Week 5 — Protein Design Part II
Part A: SOD1 Binder Peptide Design
Goal: Design short peptides that bind mutant SOD1 and then decide which ones are worth advancing toward therapy
Part 1: Generate Binders with PepMLM
The SOD1 Protein0 has base sequence:
To obtain the functional and mutated protein we need to do the removal of the start codon Methionone and mutate the fourth A to a V.
Using PepMLM in my edited copy of the notebook I got the following pepties of length 12:
I also computed the perplexity of the known SOD1 binding peptide ‘FLYRWLPSRRGG’
It appears that the model like its peptides betters than the known binder.
Actually when I wne tot the next step I finally noticed every single one of the generated peptides had an X/unknown which alphafold barfs on. I fixed this by adjusting the top_k value to be top 9 to allow more choices besides X and got:
These are more suprising than the other ones (which isn’t too surprising given the wider latidute of to pick less likely amino acids), but still decent and similar to the known binding protein.
Part 2: Evaluate Binders with AlphaFold3
Running alphfold agains these binders give:
- Known Peptide - FLYRWLPSRRGG Results
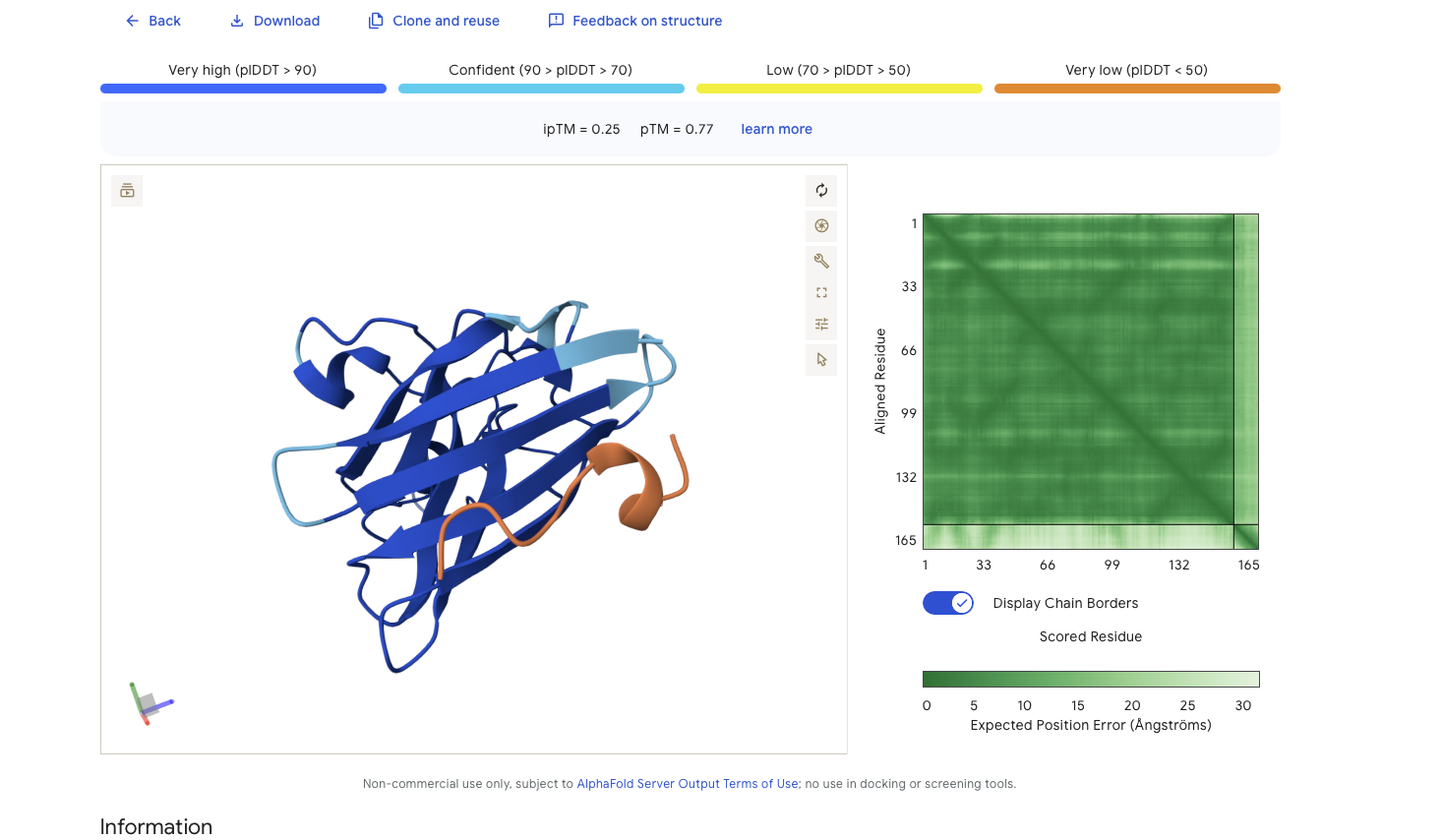
Alphafold is confident in the shape of protein. It is intersting while that while alphafold does identify it as binding to the N-terminus it doesn’t feel very confident about that binding 0.25
- Peptide Binder 0 - WDNVGYAIYSGK Results
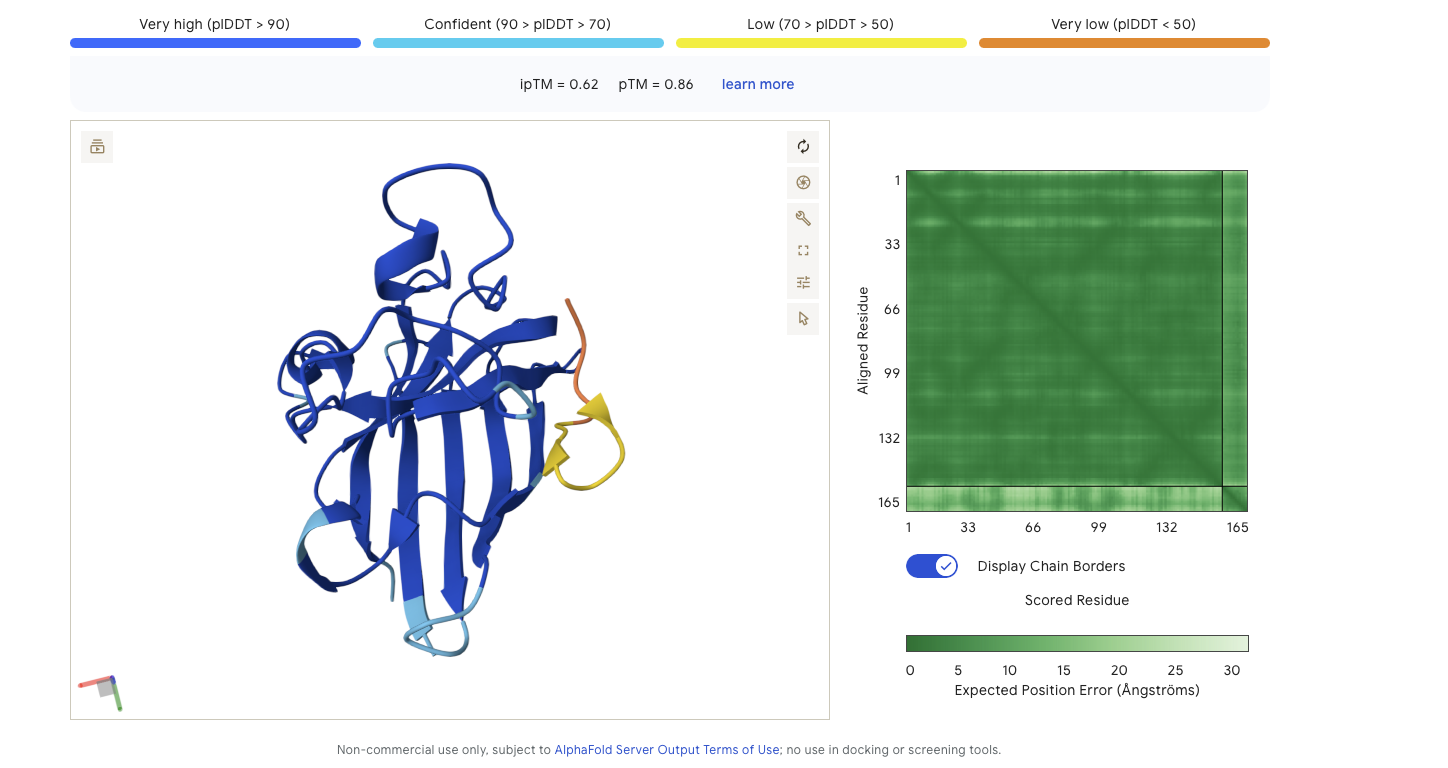
Alphafold is confident in the shape of protein. The binding certainity (ipTM) is only 0.62 also it is binding to the beta barallel not the N terminus.
- Peptide Binder 1 - HDWVGQGIDQGE Results
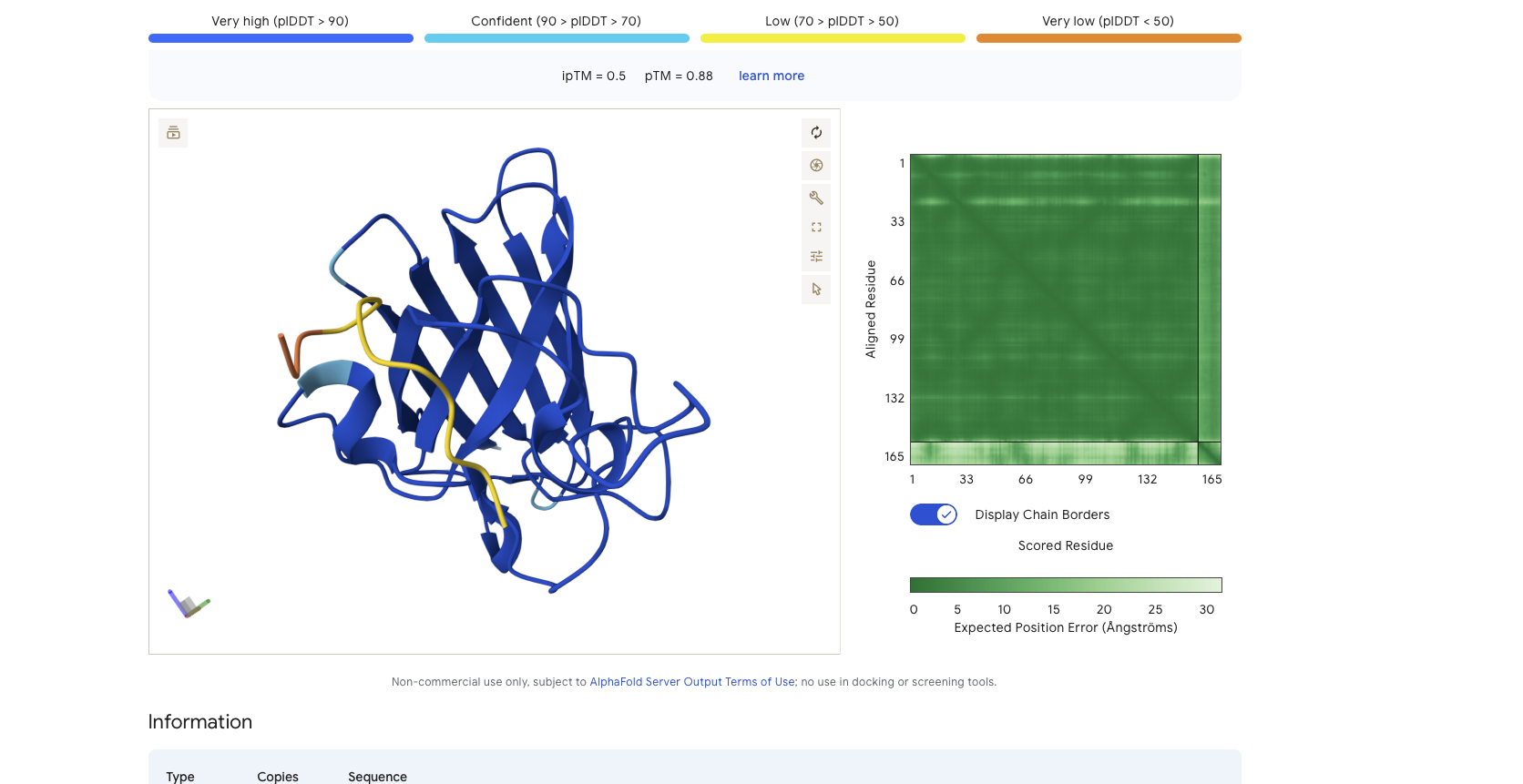
Alphafold is confident in the shape of protein but the binding certainity (ipTM) is only 0.5 also it is binding to the alpha helix not the N terminus.
- Peptide Binder 2 - ESYYDQAVDQLE Results
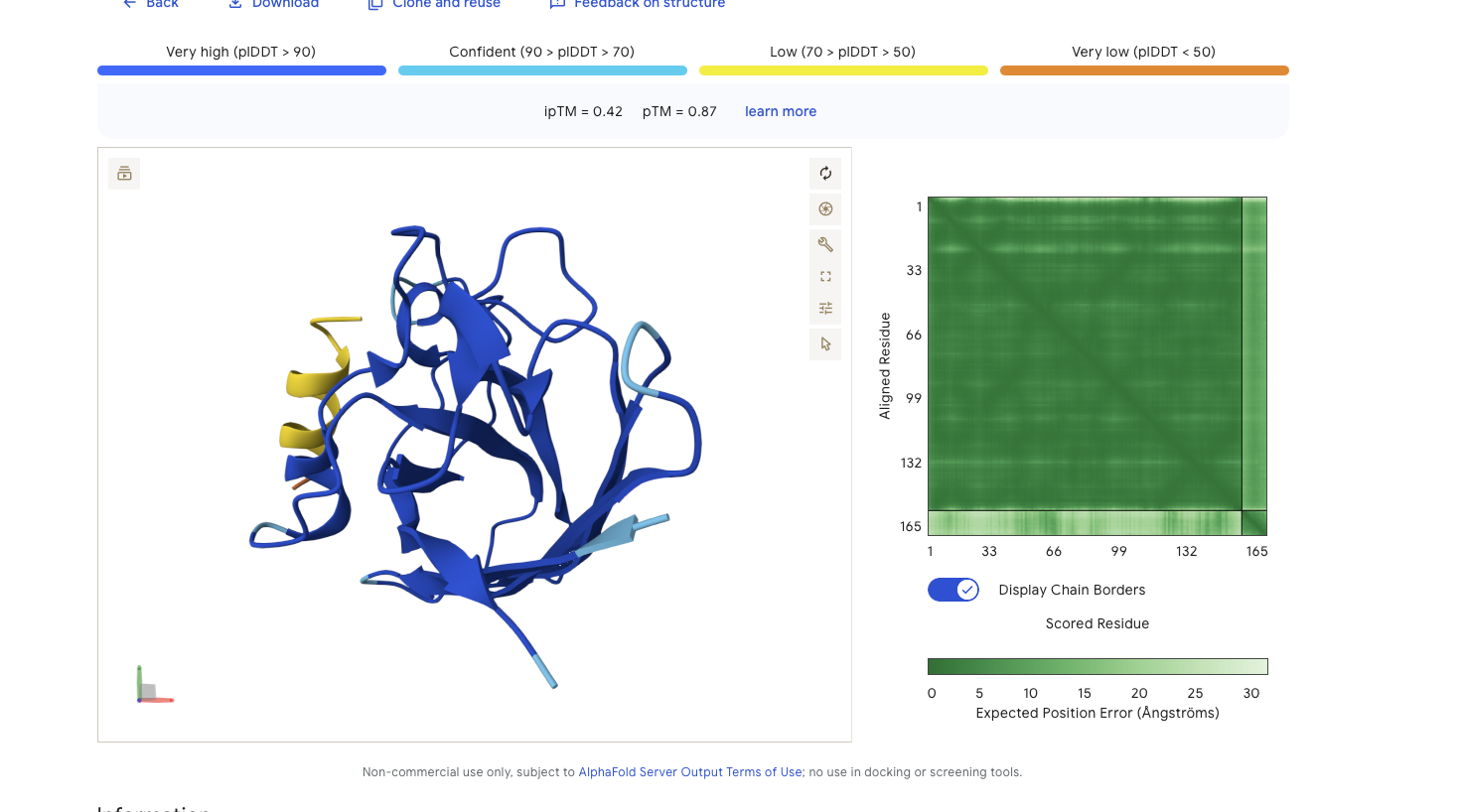
Alphafold is confident in the shape of protein. The binding certainity (ipTM) is low 0.42 and it is predicted to bind to the N-terminus.
- Peptide Binder 3 - VSYPGQVVGHLP Results
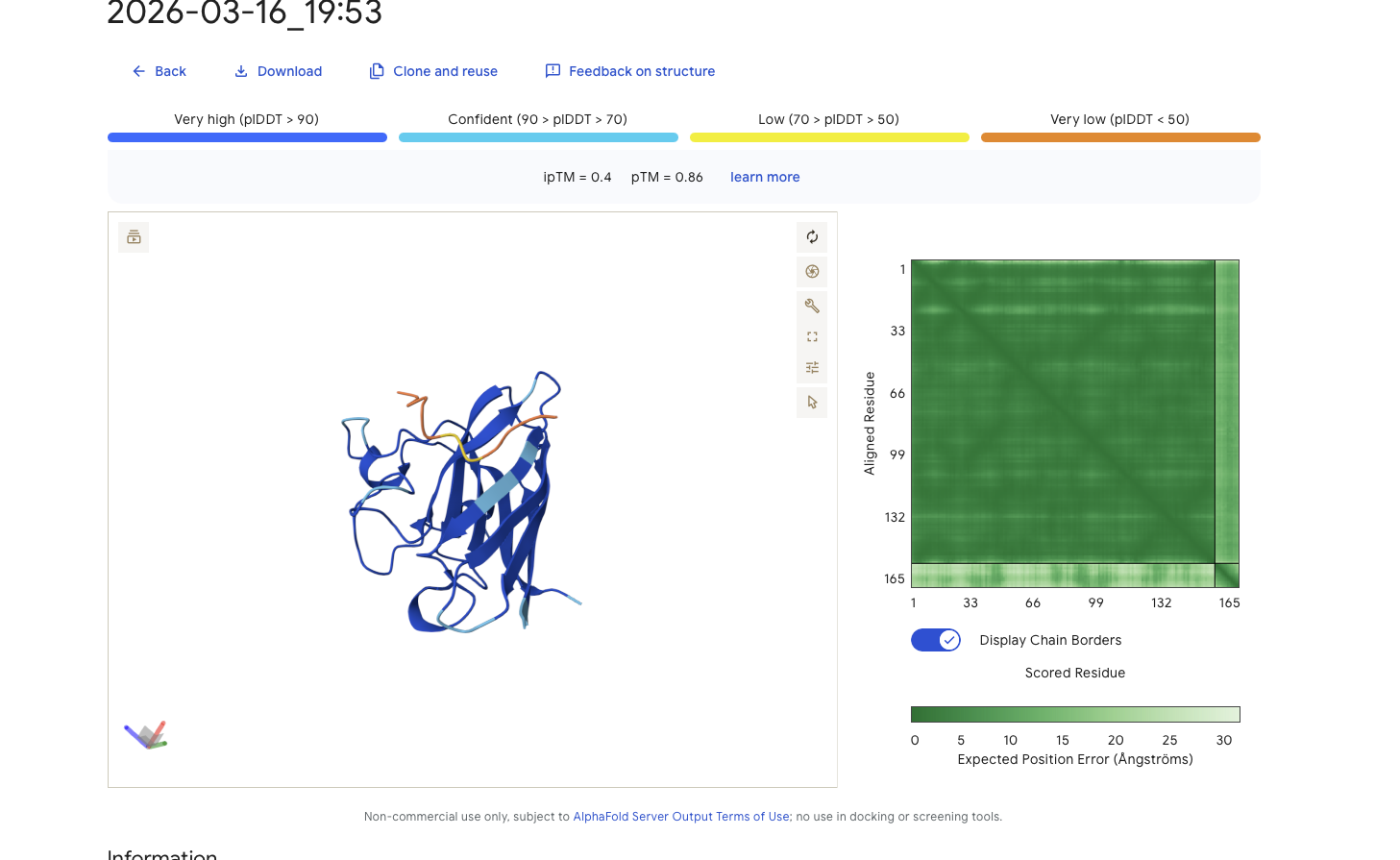
Alphafold is confident in the shape of protein. The binding certainity (ipTM) is only 0.4 also it is binding to the beta barallel not the N terminus.
Overall given that Peptide Binder 2 seems to be more confident in the binding to the N-Terminus than it is for the known peptide. Also looking at the grids for the various results it doesn’t seem like alphafold is really the confident for where the peptides are binding even for the known peptide?
Part 3: Evaluate Peptide Properties
Using Peptiverse I got the following for the basic properties
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| FLYRWLPSRRGG | 💦 Hydrophobicity (GRAVY) | Non-hemolytic | 1507.7 | Probability |
| FLYRWLPSRRGG | 🔗 Binding Affinity | Weak binding | 5.962 | pKd/pKi |
| FLYRWLPSRRGG | 📏 Length | 12 | aa | |
| FLYRWLPSRRGG | ⚖️ Molecular Weight | 1507.7 | Da | |
| FLYRWLPSRRGG | ⚡ Net Charge (pH 7) | 2.76 | ||
| FLYRWLPSRRGG | 🎯 Isoelectric Point | 11.71 | pH | |
| FLYRWLPSRRGG | 💦 Hydrophobicity (GRAVY) | -0.71 | GRAVY | |
| WDNVGYAIYSGK | 🩸 Hemolysis | Non-hemolytic | 0.050 | Probability} |
| WDNVGYAIYSGK | 🔗 Binding Affinity | Weak binding | 5.994 | pKd/pKi |
| WDNVGYAIYSGK | 📏 Length | 12 | aa | |
| WDNVGYAIYSGK | ⚖️ Molecular Weight | 1372.5 | Da | |
| WDNVGYAIYSGK | ⚡ Net Charge (pH 7) | -0.24 | ||
| WDNVGYAIYSGK | 🎯 Isoelectric Point | 5.83 | pH | |
| WDNVGYAIYSGK | 💦 Hydrophobicity (GRAVY) | -0.46 | GRAVY | |
| HDWVGQGIDQGE | 🩸 Hemolysis | Non-hemolytic | 0.043 | Probability |
| HDWVGQGIDQGE | 📏 Length | 12 | aa | |
| HDWVGQGIDQGE | ⚡ Net Charge (pH 7) | -3.14 | ||
| HDWVGQGIDQGE | 🎯 Isoelectric Point | 4.31 | pH | |
| HDWVGQGIDQGE | 💦 Hydrophobicity (GRAVY) | -1.18 | GRAVY | |
| HDWVGQGIDQGE | 🩸 Hemolysis | Non-hemolytic | 0.043 | Probability |
| HDWVGQGIDQGE | 📏 Length | 12 | aa | |
| HDWVGQGIDQGE | ⚖️ Molecular Weight | 1340.4 | Da | |
| HDWVGQGIDQGE | ⚡ Net Charge (pH 7) | -3.14 | ||
| HDWVGQGIDQGE | 🎯 Isoelectric Point | 4.31 | pH | |
| HDWVGQGIDQGE | 💦 Hydrophobicity (GRAVY) | -1.18 | GRAVY | |
| VSYPGQVVGHLP | 🩸 Hemolysis | Non-hemolytic | 0.024 | Probability |
| VSYPGQVVGHLP | 📏 Length | 12 | aa | |
| VSYPGQVVGHLP | ⚖️ Molecular Weight | 1252.4 | Da | |
| VSYPGQVVGHLP | ⚡ Net Charge (pH 7) | -0.18 | ||
| VSYPGQVVGHLP | 🎯 Isoelectric Point | 6.71 | pH | |
| VSYPGQVVGHLP | 💦 Hydrophobicity (GRAVY) | 0.30 | GRAVY | |
| ESYYDQAVDQLE | 🩸 Hemolysis | Non-hemolytic | 0.052 | Probability |
| ESYYDQAVDQLE | 🔗 Binding Affinity | Weak binding | 6.105 | pKd/pKi |
| ESYYDQAVDQLE | 📏 Length | 12 | aa | |
| ESYYDQAVDQLE | ⚖️ Molecular Weight | 1459.5 | Da | |
| ESYYDQAVDQLE | ⚡ Net Charge (pH 7) | -4.15 | ||
| ESYYDQAVDQLE | 🎯 Isoelectric Point | 4.05 | pH | |
| ESYYDQAVDQLE | 💦 Hydrophobicity (GRAVY) | -1.22 | GRAVY |
Checking the binding affinity separatrely it doesn’t seem like any of them bond strongly including the known binding peptide.
| Input | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| FLYRWLPSRRGG | 🔗 Binding Affinity | Weak binding | 5.962 | pKd/pKi |
| WDNVGYAIYSGK | 🔗 Binding Affinity | Weak binding | 5.994 | pKd/pKi |
| HDWVGQGIDQGE | 🔗 Binding Affinity | Weak binding | 5.216 | pKd/pKi |
| ESYYDQAVDQLE | 🔗 Binding Affinity | Weak binding | 6.105 | pKd/pKi |
| VSYPGQVVGHLP | 🔗 Binding Affinity | Weak binding | 5.524 | pKd/pKi |
None of my peptides (and even the known binder?) seem like great candidates. Not sure if I am doing something wrong? Given this I guess I would go with ESYYDQAVDQLE for further work since it seems to be the closest binding candidate.
Part 4: Generate Optmized Peptides
I made a copy of the notebook and tried to do the optimization computations there. Unfortunately I ran into two problems I wasn’t able to resolve:
- I wasn’t able to get the recommended GPU and could only get a T4
- I repeatedly got (what appears to be an off by one error) pretty deep in the moPPit code. I tried running with a bunhc of different parameters. I did determine that the probably seems to be with something indexed by the sample parameters since the error message changes when I change the sample count. The error I am getting is below:
Part B: BRD4 Drug Discovery Platform Tutorial
Part 1 Structural Predictions
Results
| Compound | Binding Confidence | Optimization Score | Structure Confidence |
|---|---|---|---|
| Hit | 0.76 | 0.26 | 0.90 |
| Lead | 0.44 | 0.23 | 0.88 |
| JQ1 | 0.96 | .044 | 0.98 |
Discussion
- Does Binding Confidence increase as you move from hit to clinical candidate? What would you expect, and why might it deviate?
I am not sure I understand the terminology but I am assuming that the order is Lead -> Hit -> Clinical Candidate (JQ1). As we go through this sequence I would expect binding condidence to increase in most scores. I can see situations where there could be experimental evidence that overrides the modeling results and perhaps give an inversion of scores?
- Inspect the predicted binding pose for JQ1. Can you identify potential key binding interactions.
It looks like JQ1 is binding in the top/interior of the a barrel made for the 4 helix barrel that is the primary teririary structure in the protein.
• Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead.
The optimization score for JQ1 is higher.
Part 3 Generative Virtual Screen
Ran virtual screen with recommended parameters
Part 4 Analysis
I got no results that had both an optimization score > 0.4 and binding score > 0.8 which I think means I got no “High Confidence” candidates. If I relax that to an high binding OR high optimization I get
- 2 compounds that binding scors of 0.8, with optimization scores of 0.27 and 0.34. Of these two SM-5S4NDA3E has the highest optimizatoin score and is my best candidate.
- 12 candidates with an optimization score of greater than 0.4. The three best candidates are (the rest of binding condience of less than 0.7)
- SM-279VMWMP has binding confindence of 0.78 and optimization score of 0.4
- SM-QAZ4MM4W has binding condidence of 0.77 and optimization score 0.45
- SM-2F86NWEQ has binding condence of 0.72 and optimization score 0.42
- JQ1 appears to be a much better candidate it has Binding Confidence of 0.99 and Optimization Score 0.55.
- The binding againstBRD2 for my top 4 candidates was still quite strong, between 0.8 and 0.77 so these binders are not very selective.
Part C: Final Group Project: L Protein Mutants
I did a variation of option 3 which was to generate random mutations using the information in this spreadsheet on mutants. However inspired by Professor’s Sahas mention of the constraints because the virus is encoding other proteins in different frameshifts, I did a search for the point mutations that preserve the structures of the proteins and put the results in this spreadsheet. The entire journey and the code that generates the final results is captured in this python code which is the downloaded python code from the Marimo notebook I used. Given we are trying to engineer this protein for phage therapy using modified MS2 (I think) it seems like you need to take this into account and either select mutations from this list or do your mutational analysis on both the lysis protein and frameshifted protein at the same time, which it doesn’t seem like is being done? In particular, most of the mutations in the spreadsheet are not in the list of mutations that preserve the frameshifted proteins.