| Please follow the skill instructions below and begin Phase 1: | Claude |
| Q: What biological problem are you most interested in solving? A: I’m interested in bacteriophage engineering, specifically taking microgravity-derived phage fitness insights from an International Space Station (ISS) experiment and proactively applying these insights to a non-Escherichia coli terrestrial bacteriophage Q: What system or organism would you prefer to work in? A: Thinking a Mycobacterium smegmatis (M. smegma) , but I’m not sure Q: Which synthetic biology approaches interest you most? (Select all that apply) A: ORACLE (Optimized Recombination, Accumulation, and Library Expression), In-cell recombination, and Combinatorial Library Synthesis (although I’m not sure and am open other synthetic biology approaches for this research) | Claude |
| Q: The ISS microgravity experiments you’re referencing — do you have a specific phage or dataset in mind? A: Yes — a specific phage/study (I’ll describe below) Q: For your M. smegmatis host system, which mycobacteriophage are you thinking of engineering? A: Open to suggestions based on what’s best for the project. Please note that I’m working in a low-biosafety level (BSL) lab, so that’s the only major consideration that comes to mind here Q: What is the core hypothesis — what ‘fitness’ trait from microgravity are you trying to engineer in? A: The desired fitness traits would most likely be ‘Broader host range’ and ‘Enhanced infectivity / adsorption rate’ in that order of priority | Claude |
| Q: What type of phage genes do you expect carry the microgravity fitness signal? (This shapes your DNA construct design) A: Tail fiber / receptor binding protein (RBP) mutations Q: For measuring success, which primary assay makes most sense to you? A: Combination of the above Q: You mentioned ORACLE / in-cell recombination / combinatorial library synthesis — are you set on using all three, or open to a focused recommendation? A: I’d like your recommendation given the BSL constraint and M. smeg system | Claude |
| Microgravity reshapes bacteriophage–host coevolution aboard the International Space Station (title); Phil Huss, Chutikarn Chitboonthavisuk, Anthony Meger, Kyle Nishikawa, R. P. Oates, Heath Mills4 Olivia Holzhaus, Srivatsan Raman (authors); 2026 (year) | Claude |
| Q: Does D29 with focus on the gp80 tail spike RBP sound right for your project? A: I don’t know enough to know enough so will defer to these findings Q: What host range panel would you like to test your engineered D29 variants against? A: Multiple M. smeg strains only (keep it BSL-1) Q: The paper used cell-free approaches for DMS. Do you want a cell-free component in your project? A: Do whatever will likely be logistically easier given the timeframes to complete the research (essentially 4 to maybe 6 weeks max.) | Claude |
| “generate proposal” | Claude |
| Take a look at the following quote from the URL below: “Strain-specific phage chassis to target bacteria that commonly cause infections during space flight.” What is the difference between a phage and a phage chassis? In general? In a biotechnological context? Do NOT hallucinate when answering these questions https://roadmap.ebrc.org/engineering-biology-for-space-health/ | Perplexity |
| Take a look at the following quote from the URL below: “Capability to produce novel phages on space missions for rapid control of evolved biofouling microbes. What are ’evolved biofouling microbes’? What is biofouling? I assume biofouling indicates something bad/undesirable, but I don’t know what the term actually means beyond my assumption Do NOT hallucinate when answering these questions https://roadmap.ebrc.org/engineering-biology-for-space-health/ | Perplexity |
| I understand how a phage can insert itself into a cell. Not exactly understanding if or how phages’ abilities contribute at all to personalized medicine developments (i.e., is there something about phage properties that make them particularly good candidates for personalized medical interventions)? Do NOT hallucinate when answering this question | Perplexity |
| What are the technical subcomponents of a biotechnology intervention or treatment using phage chassis? What do the supply chains look like, if any? Do NOT hallucinate when answering these questions. If you don’t know the answers to these questions, say so | Perplexity |
| “phage chassis synthetic biology manufacturing pipeline” search results | Perplexity |
| Are there any existing ways a biotechnology solution (let’s say a custom developed chassis) can proactively prevent itself from malicious dual-use? Analogous to large language model (LLM) safety refusal, are there any mechanisms that can be pre-built into a biotechnology solution to proactively prevent malicious dual-use? Do NOT hallucinate when answering these questions | Perplexity |
| How exactly do phages interact with genetic code information within a given cell? How do cell-based bacteria defend against unwanted phages? | Perplexity |
| I’m high-level aware that there are certain ’no-go’/‘do not edit’ pieces of genetic code. How are phages traditionally prevented from editing these ’no-go’/‘do not edit’ pieces of genetic code? Is that a thing? If I’m off in any way/if my conceptual underpinnings seem shaky, let me know Do NOT hallucinate when answering these questions | Perplexity |
| Tell me about about engineered synthetic biology kill switches | Perplexity |
| Have any engineered synthetic biology kill switches been implemented as part of phage therapies? Do NOT hallucinate when answering this question | Perplexity |
| If I’m making a novel phage-related therapy for astronauts, and I live in the United States, the Food and Drug Administration (FDA) would need to approve this therapy, correct? My assumption is yes. How does approval of a drug used outside of Earth’s atmosphere work from a regulatory perspective? Do NOT hallucinate when answering these questions | Perplexity |
| Are there any space health-related consortia specifically or explicitly aimed at making space medicine advancements as broadly accessible as possible to both spacefaring and terrestrial populations? If so, share information regarding said consortia Do NOT hallucinate. If you don’t know the answer to this question, say so | Perplexity |
| In medicine, what do we usually mean by ‘point of care’? What do we mean when we say that? | Perplexity |
| Do space medicine point of care guides exist? If so, are there any for commercial space tourists, astronauts, or future groups of spacefarers, including workers, etc.? | Perplexity |
| What is applied biomedicine? | Perplexity |
| What is the TRISH POCUS training referred to in the answer to the last prompt? What does POCUS refer to? | Perplexity |
| Tell me how to add a Promoter to a sequence in Benchling | Perplexity |
| Found this information from the Registry of Standard Biological Parts: BBa_J23106 Can you break down what this naming convention means and how I can find the relevant Promoter information in a sequence based on this naming convention? Do NOT hallucinate. If you don’t know the answer, say so | Perplexity |
| What is an alignment in Benchling? In Benchling, how do I put a codon optimized sequence under or next to a sequence I originally imported? Do NOT hallucinate when answering this question | Perplexity |
| How do I replace a sequence in Benchling with a codon-optimized sequence? | Perplexity |
| Bit confused regarding how to find a Promoter in a sequence in Benchling. I tried Auto-Annotate and it doesn’t seem to be working. Where should I go from here? | Perplexity |
| What is an RBS in Benchling? | Perplexity |
| What is a 7x His Tag? What is a Terminator? How do I find these in Benchling? Where are these traditionally inserted into a sequence in Benchling? | Perplexity |
| How do I paste sequences into a Benchling file? | Perplexity |
| How do I know where to insert a Promoter into a given sequence in Benchling? | Perplexity |
| Not totally understanding. If the start codon (the ATG) represents the start of the sequence, how do I insert something before that in Benchling? | Perplexity |
| What is an RBS? Where are they traditionally inserted into a sequence? | Perplexity |
| What do spacers look like in Benchling? Is it literally just empty space with no letters/codons? Something tells? | Perplexity |
| Where is a coding sequence traditionally inserted in a codon optimized sequence in Benchling? If there’s something off in what I’m saying, let me know | Perplexity |
| Where is a C-terminus in a protein in Benchling? | Perplexity |
| How do I find an amino acid view for a sequence in Benchling? | Perplexity |
| In Benchling, if I’m inserting a 7x His Tag and a Terminator, and I have a stop codon in my sequence, what is the traditional sequence? Is it 7x His Tag, stop codon, Terminator? Something else? | Perplexity |
| Any way I can add a Schema to a sequence after the fact in Benchling? | Perplexity |
| What is horizontal gene transfer? A separate question (perhaps): What is the technical term in biotechnology for transferring the abilities of one organism to another (ex. if I wanted to actually give a lizard the ability to fly like a bird by importing genetic properties that allow for the creation of wings for example)? | Perplexity |
| If I want to perform transgenesis in a biotechnological context (i.e., introduce a foreign gene into a new organism to confer a desired trait), and I want to start this process by sequencing the original foreign gene, what is considered the best practice in modern biotechnology for sequencing this original foreign gene? Is this sequencing method first, second, or third generation in the history of biotechnology? From some other period? What essential steps does it involve and how does it decode the bases of the original foreign gene? What is its output? Do NOT hallucinate when answering these questions. If you don’t know the answer to any of these questions, say so | Perplexity |
| How is Next-Generation Sequencing (NGS) considered second generation? Do NOT hallucinate when answering this question | Perplexity |
| If I want to perform transgenesis in a biotechnological context (i.e., introduce a foreign gene into a new organism to confer a desired trait), and I want to start this process by sequencing the original foreign gene via Next-Generation Sequencing (NGS), what is my input at the very beginning of the sequencing process? How is that input prepared for sequencing? Do NOT hallucinate when answering these questions. If you don’t know the answer to any of these questions, say so | Perplexity |
| What do phage isolation experiments usually entail? Are any elements of phage isolation experiments dangerous to human health and safety, and if so, why? | Perplexity |
| Take the steps in the “What phage isolation usually involves” section of the last prompt and break down the tools traditionally used for each step Do NOT hallucinate when answering this question | Perplexity |
| What is supernatant? | Perplexity |
| What are plaques in a phage isolation experiment context? | Perplexity |
| What does it mean to ‘pellet’ bacteria? | Perplexity |
| Do phages contain or have DNA? | Perplexity |
| Within the context of Gibson Assembly (biotechnology DNA assembly method), why exactly are molar ratios (apparently they need to be 2:1, insert:vector) important? What are molar ratios? Do NOT hallucinate when replying to this prompt | Perplexity |
| What exactly is the insert and what exactly is the vector within the context of the Gibson Assembly DNA Assembly method? Do NOT hallucinate when replying to this prompt | Perplexity |
| In the context of biotechnology and synthetic biology, what exactly is a plasmid backbone? Explain this to me as if I were a reasonably educated 16-year old Do NOT hallucinate when addressing this prompt | Perplexity |
| Tell me about the Phusion High-Fidelity (HF) Polymerase Chain Reaction (PCR) Master Mix. What is it? What are its subcomponents and what do they do? Do NOT hallucinate when addressing this prompt | Perplexity |
| Within the context of a Polymerase Chain Reaction (PCR), I believe primers are the pieces of DNA that get copied nth number of times, correct? If I’m mistaken, indicate as such, and the error in the initial reasoning. Do NOT hallucinate when addressing this prompt | Perplexity |
| So based on the answer to the last prompt: –Primers essentially define the space in the DNA sequence that will be copied? –What is a free 3′‑OH end? Explain this to me as if I were a relatively educated 16-year old Do NOT hallucinate when answering this prompt | Perplexity |
| Do primer pairs always need to have a temperature difference of 5°C from each other? If so, why? Do primer pairs always need to at a temperature of between 52–58°C before annealing? If so, why? What factors determine ideal primer annealing temperatures, and why? Do NOT hallucinate when addressing these prompts | Perplexity |
| What does phage therapy look like clinically? | Perplexity |
| In the answer to the last prompt, what exactly does the creation of the phage cocktail entail? Do NOT hallucinate when replying to this prompt | Perplexity |
| Take the attached .pdf file and explain to me the components to the plasmid and why they were chosen/why they make sense given the Aims and Experimental Goals of the research project outlined in the attached .md file Do NOT hallucinate/make things up when replying to this prompt. If things don’t make sense to you, or you don’t have additional context regarding the respective .pdf and .md file content, say so | Perplexity |
| I already went in and changed the amino acids myself in each of the Variants, so I’m actually not tremendously concerned about being able to pinpoint which amino acid residues changed across Variants. I care more about the 4th Caveat mentioned (DE3 lysogen status of CFT073, UTI89, MG1655). How would I verify this? Also, my Varian 1 and 2 plasmids are 2,429 kb respectively, and they’re pET-28a constructs (believe they might be meant to be expression cassettes). If there are any issues you see with this, let me know Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Want to understand the utility of Gibson Assembly in genomics. If I have a mutated fragment of E. coli bacteriophage, and I want to see how that mutated fragment interacts with (specifically how it lyses) multiple strains of E. coli. (ex. CFT073, UTI89), how can Gibson Assembly help me accomplish this goal? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Precision. It allows me to insert the precise mutated bacteriophage gene fragment into nth novel backbones to see their results, as opposed to passing phage on to new hosts | Perplexity |
| I’ve actually re-created the original Gene 17 Variant 1 and Variant 2, with corresponding amino acid mutations. Wondering how to appropriately create the primers for Gibson Assembly. How do I make sure they have the appropriate 5’ –> 3’ orientation? Also wondering if this is something I should go to Perplexity Computer mode to assist with Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Ok, so help me understand this. To test the gp17 Variants against CFT073, UTI89, and MG1655 E. coli. strains, I know I need an expression vector. I know I need primers for my Variants to allow the mutated gp17 to express itself. Taking pET-28a off the table, not understanding the connective tissue between the Variant primers and testing the primers for lysing (Pfu/mL) against CFT073, UTI89, and MG1655 E. coli. strains. In terms of an experimental protocol, am I supposed to make backbones for each of the strains, the essentially create ‘cuts’ in the backbones so the primers can be inserted? Also not understanding how all this dovetails into the idea of ‘rebooting a phage’ to test phage fitness Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Path B | Perplexity |
| So based on the results in the last couple prompts, it would be fair to say that my fragments are the mutated gp17 Variants and the phage backbone is a ’normal’ T7 phage genome (like BL21) correct? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Apologies if this is a dumb question. In the context of bacteriophage lab protocol workflows, when someone says they are going to ‘reboot the phage’, what exactly does that mean? Is it the same as Gibson Assembly? If it’s not, what comes first (pretty sure it goes Gibson Assembly –> phage reboot but not sure)? What does this look like in terms of a lab protocol, in simple terms? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| It seems like 2A is the equivalent of a transformation step from a lab protocol perspective yes? If not, or if it’s different somehow, why? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| What do Gibson Assembly protocols typically entail? What is needed to get the reaction properly set-up? What types of wet lab lab plates (if any) and what type of automation is used as part of standard Gibson Assembly protocols? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Want to understand if or how wet lab automation equipment is ever used in the context of phage rebooting. Also, what types of wet lab plates, if any, are used? Including incubation, how long does it usually take to execute a cell-free phage reboot? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| How long does it traditionally take to see plaque assays in a bacteriophage wet lab experiment? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| How is PFU/mL traditionally counted in standard bacteriophage wet lab protocols? Is this done manually in the wet lab or by some form of automated lab equipment? A bit of both? Also, what is an ANOVA statistical test? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| If I want to say a bacteriophage demonstrated greater fitness/lysing, what’s the appropriate terminology/acronym to use? Is it Multiplicity of Infection (MOI), Efficiency of Plating (EOP), or some other term? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Taking a look at this Draft Protocol, I want to understand the following: –Beginning with the end in mind, how much Variant 1 and Variant 2 (V1 and V2) DNA do I need to have by the time I reach Step 9 in order to successfully complete Steps 12 and 13? What should my quantities look like (wet lab measurements across Steps 4 - 12? –Should Step 5 be cut? I think the answer’s yes –Is a cell-free approach traditionally used for Phage Reboot (see Step 11)? Reference the attached document and the open literature on bacteriophage plaque assay/lysing to address this prompt. Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Ok, looking over the first part of the answer to the last prompt, just confirming the ‘Wet Lab Measurement Targets’ table indicate measurements to implement at each relevant Wet Lab protocol step so there’s enough V1 and V2 DNA concentrations to get appropriate PFU/mL readouts at the Plaque Assay/final read-out portion of the protocol, correct? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Apologies if this is a dumb question. Given the open academic literature on bacteriophage plaque assay/lysing, does the Step 9 concentration amount (~30 µg/mL) give us enough to work with or is it optimal relative to the expected PFU/mL output later in the protocol? Does it set things up well? Is it optimal, suboptimal, or nominal? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Wonderful – would you mind please re-doing the table that was outputted 2 prompts ago accordingly? I want all the Wet Lab Measurements in the protocol in adequate/sensible ranges based on the variance identified in the last prompt. In short, I want wet lab measurements corrected if needed across all wet lab steps in the protocol If you don’t know the prompt that’s being referenced from 2 prompts earlier in this thread, say so. Otherwise, please redo the table accordingly Do NOT hallucinate/make things up when replying to this prompt. If you don’t know something, say so | Perplexity |
| Based on this protocol, create a hypothetical PFU/mL graph showing results for Variant 1, Variant 2, and wild-type T7 bacteriophage against the intended tested E. coli. strains (CFT073, UTI89, MG1655) Explain the hypothetical results, why they are what they are, and do NOT hallucinate/make things up that aren’t sensible based on what exists in the literature on bacteriophage lysing research | Perplexity |
| Based on this protocol, create a hypothetical PFU/mL graph showing results for Variant 1, Variant 2, and wild-type T7 bacteriophage against the intended tested E. coli. strains (CFT073, UTI89, MG1655) Explain the hypothetical results, why they are what they are, and do NOT hallucinate/make things up that aren’t sensible based on what exists in the literature on bacteriophage lysing research | Perplexity |
| I made the following Variant Gibson Assembly Constructs in a standard E. coli. T7 bacteriophage backbone (NCBI Accession: V01146) based on the Variants created in the attached PDF. Want to understand the essential components of each Gibson Assembly Construct in 2nd/3rd-level detail (i.e., I want to explain the important pieces of each construct and why they’re there), but do not know how exactly where to begin. I know I have things in the Construct like primers, a Coding Design Sequence (CDS) for the mutant variant genetic fragment of interest (Gene 17), as well as T7 promoters and terminators, but not exactly sure how to piece all of this together into a cohesive narrative regarding what I constructed (the important pieces), their intended functions, and why they’re there How can we go about getting to this goal? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Let me answer your questions to the best of my ability in order: Question 1 Attempted Answer: If you look at the attached PDF, you’ll see a variety (I believe approx. 5) of amino acid substitutions for each Variant. When you look at Variant 1 and Variant 2 constructs, what you don’t see purely based on the screenshots is the fact that the respective CDS inserts per each Variant were crafted by manually changing the relevant Gene 17 amino acids per instructions in the attached PDF (i.e., reading what amino acid mutations took place and then copying them in Benchling). Then those Gene 17 fragments with the amino acid mutations were turned into their own respective fragments separate from their larger original sequence for testing within Gibson a-la the attached pdf (I think – I my memory’s a bit fuzzy on the last part). Hopefully that helps answer the question Question 2 Attempted Answer: Gibson Assembly helps express or amplify a specific genetic mutation within a novel construct. We insert primers into an expression backbone and then genetic circuit components like promoters and terminators help express said mutation. That’s my high-level understanding Feel free to ask further questions or elaborate in response to any answers. DO NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Here are the answers to your questions: –Yes, the rephrasing matches how I see my constructs –Variant 1 Fragment gp 17 CDS Amino Acid List/Sequence: MANVIKTVLTYQLDGSNRDFNIPFEYLARKFVVVTLIGVDRKVLTINTDYRFATRTTISLTKAWGPADGYTTIELRRVTSTTDRLVDFTDGSILRAYDLNVAQIQTMHVAEEARDLTTDTIGVNNDGHLDARGRRIVNLANAVDDRDAVPFGQLKTMNQNSWQARNEALQFRNEAETFRNQAEGFKNESSTNATNTKQWRDETKGFRDEAKRFKNTAGQYATSAGNSASAAHQSEVNAENSATASANSAHLAEQQADRAEREADKLENYNGLAGAIDKVDGTNVYWKGNIHANGRLYMTTNGFDCGQYQQFFGGVTNRYSVMEWGDENGWLMYVQRREWTTAIGGNIQLVVNGQIITQGGAMTGQLKLQNGHVLQLESASDKAHYILSKDGNRNNWYIGRGSDNNNDCTFHSYVHGTTLTLKQDYAVVNKHFHVGQAVVATDGNIQGTKWGGKWLDAYLRDSFVAKSKAWTQVWSGSAGGGVSVTVSQDIRFRNIWIKCANESWNFVRTGPDGIYFIASDGGWLRFQIHSNGKGFKNIMDSRSVPNAIMVENE* –Variant 2 Fragment gp 17 CDS Amino Acid List/Sequence: MANVIKTVLTYQLDGSNRDFNIPFEYLARKFVVVTLIGVDRKVLTINTDYRFATRTTISLTKAWGPADGYTTIELRRVTSTTDRLVDFTDGSILRAYDLNVAQIQTMHVAEEARDLTTDTIGVNNDGHLDARGRRIVNLANAVDDRDAVPFGQLKTMNQNSWQARNEALQFRNEAETFRNQAEGFKNESSTNATNTKQWRDETKGFRDEAKRFKNTAGQYATSAGNSASAAHQSEVNAENSATASANSAHLAEQQADRAEREADKLENYNGLAGAIDKVDGTNVYWKGNIHANGRLYMTTNGFDCGQYQQFFGGVTNRYSVMEWGDENGWLMYVQRREWTTAIGGNIQLVVNGQIITQGGAMTGQLKLQNGHVLQLESASDKAHYILSKDGNRNNWYIGRGSDNNNDCTFHSYVHGTTLTLKQDYAVVNKHFHVGQAVVATDGNIQGTKWGGKWLDAYLRDSFVAKSKAWTQVWSGSAGGGVSVTVSQDIRFRNIWIKCANESWNFFRTGMDGIYFIASDGGWLRFQIHSNGKGFKNIMDSRSVPIAIMVENE* –To the best of knowledge, I didn’t alter any promoter/terminator regions near gene 17 when building the fragments Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Let’s go with option a) for now Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| I think this is enough for me to work with post-export, so there’s no need to pause and tweak Variant 1 wording (that will be done by me off-line after the fact). Let’s now create the same 1–2 paragraph write‑up for Variant 2 now Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| The key conceptual difference is seeing which tail-fiber tip substitutions lead to greater Plaque-Forming Units/milliliter (PFU/mL) (i.e., greater bacteriophage fitness against multiple strains of E. coli. bacteria) If this seems off or incorrect in any way, say so | Perplexity |
| Let’s do something similar to what we did in the Construct Thread (see .docx file), and do something similar for the PFU/mL chart. Want to be able to clearly explain why the bacterial E. coli. strains were chosen, what expected results we expected to see across the Variants and the wild-type control, and the implications for the larger experimental aims of this research (i.e., we want to see if the bacteriophage gp17 mutations in the attached PDF unlock PFU/mL improvements against other strains of E. coli bacteria beyond the strain tested in the paper) Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| MG165 is the baseline, CFT073 for V1, and UTI189 for V2 | Perplexity |
| We would lean more towards ’narrow but strong’ it would seem like based on the fact that only one variant improve PFU/mL | Perplexity |
| We’d say it has broader host range | Perplexity |
| Find me a standard/well-cited bacteriophage lysing paper containing a Plaque-Forming Unit/milliliter (PFU/mL) read out. Want to understand how PFU/mL findings are traditionally graphically represented in the relevant academic literature Do NOT hallucinate/make things up when replying to this prompt. Let’s keep the focus on cited academic literature | Perplexity |
| Take a look at Step 8 of the attached protocol, specifically where it describes the ~70 ug/mL DNA concentrations that could be confirmed via a spectrophotometer like a Nandrop. Want to create the equivalent of the graphs displayed in Nandrop that show the intended ~70 ug/mL DNA concentration Do NOT hallucinate/make things up when replying to this prompt. Use available information on Nanodrop (or equivalent spectrophotometer) read outs to address this prompt | Perplexity |
| Take this protocol and convert it to Markdown Hugo Relearn Theme style Do NOT hallucinate/make things up when doing this. If you cannot do this, say so | Perplexity |
| So the constructs in the screenshots at the beginning of this thread are showing a bacterial expression system for a DNA construct, correct? Even if the protocol later uses a cell-free phage reboot, the Dry Lab Gibson Assembly step (the constructs) are showing a bacterial expression system for a DNA construct (the respective Variants), correct? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Taking a look at this attached protocol, what expression system am I using for the DNA construct in question (the expression of the Variants in the experiment)? Is it cell-free? Bacterial? Yeast? Pretty sure it’s cell-free based on the ‘Phage Reboot’ protocol step, but want to confirm Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Quick question: The E. coli UTI189 bacteria strain is different from the UT1 and UTI2 E. coli UTI189 bacteria strains correct? Seems like a bit of a dumb question, but am not sure Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Looking at the attached protocol, what are some common things that can/might go wrong traditionally when performing one of these (by these I mean bacteriophage PFU/mL) protocols? Give me 3-5 contingencies and mitigations and keep everything to max. 3 bullet points (specifically have a headline per contingency with maximum 3 mitigation bullet points underneath it) Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Thank you. What does the log₁₀ scale mean in simple terms? What does the ±0.3 log₁₀ SD stand for? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Another question about the graphs created in response to the previous prompt. For MG1655, why does Wild-Type outperform Variants 1 and 2? Does that make sense? If so, why? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Can you tell me how Gibson Assembly works and why it’s useful within the context of the Draft Protocol in 3-4 sentences? Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| And to be clear, based on what we know about Gibson Assembly and how it works, the exonuclease chews back, the polymerase multiplies the desired DNA fragment, and the ligase seals the multiplied fragment back into the backbone right? Correct any of my misperceptions about Gibson and do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Take the protocol at the beginning of this thread and give me a supply list of all relevant items and dollar amounts (USD). Have the final bullet list ‘Total Supply Cost:’. Have all this be in Markdown Hugo Relearn theme format Do NOT hallucinate/make things up when replying to this prompt | Perplexity |
| Here’s what I’m working on for my final project: The first aim of this project is to demonstrate if/how microgravity-induced BL21 E. coli strain bacteriophage mutations manifest in multiple different strains of E. coli. This will help discern how to increase terrestrial E. coli. bacteriophage fitness and lysing | HTGAA Claude AI Tutor |
| It has already acquired mutations. I want to apply these mutations to other E. coli bacteriophage strains to see if/how they increase fitness and lysing | HTGAA Claude AI Tutor |
| It’s the T7 bacteriophage (ATCC BAA-1025-B2). The downselected variants exhibiting the best performance against Urinary Tract Infection (UTI) bacteria showed mutations in gene 17, which seems to code for portions of the tail fiber protein. Additional mutations were found in gene 7.3, which might code for a scaffolding protein, as well as gene 11, which codes for an adaptor protein in the T7 tail. These were not discussed/referred to as the primary source of the Variants, which seemed to be gene 17 | HTGAA Claude AI Tutor |
| Believe the plasmid-based system is the intention, because I want to test the gene 17 mutation against other E. coli strains | HTGAA Claude AI Tutor |
| I’m frankly not sure. I’m doing the project in dry lab and I’m certainly not a bio SME–my experience is rather limited. Which would be easier to do or make more sense for a beginner to do, given the intention of the project? | HTGAA Claude AI Tutor |
| Plaque assay, although I don’t know how I’d do that in dry lab (except perhaps refer to it in analogous/similar diagrams with TBD wet lab validation in my final presentation). Also not entirely sure/not seeing the connective tie between using a T7 promoter with a BL21(DE3) host and how I would then test this in non-BL21 strains of E. coli? What fundamentals am I missing/not understanding here in terms of how to go from A to B given my project intention? | HTGAA Claude AI Tutor |
| Not really sure. UPEC sounds good, but I’d also be down for testing against other strains that make sense. My NCBI Reference Sequence is NC_001604.1 for Gene 17 referred to in the original University Wisconsin Madison paper on which this research is based (https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003568#sec009). Been told by my Node leaders to think about multiple strains. Think maybe testing against 3-5 strains sounds good. Just not sure what makes sense in terms of relevance and/or high BLAST with the Gene in question. | HTGAA Claude AI Tutor |
| I’ve done BLAST of NC_001604.1 gene 17 sequence sans comparison with published UTI phage therapy studies. Not sure about if/how I would confirm whether my dry lab setup gives me access to any published lysis efficiency data for T7 against UPEC strains Down for a suggested 3-5 strain panel now, along with justification/indications of non-hallucination/source validation for why said strains experimentally make sense | HTGAA Claude AI Tutor |
| This seems good for now/to start | HTGAA Claude AI Tutor |
| 96-well plate format | HTGAA Claude AI Tutor |
| Embarrassingly, I don’t know enough to know enough. What reporters or selection markers are conventionally used for projects like this? Why and how are reporters or selection markers conventionally used for projects like this? Open to your feedback on this | HTGAA Claude AI Tutor |
| It makes sense. Just not exactly sure if I’ll be ordering from Twist, as I’m a Committed Listener who attends a node ad hoc in-person and has rather limited wet lab exposure experience. Down for constituting a hypothetical Twist order (i.e., as part of my final presentation, saying ‘If I was going to do wet lab extensions of this work, I’d…’), but what I really want/need help with is designing plasmid Benchling mock-ups and any all genetic circuit diagrams in a program like Asimov in a way that makes sense for final project review/the final project presentation and is also explainable/understandable by yours truly. Any help with that would be greatly appreciated (again, not to say I don’t want assistance with a hypothetical Twist order…it just may not be 1st priority) | HTGAA Claude AI Tutor |
| Yes to both. I have a Benchling account and access to the gene 17 NCBI FASTA | HTGAA Claude AI Tutor |
| It’s asking for a species name for pET-28a in Benchling. What is appropriate here? | HTGAA Claude AI Tutor |
| Ok – I can now answer the question asked 2 prompts ago (i.e., whether the Gene 17 file [which in this case is a GenBank file] contains annotations). It does contain annotations. I also have downloaded a fully annotated pET-28a fully annotated sequence and see the T7 terminator and promoter tags. Not sure exactly how to proceed. Do I literally just go next to the T7 terminator and paste the all of Gene 17’s content between it and the T7 promoter? | HTGAA Claude AI Tutor |
| When I go to the colored block on the linear I see only the following lines with the following colors: –T7p52 (light blue color) –source (light blue color) –T7p52 (yellow color) There’s no circular map | HTGAA Claude AI Tutor |
| In the Benchling Linear map, it says the Gene 17 fragment’s 1662 bp long. It extends from 34624 to 36285 in the larger NC_001604 BL21 E. coli. bacteriophage genome | HTGAA Claude AI Tutor |
| Little bit confused. When I go to the Linear Map view, I just see 3 bars spanning the entire sequence fragment left to right. Their respective colors and labels are listed below: –T7p52 (light blue color) –source (light blue color) –T7p52 (yellow color) Based on what I’m seeing, I’m not sure where to add the annotation. When I go into Sequence Map view, I see a stop codon at the very end of the sequence fragment (TAA). I also see that the sequence fragment begins with ATG and after this light blue box that reads ‘T7p52’ (which I think might mean a promoter) there’s an AAC sequence of letters. The ‘T7p52’ light blue box contains the following letters listed above it: ATGGCT Not sure if any of this is helpful for helping me isolate what part of the sequence fragment I should annotate. Trying to share what I see so I can receive appropriate guidance/feedback | HTGAA Claude AI Tutor |
| Yes. It’s the entire sequence fragment. It begins at nucleotide position 1 (the beginning of the ATG start codon) and ends at position 1662 (the end of the TAA stop codon). So from here just annotate the entire fragment as the gene 17 CDS. Also could you please confirm that CDS stands for Coding Design Sequence? | HTGAA Claude AI Tutor |
| When I create the Annotation, I don’t have the option to select for a Feature type. I have the following options: –Annotation Type: Select ‘gene’ or ‘source’ –Strand: Select ‘Forward’, ‘Reverse’ or ‘None’ –Custom fields: Select ‘db_xref’, ‘organism’, ‘moi_type’, or ‘host’ To accurately label this annotation as a CDS how should I proceed? How do I accurately label this annotation period/in general? | HTGAA Claude AI Tutor |
| Yes, I see a colored block labeled ‘Gene 17 CDS’ appearing on my linear map | HTGAA Claude AI Tutor |
| Yes I do | HTGAA Claude AI Tutor |
| In Sequence Map view, I see the following between the T7 terminator and promoter: –6xHis –A light blue MCS –T7 tag –thrombin site –6xHis –A maroon ATG mark –A grey RBS mark –A lac operator that’s shaded a light-ish green Not sure where I paste the sequence based on this information. I don’t see the cluster of restriction site annotations referred to in the answer to the previous prompt | HTGAA Claude AI Tutor |
| The beginning of the marker or the end of the marker? | HTGAA Claude AI Tutor |
| It starts at position 298 | HTGAA Claude AI Tutor |
| The position number to the immediate left of the T7 terminator is position 25. It (and the rest of the T7 terminator) shows up before the ATG marker in the sequence. Does that make sense? Is anything out of the ordinary? How should we proceed? | HTGAA Claude AI Tutor |
| Before I answer the last question, at this point, I still have not inserted gene 17. Do I need to do that before answering your last question? | HTGAA Claude AI Tutor |
| The last position number shown in my pET-28a sequence is position number 5369 | HTGAA Claude AI Tutor |
| The gene 17 sequence is 1662 bp long. The bp notation is synonymous with the number of nucleotides, correct? | HTGAA Claude AI Tutor |
| Ok I have both sequences open and selected the region from position 298 to position 5369 in my pET-28a file. How should we proceed? What should I click on or do next? | HTGAA Claude AI Tutor |
| When I try to paste I get a reading that say ‘You’re about to paste a sequence that includes 0 annotations, 0 translations, 0 primers, 0 parts’. That’s not correct based on what I see in the Gene 17 fragment file. How should I or what type of pasting should I do from the Gene 17 fragment file? | HTGAA Claude AI Tutor |
| The paste succeeded. The new total length of the plasmid is now 1960 bp. How should we proceed? Pretty sure we’re going to need to re-annotate the new plasmid sequence at some point in this workflow | HTGAA Claude AI Tutor |
| There is something that has a ‘T7 tag’ label located before the ATG marker and before the T7 terminator. Unlike the T7 terminator, it is not a shade of light green (it’s grey). I don’t see anything explicitly labeled ‘T7 promoter’ anywhere in the sequence (this includes near the beginning of the sequence) | HTGAA Claude AI Tutor |
| No it does not | HTGAA Claude AI Tutor |
| Ok – I just restored everything in the original 5369 bp pET-28a back to its original version. Issue seems to be that the ATG marker in this sequence sits before the T7 promoter so when the Gene 17 sequence was added, it removed the T7 promoter. How should we proceed with the Gene 17 sequence re-insertion based on this information? | HTGAA Claude AI Tutor |
| In my pET-28a, the T7 promoter sequence ends at position 387. Proceed with the copy and paste job by selecting everything in the pET-28a from position 387 to the end, deleting it, then re-inserting the Gene 17 CDS fragment, correct? | HTGAA Claude AI Tutor |
| After selecting from position 388 to 5369, deleting that region, and then pasting in my Gene 17 CDS, the new total plasmid length is 2049 bp | HTGAA Claude AI Tutor |
| Yes I visually see the T7 promoter, then double checked via Ctl + F search and the ‘TAATACGACTCACTATA’ T7 promoter nucleotide sequence appears this time | HTGAA Claude AI Tutor |
| If I look at the sequence in Sequence Map view here’s what I see going from the beginning of the sequence downwards (i.e. from the 1st nucleotide position downward): 1. T7 terminator 2. T7 promoter 3. Gene 17 CDS If there’s anything wrong or incorrect here, say so | HTGAA Claude AI Tutor |
| Yes I can confirm the Gene 17 CDS annotation starts immediately after the T7 promoter in the sequence (i.e., the CDS begins right at or just after position 387). It begins at bp position 388 in the sequence | HTGAA Claude AI Tutor |
| My annotation panel shows the T7 promoter, Gene 17 CDS, and the T7 terminator, but it also contains legacy pET-28a annotations from the original sequence. Is that a major issue or can we move on/proceed? | HTGAA Claude AI Tutor |
| Do I need to clean up the existing annotations before we move on? | HTGAA Claude AI Tutor |
| So if I look to Turns 7 and 8 earlier in our exchange, we were discussing panel of multiple strains to test the T7 bacteriophage Gene 17 against. Right now we’ve inserted the T7 bacteriophage Gene 17 into a pET-28a plasmid. Not sure where to go from here based on that previously stated intention (which I still want to carry out). Believe we might need to create multiple plasmids before the final project proposal can be crafted | HTGAA Claude AI Tutor |
| Reference the content in response to Turns 7 and 8 earlier in this thread. The strains to test against were the following: – BL21(DE3) | Your baseline/control — the original host background for the microgravity phage experiment –CFT073 (UPEC) | Well-characterized uropathogenic E. coli strain; genome fully sequenced (Welch et al., 2002, PNAS); directly relevant to UTI therapeutic motivation –UTI89 (UPEC) | Second UPEC clinical isolate; used extensively in phage-host range studies (Dhakal & Mulvey, 2012); LPS structure differs from lab strains –MG1655 (K-12) | Standard lab K-12 strain; T7 infects it but with different efficiency than B-strains — good for seeing how LPS variation affects your mutant tail fiber Based on this information above, the fact that we’ve inserted the T7 bacteriophage Gene 17 into a pET-28a plasmid, and the fact we need plasmid tested across multiple strains, how should we proceed? How do we go from here based on the existing plasmid? | HTGAA Claude AI Tutor |
| What is IPTG? Can I do that purely dry lab in Benchling? If not, tell me how we test the strains against the existing T7 bacteriophage Gene 17 pET-28a plasmid Do not currently know the answer regarding whether BL21(DE3), CFT073, UTI89, and MG1655 are all DE3 lysogens | HTGAA Claude AI Tutor |
| Before we proceed, can I ask if you can ingest/extract insights from the University of Wisconsin, Madison paper on which this research is based (see URL below)? https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003568#sec009 If you can functionally do this, let me know | HTGAA Claude AI Tutor |
| Ok. Let me paste content from the paper into this chat for ingestion. My desire for doing this is the following: in most of this chat so far, we’ve focused on Gene 17, as this is the gene responsible for creating the microgravity-based Variants that were tested for and demonstrated stronger than normal fitness/lysing potential against terrestrial UTI bacteria. I want to make sure that as we craft Aim 1, we don’t miss any other Genes/relevant mutations that should also be tested against. If this means the pET-28a + Gene 17 plasmid needs to be changed somehow, or in some way, or if another plasmid needs to be created, that’s OK. I want to make sure we’re thorough in Aim 1 based on the intention stated at the beginning of this chat (if/how microgravity-induced BL21 E. coli strain bacteriophage mutations manifest in multiple different strains of E. coli. This will help discern how to increase terrestrial E. coli. bacteriophage fitness and lysing). The items in the ** ** in the parentheses seem to be of importance. So with that, here are the relevant excerpts: Section (Enriched mutations are distributed broadly in T7 phage): Next, we sought to identify mutations in the phage or bacterial genome that influenced phage-host interactions under microgravity. We performed whole-genome sequencing (WGS) of T7 and E. coli BL21 before and after incubation, using pre-incubation genomes as references to identify de novo mutations in the 23-day samples from each condition to ensure both phage and bacterial populations had ample time to propagate. To determine whether de novo non-synonymous substitutions or frameshifts in T7 were significantly enriched, we compared the pooled frequencies of abundant non-synonymous mutations to the distribution of synonymous de novo substitutions in each condition (Mann–Whitney U test, FDR-adjusted p < 0.05; Fig 3A). To assess whether specific genes had significantly more non-synonymous substitutions than other genes, we calculated this mutation density for each gene and compared it to the average mutation density per condition (one-tailed t test, FDR-adjusted p < 0.05, one-sided 95% CI; Figs 3B and S1). Finally, we compared the gene-level distribution of non-synonymous mutations between microgravity and terrestrial conditions (Mann–Whitney U test, FDR-adjusted p < 0.05) to identify genes with condition-specific enrichment of these mutations (Figs 3C and S2A). Significantly enriched (p < 0.05) phage substitutions were found across both structural and non-structural proteins under terrestrial and microgravity conditions (Fig 3B). In microgravity, gene product (gp) 7.3 and gp11 exhibited significantly more de novo non-synonymous substitutions than other genes (Figs 3B and S1A). Mutation density was overall higher terrestrially and no gene showed significant enrichment compared to others terrestrially (S1B Fig). Although gp7.3 is not fully characterized, it is considered essential for T7 infectivity in E. coli BL21 under terrestrial conditions [38]. This small 99-amino-acid protein may function as a scaffolding protein or contribute to host adsorption, though its role in the mature virion remains uncertain [39–41]. gp7.3 harbored seven significantly enriched substitutions in microgravity, the highest number observed in any gene under that condition. These substitutions were distributed throughout the protein (Figs 3D and S3), with four notable changes (E48K, E61K, D68Y, D68A) involving substantial shifts away from negatively charged residues. The only significantly enriched mutation in gp7.3 terrestrially was a six-amino-acid deletion spanning G42 to V47. The region from G39 to Q50 contained a dense cluster of substitutions and in-frame deletions, including a 3-amino-acid deletion (G39-T41) terrestrially and a deletion from M46 to Q55 in microgravity, all occurring in a region of the protein predicted to be unstructured (S3 Fig). The high number of enriched substitutions and recurring in-frame deletions in this small protein suggest that gp7.3 is both structurally flexible and critical for phage activity in both environments. gp11 is an adaptor protein within the T7 tail that connects the portal protein gp8, the nozzle protein gp12, and the six subunits of the tail fiber protein gp17 (Fig 3E) [38,40,42]. Enriched substitutions were distributed throughout gp11, spanning both exposed and buried residues (Figs 3F, S4A, and S4B). One significantly enriched substitution, R2C, arose independently twice in microgravity and is located in a flexible region capable of directly interacting with gp17 tail fibers (S4C and S4D Fig). These findings suggest that the substitutions may influence phage fitness by altering gp11’s structure or stability rather than through direct interaction with the bacterial host. Comparison of mutation abundance revealed that de novo non-synonymous substitutions were significantly more prevalent in the nozzle protein gp12 after incubation in microgravity than under terrestrial conditions, suggesting a more prominent role for this protein in microgravity (Fig 3C). Of the six individually enriched non-synonymous substitutions identified across both conditions, five involved changes toward positively charged residues (Q184R, R205H, Q242R, K404R, and W707R). These substitutions were distributed throughout the protein, with three more likely contributing to host interactions (S5 Fig). Specifically, R205 is surface-exposed and positioned near the host, Q242 lies close to the terminus of the DNA delivery channel, and Q184 faces directly toward the host. The charge shifts and spatial distribution of these substitutions highlight the functional importance of gp12 in enhancing phage fitness under both terrestrial and microgravity conditions. Several other significantly enriched substitutions were particularly notable. In microgravity, the V26I substitution in gp0.5 was the only mutation to sweep the entire phage population—and did so independently in two replicates—indicating a strong fitness advantage. gp0.5 is an uncharacterized class I gene, potentially associated with the host membrane due to the presence of a putative transmembrane helix [22]. Under terrestrial conditions, the T115A substitution in gp4.7 was significantly enriched and highly abundant across all three replicates. No mutations were detected in this gene under microgravity, suggesting selection pressure may be unique to terrestrial conditions. Although the function of gp4.7 remains unknown, BLASTP analysis identified homologs with ~40% similarity to putative HNH endonucleases in Klebsiella and Pectobacterium phages [43]. Lastly, numerous significantly enriched substitutions were found in the tail fiber gp17, particularly under terrestrial conditions (Fig 3B). In both environments, substitutions were concentrated in the C-terminal tip domain, with repeated mutations at D540 and neighboring residues. This region is a known determinant of host range and infectivity in terrestrial E. coli strains [26], and these results suggests continued importance during prolonged incubation in both gravity conditions. Section: Deep mutational scanning profiles beneficial substitutions in microgravity Bacteria often resist phage predation by mutating or downregulating surface receptors essential for phage adsorption [26,63–65]. Microgravity-induced stress may amplify this response, altering the bacterial proteome, including phage receptor profiles [15,25,66]. Such changes can drive adaptive mutations in the phage RBP. To investigate these interactions, we examined how individual substitutions in the tip domain of the T7 RBP affect phage viability in microgravity. The T7 RBP consists of six short non-contractile tails that form a homotrimer composed of a rigid shaft ending with a β-sandwich tip domain [67]. This domain is a key determinant of host recognition and interacts with host receptor LPS to position the phage for successful, irreversible binding [27–32,68]. We conducted comprehensive single-site saturation mutagenesis of the RBP tip domain, generating a library of 1,660 variants spanning residues 472–554 (based on PDB 4A0T). We then sequenced and compared mutational enrichment profiles following the 23-day selection under terrestrial and microgravity conditions. We recovered phage DNA from each sample and scored each variant based on its relative abundance before and after selection (functional score, F) normalized to wildtype (normalized functional score, FN). Scores were averaged across replicates, and only variants present in at least two replicates were retained for analysis. Although significant dropout of low-performing variants was expected due to the extended incubation, we successfully determined scores for 51.2% (880) of variants in microgravity and 39% (648) in terrestrial conditions (Figs 5A, 5B, and S7A). Variant scores correlated well across replicates despite differences in phage titer and reflected multiple rounds of replication over the 23-day incubation period, suggesting that lower-titer samples underwent selection but subsequently lost viability (S7B–S7D Fig). On average phage variants were significantly more enriched after terrestrial incubation compared to microgravity (two-sample t test, Mann–Whitney U, p < 0.001) (S8A Fig). The wild-type phage was significantly depleted terrestrially compared to microgravity (terrestrial F = 0.58, microgravity F = 3.5, p < 0.01). While variants that performed worse than wildtype (FN < 0) tended to perform similarly between microgravity and terrestrial conditions (S8B Fig), enriched variants (FN > 0) were highly divergent with no correlation between conditions (S8C Fig). Variants enriched in microgravity frequently contained methionine and isoleucine substitutions at interior positions facing the phage (Figs 5A and S8D), in contrast to our previous terrestrial results on this host [26]. Substitutions in these areas could influence the tip domain structure to facilitate adsorption with the host receptor in microgravity. Under terrestrial conditions, top-scoring variants included positively charged substitutions facing the host, consistent with our previous findings on E. coli BL21 [26]. Additional enriched variants featured negatively charged substitutions (e.g., Q488E, G521D) and glycine substitutions (e.g., G480W, G522P) that may induce structural changes in the tip domain (Figs 5B and S8B). These variants were enriched only after prolonged incubation with E. coli BL21, suggesting that such substitutions may contribute to long-term infectivity on stationary-phase hosts—an effect not observed in shorter, nutrient-rich conditions. Because variants enriched in microgravity were highly distinct from those identified under terrestrial conditions—both in this study and in our previous work—we next evaluated whether these substitutions could enhance phage activity terrestrially. If successful, these substitution patterns could be used to improve phage performance without exhaustively sampling the full combinatorial space of the gene. We constructed two combinatorial libraries, each comprising all possible combinations of 13 top-performing substitutions identified in microgravity (L490I, N502E, F506M, F506Y, F507V, F507Y, P511M, I514M, N531Q, L533K, L533M, A539M, N546I) or under terrestrial conditions (G521H, Q488A, Q488E, G521K, G522P, A547S, G521D, G521E, N502S, I495L, R542H, L533T, F506S). This strategy reduced a potential search space of over 10²¹ variants to fewer than 5,000 per library. Variants were synthesized in an oligo pool, assembled into an unbiased phage library using ORACLE, and passaged terrestrially on two clinically isolated E. coli strains (UTI1 and UTI2) that are resistant to wild-type T7 and are associated with urinary tract infections [69]. We evaluated these pools in efficiency of plating (EOP) experiments and compared their plaquing capability versus wildtype. The combinatorial pool from microgravity showed significant improvement in plaquing efficiency compared to wildtype and had substantially larger plaques, indicating the pool contained variants capable of significantly improving activity on these hosts (S9A and S9B Fig). The terrestrial library performed significantly worse or no better than wildtype. To confirm these results, we isolated individual plaques from the microgravity pool. From UTI1, we recovered a five-substitution variant (L490I, N502E, F507V, L533K, A539M; Variant 1), and from UTI2, a six-substitution variant (L490I, N502E, P511M, L533M, A539M, N546I; Variant 2). These variants demonstrated significantly higher EOP and produced significantly larger plaques on both UTI strains (Fig 5C and 5D). These findings support our hypothesis that microgravity-enriched substitutions can improve phage performance on terrestrial hosts. The extended incubation in microgravity revealed new mutational hotspots, enabling efficient navigation of sequence space to identify complex variant combinations with enhanced infectivity. Open to thoughts/feedback on how to proceed based on all of this | HTGAA Claude AI Tutor |
| Yes I want to express Variant 1 and Variant 2 of the gp17 tip domain (the microgravity-enriched multi-substitution variants) and test them against your four-strain panel, using wildtype Gene 17 as the control. Before we do that, want to get clear on if any changes will need to be made to the pET-28a + Gene 17 plasmid we just made. If yes, what changes will likely need to be made or shown? If not, can we get clear on what the function of the pET-28a + Gene 17 plasmid is? What is it showing in its current form and what should I be able to speak to about it in a final project presentation? | HTGAA Claude AI Tutor |
| Yes that makes sense. Not sure where in the process this goes (now or sometime later) but definitely want assistance making the pET-28a + gp17-Variant1 and pET-28a + gp17-Variant2 plasmids in Benchling before the full project markdown writeup is created. Not exactly sure how to do that (specifically how to verify and determine the specific Variant amino acid substitutions engineered into the Gene 17 coding sequence for re-creation in Benchling). Based on all this information and the last paragraph in your response to the last prompt, let me know how we should proceed | HTGAA Claude AI Tutor |
| Ok, so I have the original Gene 17 CDS fragment, as well as the pET-28a + Gene 17 plasmid open in Benchling. Believe I also understand how to make exact copies of things in Benchling. Bit unsure how we will do about editing codons for encoding the appropriate amino acids for each of the Variants. Tell me how we should proceed based on this information | HTGAA Claude AI Tutor |
| Not seeing an ‘Edit Sequence’ tool in my version of Benchling (could be wrong, just not seeing it). Also, shouldn’t I make a clone/exact copy of the pET-28a + Gene 17 plasmid just so the original is maintained/no unnecessary changes are made (i.e., shouldn’t a clone/exact copy be made as a sort of ‘sandbox’ for future work, so progress to date isn’t compromised)? To answer your question, yes when I look at the original Gene 17 CDS fragment, I see a string of multiple colored letters beneath the DNA sequence, which appears to be the protein sequence | HTGAA Claude AI Tutor |
| Is amino acid position the same as nucleotide/bp position? Let’s clarify that before we proceed | HTGAA Claude AI Tutor |
| I see it! I see the 490th colored letter! It’s sitting between bp 1460- and 1480 in the Linear Sequence Map (corresponding roughly to the nucleotide 1468–1470 range described in the answer to the previous prompt). It’s a green L (believe this stands for the Leucine protein). Tell me if the L stands for Leucine, and more importantly, tell me how we should proceed | HTGAA Claude AI Tutor |
| Think I’m seeing a CTC nucleotide sequence. If that doesn’t align with the standard Leucine nucleotide sequence, say so. Want to make sure this is correct before editing begins | HTGAA Claude AI Tutor |
| Before we move onto the editing, I’m pausing and realizing I still have the original Gene 17 fragment. I should make a copy and work off the copy before we move on to editing, correct? | HTGAA Claude AI Tutor |
| Think we might be talking past each other/things might be getting lost in translation. For the past couple Turns/exchanges, I’ve been looking at the original Gene 17 fragment in Benching, NOT the pET-28a_gp17_Variant1, which I created based on previous instructions. Should I be working off the pET-28a_gp17_Variant1 sequence instead? | HTGAA Claude AI Tutor |
| Ok, gotcha – in the right place. The only issue now is that there’s no amino acid translation overlay within the Gene 17 CDS annotation portion of the plasmid sequence (or the plasmid sequence as a whole). What can we do about this? How can we change this, if possible? | HTGAA Claude AI Tutor |
| When I right click there’s a translation-related option that reads ‘Analyze as translation’. Is that good to work with? | HTGAA Claude AI Tutor |
| When I click on ‘Analyze as translation I see the following options: –Strand: Select ‘Forward’ or ‘Reverse’ options (its default is Forward) –Genetic Code: There’s a ‘Standard’ option plus many more options based on organism type (they are numbered 1. up to 33. but there are sometimes jumps in the numbering) –A ‘Translate start codon as methionine’ check box Not sure what I should do here or if I’m in the right place. If none of this makes sense and you recommend I just click out of this option, say so | HTGAA Claude AI Tutor |
| Yes, I have done that. I see the green L in position 490 in the Gene 17 CDS fragment. I have now completed the first substitution changing CTC to ATC (Isoleucine). Tell me how we should proceed with the next Variant 1 substitution | HTGAA Claude AI Tutor |
| I see a GAT 3-nucleotide codon with a D under it | HTGAA Claude AI Tutor |
| Double checking, and yes, seeing a GAT 3-nucleotide codon with a D under it | HTGAA Claude AI Tutor |
| Used accession number NC_001604.1 as the Gene 17 source sequence. The paper said they used ATCC:BAA-1025 as their E. coli. sequence. Believe these are the same, but am not sure | HTGAA Claude AI Tutor |
| Here’s the paper link: https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003568#pbio.3003568.ref026 | HTGAA Claude AI Tutor |
| Here’s the paper’s complete Methods section: Methods Phage and bacterial strains T7 bacteriophage was obtained from ATCC (ATCC BAA-1025-B2). The T7 DMS library used in this study was the same library stock generated in our previous work [26]. T7 acceptor phages used for ORACLE-based construction of the combinatorial libraries were also created as previously described [26]. Escherichia coli BL21 was sourced from laboratory stocks. Uropathogenic E. coli strains UTI1 and UTI2 were provided by Dr. R. Welch (University of Wisconsin, Madison) and originate from a urinary tract infection isolate collection [69]. T7 phage was initially propagated on E. coli BL21 following receipt from ATCC and subsequently on appropriate hosts as described in specific experimental sections. All phage experiments were performed using Luria-Bertani (LB) media and the same culture conditions used for bacterial hosts. Phages were stored in LB at 4 °C for short-term use. For long-term storage, microbial samples were frozen at −80 °C in 100% LB media. Media and culture conditions All bacterial strains were cultured in LB media consisting of 1% tryptone, 0.5% yeast extract, and 1% NaCl in deionized water. LB plates were supplemented with 1.5% agar, while top agar used for phage plating contained 0.5% agar. LB media was used for all experiments, including bacterial recovery and phage propagation. All incubations were carried out at 37 °C without shaking, in either terrestrial or microgravity environments as appropriate. These samples were incubated directly in cryovials and not transported to another container for incubation. Sample preparation and handling Phage and bacterial stock titer were confirmed and samples were prepared by mixing 4 mL of E. coli BL21 in exponential phase (~1 × 108 CFU/mL) with the appropriate amount of T7 phages in Rhodium Cryotubes. Samples were immediately frozen at −80 °C and shipped to NASA as described. Asynchronous microgravity and terrestrial experiments are the norm for ISS experiments due to the uncertainty of scheduling. The initial planned time points for incubation were 1, 2, and 3 hours, and 25 days; however, actual time points were adjusted on the ISS to accommodate astronaut scheduling. Final incubation time points were 1, 2, and 4 hours, and 23 days. The duration of incubation aboard the ISS was recorded precisely, and terrestrial control samples were incubated for matching durations, based on the actual timepoints rather than the proposed schedule. This approach was necessary because real-time tracking of the samples was not possible, so microgravity and terrestrial samples could not be incubated in parallel accurately. Terrestrial samples are thus frozen for a longer duration than microgravity samples. After incubation samples were refrozen, shipped to our laboratory, and then thawed at 37 °C and immediately split for genomic DNA extraction, PCR for DMS, and titering of both phage and bacteria. Titering phage For samples returned for processing, 1 mL of each sample was centrifuged at 16g for 1 min, and the supernatant was filtered through a 0.22 μm filter. To determine phage titer, titer was first estimated by spot plates and then confirmed by whole plate EOP assays. Samples were serially diluted (1:10 or 1:100) in LB to a final dilution of up to 10−8 in 1.5 mL microcentrifuge tubes. Spot assays were performed by mixing 250 μL of stationary-phase bacterial host with 3.5 mL of 0.5% top agar. The mixture was briefly vortexed and plated onto LB agar plates pre-warmed to 37 °C. Once the top agar solidified (~5 min), 1.5 μL of each phage dilution was spotted onto the plate in series. Plates were incubated at 37 °C and checked after 20–30 hours to estimate titer. Titers were then confirmed via full-plate plaque assays. For whole-plate EOP assays, 400 μL of exponentially growing bacterial culture was mixed with 5–50 μL of diluted phage, aiming to achieve ~50 plaque-forming units (PFUs) per plate after overnight incubation. For phage susceptibility on the pre-incubation and 4-hour samples, bacteria was incubated after being directly sampled from the frozen stock for that sample. The phage–host mixture was briefly vortexed and centrifuged, then combined with 3.5 mL of 0.5% top agar. After a brief vortex, the mixture was immediately poured onto LB plates pre-warmed to 37 °C. Plates were allowed to solidify (~5 min), inverted, and incubated overnight. PFUs were counted after 20–30 hours, and final phage titers were calculated from these counts. Titering bacteria Bacterial concentrations were determined via serial dilution (1:10 or 1:100 in LB) and plating. From each dilution, 100 μL was plated and spread using sterile beads to target ~50 colony-forming units (CFUs) per plate. Plates were incubated overnight at 37 °C and counted the following day. For E. coli BL21, three independent dilution series were performed to correlate OD600 values with CFU/mL and ensure accurate bacterial concentrations during phage mixing for experimental sample preparation. PCR and sequencing All PCR reactions were performed using KAPA HiFi DNA Polymerase (Roche KK2101). The combinatorial library was generated using the ORACLE method, as previously described [26]. Cloning procedures followed manufacturer instructions unless otherwise specified. For WGS, phage genomes were extracted using the Norgen Biotek Phage DNA Isolation Kit (Cat. 46800), and bacterial genomic DNA was extracted using the Norgen Biotek Bacterial Genomic DNA Isolation Kit (Cat. 17900). Genomic DNA libraries were prepared using the Illumina DNA Prep kit (Cat. 20060060) and sequenced on an Illumina NextSeq 1000 platform. PCR reactions for amplification of the DMS and combinatorial libraries used 1 μL of undiluted phage lysate directly as template (DNA isolation is not required), with an extended denaturation step of 5 min at 95 °C. For low phage titers in DMS samples, PCR and next-generation sequencing failed using this approach, presumably because of reduced template in these samples. To overcome this, we concentrated all of the remaining volume of each sample (~2 mL) approximately 100-fold using Pierce Protein Concentrators PES, 10K MWCO (Cat. 88513) and used 3 μL of the concentrated sample per PCR reaction to enabling successful amplification and analysis. For plaque analysis on UTI strains, small plaque samples were picked directly and used as PCR template. Detailed cloning protocols are available upon request. General data analysis Multiplicity of infection (MOI) was calculated by dividing the phage titer by the corresponding bacterial concentration. Initial MOI is calculated based on the bacteria and phage titer before being frozen for transit to the ISS. The MOI for the T7 DMS library was estimated using a helper plasmid, as described previously [26]. EOP values were calculated using E. coli BL21 as a reference host. EOP was defined as the phage titer on the test host divided by the titer on the reference host, followed by log₁₀ transformation. Values are reported as mean ± standard deviation (SD). Deep sequencing was performed to evaluate phage populations as described previously [26]. Phage sequencing achieved an average depth of ~49,000× per base across the genome, enabling detection of low-abundance mutations. Bacterial sequencing depth averaged ~250× per base in phage-mixed samples and ~1,300× in phage-free samples, limiting mutation analysis in the former to more abundant variants. WGS mutations were identified using Breseq [73]. For Fig 3, genes were grouped based on GO classifications [44,45]: Membrane-associated genes: GO:0016020 (Membrane), GO:0009103 (LPS biosynthesis), GO:0030288 (Outer membrane bound periplasmic space), GO:0042597 (Periplasmic space). Metabolism-associated genes: GO:0008152 (Metabolic process), GO:0019222 (Regulation of metabolic process). Statistical analysis To evaluate whether non-synonymous de novo substitutions and frameshift mutations were significantly enriched compared to synonymous substitutions, we compared the frequency of each non-synonymous substitutions and frameshift (phage: >1% abundance; bacteria: >25% abundance) to the distribution of synonymous mutations using a one-sided Mann–Whitney U test with Benjamini–Hochberg false discovery rate (FDR) correction (scipy.stats.mannwhitneyu, statsmodels.stats.multitest.multipletests, method = ‘fdr_bh’,scipy V1.10.1, statsmodel v 0.14.0) [74]. Adjusted p-values < 0.05 were considered significant. This approach assumes that after 23 days of selection the distribution of synonymous substitutions approximates either a neutral baseline or reflects minimal selective pressure, with the benefit that if there is positive selection for synonymous substitutions there would be no increase in false positives using this method. To determine whether non-synonymous de novo substitutions and frameshift mutations were more abundant in bacterial samples exposed to phage, we applied the Mann–Whitney U test (scipy.stats.mannwhitneyu, scipy V1.10.1) to compare mutation frequencies across groups [74,75]. Due to high detection limits in phage-mixed samples, we also performed left-censored data analysis using Kaplan–Meier survival curves (lifelines.KaplanMeierFitter, lifelines V0.27.8) and applied a log-rank test (lifelines.statistics.log-rank_test, lifelines V0.27.8) to assess significant differences in mutation distributions between groups [76–78]. To assess if the 4-hour bacterial population of mutations was significantly different from the pre-incubation condition, we performed Jenson–Shannon divergence (scipy.spatial.distance, jensenshannon, V1.10.1) and correlated results using Pearson R (scipy.stats, pearsonr, v1.10.1). To determine if the titer of wild-type T7 phage was significantly different between the pre-incubation and 4-hour bacterial population, we used a Welch’s t test (scipy.stats, ttest_ind_from_stats, v1.10.1). Mutation density in phage genes was calculated by dividing the number of non-synonymous de novo substitutions and frameshift mutations by the length (in amino acids) of each protein product. To assess whether any gene had significantly higher mutation density, we compared individual gene densities to the condition-specific average using a one-tailed t test with Benjamini-Hochberg FDR correction (scipy.stats.ttest_1samp, statsmodels.stats.multitest.multipletests, method = ‘fdr_bh’, alternative = ‘greater’, scipy V1.10.1, statsmodel v 0.14.0). Additionally, one-tailed 95% confidence intervals were calculated using scipy.stats.t.ppf (scipy V1.10.1) and visualized in volcano plots in python [74]. Structural visualization Structural model images were generated using the PyMOL Molecular Graphics System, Version 3.0 (Schrödinger, LLC). Gp7.3 structure was predicted using AlphaFold2 and ColabFold with MMseqs2, using the predicted structure with the highest confidence [79–81]. Electron Microscopy images were based on PDB 7BOX (gp11) and PDB 7BOY (gp12) [41]. A composite structure of the T7 portal–tail complex is shown from PDB 9JYZ [82]. Numbering for DMS and combinatorial library positions are based on PDB 4A0T [67]. | HTGAA Claude AI Tutor |
| The paper is about T7 phage evolution under microgravity. The Variants were then tested terrestrially. Those Variants were based off Gene 17 modifications. I want to make similar mutations to other E. coli. bacteriophage strains to see if we can improve their fitness/lysing. I thought I had the correct source sequence from NCBI (NC_001604.1), but am not sure. | HTGAA Claude AI Tutor |
| Apologies, think there might be some hallucinating going on. NC_001498.1is a measles virus genome, not a T7 genome | HTGAA Claude AI Tutor |
| The accession number of the top result I see is NC_001604.1, which is the sequence I chose, which does have 39,937 bp | HTGAA Claude AI Tutor |
| I counted from the start of the gp17 CDS, not the full genome | HTGAA Claude AI Tutor |
| I see a T nucleotide at position 490. I see a G nucleotide at position 488. There’s still a D (Aspartate/GAT) below it. Interestingly enough, positions 484-486 have a 3-codon nucleotide sequence (ACT) and a T below it. Not sure if that’s relevant whatsoever | HTGAA Claude AI Tutor |
| May I ask why I’m being asked to divide by 3 if the result is not a whole number (the result is 161.333333 indefinitely–not sure if that’s useful). | HTGAA Claude AI Tutor |
| Position 388 | HTGAA Claude AI Tutor |
| Seeing a GAT 3-nucleotide sequence coding for D (Aspartate/GAT) at nucleotide position 1852-1854 | HTGAA Claude AI Tutor |
| Seeing a GAT 3-nucleotide sequence coding for D (Aspartate/GAT) at nucleotide position 1852-1854 | HTGAA Claude AI Tutor |
| Know they used E. coli. BL21. The closest things I think I could find that relates to gp17 and its mutations are the DOI links below. Not sure if these are helpful in response to answering your question https://doi.org/10.1371/journal.pbio.3003568.s005 https://doi.org/10.1371/journal.pbio.3003568.s009 | HTGAA Claude AI Tutor |
| Here you go: Mutation,Variant(s),WT Amino Acid,WT Codon,Mutant Amino Acid,Mutant Codon L490I,V1 & V2,Leucine (L),CTT,Isoleucine (I),ATT N502E,V1 & V2,Asparagine (N),AAC,Glutamic Acid (E),GAA F507V,V1 only,Phenylalanine (F),TTC,Valine (V),GTT P511M,V2 only,Proline (P),CCG,Methionine (M),ATG L533K,V1 & V2,Leucine (L),CTC,Lysine (K),AAA A539M,V1 & V2,Alanine (A),GCT,Methionine (M),ATG N546I,V2 only,Asparagine (N),AAC,Isoleucine (I),ATC | HTGAA Claude AI Tutor |
| No idea | HTGAA Claude AI Tutor |
| Well, we previously changed the Leucine to Isoleucine. So that’s already been done. Should we proceed with something else. | HTGAA Claude AI Tutor |
| I think I need to continue inducing all the mutations for both the Variants based on the table listed above and then create a project design to show how these variants would test against other strains of E. coli mentioned previously in this conversation (Turn 44, and the strains were CFT073 (UPEC), UTI89 (UPEC), and MG1655 (K-12)). How should we go from here. Think I actually might be able to re-create the mutations in separate plasmids, but am unsure about the rest of the project design | HTGAA Claude AI Tutor |
| Do you remember the entire context of this conversation? I created a pET-28a + Gene 17 plasmid with your assistance, and the entire project will be dry lab. Not sure a Twist Bioscience order will be done, or if it’s first priority. Introducing the mutations into an existing plasmid backbone. Let me know how to proceed with the project design (moving past the Variant plasmids to testing the separate strains in dry lab/writing out how I would do this in wet lab) Let’s get clear on what I’d measure, how I’d be measuring, etc. | HTGAA Claude AI Tutor |
| Measure the plaque assay I believe. | HTGAA Claude AI Tutor |
| I think the original wild-type strain, right? If I’m mistaken, and there would be another more optimal control(s), let me know | HTGAA Claude AI Tutor |
| So I’ve created plasmids in Benchling where I re-created the original Gene 17 CDS amino acid mutations across Variants 1 and 2. I now want to make sure the proposal shows how I would test their activity against the other E. coli. strains mentioned above (CFT073 (UPEC), UTI89 (UPEC), and MG1655 (K-12)). Can we just summarize how this would occur or be written out in the project proposal, and how we’d take into account that the project in its current form is dry lab? | HTGAA Claude AI Tutor |
| Apologies if this is a dumb question. We already expressed gp17 in a pET-28a plasmid multiple times (first as the basic plasmid with no mutations, and then as two separate plasmids showcasing respective Variant 1 and 2 mutations). So what is the nature of the question you just asked? Is there hallucinating going on, or is there something unaccounted for that you think we should delineate in the proposal? | HTGAA Claude AI Tutor |
| Ok, thanks for the clarification. Yes, let’s add the extra level of detail and specify IPTG induction details. If you could also let me know what the IPTG acronym stands for in this context, that would be greatly appreciated also | HTGAA Claude AI Tutor |
| Generate proposal | HTGAA Claude AI Tutor |
| Apologies, but saw reference to T4 in the proposal. Believe we meant to say T7. Mind editing the proposal accordingly? If this notion is incorrect, say so | HTGAA Claude AI Tutor |
| In the source paper on which this project is based, the researchers say “Here, we report the dynamics between T7 bacteriophage and Escherichia coli in microgravity aboard the International Space Station (ISS)”. So think the intention is that we’re working with T7. There’s no mention of bacteriophage T4 anywhere in the original paper (see link below): https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003568#sec009 | HTGAA Claude AI Tutor |
| Yes, regenerate the full corrected proposal now with T7 substituted throughout | HTGAA Claude AI Tutor |
| Apologies, but the fully corrected proposal with the T7 substitutions appears as one continuous scroll/blob of Markdown. Do you mind re-formatting the content so it looks like the format of the Turn 86 response, with similar spacing and item verification? The content itself does not and should not change – just how it’s formatted/presented | HTGAA Claude AI Tutor |
| Ok. Thank you for creating this. I’ve read through the project proposal and have some questions. If you can’t address them or respond to them, all good. –Can you remind me what the pET-28a plasmid vector stands for? Why is it useful or necessary? Is it right to call it a plasmid vector or is another term more technically appropriate? –Would you mind explaining each step of the Experimental Design in relatively simple terms, as if I was a reasonably educated 17 year old with some bio knowledge, but who is still a bio n0ob in the grand scheme of things. Explain the following as part of this ask: -the why(s) behind each step -the logical flow/progression of one step to the next -what we’re measuring as part of this Experimental Design, how we’re measuring it, and what types of graphs or images we’d traditionally use to do said measuring -how we’re ultimately testing the fitness of the microgravity-based Variants against the CFT073, UTI89, and MG1655 E. coli. strains | HTGAA Claude AI Tutor |
| It looks like the answer to the first question got cut off a bit | HTGAA Claude AI Tutor |
| Ok. Thank you for describing this. Before we move on, can we describe why we specifically chose a pET-28a expression vector for this specific Experimental Design? What is the logic behind it? If we want to test microgravity-based Variants against the CFT073, UTI89, and MG1655 E. coli. strains, why must we put the T7 Gene 17 CDS into a plasmid first? Do NOT hallucinate/make things up when replying to this prompt | HTGAA Claude AI Tutor |
| Thanks – helpful. Let’s move on to the second question (as a reminder, this second question is to explain each step of the Experimental Design in relatively simple terms, as if I was a reasonably educated 17 year old with some bio knowledge, but who is still a bio n0ob in the grand scheme of things). As part of this ask, explain the following: -the why(s) behind each step -the logical flow/progression of one step to the next -what we’re measuring as part of this Experimental Design, how we’re measuring it, and what types of graphs or images we’d traditionally use to do said measuring -how we’re ultimately testing the fitness of the microgravity-based Variants against the CFT073, UTI89, and MG1655 E. coli. strains | HTGAA Claude AI Tutor |
| Taking a look at Turn 90. It says that it outputted 15 Experimental Steps, but it only outputted 12. Complete the rest of the Experimental Steps by listing out the remaining 3 steps. Reference previous content in this chat to construct the Experimental Setup with all 15 steps Do NOT hallucinate/make things up when replying to this prompt | HTGAA Claude AI Tutor |
| Looking at Steps 14 and 15 in the last prompt, approximately how long do we think it should take to complete these Steps? Do NOT hallucinate/make things up when replying to this prompt | HTGAA Claude AI Tutor |
| There are multiple types of T7 bacteriophage right? In this paper, the researchers only used a specific type of T7 bacteriophage that infects E. Coli, correct? Indicate any areas where my reasoning/thinking is off and do NOT hallucinate/make anything up when replying to this prompt | Gemini |
| Just so we’re clear, the type of E. Coli. T7 bacteriophage studied in this paper is NOT the only type of E. Coli. T7 bacteriophage out there, correct? Do NOT hallucinate/make things up when replying to this thread | Gemini |
| Liking these results, but would like to find another E. Coli. T7 bacteriophage, potentially one that’s a bit unorthodox/out of the box to BLAST, that would likely still have a 95% match. Any thoughts about what these might be/how to proceed? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Going off the last prompt, how would I run a comparative BLAST against this specific gene and analogous examples listed above? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| After running the BLAST (including Blastn) the results are less than < 95%. The highest % was 89% for T3 (NC_003298.1) via Megablast. Any other thoughts about where/how to find somewhat unorthodox E. Coli. T7 bacteriophage candidates in the 95%+ range? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Looking over the paper again, confirming that the gene associated with the respective 5 and 6 substitution variants referenced in the paragraph before the ‘Discussion’ section is Gene 17, correct? Slightly confused about how that works or relates to the following phrase in the ‘Discussion’ section: “In microgravity, structural genes gp7.3, gp11, and gp12 emerged as particularly important,”. Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Help me understand what I’m seeing. What does the ‘L’ in matches with an ‘L’ at the end (ex. ‘Mutant Escherichia phage T7 clone T7_90L’) mean? What does the ‘S’ in matches with an ’s’ at the end (ex. ‘Mutant Escherichia phage T7 clone T7_50S’) mean? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Want to understand which of these results derive from a different strain of E. Coli. than the one referenced in the paper (ATCC BAA-1025-B2). Is it all of them? None of them? A bit confused. Tell me what I should be looking for or if my thinking is off here. Do NOT hallucinate/make things up when replying to this thread | Gemini |
| For the result for Sequence ID: MZ375271.1, it says Range: 32650 to 34311. How do I find this in the raw FASTA file for this sequence? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| How far back in this chat can you go? Want to re-surface the results of the prompt that asked about the specific gp17 mutations that took place in each of the Variants. Want to understand which nucleotides and amino acids to change to re-create the mutations found in Variants 1 and 2. Do NOT hallucinate/make things up when replying to this thread | Gemini |
| Based on the paper in the shared tab, can you tell me what strain of E. Coli. the University of Wisconsin, Madison researchers tested their bacteriophage against? Believe it was BL21 but want to confirm. Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Thank you. While I understand some combinatorial work was used to create Variants 1 and 2 in the paper in the tab, can you explain how I would simply explain how the Variants were created in a phrase approximately the length of a Tweet? Would something like, ‘Variants 1 and 2 were created via a combinatorial approach deriving from nth mutations, with down selection for the highest PFU/mL’ technically make sense? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Why did the Variant 1 and Variant 2 bacteriophage Gene 17 T7 tail fiber substitutions seem to have higher infection rates against terrestrial UTI? What exactly about Gene 17 and the tail fiber substitutions specifically seemed to be so special/useful in achieving this higher infectivity rate? What other microgravity-derived mutations outside of Gene 17 might also help improve E. Coli. bacteriophage infectivity rate going forward (i.e., if I was going to test different microgravity derived genetic mutations/amino acid substitutions, what would make sense)? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| In the E. Coli. bacteriophage genome, what genes are gp7.3, 11, and 12 associated with? Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| Tell me how the researchers for this paper created the Variants in 2-3 sentences. Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| When we say microgravity unlocks synergistic mutations in the E. Coli bacteriophage, what exactly do we mean by this? Explain in 3-4 sentences. Do NOT hallucinate/make things up when replying to this prompt | Gemini |
| If i want to locate the position of gene 17 in e coli t7 bacteriophage, how do I do that?Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Can you point me to or create an image showing the ’tree of life’ of the phage kingdom of life? Want to visually see the common genetic ancestor(s) or T7 bacteriophage and D29 mycobacteriophage. Want to see a pictorial overview of the breakdown of life in the phage kingdom. If this does not exist/if this is not grounded in scientific fact, say soDo NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Give me a visual for the last bullet point to drive home the point in basic terms. Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Not seeing the transduction visual | Google AI Mode |
| Working on some research to show similar Gene 17 mutations (the ones that created Variants 1 and 2 in the attached paper) in a different yet similar bacteriophage based off the bacteriophage in this paper (ATCC BAA-1025-B2). Been told to look for a T7 look-alike phage lysate for Gene 17 of the following E. coli strain which I believe I’ve done via BLAST. Found GenBank: MZ375271.1 as a potential candidate. I’ve also been told to go look for phages for DNA extraction, to use AlphaFold for validation of the relevant structural mutations, and I ultimately need to design primers for polymerase chain reaction (PCR) for rebooting the phage.Where does cphages fit into all this? What does it mean to extract DNA based from cphages? How does all this (cphages, AlphaFold) inform the designing of primers for PCR?Do NOT hallucinate/make things up when replying to this prompt. If my thinking is unclear, wrong, or doesn’t make sense in any part of the prompt, say so | Google AI Mode |
| phage dna database | Google AI Mode |
| If I have a fragment of a sequence in NCBI (i.e., a specific gene that’s part of a larger NCBI sequence), how do I import that specific fragment into Benchling in such a way that I preserve any and all existing annotations? Do NOT hallucinate/make anything up when replying to this thread | Google AI Mode |
| Can the Cocktail program referred to in the second category (Simulation of Lysing Rates and Kinetics), work on a Mac? If the answer’s no, all good. Just let me know. Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Is a bacteriophage a virus or an actual living organism? If it is one of these two things, why is this the case? Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Are there any websites out there for verifying a plasmid relative to a designated function (ex. if I want a plasmid to do X function, can I upload a file and have the web service determine whether I have the right number of base pairs or parts for that function)? Do NOT hallucinate/make things up when replying to this thread | Google AI Mode |
| If I want to test a fragment of mutated Gene 17 T7 bacteriophage phage DNA against several different strains of E. coli bacteria, what are the core pieces I need in my plasmid? Right now I have the following:–T7 Terminator–T7 Promoter–Ribosome Binding Site (RBS)–Multiple Cloning Site (MCS)–ATG start codon annotation–Lac operator–Thrombin site–2 6xHis tags–Gene 17 Coding Sequence (CDS)–Gene 17 Amino Acid TranslationIs there anything I’m glaringly missing? Anything(s) that seem off relative to my goal? Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Helpful. The aim here is to test for a phage complementation/infection/lysing assay against CFT073, UTI89, and MG1655 E. coli strain. sDo NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| See 1st prompt.The aim here is to test for a phage complementation/infection/lysing assay against CFT073, UTI89, and MG1655 E. coli strains. Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| So when I’m replacing these components, do I essentially look up their FASTA files, insert them into the plasmid sequence as needed, and annotate them accordingly? Is there any location-specific information I should know about where to place some of the items included in the list in the response to the last prompt? Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Explain how I se the strict 5’ –> 3’ order in a Benchling sequence. Also explain to me how I can discern the orientation of my Backbone in a Benchling sequence. Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| Tell me what the ‘Run Primer3’ option in Benchling does. Do NOT hallucinate/make things up when replying to this thread | Google AI Mode |
| Show me publicly accessible examples of E. coli. bacteriophage genome fragment primers assembled in Benchling for Gibson Assembly. Want to understand how these are constructed.Do NOT hallucinate and/or make things up when replying to this prompt | Google AI Mode |
| What is a wordlist in Benchling? What does it do/what’s its function?Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| When creating a Gibson Assembly Construct in Benchling both the Backbone and Insert should have the same orientation, correct? Believe this is the case (or usually the case), but just want to verify. Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| if i’m growing bacteria on agar (example e. coli strains) how do I know when they’ve reached log phase? | Google AI Mode |
| pfu/ml graph | Google AI Mode |
| It’s true that if I just write out a string out letters representing an amino acid sequence, I cannot determine the exact amino acid sequence based on those letters because a single letter can stand for multiple amino acids, correct? Isn’t additional context needed?. Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |
| The Nanodrop tool for measuring DNA quantities in biology is a type of spectrophotometer, correct? Do NOT hallucinate/make things up when replying to this prompt. Reply to this prompt based on the knowledge at hand about the Nanodrop tool | Google AI Mode |
| ug/mL | Google AI Mode |
| In the context of E. coli. bacterial strains, what does the acronym UPEC stand for? Believe it stands for Uropathogenic E. coli., but want to confirm. Moreover, when testing bacteriophage fitness/lsying (PFU/mL), why does testing against UPEC E. coli. bacteria strains matter?Do NOT hallucinate/make things up then replying to this prompt | Google AI Mode |
| if i’m growing bacteria on agar (example e. coli strains) how do I know when they’ve reached log phase? | Google AI Mode |
| Remind me again what log phase means within the context of growing bacteria on agar (example E. coli strains). Do NOT hallucinate/make things up when replying to this prompt | Google AI Mode |



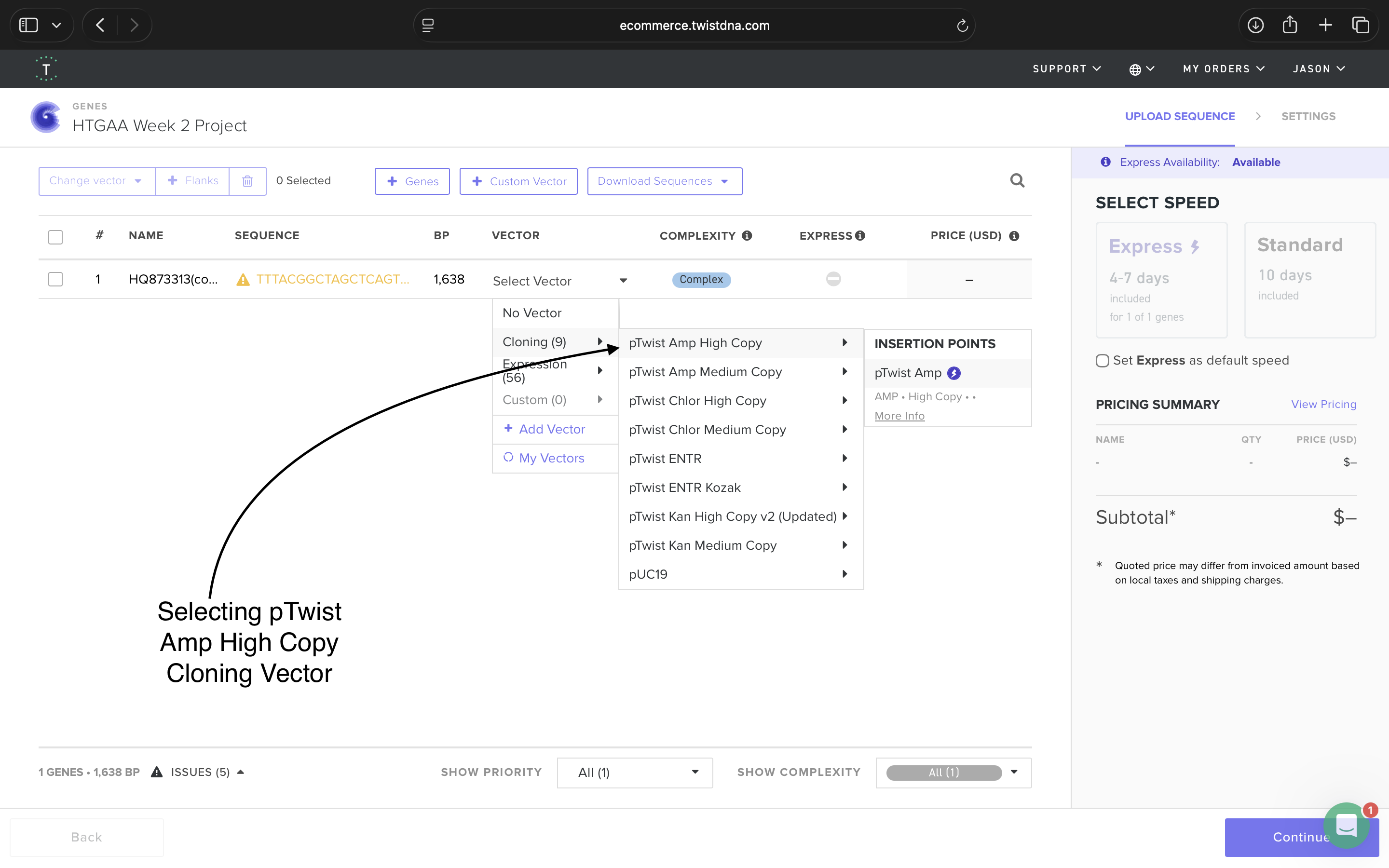
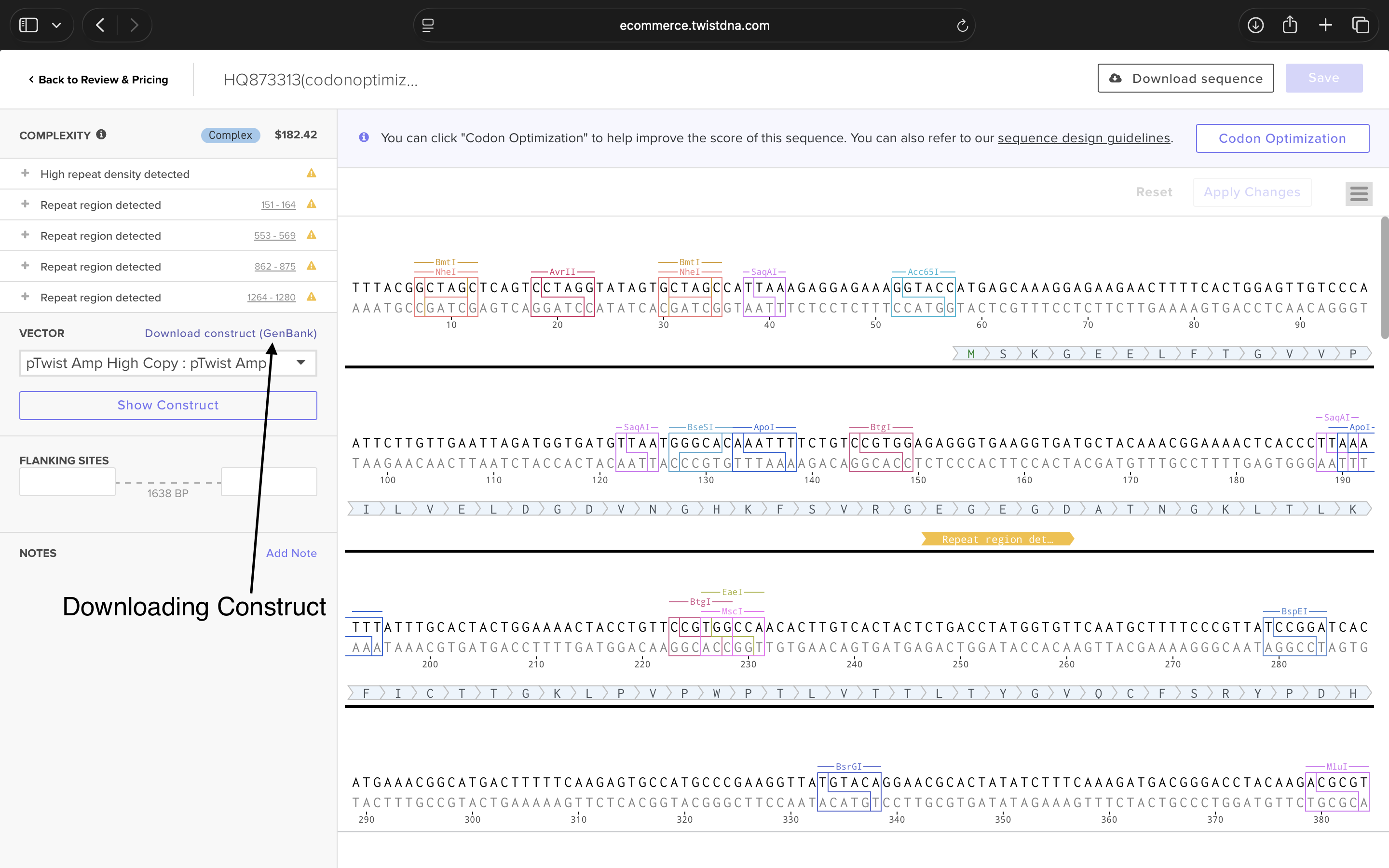

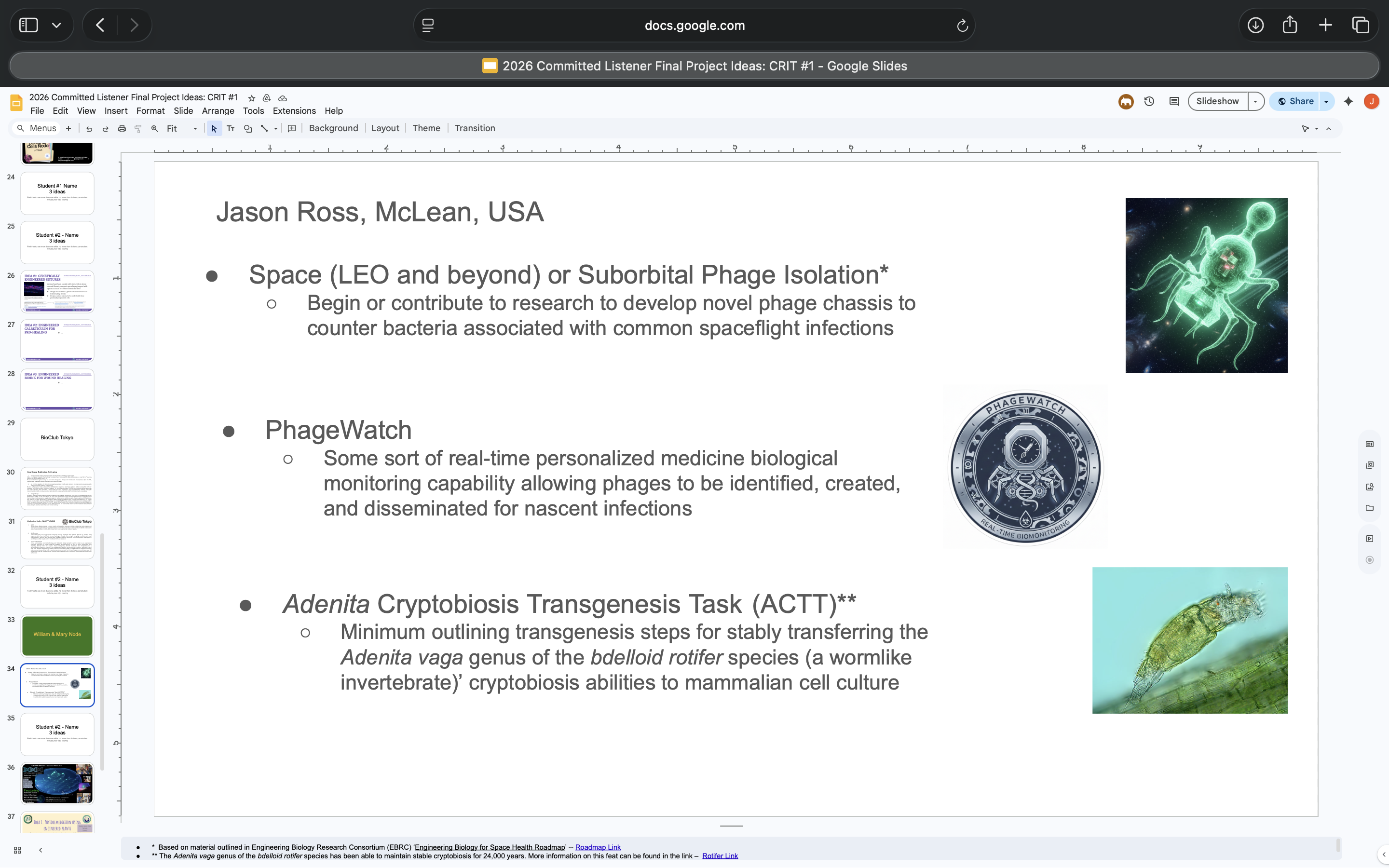

.png)
.png)
.png)
.png)
.png)
.png)
.png)

.png)
.png)
.png)














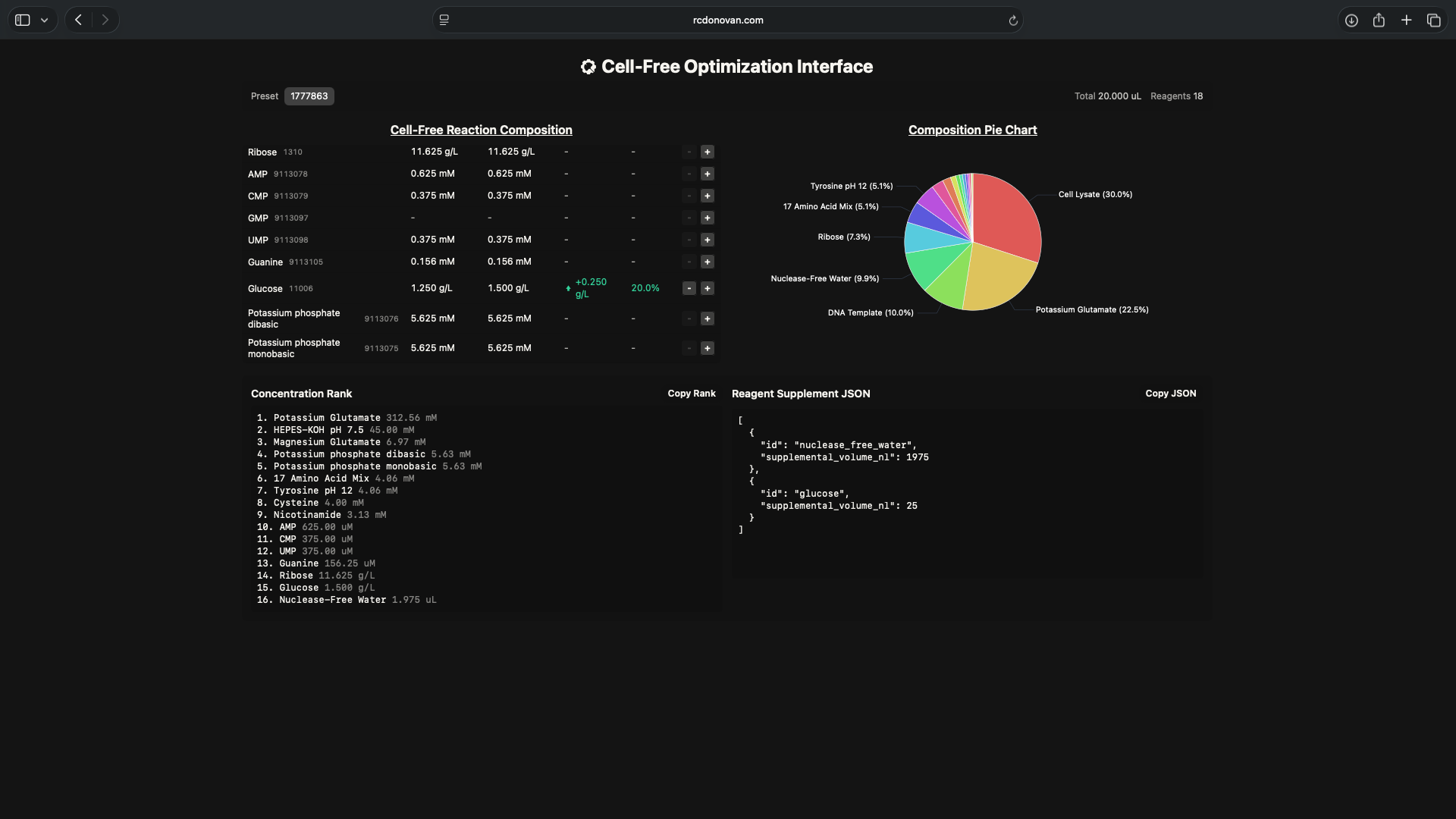

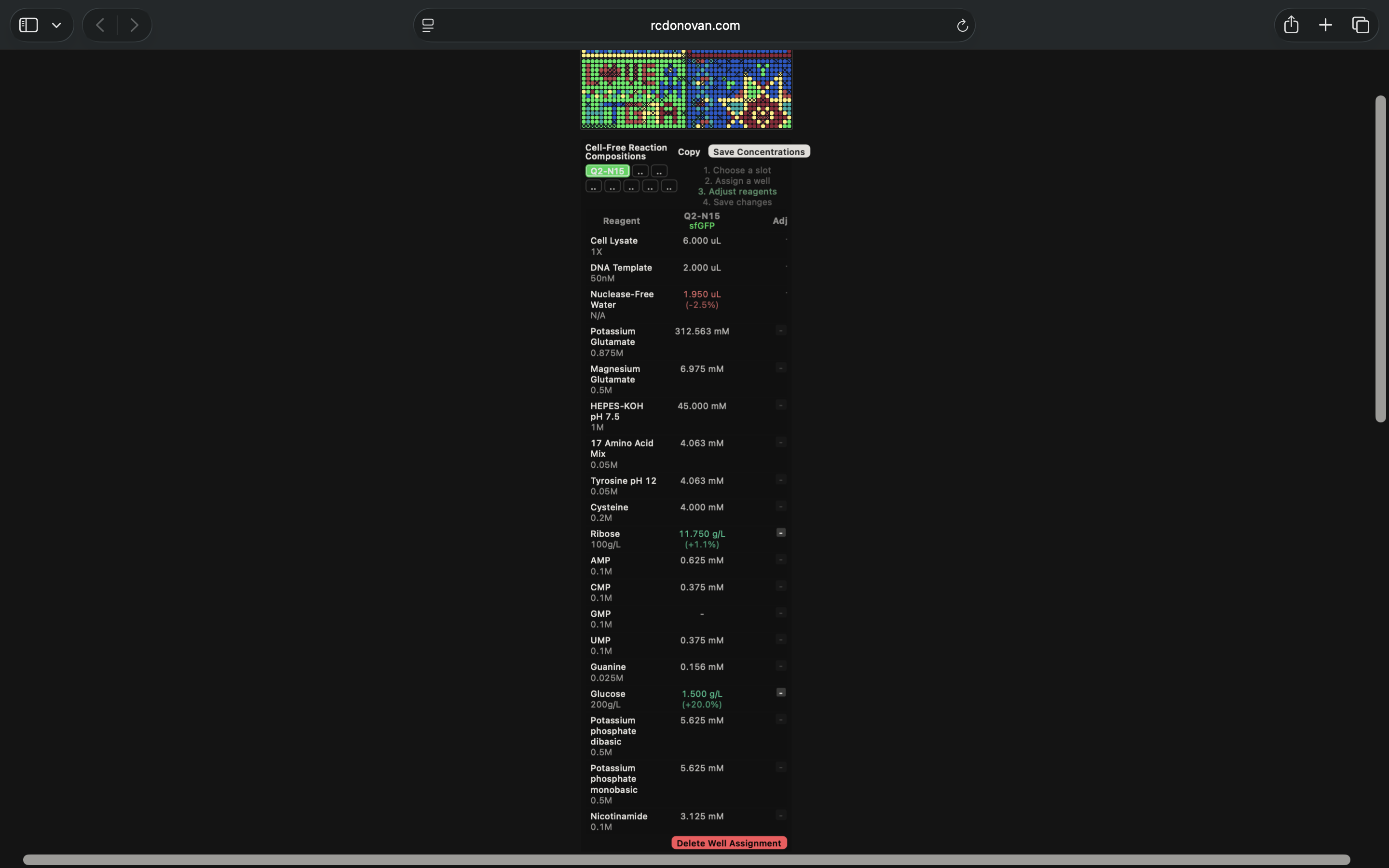













![virtual_digest_sequence_Mycobacterium-phage-Kampy-complete-sequence-51378-bp-including-10-base-3-overhang-CGGCCGGTAA-Cluster-A4_[HiddenHello]](/2026a/jason-ross/labs/week-02-lab-dna-gel-art/virtual_digest_sequence_Mycobacterium-phage-Kampy-complete-sequence-51378-bp-including-10-base-3-overhang-CGGCCGGTAA-Cluster-A4_[HiddenHello].png)





































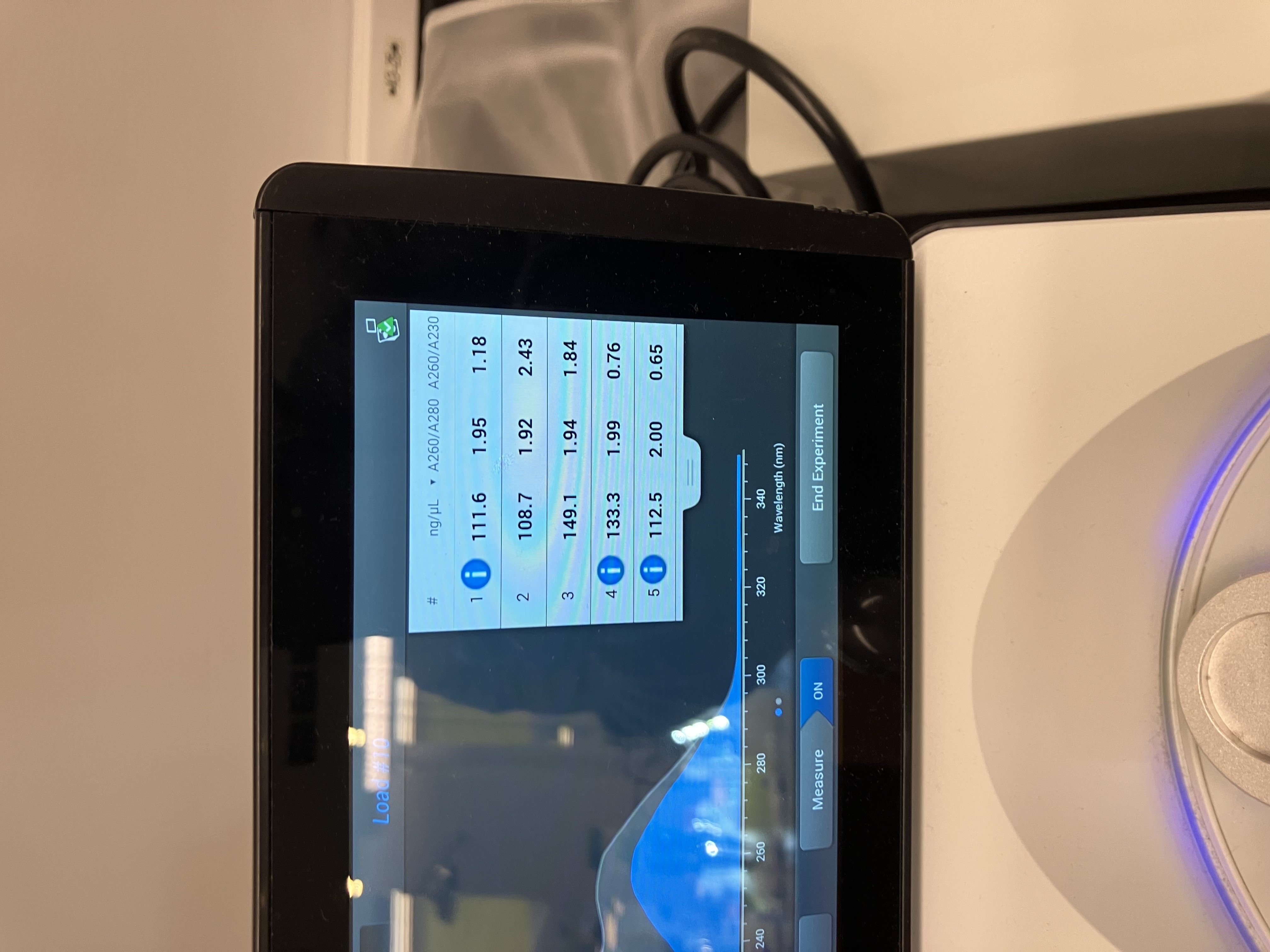




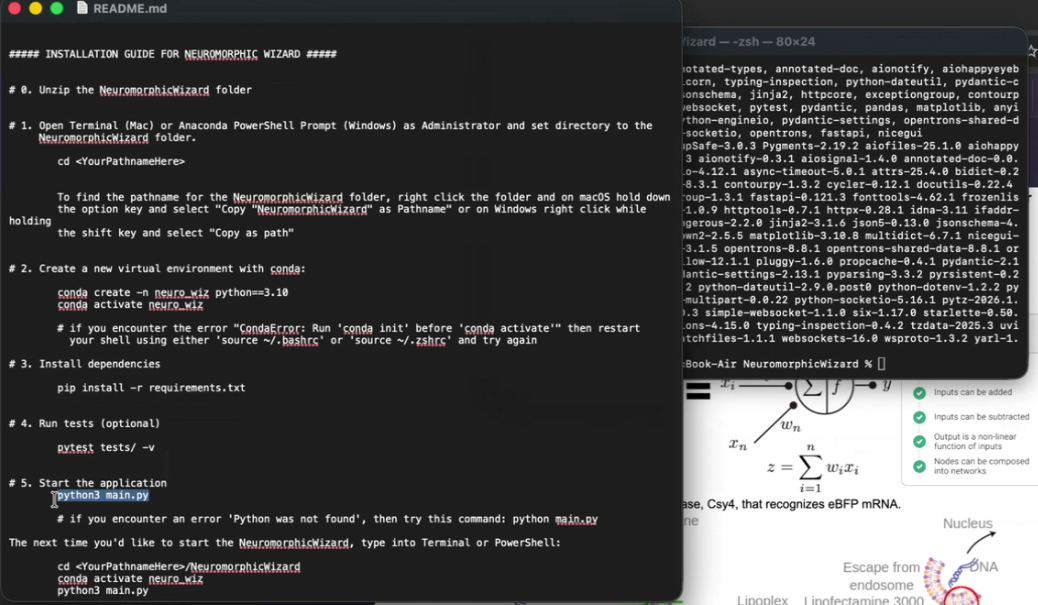
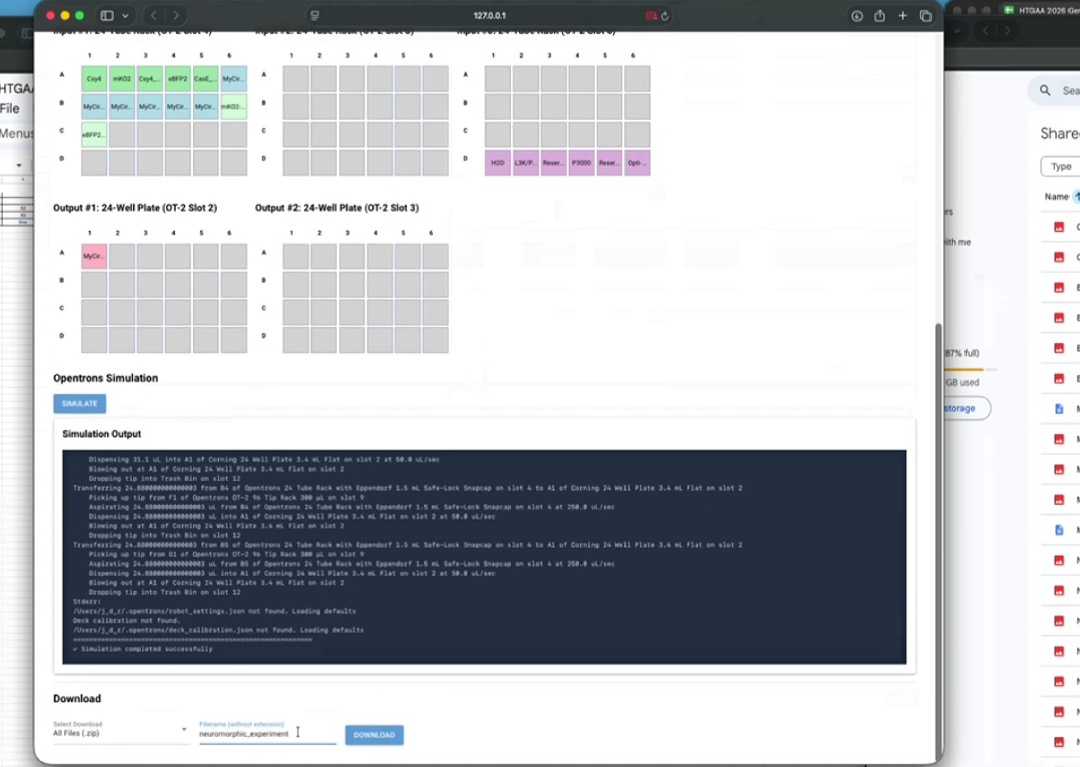
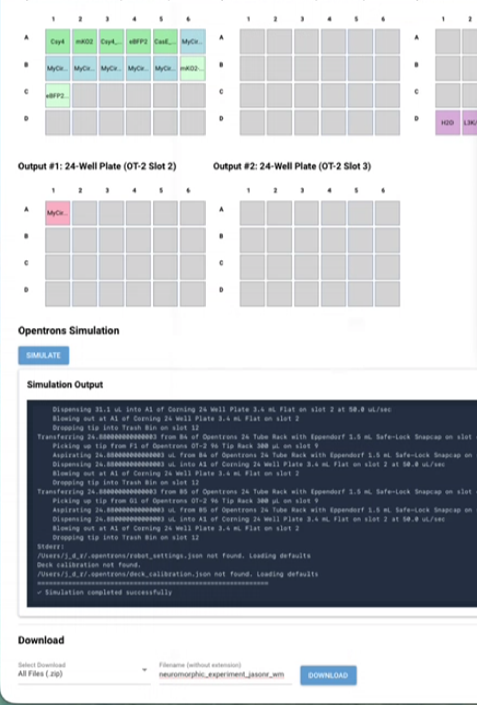
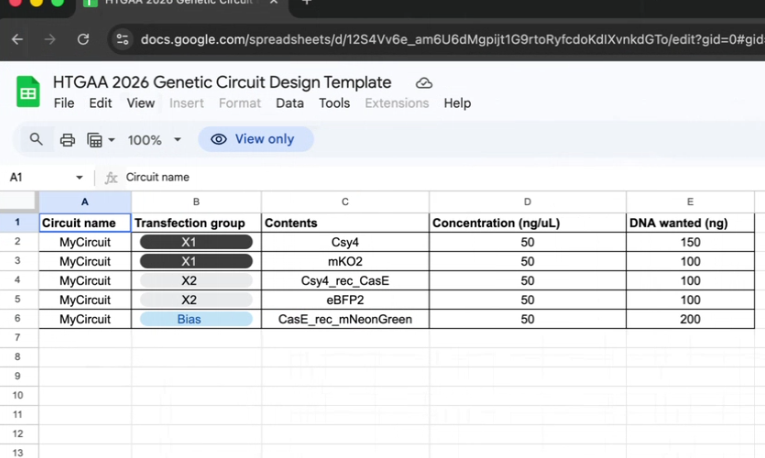





{kind=link}