Week 4 HW: Protein Design Part I
Part A: Conceptual Questions
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
- Why are there only 20 natural amino acids?
- Can you make other non-natural amino acids? Design some new amino acids.
- Where did amino acids come from before enzymes that make them, and before life started?
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
- Can you discover additional helices in proteins?
- Why are most molecular helices right-handed?
- Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
- Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
- Design a β-sheet motif that forms a well-ordered structure.
Okay I am going to take a first pass through here just going off the lecture, wikipedia, and background knowledge I already have and then go back and try with AI assistance for the ones I have no answer for.
- A Dalton is another name for atomic mass unit. $6\times1023$ hydrogen atoms/dalton/atomic massunits is roughly $1g$ of mass, so $1g$ of amino acids is roughly $6\times1021$ amino caid molecues and 500g of meat should contain at most $3\times1024$ amino acid molecues. In reality, a good fraction of the mass of meat is water, fat, and other non-protein so the number will be less than that, probably between 10-30% of the max depending ont he meat involved, which gives a range of $3\times1023$ to $1\times10^24$ molecues.
- The state of being a cow is a complex relationship between a cow’s cells. Being a cow cell is a complex relationship between the DNA, lipids, and proteins in that cell as well as the cell’s history and its relationship to other cow cells. IN other words being a cow is a delicate state. Eating a cow (especially if we cook it first) involves destruction of the cells, relationships, and molecues breaking them down into component parts at a molecular level, in particular protein is broken down into its component amino acids so all the relationship,s structure, and patterns are lost and re-assembled into the patterns and structure of whatever is eating the cow by the machinery of the organisms and its cells (plus the injection of energy). You can acquire the molecular shadow of what you eat in the form of the isotope concentration of your components, e.g. if you eat at lot of corn (or things that eat corn) you will have the isotope ratio of a C4 plants like corn.
- Not sure there is a strict answer to this, because it seems like there is something of a historical contigency here. In fact my past seemed to indicate that different people cite differen numbers of “natural amino acids”, e.g. wikipedia says 22 amino acids instead of 20. Given that I think the best answer is something like early life must have existing in an environment where the current naturally occuring amino acids were being manufactured by some abiogenic process and ended up being incorporated into the structure of early proto-life. In addition to requiring that the amino acids was created by some abiogenic proces in decents amounts, the current amino acids of life are also the ones that life figured out how to internally synthesize. I can imagine there may have been a commonly occuring abiogenic amino acid that some proto-life started using, but proto-life didn’t figure out how to self-synthesize. As the abiogenic source of a hard to biologically synthesize amino acid waned, proto-life that used that amino acid would have been very strongly selected against, so even a common abiogenic amino acid may not show up as current “natural” amino acid.
- An amino acide is an organic compound that has both carboyxl and an amine group. This means that the to design an amino acid you can attach those groups to any organic/carbon backbone. For example, you could take octane (eight carbon atoms in a chain with hydrogen atoms) and attach a carboxyl and amine group to the last atom in the chain to make an amino acid (octine?). It is hard to tell if this is a “new” amino acid because there are 500+ amino acids just in nature according to wikipedia without a web/AI search.
- There are abiogenic processes that naturally create amino acids. One famous experiment put methane, nitrogen, etc in a jar and passed electricity through and ended up creating many organic compounds including amino acids. We have also detected amino acids and other organic compounds on remote comets/asteroids presumably created abiogenically by heating/cooling/light energy impacting on the frozen components like methane ice.
- I have no idea, but seems like D should be right-handed?
- I am not sure what this question is asking? Additional relative to what?
- Again, I am not sure what this question is asking? I assume they mean most biological molecular helices, because I don’t know that molecular helices have a preferred direction in general. If it is biological, I guess this is because some organic compounds are chiral and biology (because of historical accident?) selects/builds only one chirality of that organic compound. The chirality then impacts the shape formed when repeating units bind together leading to helices with a certain direction also.
- Not sure, but if I had to guess it would be hydrogen bonding between the parts of amino acids that are perpendicular to the direction of th sheet?
- No Idea
- No idea.
Ok, now I will check/redo using outside research and AI especially for the last questions. Below is a record of main prompts I asked Gemini:
- “What governs whether the peptides bond 4 forward or to a completely different segment (beta sheet).”
- “What can you tell me about which way the helix turns on how it relates to chirality and the direction that DNA helix turns?”
- “What is protein tertiary structure and what governs it? Also hydrogen bonds?”
- “Which R group interactions stack beta-sheets on top of each other?”
- “What are amyloid beta sheets and how do they form?”
- “How would I go about designing a beta-sheet motif and what does that even mean?”
- “What are examples of amino acids that are part of the beta-sheet vs the flexible turn between strands (proline & glycine) or is it just that any amino basic except proline and glycine can be in the beta-sheet part of the strand? Or maybe even proline and glycine can be in the beta strand as long as there aren’t 3-4 flexible amino acids in a row?”
With this info I revise/add answers
- My guess above was wrong. For amino acids while they are right handed chirality this apparently creates a left handed helix. Strangely enough for sugars a right hand chirality creates a right handed helix which is what I was remembering when I guessed.
- Still don’t understand this question.
- I think I have a better feeling for this now. Beta sheets are formed by the hydrogen bounds between adjacent amino acid chains. This formation of the beta-sheet with hydrogen bonds exposes the R-groups of the amino acids in the sheet in an up and down direction/pattern relative to the sheet. The R groups that are exposed goven the binding between sheets. For example, some amino acids have R groups that can allow hydrogen bonding between sheets. Apparenly the tightest binding between sheets happens when the R groups are such that they can expel the water between the sheets and sterically snap together because the exposed R-groups line up shape-wise and have the ability (hydrophobic) to expel water between sheets.
- As far as I can tell amyloid proteins have an autocatalytic tendency to form the tight fitting beta-sheets mentioned in #9, so if some condition (heat, disease, genetics) leads to creation of 2-4 tight fitting beta-sheets in an amyloid-protein it will eventually capture other amyloid proteins and recruit them into the same tight-fitting beta-sheets. Since it is very hard to pry these sheets apart, these clusters tend to grow slowly over time intefering with normal function of protein and surrounding tissue.
- Not sure exactly what we are looking for here still? A DNA/amino acid sequence? I do understand the basic structure of beta sheets to know that I can make a beta-sheet by picking runs of 5-10 amino acids with mostly bulky R groups, like valine or tyrosine joined together with a proline + glycine loops that allow the protein strand to bend back on itself so the bulky runs can hydrogen bond each other. If you put enough runs like this together you will get a very wide beta-sheet which will then tend to wrap around and make a barrel or cylinder. So if I wnated to “design” a beta-sheet I would just repeat pattern like TVTVTVTCPG a bunch of times?
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I chose casein which is the major protein in milk, because I like dairy products and it would be interesting to see how the common properties of something I interact with every day relates back to the protein structure and DNA sequence. I also remember seeing a video about using milk/caesin to make a fiber that you can make clothese from which points to a protein that potentially has a lot of uses.
Identify the amino acid sequence of your protein
It looks like there are actually several different casein protein ($\alpha S1, \beta, and \kappa$) variants that appear in most mammal milk. I think it will be interesting to compare them and see how they different so I will look at sequence for all of them in as well as comparing across species.
I collected the sequences from uniprot and did work in this colab notbook. I used NCBI Protein Blast for the homolog searches. For family search I used Interpro. For structures I looked in RCSB Protein Data Bank
{‘humans’: {‘alpha’: (185, [(‘E’, 20)]), ‘beta’: (226, [(‘P’, 39)]), ‘kappa’: (182, [(‘P’, 29)])}, ‘cows’: {‘alpha’: (214, [(‘E’, 25)]), ‘beta’: (226, [(‘P’, 39)]), ‘kappa’: (190, [(‘P’, 21)])}}
$\alpha S1$:
185 amino acids, most common is ‘E’ 20 times (glutamic acid)
The closest homologs to this (not too surprising) were in the great apes (100 percent match). A little more suprising was how much change has happened in mammalian milk and that human alpha protein is closer to bats and seals than sheep (only 37 percent identical).
Not too suprisingly, interpro identified the family as alpha/beta caseins
Very suprisingly there don’t seem to be any solved crystallography for human alpha caesin in Uniprot, just alpha fold predictions. I checked this with Gemini via:
“I am slightly surprised that human casein alpha has not been crystallized and had crystallography done? I would have thought it would be a basic first protein people would do?”
The reply was that alpha casein is an intrinsically disordered protein which is hard to crystallize, which led me down a rabbit hole where I discovered that the unordered parts of our proteins where it is hard to predict structure are important in eukaryotes/larger animals for regulation etc, even though we don’t have great ways to understand them yet.
214 amino acids, most common is ‘E’ 25 times (glutamic acid)
Similar results happen for cows, where the closest homologs are water buffalo and other hoofed ruminants like sheep are pretty close.
Not too suprisingly, interpro identified the family as alpha/beta caseins
Uniprot did have some electron microscope image that included cow casein, but this is sort of incidental because they are feeding casein into an e. coli motor that untangles tangled proteins like casein.
I used the Gemini prompt below to understand this:
“When looking at cow alpha casein I do seem electron microscope images where there are looking at E Coli on casein substrate: https://www.ebi.ac.uk/emdb/EMD-4623. What does that mean?”
$\beta$:
226 amino acids, most common is ‘P’ (proline) 39 times
Simlar homologs to the alpha case
Again interpro identified the family as alpha/beta caseins
Here we have the same issue as with the alpha version of casein, though there is a group PED studying intrinsically disordered proteins that are collecting additional modeling results about the entire structural ensemble/deformations and someone has modeling result based on a combination of alphflex and molecular dynamics simulations.
224 amino acids most common is ‘P’ (proline) 39 times
Simlar homologs to the alpha case
Again interpro identified the family as alpha/beta caseins
Here I was suprised to see that there are PDB entries for cow casein. There are mostly of small (rigid parts?) of the entire protein? There is one entry that claims to be the full protein sequence (PDB 7TTR), but going to that PDB entry (and verifying with Gemini) shows this is another case of casein be used as an input into a test of an untangling motor.
$\kappa$:
182 amino acids, most common is ‘P’ (proline) 29 times
Simlar homologs to the alpha case
Again interpro found it was in the kappa family
Kappa only has some predictions.
190 amino acids, most common is ‘P’ (proline) 21 times
Simlar homologs to the alpha case
Again interpro found it was in the kappa family
Kappa only has some predictions.
Using the foldseek links from Uniprot structure recommendations doesn’t seem to lead to any good anlogs or structureally similar
I ran clustal omega to see how sequences aligned across species too. Alpha Casein. There was pretty tight alignment at the start and then not much. Kappa proteins alignment seemed to have been spread out across the entire protein Kappa Casein
- Alpha Casein Cross Species Alignment

- Kappa Casein Cross Species Alignment

3D Visualization
I used desktop PyMol and liberal use of “gemini” to help with this (queries like “how do I color by acid base in pymol”)
Visualize in Different Modes
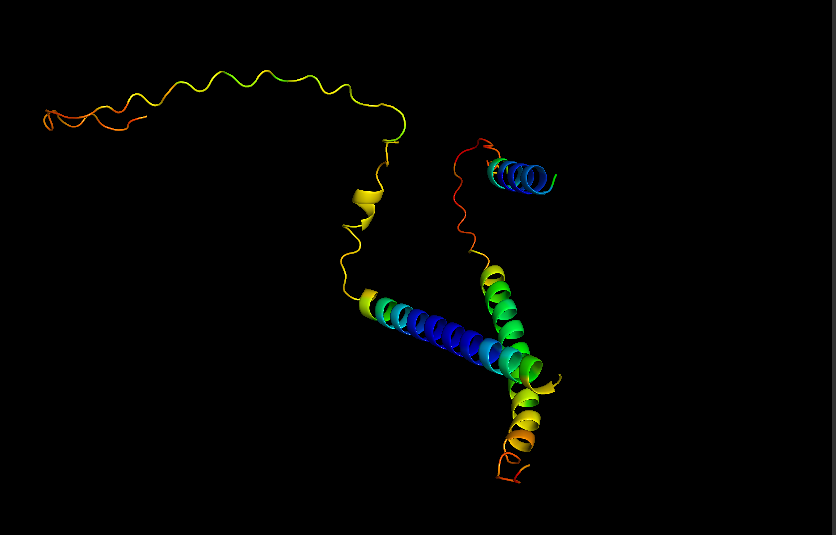
- Cartoon for Human Alpha Casein
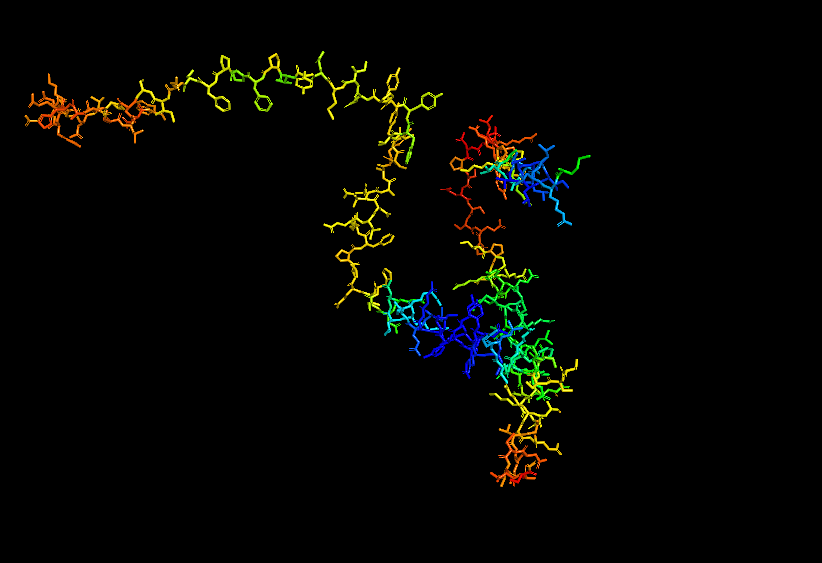
- Stick for Human Alpha Casein
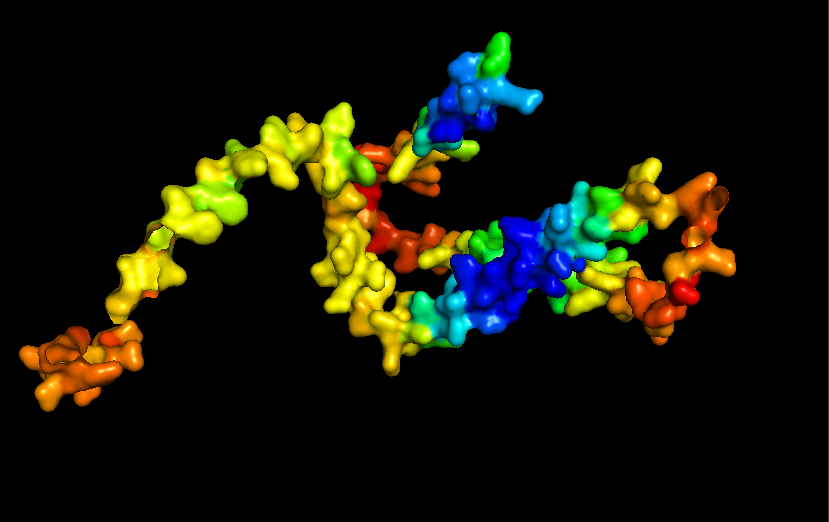
- Surface for Human Alpha Casein
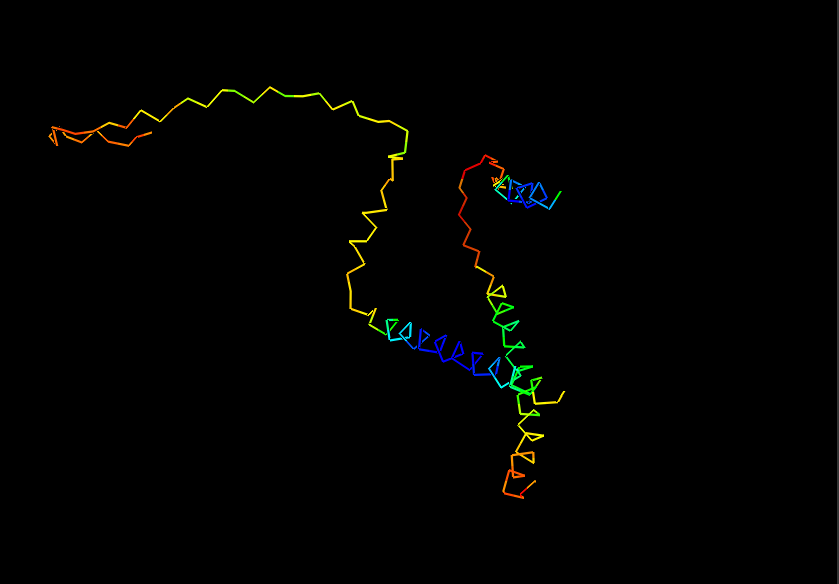
- Ribbon for Human Alpha Casein
Visualize Secondary structure
Casein alpha has 4 alpha-helices on a single strand, 2 small ones on the end and 2 larger ones in the middle. The alpha strands have long unstructured runs between them, so they probably flop around a lot.
You don’t really need to color because you can see pretty clearly in the cartoon view above.
Visualizes Residues
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? THere doesn’t seem to be any well-defined sides/regions that I can see in this view. In fact they two kinds of residues seem to be well-mixed across the entire strand. Maybe this contributes to the inherent disorder of the protein.
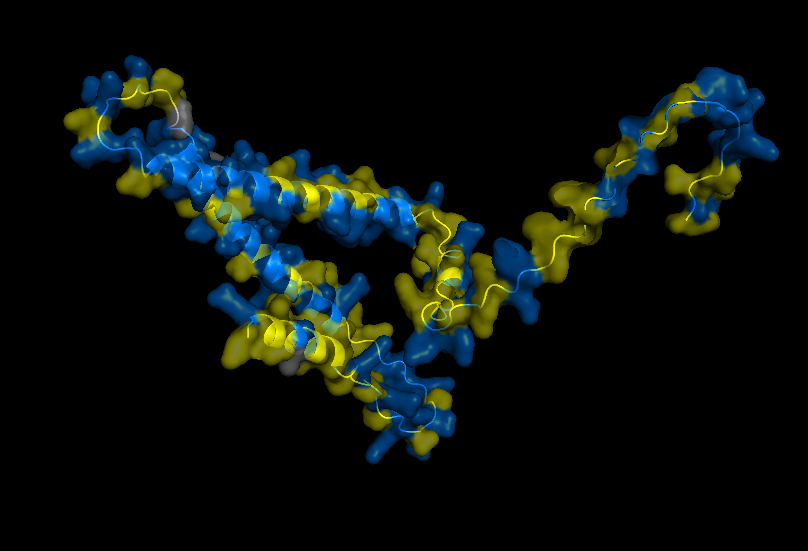
Record of Gemini Prompts for Part B
- What is the primary protein in milk? Caesin?
- What is a protein homolog?
- Where is the best place to look for protein homologs using BLAST?
- Is there a standard way to identify “protein family”?
- Any suggestions about how to search for structure information in the protein data bank if I have uniprot references? Searching for sequence or keywords in PDB doesn’t seem very fruitful
- I am slightly surprised that human casein alpha has not been crystallized and had crystallography done? I would have thought it would be a basic first protein people would do?
- So seems like alphafold predictions for intrinsically disordered may not be very good since we don’t have input data?
- What percentage of human proteins do we think are intrinsically disordered?
- So it what are the best avenues/tools for understanding the dark proteome in animals like humans? It seems like we are missing a lot of info about how proteins function and how we can design/understand them in that context.
- So the micelles structure that casein forms might be mirrored as membranes organelles/reaction chambers inside a cell?
- When looking at cow alpha casein I do seem electron microscope images where there are looking at E Coli on casein substrate: https://www.ebi.ac.uk/emdb/EMD-4623. What does that mean? It sounds like the casein is incidental here?
- What is a structure classification family for proteins.
- There are foldseek links in the structure entries in uniprot? What are those, and how do you use them?
- I guess it can find partial matches? It gave me a few viruses with partial match positions and very low probability? Are those false positives?
Part C: Using ML-Based Protein Design Tools
- My copy of the notebook
- For this part of the homework, I chose Curlin of the main components of E. Coli biofilms and is an amyloid type protein. This sequence is
C1: Protein Language Modeling
Deep Mutational Scan
Running the ESM2 model in my copy of the Colab notebook for the Curlin csgA protein gave
- Deep mutation Scan for Curlin csgA
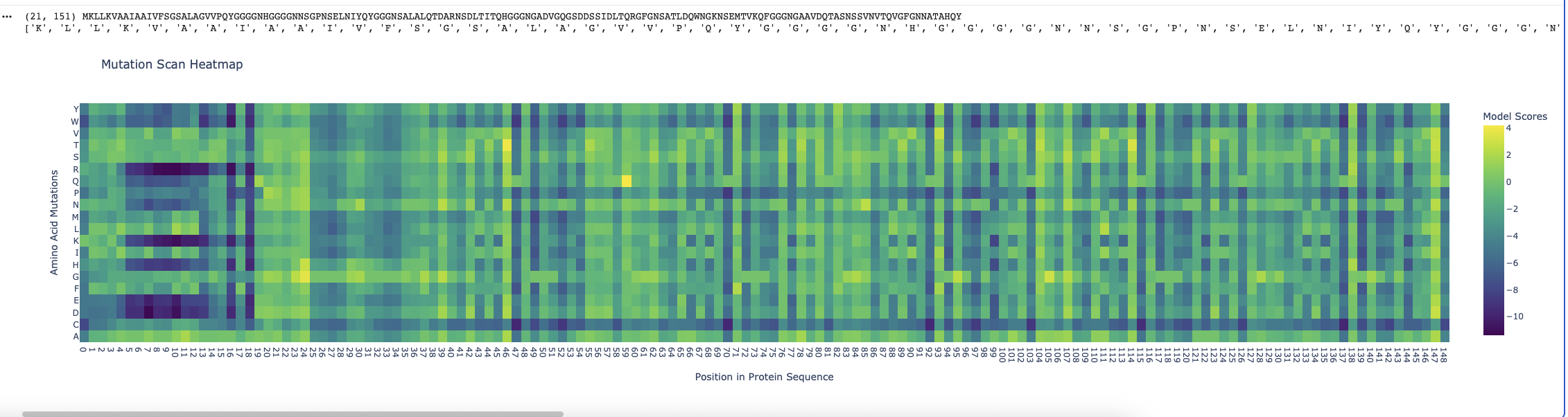
I found the coloring of this heatmap to be hard to understand because it is all shades of green instead of different colors for positive and negative mutations. Visually seeing the “no mutation” amino acids was especially hard. Also I noticed that there was a trimming problem and the first amino acid was missing from the analysis. Given this I changed the color theme and trimming to get this version
- Revised Deep Mutation Scane for Curlin csgA
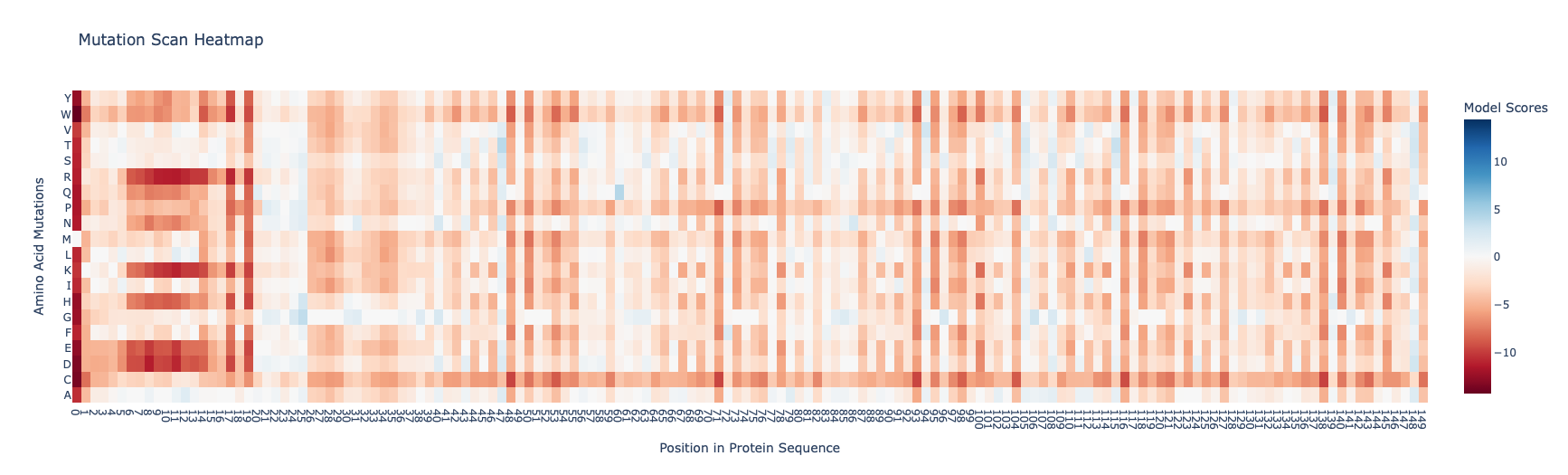
In this image the no mutation parts of the map are white, “more typical” mutations are blue, “unusual/bad” mutations are red as determined by the model score.
Some observations:
- Some amino acids are relatively interfchangable, for example in most places where the is a L (leucine) and I (isoleucine) is almost as good.
- There are periodic Qs (glutamine) spaced roughly equally far apart (48, 71, 93, 104, 116, 138) which have no other reasonable option which indicates they are crucial to the structure and function of the protein. The amide group in the gluatamine may be important for the links between the beta sheets.
- Likewide there are perodic Gs (glycines) spaced between the Qs which also don’t have good replacements. These may be important for the turns between the beta sheets in the protein.
Latent Space Analysis
I edited the script in the notebook to include my protein and then did the latent space analysis to get the below graphs
- Top Level Latent Space
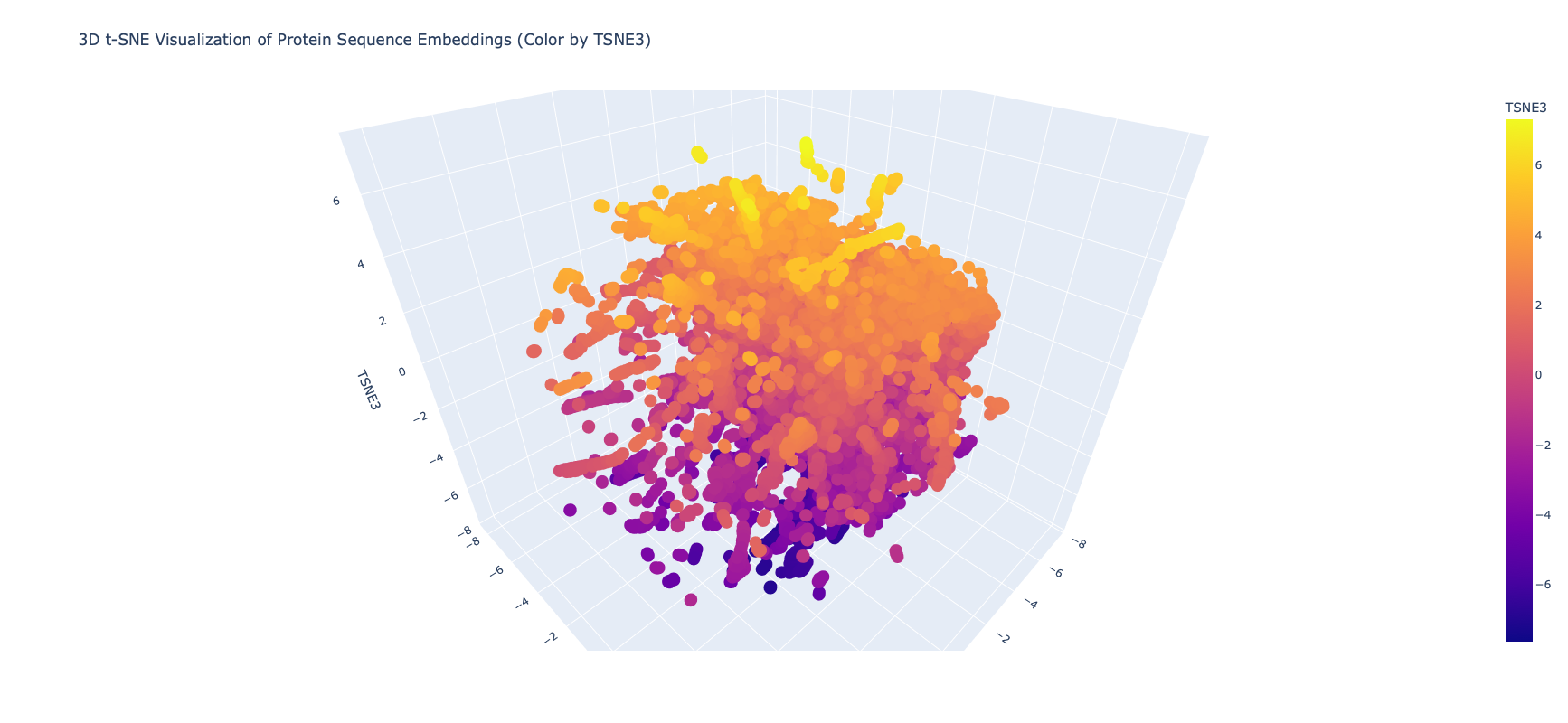
It was extrememly hard to see or find my protein, even when I knew the coordinates, because of the number of proteins and the “jump to nearest” hover feature of plotly. Given this I asked Gemini for some code on how to zoom plotly and looked at smaller neighborhoods
- Close in Latent Space Zoom

When I zoomed in to a unit cube centered on my protein (circled in red) at (1.9201405, -3.6876564, 2.1404576) there was only one other protein in that window, which was a collagen adhesin. So it is interesting how isolated and unique curlin was. This may reflect the library we are comparing against and the fact that it hard to do xray crystallography on these kind of proteins?
- Close in Latent Space Wider Zoom
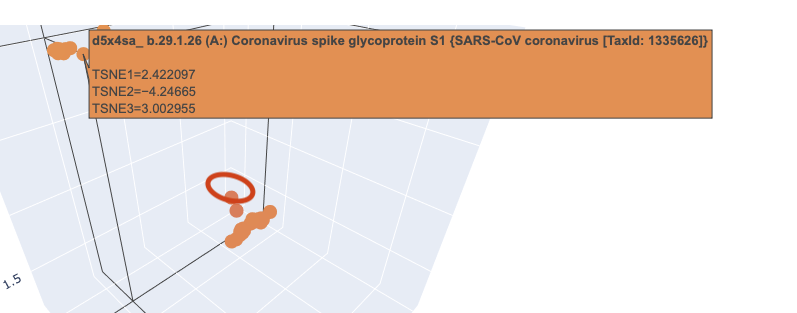
Given there was so few other proteins in the unit cube, I zoomed out to cube of size 2. This cube had more proteins (including theh covid spike protein?) but there didn’t seem to be any clustering that pointed to curlin even on this wider scale.
Latent Space Analysis Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2: Protein Folding
Fold your protein
- Refold

Refolding my protein with the original sequence gives something very close to the PDB alphafold (this sequence doesn’t have a good crystallygraphy PDB becuase it doesn’t crystaliize unfiormly).
- Original PDB alphafold
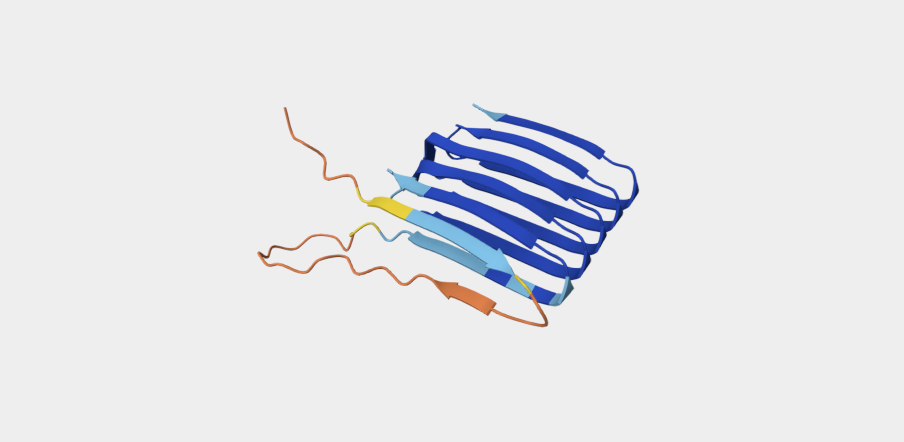
It is probably not too suprising these are very close since the alpha fold picture is also an inferred fold based on similar training data.
Mutate and Fold
Since there is no golden crystallography source here I wanted to see if the “bad” mutations identified in the first part have a dramatic inferred impact on structure. I mutated my squence by changing all of the “crucial” glutamines (Q) to W which was the “worst mutation” possible. This did have an impact on the structure (distorting the beta sheets when viewed from the side) but it wasn’t as dramatic as I was expecting. Maybe this is because the different amyloid protetins needs to stick together closely and these distortions mean that don’t fit together as well?
- Mutated Curlin Refold
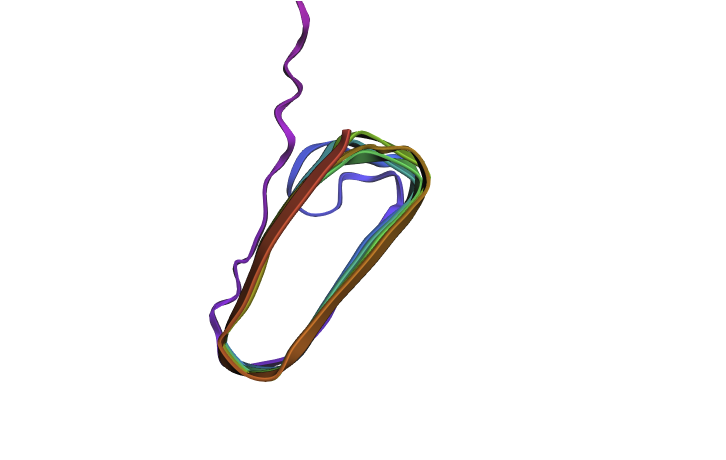
- Original Curlin From Side

with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3: Protein Generation
The code uses PDB but I don’t have entire sequence in PDB so I used large EM fragment (PDB 8ENQ) (which
PNSELNIYQYGGGNSALALQTDCRNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQCGFGNNATAHQY
New Sequence was
GATTATVTQTGTNNTATVTVSNAANSTVTVTQTGTNNTATVTHSSDNSTVTVTQTGTNNTATVTVSGSTNSTVTVTQTGTNNTATVTHTASNATVTVTQTGTNNTATATST
The refhold of the new protein basically looks the same as before.
Part D: Group Brainstorm on Bacteriaphage Engineering
We met to brainstorm on Feb 28 and are documenting results in this google doc
Part E: William and Mary Node Questions
- Be prepared to answer/discuss all 11 questions posed by Dr. Zhang. We will choose the most interesting ones to discuss in class.
- Be prepared to discuss the phage literature reading.
- A discussion of the phage literature will lead into our main discussion point: please be prepared to address and discuss the “big picture” question: how to apply these protein analysis tools to engineer a better bacteriophage. Please develop specific ideas for discussion.
- Time permitting - we will review final projects.
MKLLKVAAIAAIVFSGSALAGVVPQYGGGGNHGGGGNNSGPNSELNIYQYGGGNSALALQTDARNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQVGFGNNATAHQY
M GVVPQYGGGGNHGGGGNNSGPNSELNIYQYGGGNSALALQTDCRNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQCGFGNNATAHQY HHHHHH GVVPQYGGGGNHGGGGNNSG
PNSELNIYQYGGGNSALALQTDARNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQVGFGNNATAHQY PNSELNIYQYGGGNSALALQTDCRNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQCGFGNNATAHQY
PNSELNIYQYGGGNSALALQTDCRNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQCGFGNNATAHQY GATTATVTQTGTNNTATVTVSNAANSTVTVTQTGTNNTATVTHSSDNSTVTVTQTGTNNTATVTVSGSTNSTVTVTQTGTNNTATVTHTASNATVTVTQTGTNNTATATST