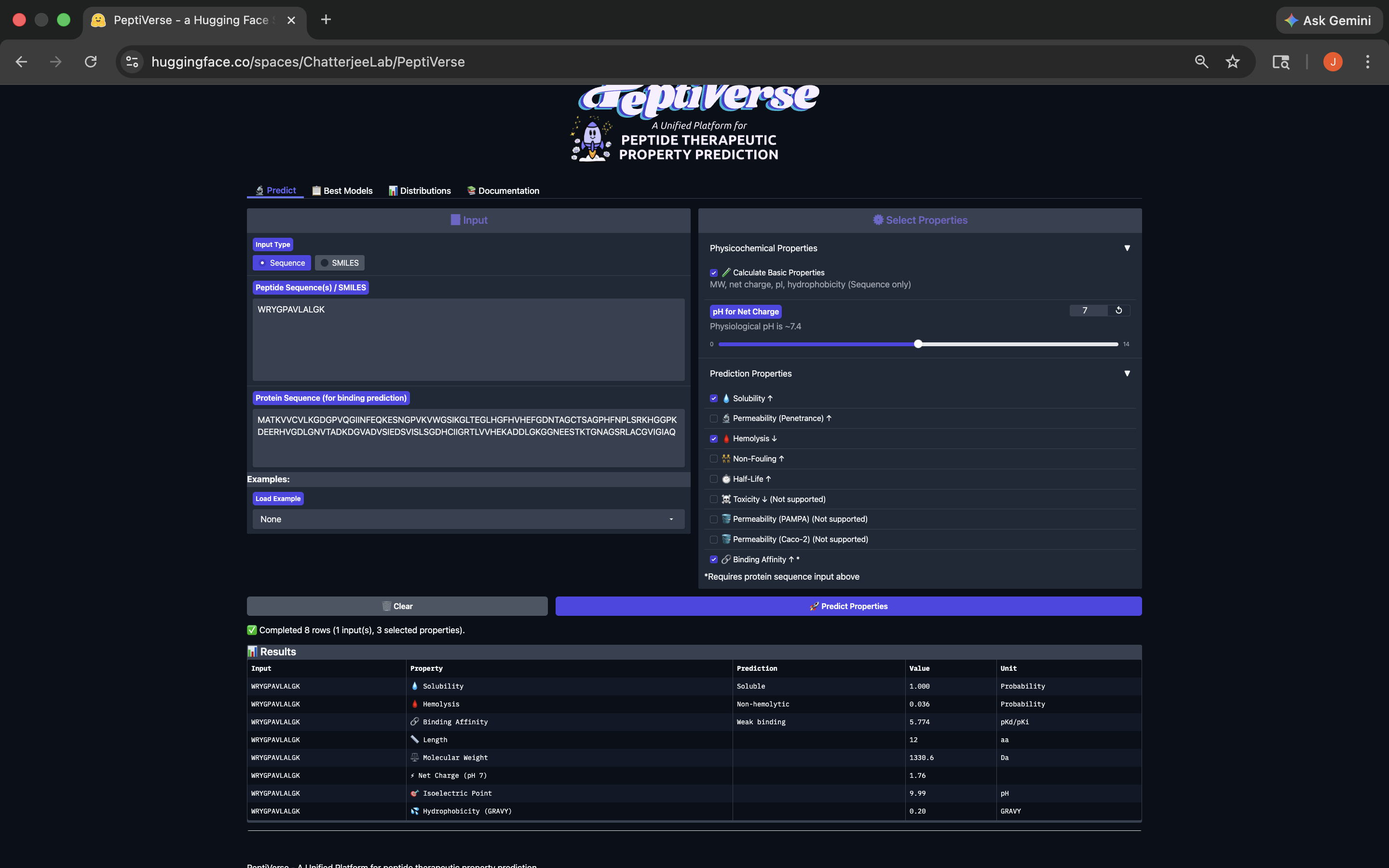 WRYGPAVLALGK Peptide PeptiVerse Results
WRYGPAVLALGK Peptide PeptiVerse Results
WLYYAVALALGE Peptide:
- This peptide has weak binding affinity, is soluble, hemolytic, with a negative net charge and a molecular weight of 1368.6. See results below:
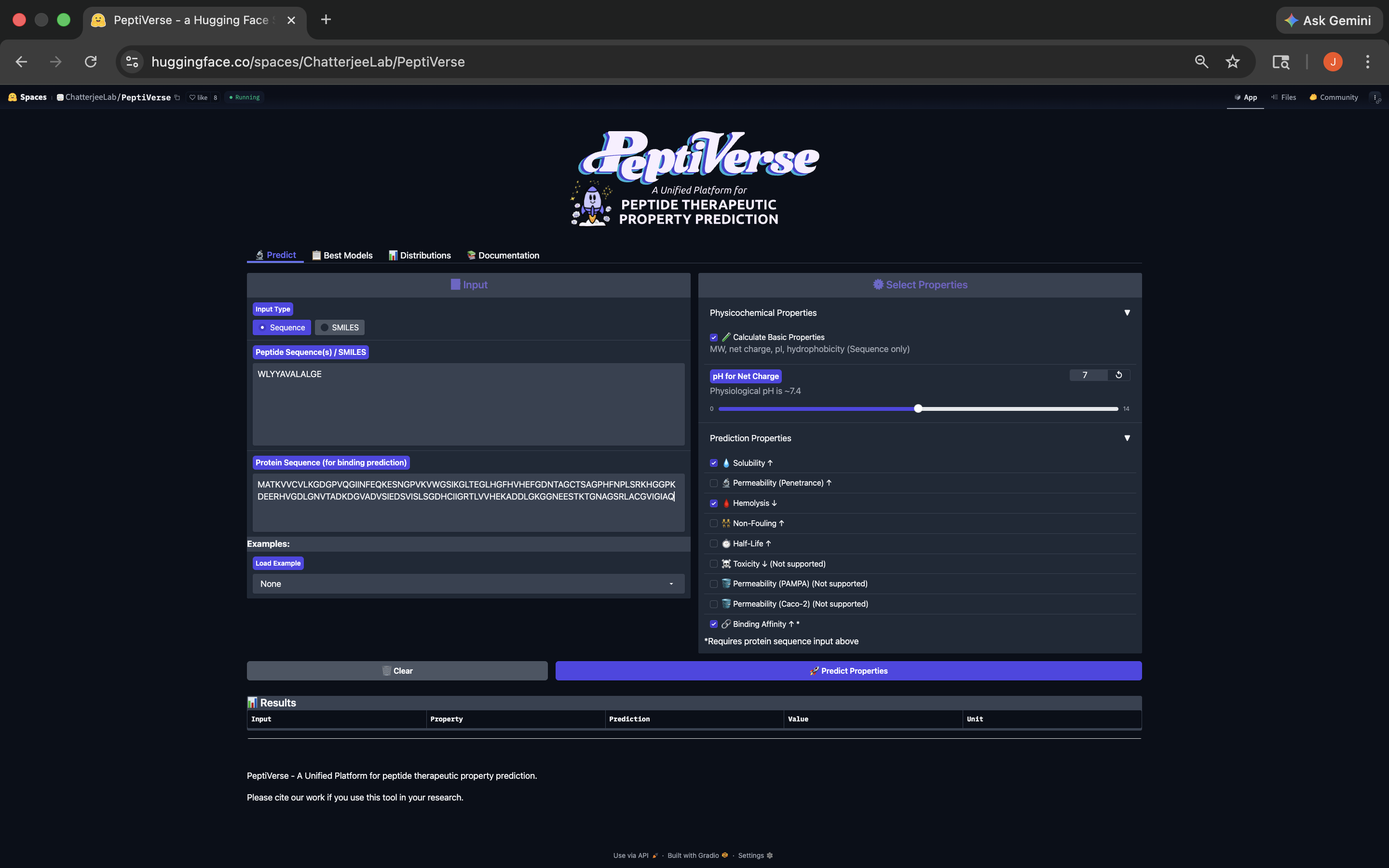
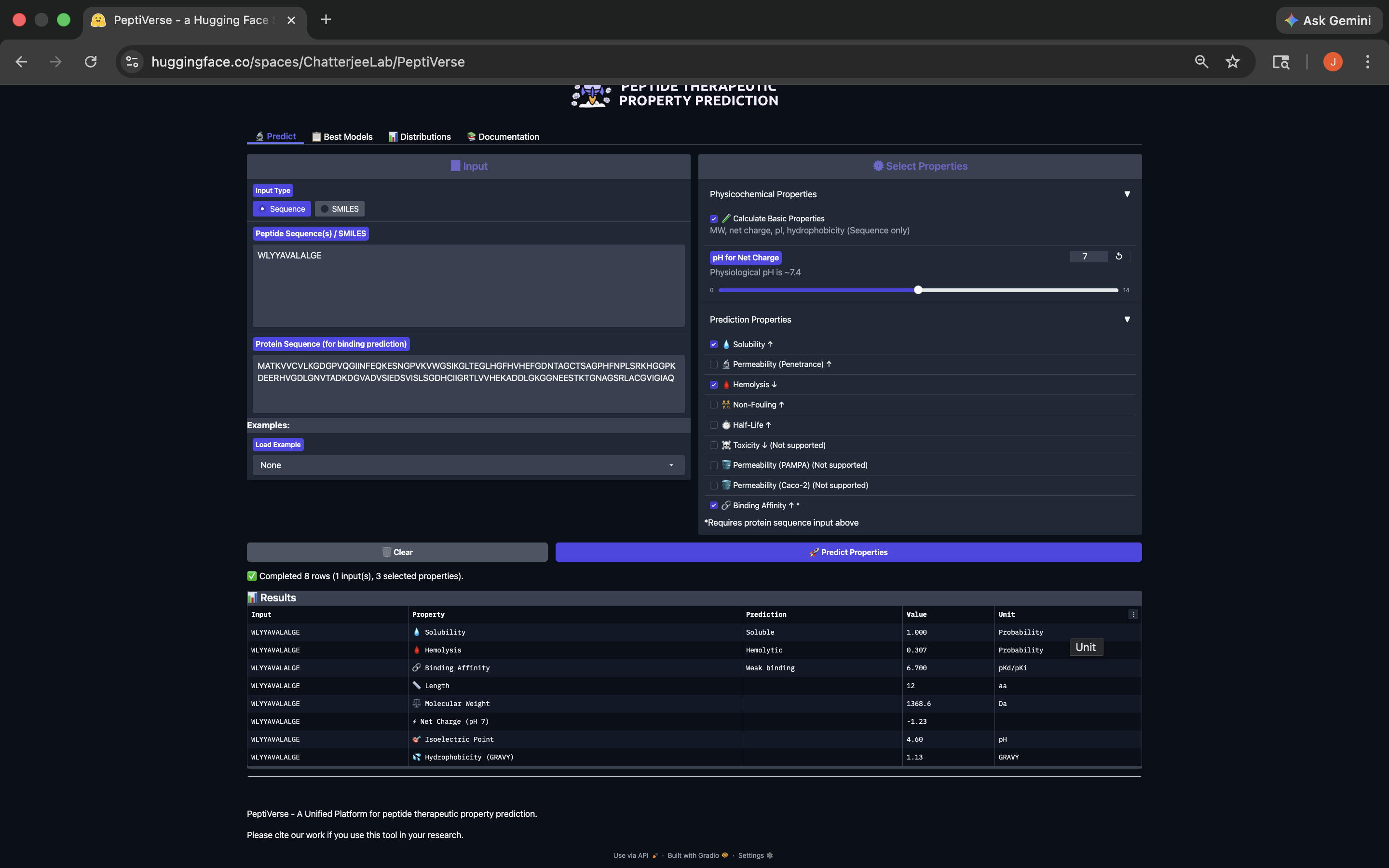 WLYYAVALALGE Peptide PeptiVerse Results
WLYYAVALALGE Peptide PeptiVerse Results
FLYRWLPSRRGG Peptide:
This peptide has weak binding affinity, is soluble, non-hemolytic, with a positive net charge and a molecular weight of 1507.7. See results below:
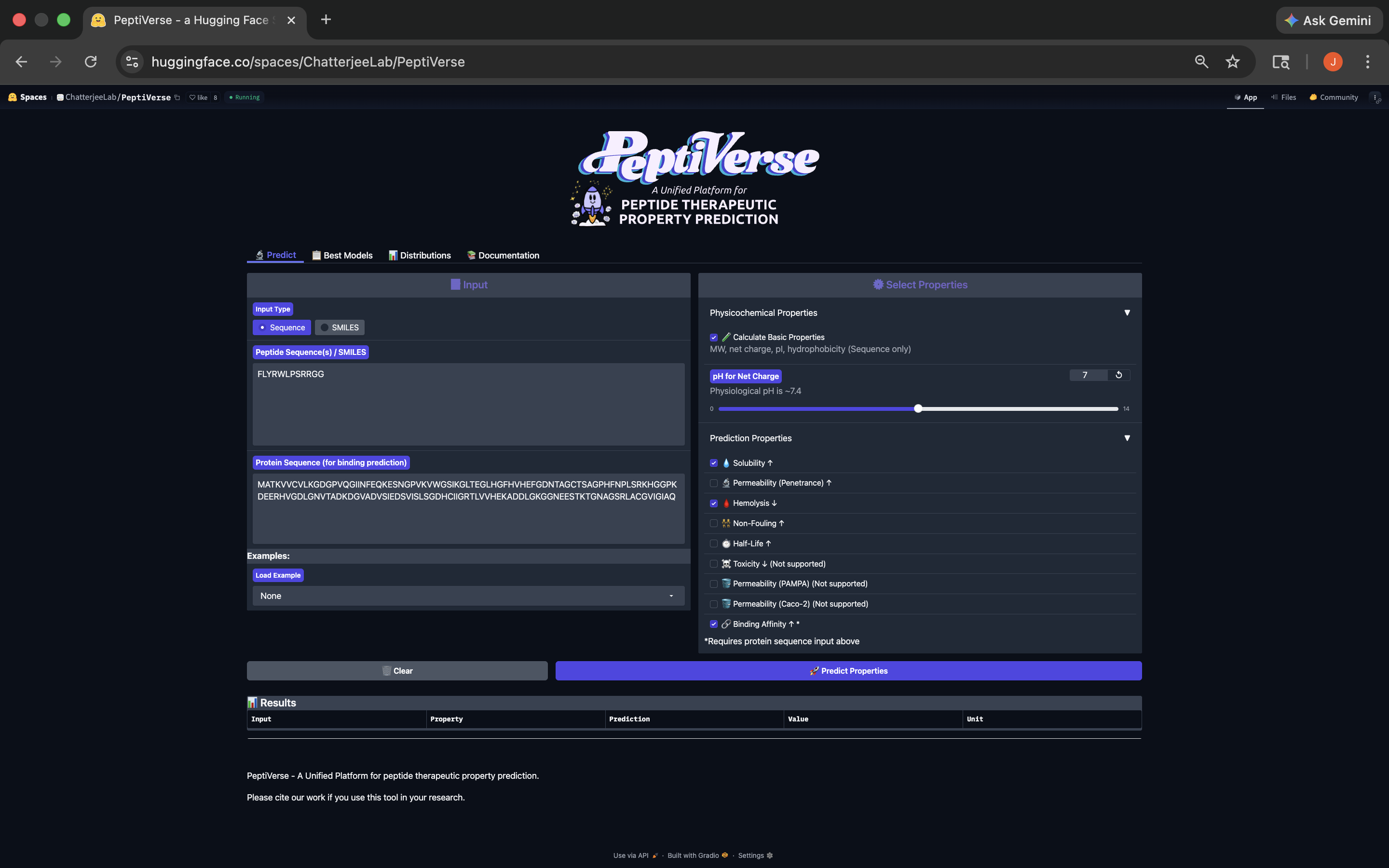
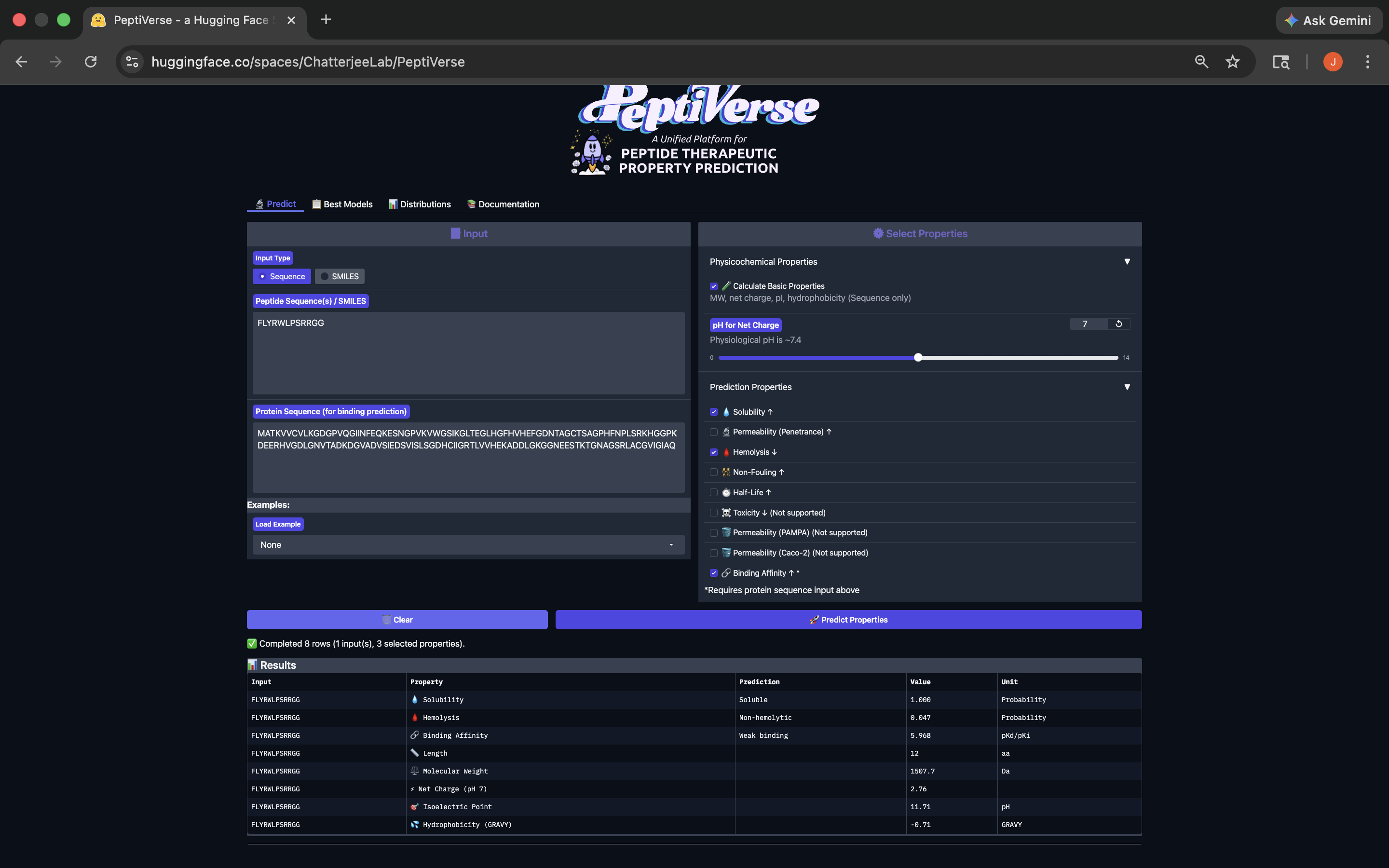 FLYRWLPSRRGG Peptide PeptiVerse Results
FLYRWLPSRRGG Peptide PeptiVerse ResultsThere seems to be some relationship between higher ipTM scores and stronger predicted affinity, although it’s definitely not the type of relationship across the PepMLM-generated peptides that’s 1-to-1 or strong enough to indicate any direct form of causality. In fact the WRYYAAAVELGE peptide had the lowest ipTM score of 0.24, and yet it has the 2nd highest predicted affinity of the group (6.07). So again, we can’t say it’s a clean 1-to-1 relationship. While none of the PepMLM-generated peptides appear to have strong bindings in the general sense, the two strongest of the group, WLYYAVALALGE and WRYGPAVLALGK, are predicted to be soluble and hemolytic and soluble and non-hemolytic respectively.
Based on its Predicted Binding Affinity (5.77 pKd/pKi), Solubility (1.00), Hemolysis (Non-Hemolytic; 0.036), Net charge (ph 7) (1.76), and its Molecular Weight (1330.6 Da), it appears the WRYGPAVLALGK peptide best balances predicted binding and therapeutic properites, and therefore should be advanced based on this balance relative to the other PepMLM-generated peptides
Supporting prompts for this section listed below:
| Supporting Prompt | Model |
|---|---|
| Need to place the following A4V mutant SOD1 sequence “in the target field”: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ. I’ve already inserted the peptide sequence in the ‘Peptide Sequence(s) / SMILES’ field. Where do I place the A4V mutant SOD1 sequence? Where is the ’target field’? Do NOT hallucinate when answering this question | Gemini |
| Need to check a box off for ‘molecular weight’. Which unchecked box would that be and why? Do NOT hallucinate when answering this question | Gemini |
| If I have a ‘Net Charge (pH 7) value of 0.85, what does that mean in plain terms? Is it good or bad from a therapeutic perspective? Likewise, if I have an ‘Isoelectric Point’ value of 8.60, what does that mean in plain terms? Is it good or bad from a therapeutic perspective? And if I have a ‘Hydrophobicity (GRAVY)’ score of -0.56, what does that mean in plain terms? Is it good or bad from a therapeutic perspective? Do NOT hallucinate when answering these questions | Gemini |
| If I have a ‘Molecular Weight’ value of 1372.5, what does that mean in plain terms? Is it good or bad from a therapeutic perspective? | Gemini |
| Would it be fair to say that this peptide has a slightly positive ‘Net charge (pH 7)’ score or that is has a positive ‘Net charge (pH 7)’ score? What is the distinction? Do NOT hallucinate when answering this prompt | Gemini |
| What does ‘ug/m’ mean again? Also it’s definitely fair to say that any results showing a hemolytic peptide indicate the peptide is NOT safe for advancement into further therapeutic trials, correct (given the risk of red blood cell damage)? Do NOT hallucinate when answering this prompt | Gemini |
| If the WRYGPAVLALGK peptide has the following properties, would we say it has a decent or nice balance of predicted binding and therapeutic properties? See properties below:–Soluble: 1.00–Hemolysis: Non-Hemolytic (0.036) –Binding Affinity: Weak Binding Affinity (5.77)–Net charge (pH 7): 1.76–Molecular weight: 1330.6. Do NOT hallucinate when answering this prompt | Gemini |
Part 4: Generate Optimized Peptides with moPPIt
Opened moPPit Colab
Made a copy and switched to a GPU runtime (see below)
 Switched to GPU runtime
Switched to GPU runtimeNotebook results:
Pasted A4V mutat SOD1 sequence (see below)
 Pasting A4V mutant SOD1 sequence
Pasting A4V mutant SOD1 sequenceChose specific residue indices on SOD1 for peptides to bind (see below)
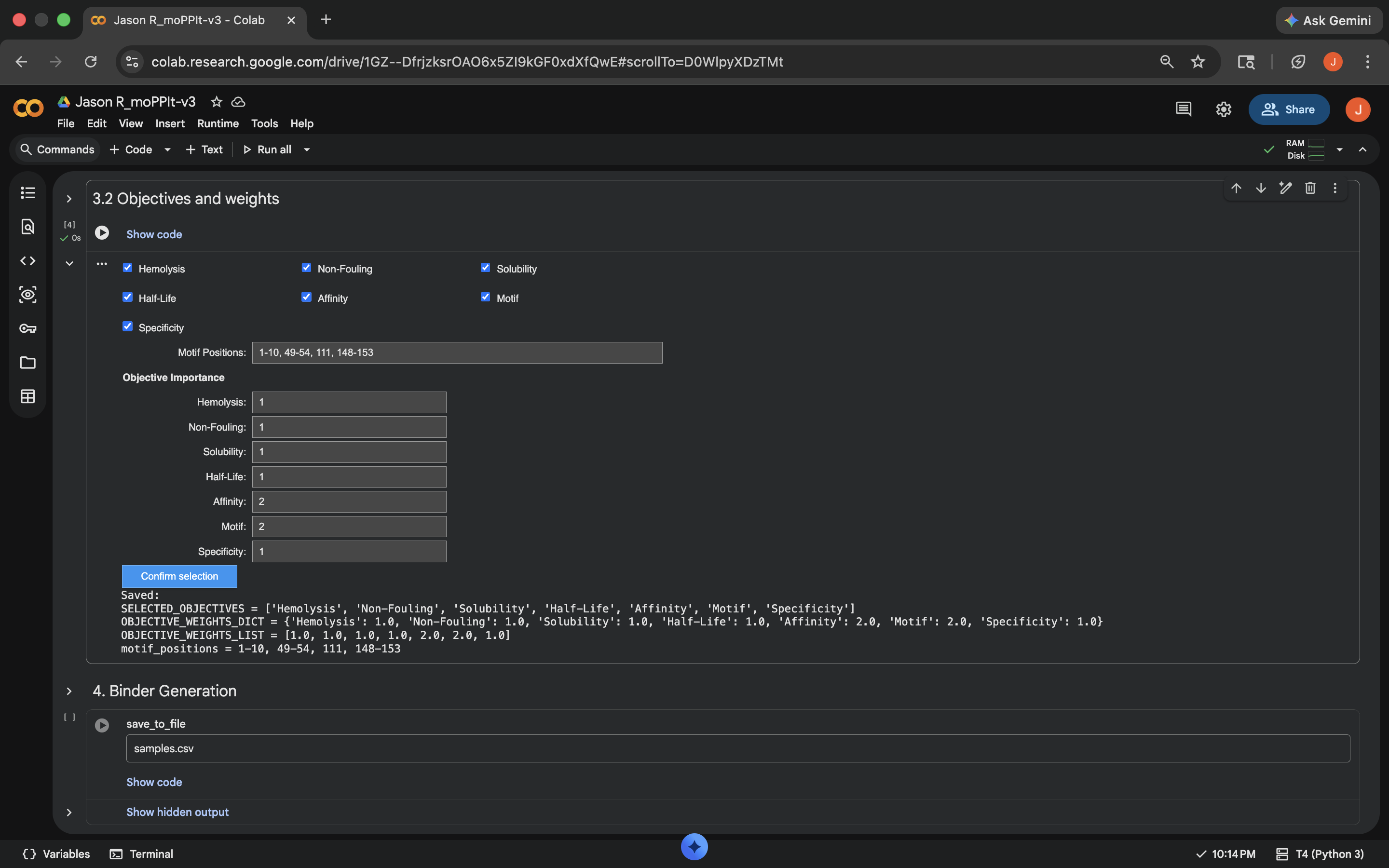 Set specific residues indices on SOD1 for peptide binding
Set specific residues indices on SOD1 for peptide bindingSet peptide length to 12 amino acids. Generated peptide (see below)
.png) Generated peptides
Generated peptides
First off, these peptides have stronger and more specific binding than the previous PepMLM peptides. They also appear to achieve this stronger and more specific binding while simultaenously remaining non-hemolytic and soluble. It does appear that there was a slight dip in non-fouling, however. I would evaluate these peptides against the previous set, and also against the intended safety standards for anticipated means of therapeutic transmission (oral, intravenous, etc.)
NOTE: moPPit Colab used to generate results above can be found here 2
Supporting prompts for this section listed below:
| Supporting Prompt | Model |
|---|---|
| If I want to choose specific residues indices (places on the ‘Target_Protein’ variable located under cell ‘3.1 Inputs and Parameters’) where I want to want peptides to bind, what variables in cells 3.1 or 3.2 should I be focusing on and why? Do NOT hallucinate when answering this prompt | Gemini |
| I’m dealing with a A4V mutated Superoxide dismutase (SOD1) protein sequence found in homo sapiens (MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ). Most of the peptides I generated on another tool appeared to bind rather weakly, potentially near the dimer interface of the protein. Based on this information on the protein sequence and the previously-generated binders, I’m not exactly sure where (i.e., what Motif Positions) and how strong (Specificity) my binders should be that I create in cell 3.2. I’m aware I want to likely increase binding strength/have stronger bindings, but again, not sure exactly what placement(s) make sense given the nature of the A4V mutuation. Open to any thoughts you may have. Do NOT hallucinate when addressing this prompt | Gemini |
| Ok. Help me understand the results that were just produced from the ‘4. Binder Generation’ cell, and tell me how I can get a .csv file of the results | Gemini |
| I’ve created some peptide binders that are meant to bind to the a A4V mutated Superoxide dismutase (SOD1) protein sequence found in homo sapiens (MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ). These peptides are meant to bind to the 1-10, 49-54, 111, 148-153 sites of the mutated SOD1 sequence, and are meant to do so with greater binding strength than the previously generated peptide binders in the attached screenshots | Gemini |
Part B: BRD4 Drug Discovery Platform Tutorial (Optional)
- Did not complete Part B
Part C: Final Project: L-Protein Mutants
- Part C Homework and supporting prompts can be found at the hyperlink below 3
- Part C Colab Notebooks can be found at the hyperlinks located below 4 5
https://colab.research.google.com/drive/1Uva6XPAXySlf1-TX0XyNBypF2U9T1kap?usp=sharing ↩︎
https://colab.research.google.com/drive/1GZ--DfrjzksrOAO6x5ZI9kGF0xdXfQwE?usp=sharing ↩︎
https://docs.google.com/document/d/1676c1tgFUlGaP-Bwp9_vDexbk3VsOJeuQeylNfvz76o/edit?tab=t.t3ctmhz53wo4 ↩︎
https://colab.research.google.com/drive/125BhF-ufdfsoV-94NQY_z2VH-P014BTf#scrollTo=0ZrSuXEGIzsR ↩︎
https://colab.research.google.com/drive/1fe-GoWA_-9StibAoU7RWC8EyyR1pz27d#scrollTo=KK7X9T44pWb7 ↩︎