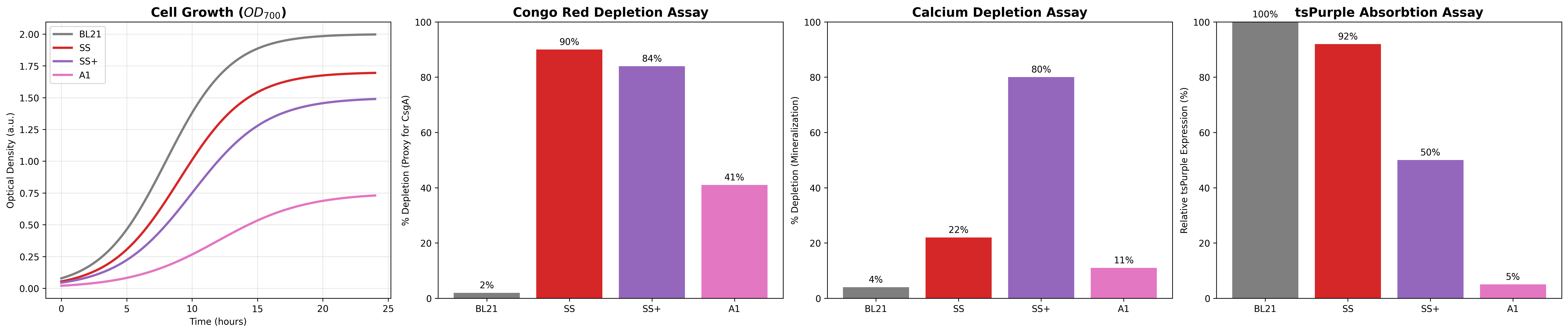I am based in the Research Triangle area of North Carolina. My background is in math and programming, but I have always been fascinated by biology. I am hoping this course and my membership in the Triangle DIY Biology group and lab will allow me to learn enough to let me contribute to interesting applications of synthetic biology and bioengineering. I am especially interested in approaches that can reduce the expense of bioengineering at scale in order to unlock the potential of biotechnology to have beneficial impacts on the world.
Final Project Frugal Benchtop Bioreactors: Editing the DNA of an organsim is more accessible than ever. Basic lab equipment and plasmid services like GenScript mean that you can dream up your own sequence and express it in an host for around a hundred US dollars. However to unlock the real world impact of gene edits you usually need to be able to scale the production up. The next step in scaling beyond the shaker flask is a bench-top bioreactor where you figure out how to actively manage and optimize your organisms growth and characterstics. This expense of the benchtop stage makes it less accessible than the edit stage even though in many ways the technology involved is simpler. For example a new benchtop bioreactor typically costs tens of thousands of dollars or more. Even used bioreactors costs thousands of dollars.
Homework Part 0: Basics Of Gel Electrophoresis Attended lecture and watched recitation video
Part 1: Benchling & In-silico Gel Art Link to Benchling Project , not sure how can see this link I asked to join HTGAA group but doesn’t seem like my invite was accepted yet? My drawing of an “E Gel Person” and associated enzymes in each lane are also in screenshot below Part 2: Gel Art - Restriction Digests and Gel Electrophoresis I don’t have access to these enzymes and DNA in my local makerspace lab.
Opentron Python Script Art Basic Idea The limited pixel resolution and colors of the petri dish reminded me of the old school bitmap monitors like the IBM PC that I grew up with. Also I wasn’t looking forward to guessing/figuring out a lot of pixel locations by hand, so I took a retro route and wrote some code to provide a terminal like API that let’s you specify a cursor location to write text to using a specific bitmap font and color.
Part A: Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Why do humans eat beef but do not become a cow, eat fish but do not become fish? Why are there only 20 natural amino acids? Can you make other non-natural amino acids? Design some new amino acids. Where did amino acids come from before enzymes that make them, and before life started? If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? Can you discover additional helices in proteins? Why are most molecular helices right-handed? Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Design a β-sheet motif that forms a well-ordered structure. Okay I am going to take a first pass through here just going off the lecture, wikipedia, and background knowledge I already have and then go back and try with AI assistance for the ones I have no answer for.
Editing the DNA of an organsim is more accessible than ever. Basic lab equipment and plasmid services like GenScript mean that you can dream up your own sequence and express it in an host for around a hundred US dollars. However to unlock the real world impact of gene edits you usually need to be able to scale the production up. The next step in scaling beyond the shaker flask is a bench-top bioreactor where you figure out how to actively manage and optimize your organisms growth and characterstics. This expense of the benchtop stage makes it less accessible than the edit stage even though in many ways the technology involved is simpler. For example a new benchtop bioreactor typically costs tens of thousands of dollars or more. Even used bioreactors costs thousands of dollars.
Substantial progress has been made toward accessible and open bioreactors with efforts like BIO-SPEC: An open-source bench-top parallle bioreactor system, but the Bill Of Materials for a BIO-SPEC system is still over 2000 Euros. Another great project is the Pioreactor which is available as a kit for only $350 US dollars. However the max volume of a Pioreactor is 40mL and it only has a single tank, which limits the ability to do things like media optimization unless you buy multiple Pioeractors.
The goal of my final project would be able to take advantage of fabrication tools like 3d printing to design and build a simple benchtop bioreactor with a bill of materials of around a hundred US dollars, demonstrate the reactors works by scaling up and optimizing production for at least one simple engineered microbe, and then release the plans and software with an open license. The bioreactor should support at least up to 1L of total culture and have multiple reaction tanks to support environmental optimization. A key aspect of benchtop scale up in monitoring and optimizing both the biomass and level of expression, which can be difficult to monitor cheaply. To prove out the concept and provide a demo project for the system the project will also edit an organism like a microbe to express a colored protein so that monitoring and optimizing is easier using inexpensive technology like digital cameras. Eventually a library of scaffold organisms could be developed that allow uses to insert their edits into a the organism in a way that their protein will be co-expressed with the easy to monitor protein like color to enable inexpensive and cheap scale up.
Governance Policy and Actions
Equity And Automony
In line with the desire for to giving as many people as possible the opportunity to learn how to scale up thier bio-engineering projects the primary policy concern is promoting ongoing equity and autonomy.
Actions
The project can release all CAD designs, Documentation and, software is released with open licenses that allow people to use and extend. (Most Effective - 1 )
The project can ensure the bill of materials only includes items that are broadly available and/or have open licenses. (Minimally Effective - 3)
Fab labs and service manufactures can sign up to print and mail kits of 3d printed parts to people who don’t have access to 3d printers. (Minimally Effective - 3)
Schools and DIY labs can incorporate this into their curriculum or sponsor people to build them locally in order to expand the number of people who have experience with the process of scaling up synthetic organisms. (Mininimally Effective - 3)
An existing organization, like Neosynbio or Frugal Science Academy which support open and frugal biological tools, can sponsor the project and manage the licenses and copyrights. In addition to providing exposure, if the project is successful this allows the project to exist beyond a single person and prevents license and copyright changes that restrict access. (Most Effective - 1)
Biosafety
While a bench-top reactor does not introduce new biological risks it magnifies the ones that already exist in the bio-engineered organism, so a secondary policy goal needs to make be managing this magnified risk.
Actions
The project can provide documentation and training materials can reference existing training on bioethics and biosafety. (Moderately Effective - 2)
The project and labs can provide known safe demo projects that demonstrate the principles of scale up and optimization with minimal biological risk. (Minimally Effective - 3)
Schools and Labs can extend their existing bioethics and biosafety training to explictly discuss how to manage the magnified risk of scaling up a biological process. (Moderately Effective - 2)
Governments and regualtors can extend thier existing regulations and logging practices to cover specfic requirements around scaling up or just regulate edits with the assumption that scale up will happen (Moderately Effective - 2)
Homework Questions
Homework Questions from Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate for polymerase is 1 in 10^6 according to slide #8. Slide #10 indicates the human genome is 3.2 Gbp, so a single copy of the genome is likely to have ~1000 errors which is pretty high especially for fast growing cells that replicate once a day, since errors will accumulate with each replication. Biology deals with this by having many layers of error correction and handling beyond the already excellent ones built into the polymerase. One example is the Lamers et al work on MutS from the slides which correct higher level structural errors in the DNA. Beyond error corrections, there are also mechanisms that cause cells with serious errors to self-destruct or be marked for destruction by other cells. This fail-safe removes cells with serious errors from the population so that don’t replicate more.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Slide 6 indicates that the average human protein is about ~1000 base pairs and ~330 amino acids. While the exact number of alternate codings depends on the specific amino acid sequence of the protein, on average each amino acid has ~3 codons that map to it (64/21), so there ~3^330 alternate codings for an average human protein. In practice all of these encodings may not actually work because at the end of the day different codons are still physically and chemically different from other codons which can create differences in the structure of the DNA and translated RNA as indicated in later slides. For example structural differences in RNA chains can impact ribosome translational efficiency, which means a given DNA chain might code for the same protein but make too much or too little of that protein for the organism to survive.
Homework Questions from Dr. LeProust: [Lecture 2 slides]
What’s the most commonly used method for oligo synthesis currently?
From the Jacobson slides and the timeline in the LeProust slides it appears that while the details of the chemistry, level of automation, and miniaturization has been massively improved over the years most olgio synthesis methods are variants of an open loop chemical synthesis with a protection group .
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Also from the Jacobson slides it the error rate for chemical synthesis is 1 in 100 base pairs. That error rate would make it very hard to go much beyond a couple hundred base pairs without getting an error.
Why can’t you make a 2000bp gene via direct oligo synthesis?
2000 base pairs is much too large for a 1 in 100 error rate. At 2000 bps, even if you sequence the the different olgios produced and then try fo find and amplify a good olgio you have an almost zero chance of getting a good sequence of that length in the first place.
Homework Questions from George Church
I looked at question #1
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
I had to google “What is the lysine contingency” because I had forgotten that part of Jurassic Park. Looking at Wikipedia - Essential Amino Acids it looks like there are 9 amino acids that animals absolute can’t synthesize and 6 more that can’t always be synthesized in sufficient amounts. I was not able to reconcile this vs the 10 amino acids in the question. Independent of that it is clear that the “Lysine Contigency” makes no sense as a form of bio-safety for animals, since all animals already suffer from the lysine contingency and do just fine getting Lysine from the food they eat. Even knocking out the ability to produce a non-essential amino acid like Alanine would not help containment unless the dinosaur species has such a narrow diet that it couldn’t survive any any modern wild plant or animal matter. In particular carnivorous dinosaurs that are happy to eat modern mammals (the ones you most want to contain) could get the full suite of amino acids from their prey. I used Google and their AI answer to verify that all amino acids survive stomach acid and digestion.
Extra Investigation for GRO:
While reading the George Church slides on Geneticially Recoded Organisms (GRO) and the fact that they are immune to viral infection “Swapped genetic code blocks viral infections and gene transfer”, I was driven to do some additional investigation into the risks using Google Gemini knowing how important viral infection is in microbial control in the wild. Some of the main prompts were:
“Isn’t immune to natural viruses a little dangerous since that is the main predator/cause of death of most microbes?”
“It seems like if they could escape and dump the auxotrophy gene they might have enough of a survival advantage (there are many slow growth bacteria in nature) that they could climb back up the fitness curve over time.”
“what is the pragmatic outcome advantage or technology that justifies this risk”
I need to do more investigation in this area, but my first impression is that the researchers involved are thinking deeply about the bio-safety risks and developing layered countermeasures. However many of those countermeasures are fundamentally dependent on responsible actors in a high trust world and it isn’t clear that in a low trust multi-polar world these safeguards will be sufficient? The same economic forces pushing in this direction (virus free bioreactors) may tempt people to bypass the fitness safe-guards for economic benefit. Once a virus immune GRO population becomes established in the wild it isn’t clear to me how you would eradicate them. Even if they are initially slow growing or not dominant in their niche, being immune to viruses is such an incredible fitness advantage that it seems likely GRO microbes will eventually climb the fitness curve and come to dominate all ecosystems. This might take thousands or even millions of years, but seems like a very bad outcome. I guess we would have to hope that natural virus evolution figures out how to bypass the GRO defense before then?
Week 2 HW: DNA Read, Write, and Edit
Homework
Part 0: Basics Of Gel Electrophoresis
Attended lecture and watched recitation video
Part 1: Benchling & In-silico Gel Art
Link to Benchling Project , not sure how can see this link I asked to join HTGAA group but doesn’t seem like my invite was accepted yet?
My drawing of an “E Gel Person” and associated enzymes in each lane are also in screenshot below
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
I don’t have access to these enzymes and DNA in my local makerspace lab.
Part 3: DNA Design Challenge
Protein Choice
For the frugal bioreactor project I thought it would be ideal to have an organism and protein combination that are easily detected to help people (including myself) learn how to do yield optimization experiments. To that end I am choosing a protein that has a easily visible color, so intensity of color can be used to judge protein expression. To find a list of proteins I
Asked Gemini “Are there proteins that have visible color and can be expressed by e coli?”.
Selected EforRed from the list, since it will probably be easiest to measure intentisity of primary colors
Found EforRead in FPBase though common alias is apparently eforCP. There is spectrum data on FPBase also which could be useful for calibration and detection.
Encdoe the protein as a DNA string. There are lots of tools to this, but for fun I just wrote python code using a mapping from RNA to Amino Acids I took out of some old Rosalind code that mapped RNA to proteins.
# Want to find a DNA encoding of a protein# This is RNA to DNA mapping from Rosalindprot_string="""UUU F
UUC F
UUA L
UUG L
UCU S
UCC S
UCA S
UCG S
UAU Y
UAC Y
UAA Stop
UAG Stop
UGU C
UGC C
UGA Stop
UGG W
CUU L
CUC L
CUA L
CUG L
CCU P
CCC P
CCA P
CCG P
CAU H
CAC H
CAA Q
CAG Q
CGU R
CGC R
CGA R
CGG R
AUU I
AUC I
AUA I
AUG M
ACU T
ACC T
ACA T
ACG T
AAU N
AAC N
AAA K
AAG K
AGU S
AGC S
AGA R
AGG R
GUU V
GUC V
GUA V
GUG V
GCU A
GCC A
GCA A
GCG A
GAU D
GAC D
GAA E
GAG E
GGU G
GGC G
GGA G
GGG G"""# Do the reverse mapping just let the last on mapped winprot_rna_map={x.split(" ")[1]:x.split(" ")[0]forxinprot_string.split('\n')}# skip spacesprot_rna_map[" "]=""print(len(prot_rna_map),prot_rna_map)my_protein="""MSVIKQVMKT KLHLEGTVNG HDFTIEGKGE GKPYEGLQHM KMTVTKGAPL PFSVHILTPS HMYGSKPFNK YPADIPDYHK QSFPEGMSWE RSMIFEDGGV CTASNHSSIN LQENCFIYDV KFHGVNLPPD GPVMQKTIAG WEPSVETLYV RDGMLKSDTA MVFKLKGGGH HRVDFKTTYK AKKPVKLPEF HFVEHRLELT KHDKDFTTWD QQEAAEGHFS PLPKALP"""my_rna="".join([prot_rna_map[l]forlinmy_protein])print("-----")print(my_rna)my_dna=my_rna.replace("U","T")print("-----")print(my_dna)
The result I got (out of the many possible encodings) was:
For a given protein string of amino acids there are many possible DNA codes that could generate that protein. While all these codes are equivalent in an abstract sense in reality the RNA machinery like ribosomes and T-RNA in each organism is differen and the physical characterstics of equivalent codons can be different which can impact an organism’s ability to actual translate the DNA into a protein. For example the organism could be more efficient with certain T-RNA encodings than others impacting the rate or expresion or an organism could give special meanings to sequences of codons that it doesn’t use in its own proteiens. To avoid this you want to optimize your encoding for expression in your chosen organism.
I tried to optimize my sequence on twist, but I got 404 whenever I went to their codon optimization calculator. Instead I googled for codon optimization calculator and used another free one, VectorBuilder to get below optimization. I used E. Coli as my organism and didnt’ specify any restriction enzymes to avoid:
I knew GC was ratio of those bases in the sequence which varies form species to species and can impact structure, but I didn’t know what CAI was so I googled it and learned it is the “Codon Adaption Index”, which is a measure for a specific codon of how likely your codon’s are to be the organism’s preferred choice in terms of T-RNA frequency.
Now What
I still have a lot to learn about how to express proteins, but at a high level I know with cell culture
Pick an organism to express, I will asumme it is a bacteria like E. Coli
Manufacture this DNA segment (with a service like twisted) in side a plasmid in a way that
The plasmid has some other trait like antibiotic resistance that we can use to select for bacteria that have taken up the plasmid
Some promoter that we can use to make sure our protein is expressed.
Use some kind of transformation method to get the plasmid inside of the bacteria
Select for bacteria that have the plasmid by culturing and then putting on antibiotic gel and selecting survivors
Culture and grow these bacteria at scale and triggering conditions (if any) for our protein to be expressed.
Part 4: Twist Synthesis Order
Followed the homework directions pretty closely here using the same prefix/suffix sequences they recommend even the purification histadines though I probably don’t need to extract my color protein? This is my linear sequence.
After that I followed the instructions to create a twist order, downloaded in genebank format, and imported plasmid into benchling
I used Gemini freely to understand Benchling and standard practices in DNA construction and editing. Prompts where:
“IN benchlig once I create a DNA sequence, can I add more DNA to it in benchling?”
“When I right click it asks me if I want to insert bases or parts. What is the difference?”
“When we talk about DNA sequences are we typically using the 5 to 3 or 3 to 5 strand? What is each called and which one is the primary thing we are editing and viewing in benchling?”
Is it best practice to separately annotate start and stop codons on their strands?
Part 5: DNA Read, Write, Edit
DNA Read
What DNA?
If I could sequence and DNA I would sequence Valonia Ventriscosa which is an where a single cell (multiple nuclei) that can grow to be centimeters across! My local DIY bio lab has just started working on doing synthetic biology with them and the first step would be to sequence the entire genome which hasn’t been done yet.
What Sequencing?
I think to assmeble the overall structure of the genome from scratch we need the longest possible reads, so we would want to use a third generation sequencing method like nanapore for that. Once we have the overall structure we might want to use a high-bandwidth sequencing like illumini to get better resolution and coverage, including how much variation there is between indviduals.
To get started we need a decent amount of DNA. This is roughly the amount in 1 million nuclei? We have estimated (assuming that a 1mm cell has ~1000 nuclei) that we need around 1000 1mm cells. Once we have the cells we would need to separate out the DNA from the rest of the cell.
Since the lecture didn’t go into much detail on prep with a nanapore I asked gemini “What kind of preparation do you need to use a nanapore sequencer”. The summary is:
Gentle separation of DNA so you keep the strands long
High purity (non phenols/salts)
Repair the DNA ends so taht they are blunt and have a single A overhang
Attach a motor protein to DNA to move strand through the membrane.
Setup the flow cell with buffer
Put your sample in
Again I didn’t know the output format so I asked Geminin “What is the raw output format of nanapore”. Summary is
The data is large (can be a terabyte)
Nanapore recrods the raw signal as electrical voltage changes as DNA goes through pore. The wiggles are stored in in binary files in either POD4 or FAST5 format
The DNA sequence implied from raw electrical is also output as FASTQ files
DNA Write
If I was going to synthesize DNA independent of editing I would encode wikipedia as DNA. In particular, it would be fun to encode the main text of the wikipedia page about DNA as a DNA strand. Using wc shell utility on a copy of the file I see it is about 64K bytes, which is something that could be encoded as DNA.
I checked with Gemini “What are the abilities and limitations of cell-free DNA assembly? How long can strands be?” and it looks like this size is possible to do with cell-free assmbly and pushing the upper end. I think from the lecture to pull this off you would need to synthesize small pieces (in the range of 500 bps) with olgio synthesis so that they have correctly overlapping ends and then repeatedly use Gibson assembly. If you arrange the reactions you can take advantage of the fact that fragments double in size to do this in 9 rounds of repeated glueing fromt the intiail 128 segements you start with
DNA Edit
If I had the capability to do any DNA edits I wanted, I would want to try to edit a Eukaroyte like algae or yeast do genetically support symbosis with some kind of hydrogen oxidizing bacteria. This would be a massive amount of editing to both the bacteria and the host, but would unlock the ability to directly produce interesting products at scale with only water, sunglight, and atomsphere to produce proteins at scale. For example it would enable things like celluar agiculture and space habitats to functions without existing agriculture inputs.
This is such a massive edit I am not even sure what technologies would work. On the bacteria side you would probalby want to use the edit techniques used in the construction of the minimal living cell though you would want to possibly cut even deeper because you want to make sure some of the bacterias fundamental functions are produced by the host. My earlier Gemini query on cell-free assembly volunteered that the Minimal Bacterial Genom project used a combination of cell-free assembly and yeast-based (TAR Cloning). I don’t know anything about TAR cloning so I asked Gemini “Can you tell me more about TAR cloning?” and it sounds like it the pefect thing to do large scale and complex construction and edits of DNA, so it is probably what would end up being used.
Week 3 — Lab Automation
Opentron Python Script Art
Basic Idea
The limited pixel resolution and colors of the petri dish reminded me of the old school bitmap monitors like the IBM PC that I grew up with. Also I wasn’t looking forward to guessing/figuring out a lot of pixel locations by hand, so I took a retro route and wrote some code to provide a terminal like API that let’s you specify a cursor location to write text to using a specific bitmap font and color.
Result
The code to turn the petri dish into a screen was pretty straight-forward to do even by hand. I did bunch of experiments of what I should do with the API and ended up making a logo for my local bio makerspace:
With the screen API drawing this log is only a few lines of python
I think AI could have done this really quickly but keeping with the retro theme I did things by hand in my colab notebook. Even by hand this wasn’t too bad and ended up being about 100 lines of python code for the logic.
Encoded the bytes as dictionaries and binary numbers (so you can sort of see the shape), where highest value digit is upper leftmost digit
Code supports screens with different bitmap sizes.
The API uses (row, line) numbers to specify location like an old-fashioned terminal instead of (x,y).
Since the petri dish is a circle I made (0,0) the center row and line and used negative numbers for rows to left and lines below the center
The number of rows and lines a petri dish can fit depends on the size of the font and the physical pixel separation which are specified when the Petri Screen is created.
I experiemented with a bunch of different pixel separations. I ended up using 1mm, but not how this will actually look in real life.
The python code itself is NOT optimized for speed and uses string operations to manipulate the bytes.
The use of the robot is optimized because the screen computes all of the pixels before rendering begins, so
Uses minimal number of tips
Uses minimal amount of fluid
The move path is also near optimal because the tip is moved in a left to right scan for each color across the entire screen
There are a lot of sources for bitmap font data all of which use a different format. I went with class IBM 8x8 font data available in this projects header files. I converted this to my format using some simple search and replace and then running this python code outside of the the notebook.
I wasn’t able to make my node’s office hours and didn’t get a reply to if and how we are supposed to run my code, so I wasn’t able to actually run.
Post-Lab Questions
Find and Describe an Opentron Paper
I read this paper Environmental modulators of algae-bacteria
interactions at scale. This paper explores the dependence of how autotraphs and hetrotrophs interact as a combined system under various environmental conditions. The space of variables for a two organism system under a variety of environmental conditions is large (~225K), so massively parallel automation is essential. The key technology in this space is to observe nano-liter size well droplets in a microfluidic system using fluorescent bar codes to identify the conditions in each droplet and to also fluroescent intensity to measure protein growth. Given the small scale, number of possibilities, and precision required the solutions used to create the droplets where mixed by an opentron-2 robot (STAR section page e2).
Lab Automation for Final Project
My main current proposed project is building a frugal benchtop bioreactor, which is more along the lines of trying to bring simple automation and sensor control to a wider audience. Given this it isn’t clear how I could use an opentron? Even if I do another project it also isn’t clear that as a remote comitted listener I would have access to either opentron or lab automation in a way that I could do a project?
Final Project Ideas
1. Frugal Benchtop
This was what I outlined in homework #1. Would try to include demo projects with e. coli and/or yeast that have been edited to express visible spectrum colors
2. Non-sterile simple genetic modification
As an extension of sourdough in to create a 2-species system that supports genetic modification of either microbe. Sourdough environment already selects against most competitors.
3. Edit yeast or algae to symbosis with HOB
Not even realy sure how to go about this because it is a massive edit.
Extra Node Specific Questions
I imagine AI could easily answer this question, but I intentially tried to just use the notes that were sent out and my basic understanding of howo things to work to see what I could guess without help.
1. If we were given a random segment of 100,000 bases, how would we determine if it is encoding for eukaryotic or prokaryotic genes? Could we find specific “parts” - promoters, operators, enhancers, silencers?
Assuming it is protein coding section of DNA I think you would have a decent chance of finding enough patterns to make good guesses about the structure and encodiongs
Prokaryote vs Eukaryote
Some things you could look for
Circular vs linear
Ribosome Binding Sequences
Origin of Replications
Specific Parts
If you can identify patterns that you think are potential ori and RBS then you can use those guesses to orient on the strand and look for
Between Ori and RBS sequence: Common promoters and operators for the domain you think it is
After the RBS: Look for start codons and stop codons. Do the stop codons seem far enough away from start codon to encode a reasonable size protein.
If you have multiple potential RBS sites and resonable stop codons, you can than iteratively look for more promoters and operators between stop codon and the next potential RBS.
2. Be able to explain why you chose your sequencing method for HW #2 and what other options you considered. Be able to explain how you would synthesize a particular piece of DNA - is it all in one piece, or are you assembling several synthesized parts together? What editing methods did you consider, and how would you confirm your DNA editing approach worked? Basically, be able to discuss your answer to this question.
3. What published paper using Opentrons are you analyzing for HW #3? In discussing with your peers, have researchers generally approached this tool for similar uses, or for vastly different fields?
See above.
Week 4 HW: Protein Design Part I
Part A: Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Why are there only 20 natural amino acids?
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Can you discover additional helices in proteins?
Why are most molecular helices right-handed?
Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Design a β-sheet motif that forms a well-ordered structure.
Okay I am going to take a first pass through here just going off the lecture, wikipedia, and background knowledge I already have and then go back and try with AI assistance for the ones I have no answer for.
A Dalton is another name for atomic mass unit. $6\times1023$ hydrogen atoms/dalton/atomic massunits is roughly $1g$ of mass, so $1g$ of amino acids is roughly $6\times1021$ amino caid molecues and 500g of meat should contain at most $3\times1024$ amino acid molecues. In reality, a good fraction of the mass of meat is water, fat, and other non-protein so the number will be less than that, probably between 10-30% of the max depending ont he meat involved, which gives a range of $3\times1023$ to $1\times10^24$ molecues.
The state of being a cow is a complex relationship between a cow’s cells. Being a cow cell is a complex relationship between the DNA, lipids, and proteins in that cell as well as the cell’s history and its relationship to other cow cells. IN other words being a cow is a delicate state. Eating a cow (especially if we cook it first) involves destruction of the cells, relationships, and molecues breaking them down into component parts at a molecular level, in particular protein is broken down into its component amino acids so all the relationship,s structure, and patterns are lost and re-assembled into the patterns and structure of whatever is eating the cow by the machinery of the organisms and its cells (plus the injection of energy). You can acquire the molecular shadow of what you eat in the form of the isotope concentration of your components, e.g. if you eat at lot of corn (or things that eat corn) you will have the isotope ratio of a C4 plants like corn.
Not sure there is a strict answer to this, because it seems like there is something of a historical contigency here. In fact my past seemed to indicate that different people cite differen numbers of “natural amino acids”, e.g. wikipedia says 22 amino acids instead of 20. Given that I think the best answer is something like early life must have existing in an environment where the current naturally occuring amino acids were being manufactured by some abiogenic process and ended up being incorporated into the structure of early proto-life. In addition to requiring that the amino acids was created by some abiogenic proces in decents amounts, the current amino acids of life are also the ones that life figured out how to internally synthesize. I can imagine there may have been a commonly occuring abiogenic amino acid that some proto-life started using, but proto-life didn’t figure out how to self-synthesize. As the abiogenic source of a hard to biologically synthesize amino acid waned, proto-life that used that amino acid would have been very strongly selected against, so even a common abiogenic amino acid may not show up as current “natural” amino acid.
An amino acide is an organic compound that has both carboyxl and an amine group. This means that the to design an amino acid you can attach those groups to any organic/carbon backbone. For example, you could take octane (eight carbon atoms in a chain with hydrogen atoms) and attach a carboxyl and amine group to the last atom in the chain to make an amino acid (octine?). It is hard to tell if this is a “new” amino acid because there are 500+ amino acids just in nature according to wikipedia without a web/AI search.
There are abiogenic processes that naturally create amino acids. One famous experiment put methane, nitrogen, etc in a jar and passed electricity through and ended up creating many organic compounds including amino acids. We have also detected amino acids and other organic compounds on remote comets/asteroids presumably created abiogenically by heating/cooling/light energy impacting on the frozen components like methane ice.
I have no idea, but seems like D should be right-handed?
I am not sure what this question is asking? Additional relative to what?
Again, I am not sure what this question is asking? I assume they mean most biological molecular helices, because I don’t know that molecular helices have a preferred direction in general. If it is biological, I guess this is because some organic compounds are chiral and biology (because of historical accident?) selects/builds only one chirality of that organic compound. The chirality then impacts the shape formed when repeating units bind together leading to helices with a certain direction also.
Not sure, but if I had to guess it would be hydrogen bonding between the parts of amino acids that are perpendicular to the direction of th sheet?
No Idea
No idea.
Ok, now I will check/redo using outside research and AI especially for the last questions. Below is a record of main prompts I asked Gemini:
“What governs whether the peptides bond 4 forward or to a completely different segment (beta sheet).”
“What can you tell me about which way the helix turns on how it relates to chirality and the direction that DNA helix turns?”
“What is protein tertiary structure and what governs it? Also hydrogen bonds?”
“Which R group interactions stack beta-sheets on top of each other?”
“What are amyloid beta sheets and how do they form?”
“How would I go about designing a beta-sheet motif and what does that even mean?”
“What are examples of amino acids that are part of the beta-sheet vs the flexible turn between strands (proline & glycine) or is it just that any amino basic except proline and glycine can be in the beta-sheet part of the strand? Or maybe even proline and glycine can be in the beta strand as long as there aren’t 3-4 flexible amino acids in a row?”
With this info I revise/add answers
My guess above was wrong. For amino acids while they are right handed chirality this apparently creates a left handed helix. Strangely enough for sugars a right hand chirality creates a right handed helix which is what I was remembering when I guessed.
Still don’t understand this question.
I think I have a better feeling for this now. Beta sheets are formed by the hydrogen bounds between adjacent amino acid chains. This formation of the beta-sheet with hydrogen bonds exposes the R-groups of the amino acids in the sheet in an up and down direction/pattern relative to the sheet. The R groups that are exposed goven the binding between sheets. For example, some amino acids have R groups that can allow hydrogen bonding between sheets. Apparenly the tightest binding between sheets happens when the R groups are such that they can expel the water between the sheets and sterically snap together because the exposed R-groups line up shape-wise and have the ability (hydrophobic) to expel water between sheets.
As far as I can tell amyloid proteins have an autocatalytic tendency to form the tight fitting beta-sheets mentioned in #9, so if some condition (heat, disease, genetics) leads to creation of 2-4 tight fitting beta-sheets in an amyloid-protein it will eventually capture other amyloid proteins and recruit them into the same tight-fitting beta-sheets. Since it is very hard to pry these sheets apart, these clusters tend to grow slowly over time intefering with normal function of protein and surrounding tissue.
Not sure exactly what we are looking for here still? A DNA/amino acid sequence? I do understand the basic structure of beta sheets to know that I can make a beta-sheet by picking runs of 5-10 amino acids with mostly bulky R groups, like valine or tyrosine joined together with a proline + glycine loops that allow the protein strand to bend back on itself so the bulky runs can hydrogen bond each other. If you put enough runs like this together you will get a very wide beta-sheet which will then tend to wrap around and make a barrel or cylinder. So if I wnated to “design” a beta-sheet I would just repeat pattern like TVTVTVTCPG a bunch of times?
Part B: Protein Analysis and Visualization
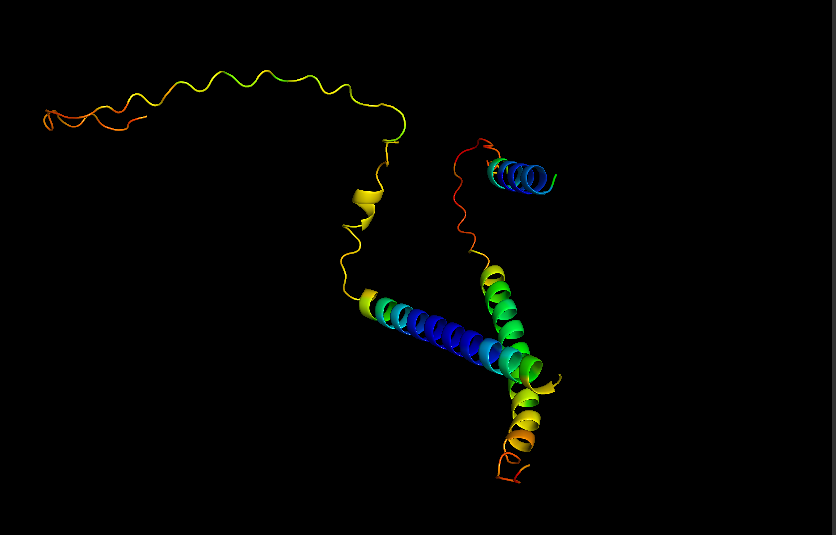
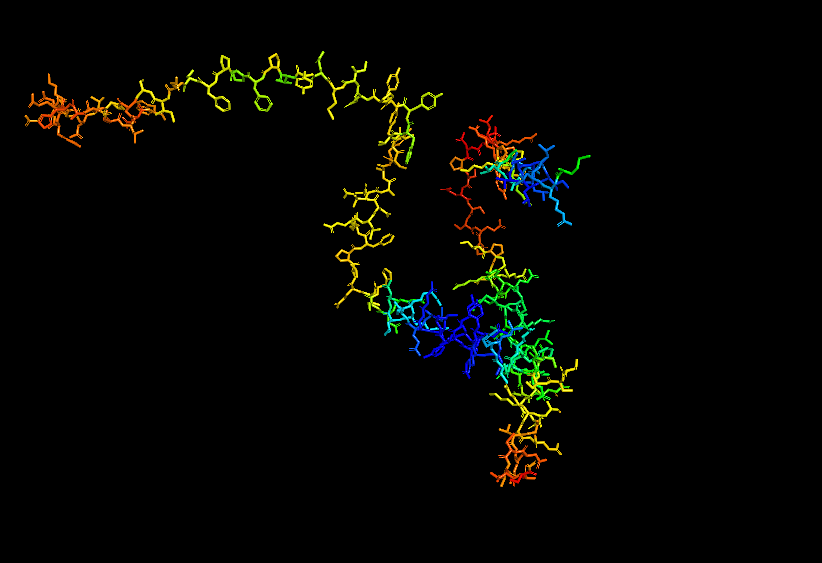
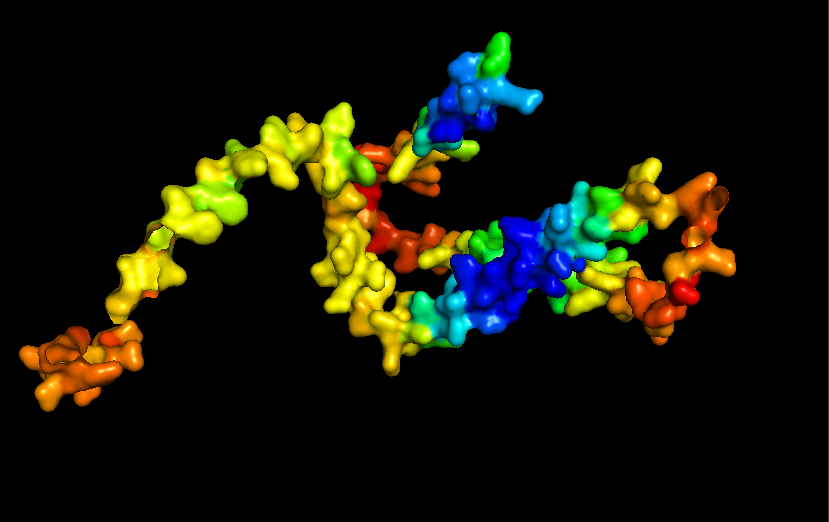
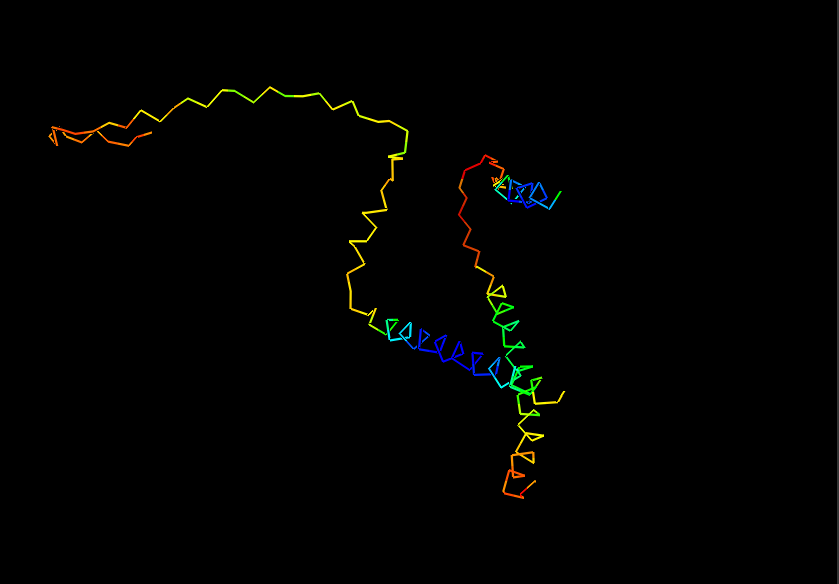
Briefly describe the protein you selected and why you selected it.
I chose casein which is the major protein in milk, because I like dairy products and it would be interesting to see how the common properties of something I interact with every day relates back to the protein structure and DNA sequence. I also remember seeing a video about using milk/caesin to make a fiber that you can make clothese from which points to a protein that potentially has a lot of uses.
Identify the amino acid sequence of your protein
It looks like there are actually several different casein protein ($\alpha S1, \beta, and \kappa$) variants that appear in most mammal milk. I think it will be interesting to compare them and see how they different so I will look at sequence for all of them in as well as comparing across species.
The closest homologs to this (not too surprising) were in the great apes (100 percent match). A little more suprising was how much change has happened in mammalian milk and that human alpha protein is closer to bats and seals than sheep (only 37 percent identical).
Not too suprisingly, interpro identified the family as alpha/beta caseins
Very suprisingly there don’t seem to be any solved crystallography for human alpha caesin in Uniprot, just alpha fold predictions. I checked this with Gemini via:
“I am slightly surprised that human casein alpha has not been crystallized and had crystallography done? I would have thought it would be a basic first protein people would do?”
The reply was that alpha casein is an intrinsically disordered protein which is hard to crystallize, which led me down a rabbit hole where I discovered that the unordered parts of our proteins where it is hard to predict structure are important in eukaryotes/larger animals for regulation etc, even though we don’t have great ways to understand them yet.
Similar results happen for cows, where the closest homologs are water buffalo and other hoofed ruminants like sheep are pretty close.
Not too suprisingly, interpro identified the family as alpha/beta caseins
Uniprot did have some electron microscope image that included cow casein, but this is sort of incidental because they are feeding casein into an e. coli motor that untangles tangled proteins like casein.
I used the Gemini prompt below to understand this:
“When looking at cow alpha casein I do seem electron microscope images where there are looking at E Coli on casein substrate: https://www.ebi.ac.uk/emdb/EMD-4623. What does that mean?”
Here we have the same issue as with the alpha version of casein, though there is a group PED studying intrinsically disordered proteins that are collecting additional modeling results about the entire structural ensemble/deformations and someone has modeling result based on a combination of alphflex and molecular dynamics simulations.
Here I was suprised to see that there are PDB entries for cow casein. There are mostly of small (rigid parts?) of the entire protein? There is one entry that claims to be the full protein sequence (PDB 7TTR), but going to that PDB entry (and verifying with Gemini) shows this is another case of casein be used as an input into a test of an untangling motor.
Using the foldseek links from Uniprot structure recommendations doesn’t seem to lead to any good anlogs or structureally similar
I ran clustal omega to see how sequences aligned across species too. Alpha Casein. There was pretty tight alignment at the start and then not much. Kappa proteins alignment seemed to have been spread out across the entire protein Kappa Casein
Alpha Casein Cross Species Alignment
Kappa Casein Cross Species Alignment
3D Visualization
I used desktop PyMol and liberal use of “gemini” to help with this (queries like “how do I color by acid base in pymol”)
Visualize in Different Modes
Cartoon for Human Alpha Casein
Stick for Human Alpha Casein
Surface for Human Alpha Casein
Ribbon for Human Alpha Casein
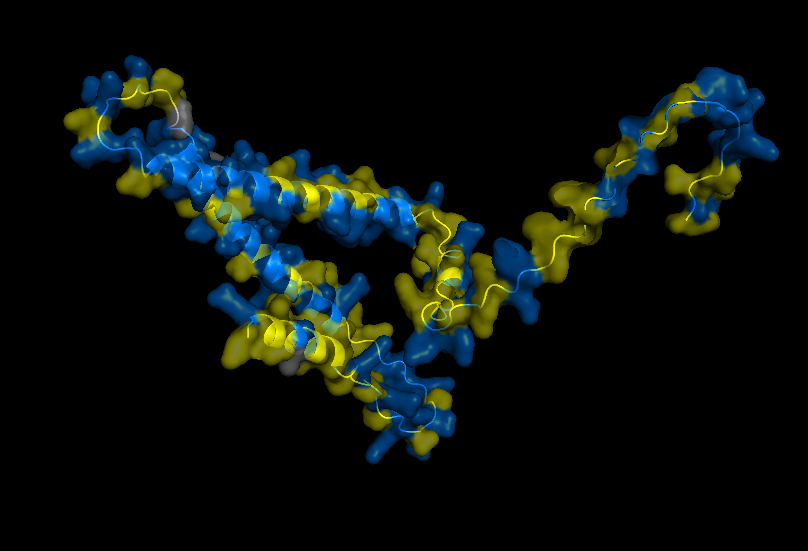
Visualize Secondary structure
Casein alpha has 4 alpha-helices on a single strand, 2 small ones on the end and 2 larger ones in the middle. The alpha strands have long unstructured runs between them, so they probably flop around a lot.
You don’t really need to color because you can see pretty clearly in the cartoon view above.
Visualizes Residues
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? THere doesn’t seem to be any well-defined sides/regions that I can see in this view. In fact they two kinds of residues seem to be well-mixed across the entire strand. Maybe this contributes to the inherent disorder of the protein.
Record of Gemini Prompts for Part B
What is the primary protein in milk? Caesin?
What is a protein homolog?
Where is the best place to look for protein homologs using BLAST?
Is there a standard way to identify “protein family”?
Any suggestions about how to search for structure information in the protein data bank if I have uniprot references? Searching for sequence or keywords in PDB doesn’t seem very fruitful
I am slightly surprised that human casein alpha has not been crystallized and had crystallography done? I would have thought it would be a basic first protein people would do?
So seems like alphafold predictions for intrinsically disordered may not be very good since we don’t have input data?
What percentage of human proteins do we think are intrinsically disordered?
So it what are the best avenues/tools for understanding the dark proteome in animals like humans? It seems like we are missing a lot of info about how proteins function and how we can design/understand them in that context.
So the micelles structure that casein forms might be mirrored as membranes organelles/reaction chambers inside a cell?
When looking at cow alpha casein I do seem electron microscope images where there are looking at E Coli on casein substrate: https://www.ebi.ac.uk/emdb/EMD-4623. What does that mean? It sounds like the casein is incidental here?
What is a structure classification family for proteins.
There are foldseek links in the structure entries in uniprot? What are those, and how do you use them?
I guess it can find partial matches? It gave me a few viruses with partial match positions and very low probability? Are those false positives?
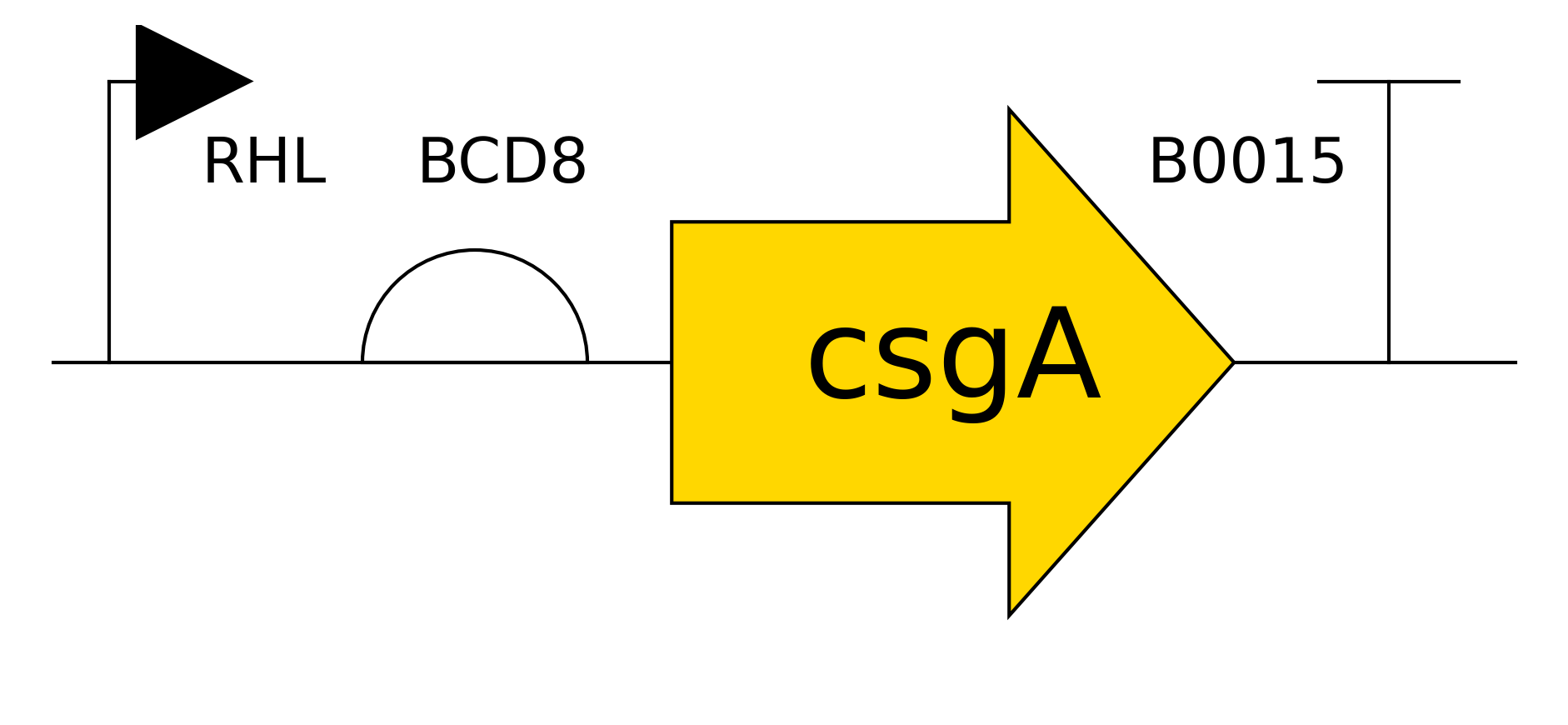
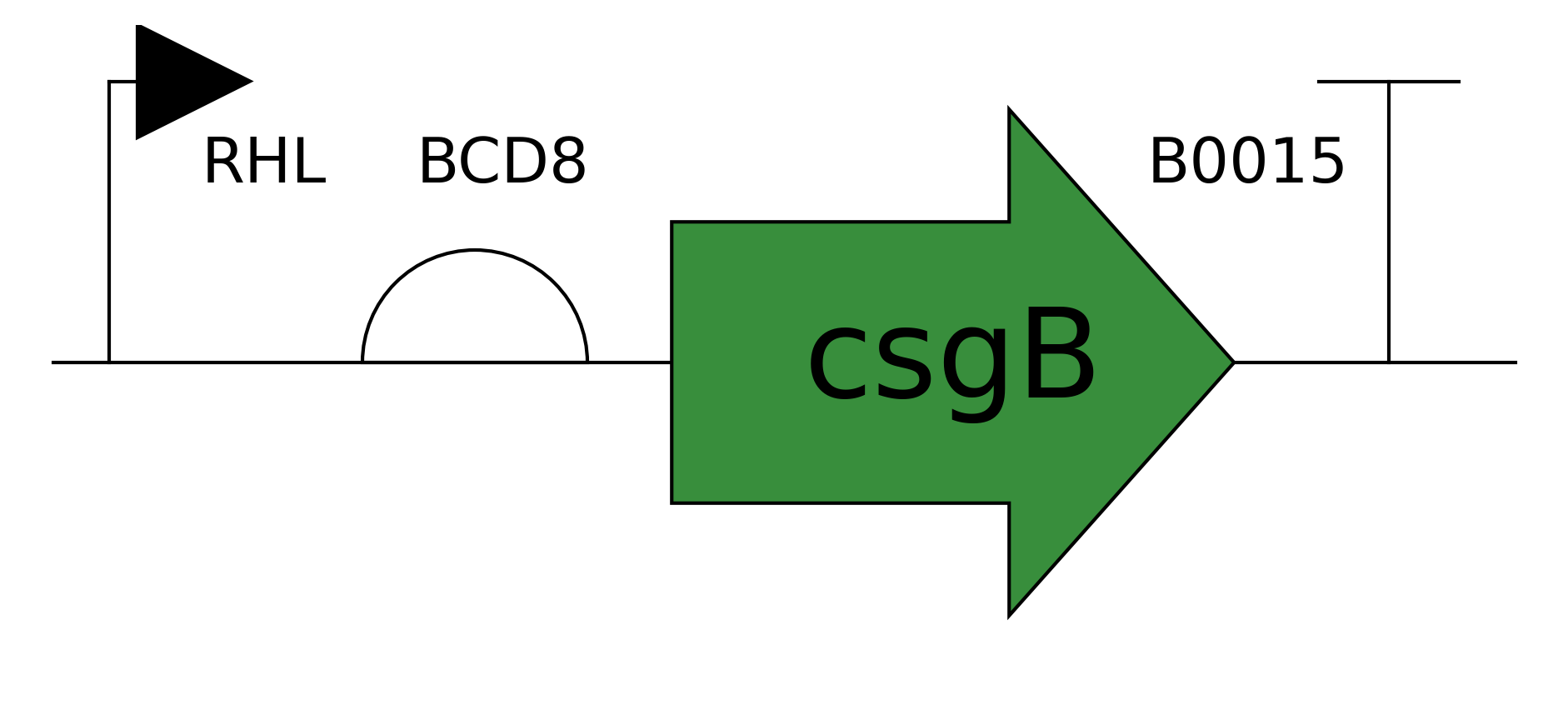
Running the ESM2 model in my copy of the Colab notebook for the Curlin csgA protein gave
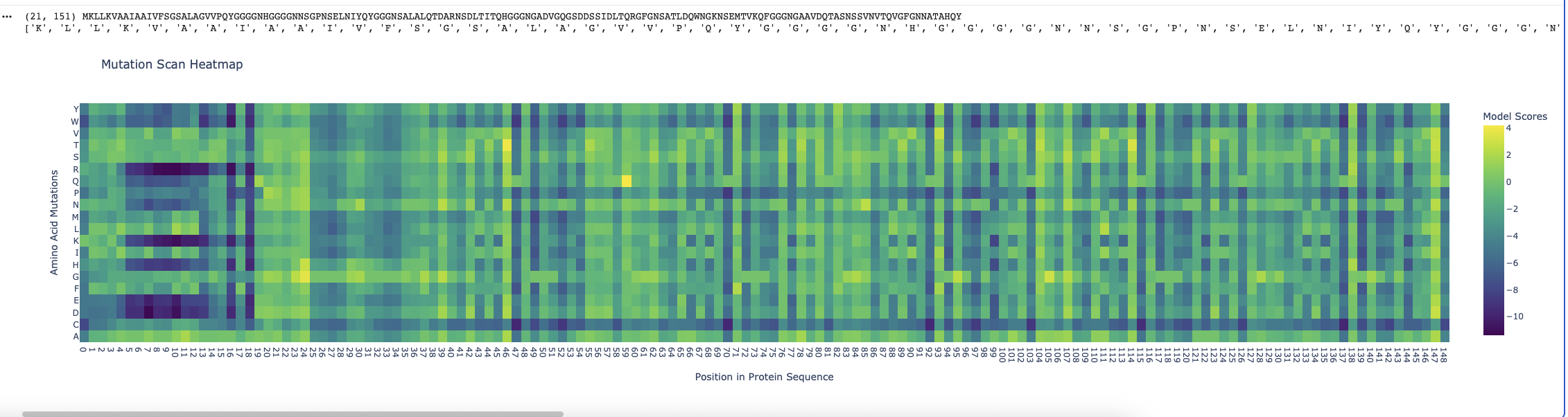
Deep mutation Scan for Curlin csgA
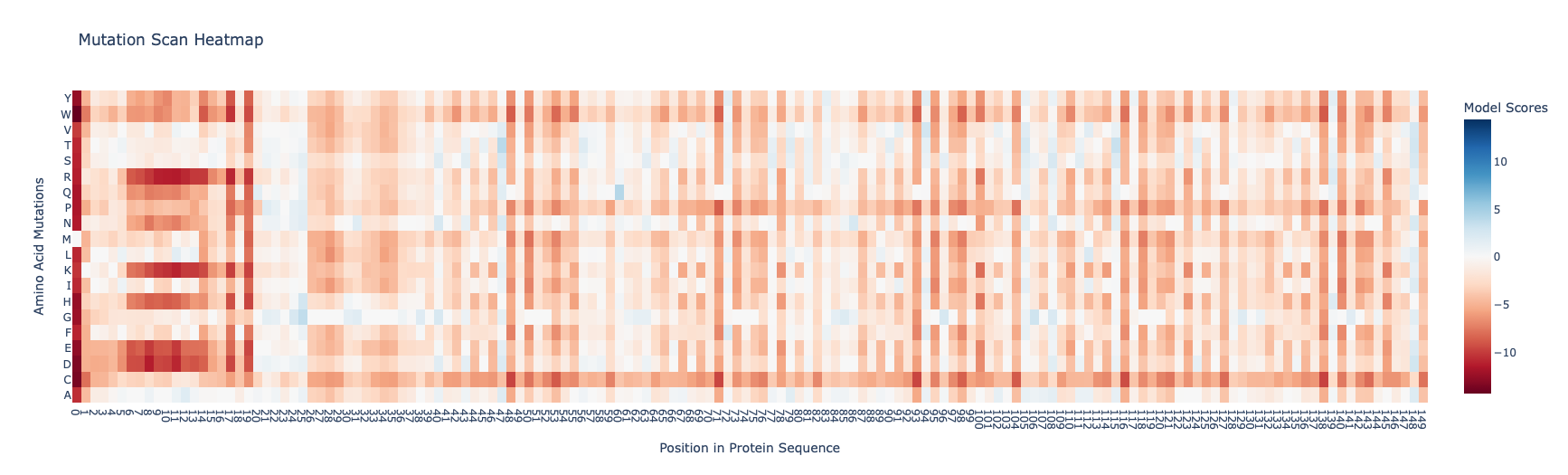
I found the coloring of this heatmap to be hard to understand because it is all shades of green instead of different colors for positive and negative mutations. Visually seeing the “no mutation” amino acids was especially hard. Also I noticed that there was a trimming problem and the first amino acid was missing from the analysis. Given this I changed the color theme and trimming to get this version
Revised Deep Mutation Scane for Curlin csgA
In this image the no mutation parts of the map are white, “more typical” mutations are blue, “unusual/bad” mutations are red as determined by the model score.
Some observations:
Some amino acids are relatively interfchangable, for example in most places where the is a L (leucine) and I (isoleucine) is almost as good.
There are periodic Qs (glutamine) spaced roughly equally far apart (48, 71, 93, 104, 116, 138) which have no other reasonable option which indicates they are crucial to the structure and function of the protein. The amide group in the gluatamine may be important for the links between the beta sheets.
Likewide there are perodic Gs (glycines) spaced between the Qs which also don’t have good replacements. These may be important for the turns between the beta sheets in the protein.
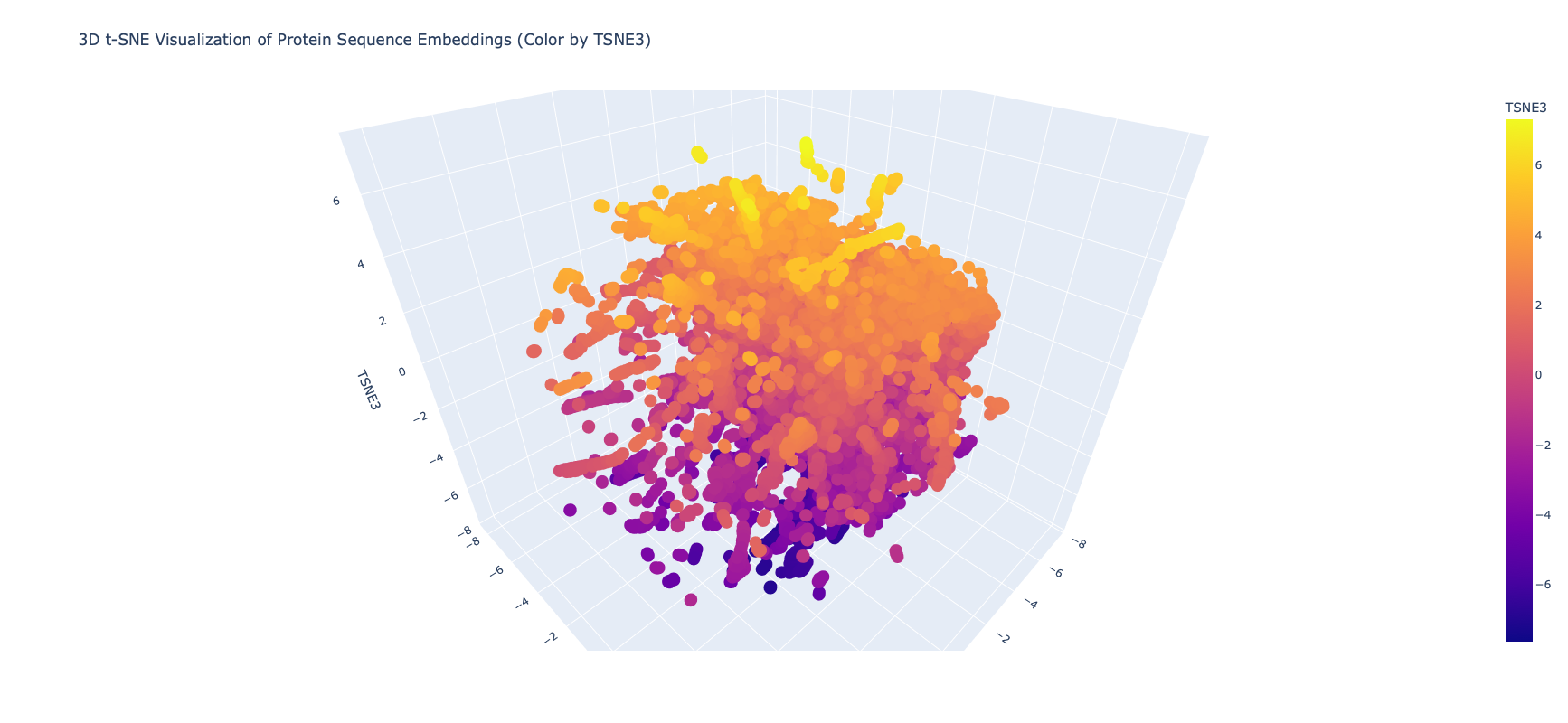
Latent Space Analysis
I edited the script in the notebook to include my protein and then did the latent space analysis to get the below graphs
Top Level Latent Space
It was extrememly hard to see or find my protein, even when I knew the coordinates, because of the number of proteins and the “jump to nearest” hover feature of plotly. Given this I asked Gemini for some code on how to zoom plotly and looked at smaller neighborhoods
Close in Latent Space Zoom
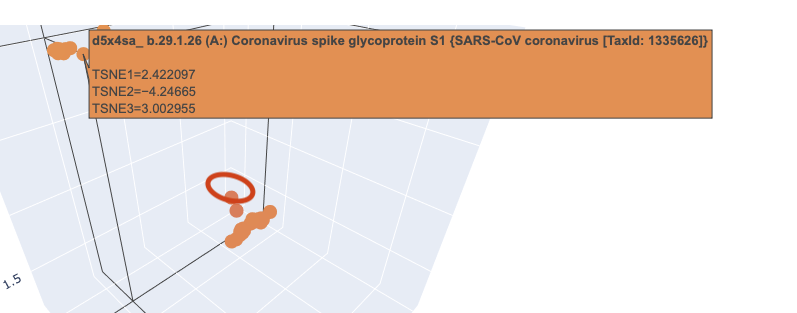
When I zoomed in to a unit cube centered on my protein (circled in red) at (1.9201405, -3.6876564, 2.1404576) there was only one other protein in that window, which was a collagen adhesin. So it is interesting how isolated and unique curlin was. This may reflect the library we are comparing against and the fact that it hard to do xray crystallography on these kind of proteins?
Close in Latent Space Wider Zoom
Given there was so few other proteins in the unit cube, I zoomed out to cube of size 2. This cube had more proteins (including theh covid spike protein?) but there didn’t seem to be any clustering that pointed to curlin even on this wider scale.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2: Protein Folding
Fold your protein
Refold
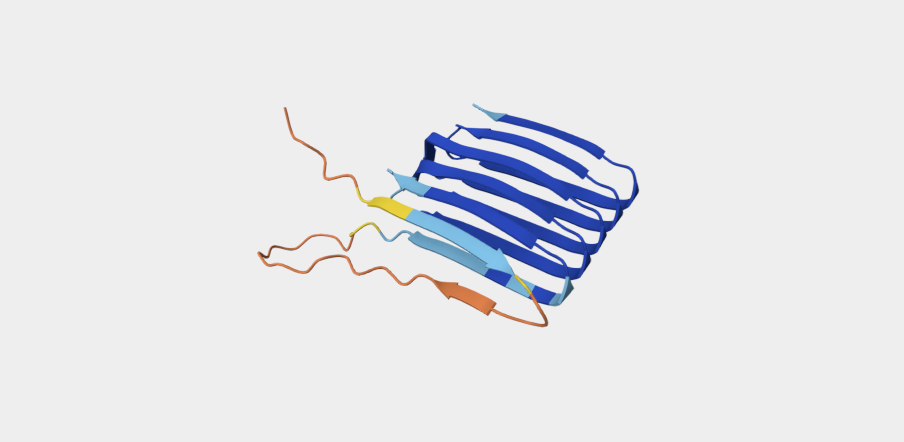
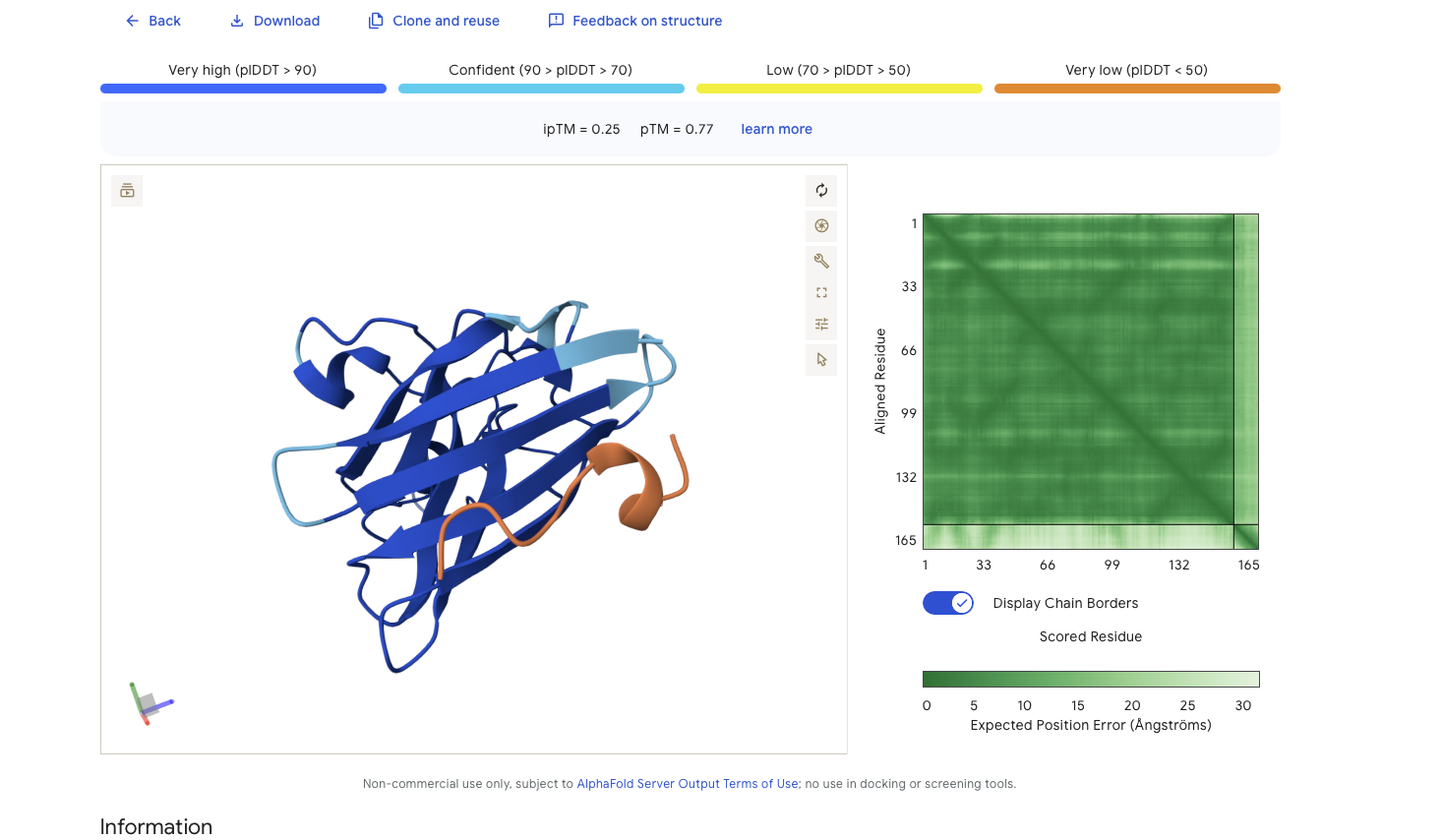
Refolding my protein with the original sequence gives something very close to the PDB alphafold (this sequence doesn’t have a good crystallygraphy PDB becuase it doesn’t crystaliize unfiormly).
Original PDB alphafold
It is probably not too suprising these are very close since the alpha fold picture is also an inferred fold based on similar training data.
Mutate and Fold
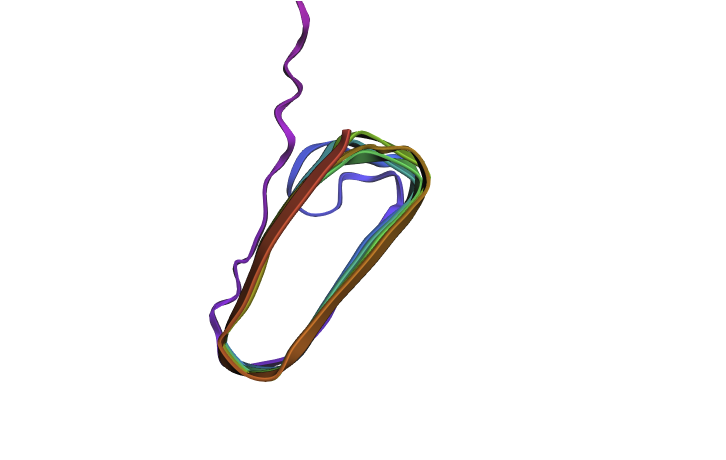
Since there is no golden crystallography source here I wanted to see if the “bad” mutations identified in the first part have a dramatic inferred impact on structure. I mutated my squence by changing all of the “crucial” glutamines (Q) to W which was the “worst mutation” possible. This did have an impact on the structure (distorting the beta sheets when viewed from the side) but it wasn’t as dramatic as I was expecting. Maybe this is because the different amyloid protetins needs to stick together closely and these distortions mean that don’t fit together as well?
Mutated Curlin Refold
Original Curlin From Side
with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3: Protein Generation
The code uses PDB but I don’t have entire sequence in PDB so I used large EM fragment (PDB 8ENQ) (which
The refhold of the new protein basically looks the same as before.
Part D: Group Brainstorm on Bacteriaphage Engineering
We met to brainstorm on Feb 28 and are documenting results in this google doc
Part E: William and Mary Node Questions
Be prepared to answer/discuss all 11 questions posed by Dr. Zhang. We will choose the most interesting ones to discuss in class.
Be prepared to discuss the phage literature reading.
A discussion of the phage literature will lead into our main discussion point: please be prepared to address and discuss the “big picture” question: how to apply these protein analysis tools to engineer a better bacteriophage. Please develop specific ideas for discussion.
M
GVVPQYGGGGNHGGGGNNSGPNSELNIYQYGGGNSALALQTDCRNSDLTITQHGGGNGADVGQGSDDSSIDLTQRGFGNSATLDQWNGKNSEMTVKQFGGGNGAAVDQTASNSSVNVTQCGFGNNATAHQY HHHHHH
GVVPQYGGGGNHGGGGNNSG
It appears that the model like its peptides betters than the known binder.
Actually when I wne tot the next step I finally noticed every single one of the generated peptides had an X/unknown which alphafold barfs on. I fixed this by adjusting the top_k value to be top 9 to allow more choices besides X and got:
These are more suprising than the other ones (which isn’t too surprising given the wider latidute of to pick less likely amino acids), but still decent and similar to the known binding protein.
Part 2: Evaluate Binders with AlphaFold3
Running alphfold agains these binders give:
Known Peptide - FLYRWLPSRRGG Results
Alphafold is confident in the shape of protein. It is intersting while that while alphafold does identify it as binding to the N-terminus it doesn’t feel very confident about that binding 0.25
Peptide Binder 0 - WDNVGYAIYSGK Results
Alphafold is confident in the shape of protein. The binding certainity (ipTM) is only 0.62 also it is binding to the beta barallel not the N terminus.
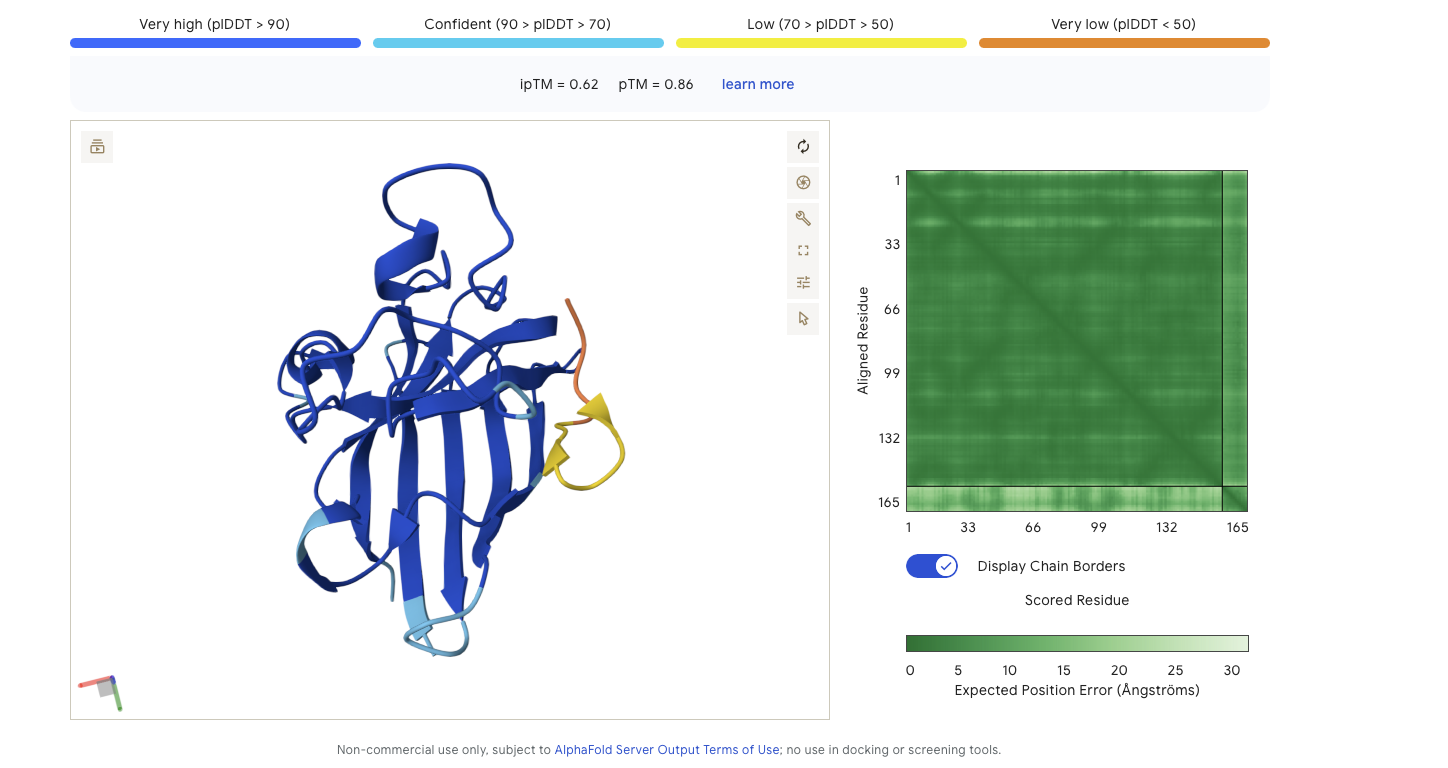
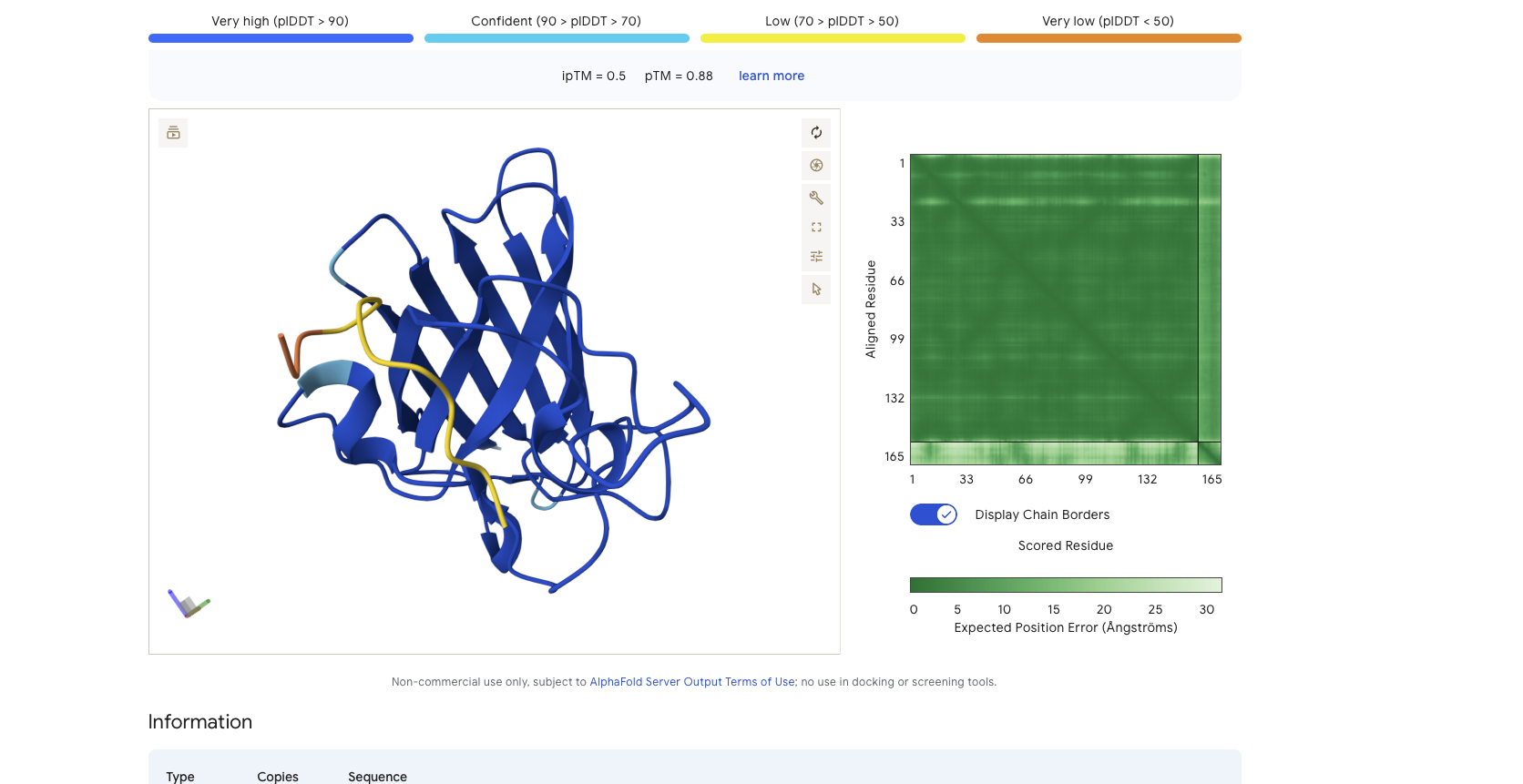
Peptide Binder 1 - HDWVGQGIDQGE Results
Alphafold is confident in the shape of protein but the binding certainity (ipTM) is only 0.5 also it is binding to the alpha helix not the N terminus.
Peptide Binder 2 - ESYYDQAVDQLE Results
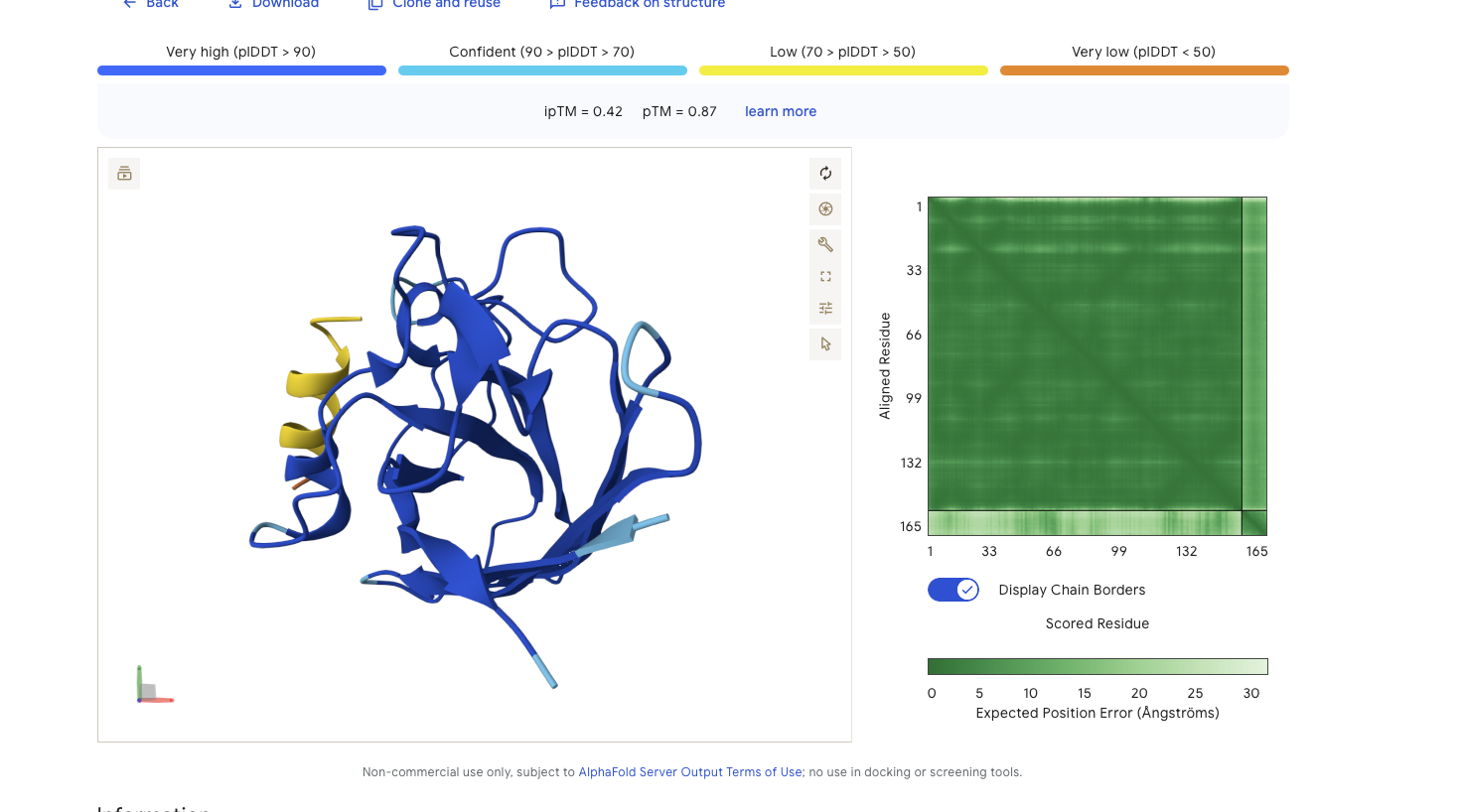
Alphafold is confident in the shape of protein. The binding certainity (ipTM) is low 0.42 and it is predicted to bind to the N-terminus.
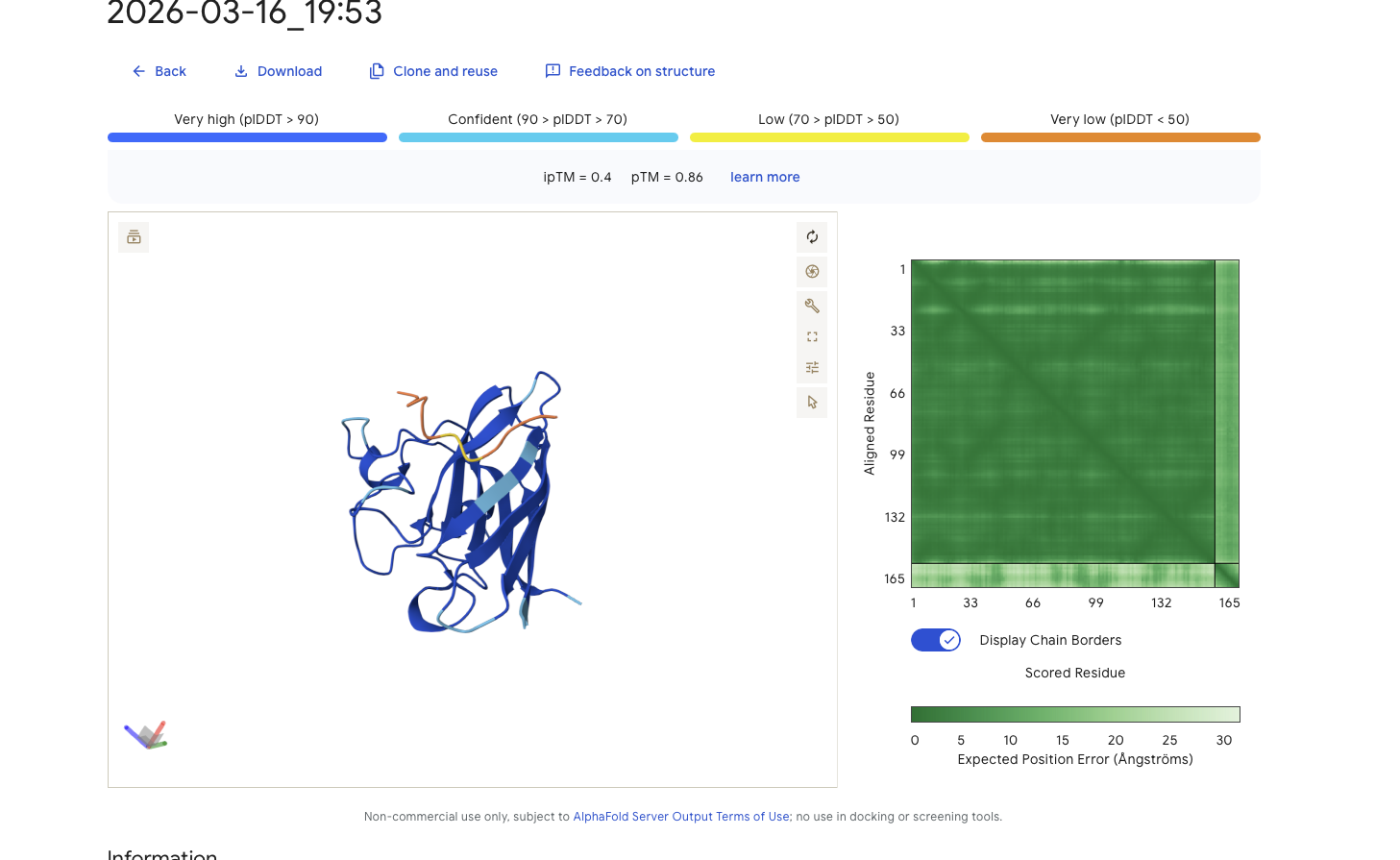
Peptide Binder 3 - VSYPGQVVGHLP Results
Alphafold is confident in the shape of protein. The binding certainity (ipTM) is only 0.4 also it is binding to the beta barallel not the N terminus.
Overall given that Peptide Binder 2 seems to be more confident in the binding to the N-Terminus than it is for the known peptide. Also looking at the grids for the various results it doesn’t seem like alphafold is really the confident for where the peptides are binding even for the known peptide?
Part 3: Evaluate Peptide Properties
Using Peptiverse I got the following for the basic properties
Input
Property
Prediction
Value
Unit
FLYRWLPSRRGG
💦 Hydrophobicity (GRAVY)
Non-hemolytic
1507.7
Probability
FLYRWLPSRRGG
🔗 Binding Affinity
Weak binding
5.962
pKd/pKi
FLYRWLPSRRGG
📏 Length
12
aa
FLYRWLPSRRGG
⚖️ Molecular Weight
1507.7
Da
FLYRWLPSRRGG
⚡ Net Charge (pH 7)
2.76
FLYRWLPSRRGG
🎯 Isoelectric Point
11.71
pH
FLYRWLPSRRGG
💦 Hydrophobicity (GRAVY)
-0.71
GRAVY
WDNVGYAIYSGK
🩸 Hemolysis
Non-hemolytic
0.050
Probability}
WDNVGYAIYSGK
🔗 Binding Affinity
Weak binding
5.994
pKd/pKi
WDNVGYAIYSGK
📏 Length
12
aa
WDNVGYAIYSGK
⚖️ Molecular Weight
1372.5
Da
WDNVGYAIYSGK
⚡ Net Charge (pH 7)
-0.24
WDNVGYAIYSGK
🎯 Isoelectric Point
5.83
pH
WDNVGYAIYSGK
💦 Hydrophobicity (GRAVY)
-0.46
GRAVY
HDWVGQGIDQGE
🩸 Hemolysis
Non-hemolytic
0.043
Probability
HDWVGQGIDQGE
📏 Length
12
aa
HDWVGQGIDQGE
⚡ Net Charge (pH 7)
-3.14
HDWVGQGIDQGE
🎯 Isoelectric Point
4.31
pH
HDWVGQGIDQGE
💦 Hydrophobicity (GRAVY)
-1.18
GRAVY
HDWVGQGIDQGE
🩸 Hemolysis
Non-hemolytic
0.043
Probability
HDWVGQGIDQGE
📏 Length
12
aa
HDWVGQGIDQGE
⚖️ Molecular Weight
1340.4
Da
HDWVGQGIDQGE
⚡ Net Charge (pH 7)
-3.14
HDWVGQGIDQGE
🎯 Isoelectric Point
4.31
pH
HDWVGQGIDQGE
💦 Hydrophobicity (GRAVY)
-1.18
GRAVY
VSYPGQVVGHLP
🩸 Hemolysis
Non-hemolytic
0.024
Probability
VSYPGQVVGHLP
📏 Length
12
aa
VSYPGQVVGHLP
⚖️ Molecular Weight
1252.4
Da
VSYPGQVVGHLP
⚡ Net Charge (pH 7)
-0.18
VSYPGQVVGHLP
🎯 Isoelectric Point
6.71
pH
VSYPGQVVGHLP
💦 Hydrophobicity (GRAVY)
0.30
GRAVY
ESYYDQAVDQLE
🩸 Hemolysis
Non-hemolytic
0.052
Probability
ESYYDQAVDQLE
🔗 Binding Affinity
Weak binding
6.105
pKd/pKi
ESYYDQAVDQLE
📏 Length
12
aa
ESYYDQAVDQLE
⚖️ Molecular Weight
1459.5
Da
ESYYDQAVDQLE
⚡ Net Charge (pH 7)
-4.15
ESYYDQAVDQLE
🎯 Isoelectric Point
4.05
pH
ESYYDQAVDQLE
💦 Hydrophobicity (GRAVY)
-1.22
GRAVY
Checking the binding affinity separatrely it doesn’t seem like any of them bond strongly including the known binding peptide.
Input
Property
Prediction
Value
Unit
FLYRWLPSRRGG
🔗 Binding Affinity
Weak binding
5.962
pKd/pKi
WDNVGYAIYSGK
🔗 Binding Affinity
Weak binding
5.994
pKd/pKi
HDWVGQGIDQGE
🔗 Binding Affinity
Weak binding
5.216
pKd/pKi
ESYYDQAVDQLE
🔗 Binding Affinity
Weak binding
6.105
pKd/pKi
VSYPGQVVGHLP
🔗 Binding Affinity
Weak binding
5.524
pKd/pKi
None of my peptides (and even the known binder?) seem like great candidates. Not sure if I am doing something wrong? Given this I guess I would go with ESYYDQAVDQLE for further work since it seems to be the closest binding candidate.
Part 4: Generate Optmized Peptides
I made a copy of the notebook and tried to do the optimization computations there. Unfortunately I ran into two problems I wasn’t able to resolve:
I wasn’t able to get the recommended GPU and could only get a T4
I repeatedly got (what appears to be an off by one error) pretty deep in the moPPit code. I tried running with a bunhc of different parameters. I did determine that the probably seems to be with something indexed by the sample parameters since the error message changes when I change the sample count. The error I am getting is below:
Target Motifs: [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12]
tensor([ 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12], device='cuda:0')
Some weights of EsmModel were not initialized from the model checkpoint at facebook/esm2_t33_650M_UR50D and are newly initialized: ['esm.pooler.dense.bias', 'esm.pooler.dense.weight']
You should probably TRAIN this model on a down-stream task to be able to use it for predictions and inference.
Some weights of EsmModel were not initialized from the model checkpoint at facebook/esm2_t33_650M_UR50D and are newly initialized: ['esm.pooler.dense.bias', 'esm.pooler.dense.weight']
You should probably TRAIN this model on a down-stream task to be able to use it for predictions and inference.
NFE: 0: 0%| | 0/0.9990000128746033 [00:00<?, ?it/s]Weight Vector: tensor([1., 1.], device='cuda:0')
NFE: 0: 0%| | 0/0.9990000128746033 [00:07<?, ?it/s]
Traceback (most recent call last):
File "/content/moPPIt/moppit.py", line 81, in <module>
x_1 = solver.multi_guidance_sample(args=args, x_init=x_init,
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/usr/local/lib/python3.12/dist-packages/torch/utils/_contextlib.py", line 124, in decorate_context
return func(*args, **kwargs)
^^^^^^^^^^^^^^^^^^^^^
File "/content/moPPIt/flow_matching/solver/discrete_solver.py", line 365, in multi_guidance_sample
guided_u_t, pos_indices, cand_tokens, improvement_values, delta_S = guided_transition_scoring(x_t, u_t, w, score_models, t, w, args)
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/content/moPPIt/flow_matching/utils/multi_guidance.py", line 69, in guided_transition_scoring
improvement *= importance[count]
~~~~~~~~~~^^^^^^^
IndexError: index 2 is out of bounds for dimension 0 with size 2
---------------------------------------------------------------------------
RuntimeError Traceback (most recent call last)
/tmp/ipykernel_1400/1413049073.py in <cell line: 0>()
38 ret = proc.wait()
39 if ret != 0:
---> 40 raise RuntimeError(f"moo.py failed with code {ret}")
RuntimeError: moo.py failed with code 1
Part B: BRD4 Drug Discovery Platform Tutorial
Part 1 Structural Predictions
Results
Compound
Binding Confidence
Optimization Score
Structure Confidence
Hit
0.76
0.26
0.90
Lead
0.44
0.23
0.88
JQ1
0.96
.044
0.98
Discussion
Does Binding Confidence increase as you move from hit to clinical candidate? What would
you expect, and why might it deviate?
I am not sure I understand the terminology but I am assuming that the order is Lead -> Hit -> Clinical Candidate (JQ1). As we go through this sequence I would expect binding condidence to increase in most scores. I can see situations where there could be experimental evidence that overrides the modeling results and perhaps give an inversion of scores?
Inspect the predicted binding pose for JQ1. Can you identify potential key binding
interactions.
It looks like JQ1 is binding in the top/interior of the a barrel made for the 4 helix barrel that is the primary teririary structure in the protein.
• Compare the Optimization Scores. How do the scores compare for JQ1 vs the Lead.
I got no results that had both an optimization score > 0.4 and binding score > 0.8 which I think means I got no “High Confidence” candidates. If I relax that to an high binding OR high optimization I get
2 compounds that binding scors of 0.8, with optimization scores of 0.27 and 0.34. Of these two SM-5S4NDA3E has the highest optimizatoin score and is my best candidate.
12 candidates with an optimization score of greater than 0.4. The three best candidates are (the rest of binding condience of less than 0.7)
SM-279VMWMP has binding confindence of 0.78 and optimization score of 0.4
SM-QAZ4MM4W has binding condidence of 0.77 and optimization score 0.45
SM-2F86NWEQ has binding condence of 0.72 and optimization score 0.42
JQ1 appears to be a much better candidate it has Binding Confidence of 0.99 and Optimization Score 0.55.
The binding againstBRD2 for my top 4 candidates was still quite strong, between 0.8 and 0.77 so these binders are not very selective.
Part C: Final Group Project: L Protein Mutants
I did a variation of option 3 which was to generate random mutations using the information in this spreadsheet on mutants. However inspired by Professor’s Sahas mention of the constraints because the virus is encoding other proteins in different frameshifts, I did a search for the point mutations that preserve the structures of the proteins and put the results in this spreadsheet. The entire journey and the code that generates the final results is captured in this python code which is the downloaded python code from the Marimo notebook I used. Given we are trying to engineer this protein for phage therapy using modified MS2 (I think) it seems like you need to take this into account and either select mutations from this list or do your mutational analysis on both the lysis protein and frameshifted protein at the same time, which it doesn’t seem like is being done? In particular, most of the mutations in the spreadsheet are not in the list of mutations that preserve the frameshifted proteins.
Week 6 — Genetic Circuits Part I: Assembly Technologies
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Endonucleases to eat away at the ends of all the DNA strands to expose single stranded ends
DNA Ligase to take matching ends and stitch them together
What are some factors that determine primer annealing temperature during PCR?
One factor is the proportion of G and C vs A and T because G and C have 3 hydrogren points they require more energy/higher temperature to break than the 2 hydrogen bonds linking A and T
3 dimensional structure like hairpins can also have an impact on the temper
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Restriction enzymes cut the DNA into a linear piece and most of them leave sticky ends which can be joined together by DNA ligase directly after they are cut which is simple because you can cut and join at the same time. The downside is that enzymes have specific recognition sites that you must use and engineer around. These sites are not that long and are often palindromic, which increases the chance of cuts where you don’t want them. On the other hand, the very fact that they recognition sites are limited makes standard like BioBrick and MoClo possible.
PCR uses custom primers and to walk the DNA in two different directions during the amplification cycles.
The primers are longer than most recognition sites and can be almost anything. This gives you a lot of flexiblity to custom match your DNA without having to look at specific sites.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
You need to make sure that your primer sequences are unique
You need to make sure your primer sequences line up so that the pieces get glued together in the order you want.
How does the plasmid DNA enter the E. coli cells during transformation?
Describe another assembly method in detail (such as Golden Gate Assembly)
Is a restriction enzyme method
Uses Type IIS enzymes which cuts to one side of the recognition site, in parituclar it doesn’t destroy the recognition site when it cuts.
Because the recognition site stays intact you can make sure that the piece you want to glue to doesn’t have the recognition site. This means that if your DNA glues back to the piece it was just cut from in stead of the piece you want it to glue to it will again be a target for the Type IIS enzyme that is being used.
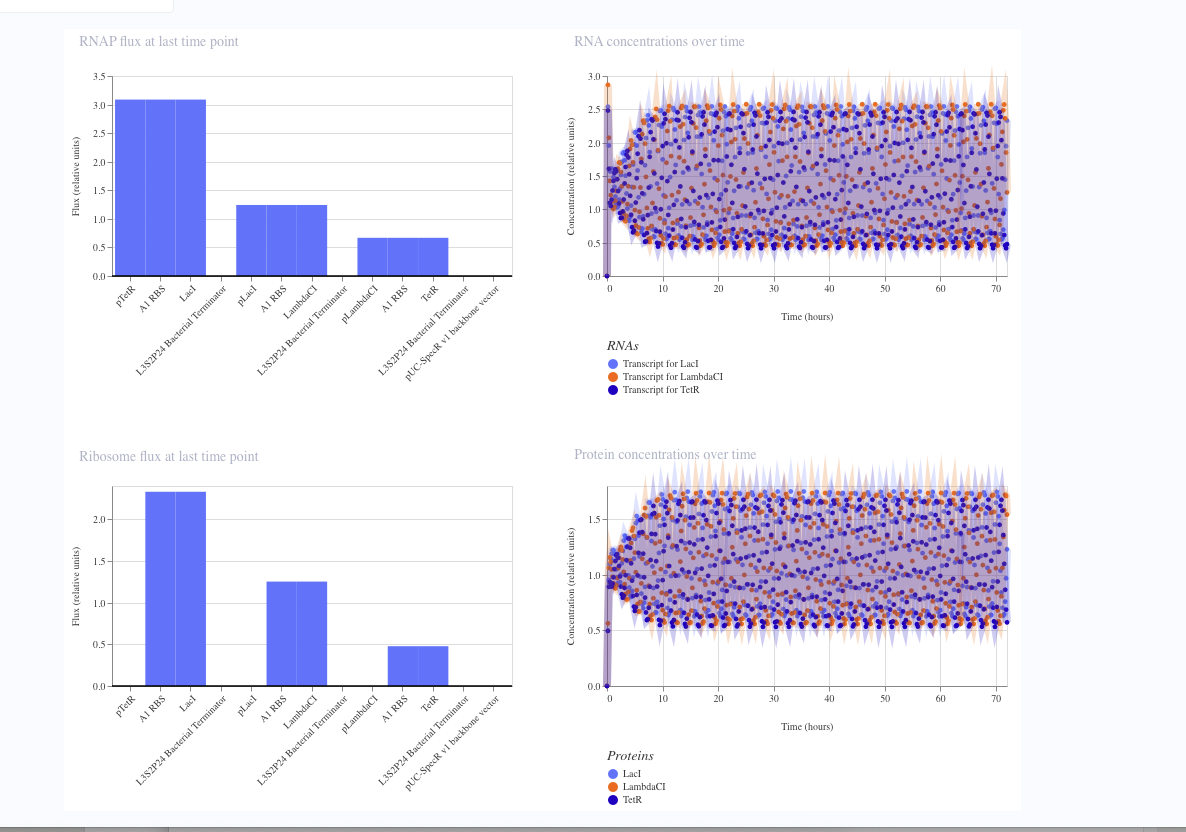
Asimov Kernel Modeling
I simulated a represillator in Aismov and got following results:
Week 7 — Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracelluar Artificial Neural Networks (IANNs)
IANNs Advantages
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
More closely mirror most natural genetic circuits where responses are naturally anolog.
In cases where you have analog input and output, which is many biological situations, the number of components required to transform to digital, do a digital computation, and then encode back in an analog output is often very large. This is not a big deal for digital circuits because we have figured out how to make each component so small and cheap that the flexiblity and debugability of boolean circuits is a huge win. For a biological circuit where even a 1000 components spread across many different cells is ambitious it is not clear this trade off is worth it in all cases.
Biological circuits are noisy and different proteins have different max and min expressoin levels, this makes it much harder to digitize the underlying analog signals than in a digital circuit where the highs and lows are same across the circuit and we can safely digitize by saying the signal is “near” high or “near” low without too much of a chance of cross over (though we still need to worry about transient states, which is why we often have a clock).
Application
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Not sure I have enough knowledge to come up with a really great application. Based on the lecture and what I know of simple neural nets I think they would be useful for siutations where you want to do multi-dimensional edge-detection. That is you have several different analog inputs, like oxygen level, light level etc and you want to have a variable response based on some complicated combination of them. For example, maybe an engineered bacteria that is added to canned/jarred food that fluoreces/turns color when conditions are favorable for botulism spores to reproduce.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Looking at the mycelium materials wikipedia page for background. It looks like there applications for mycellium leather replacements and also building materials.
A cool project with mycelium I remembered from past reading is using mycleium to grow materials/buildings on the moon or Mars that NASA is studying
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi have a very broad range of chemistries they have figured out that other domains can’t, i.e. decomposing lignin from trees.
Fungi naturally emit substantces as part of their external digestion which can make extraction easier and more scalable.
Fungi are eukaryotes so they can do complex post-translational modfifications that bacteria can’t, which could be needed in some applications, e.g. drugs for people.
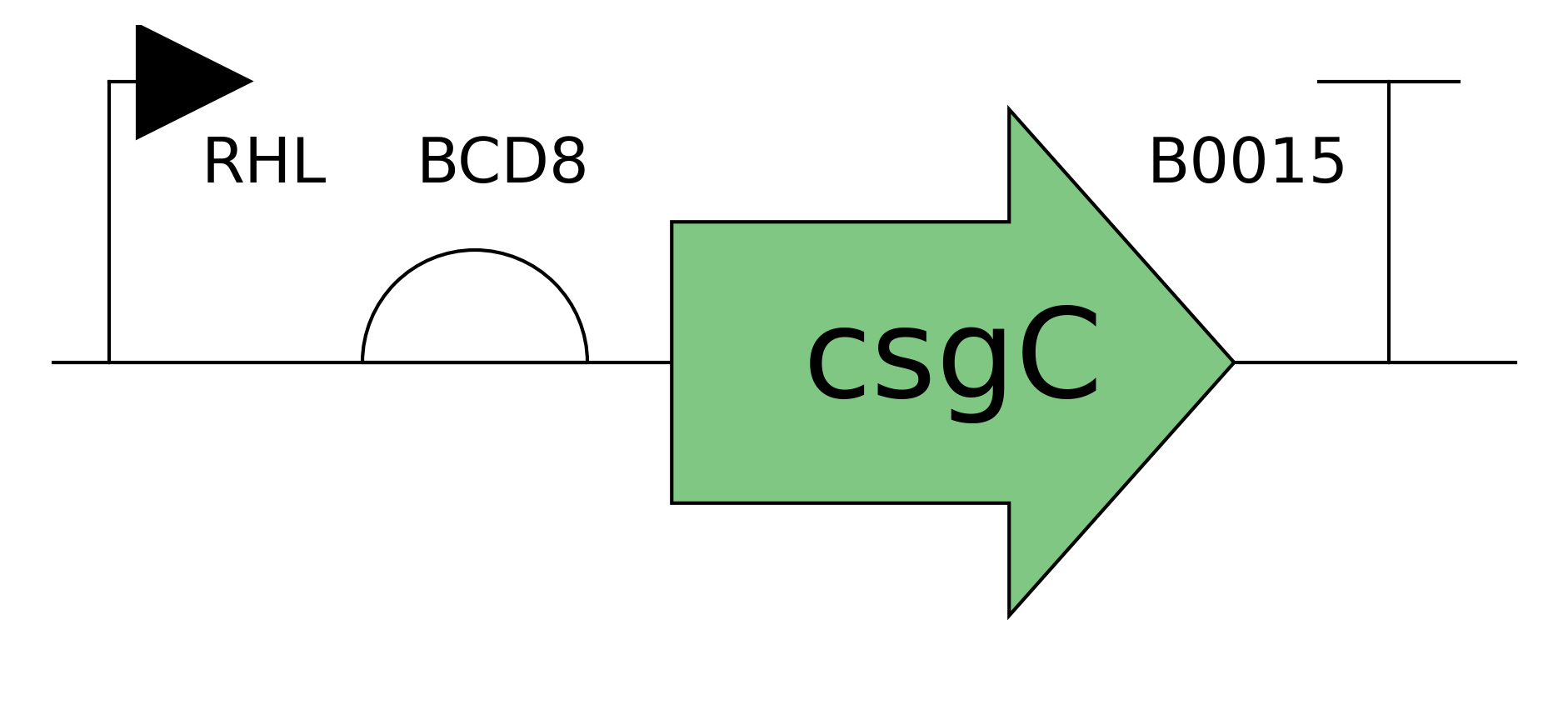
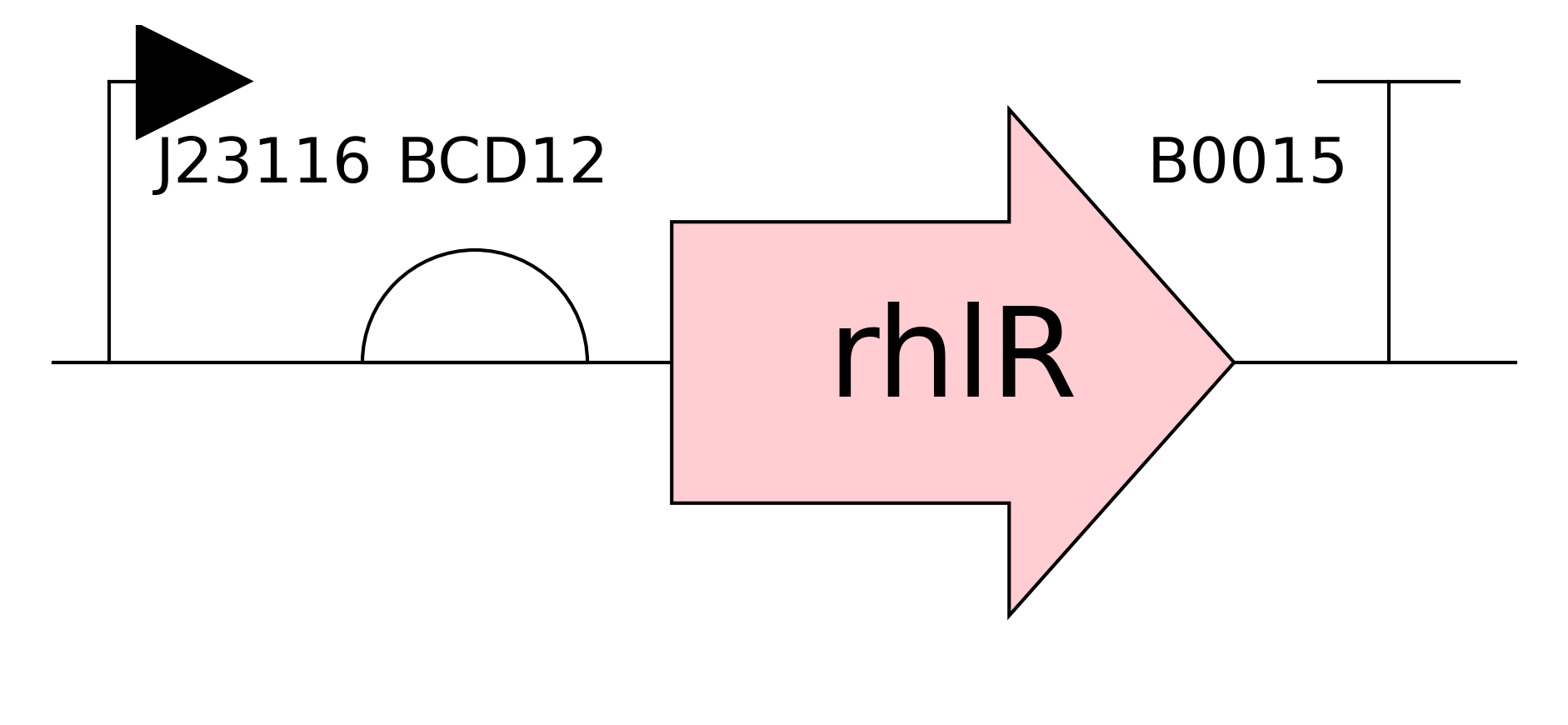
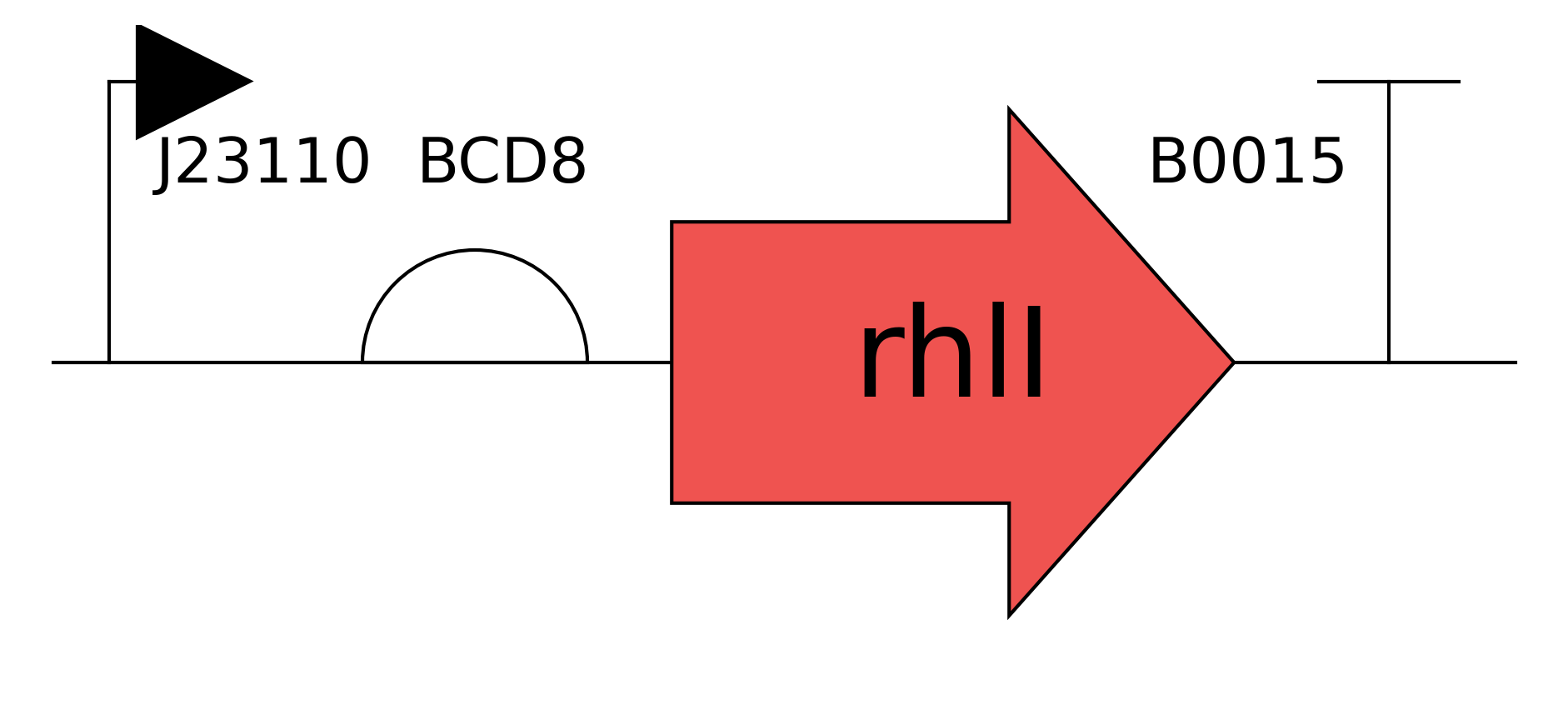
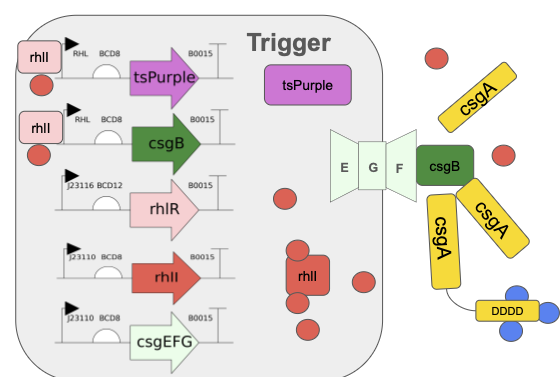
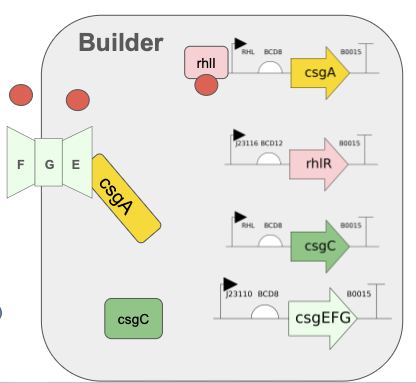
Need to characterize how much tsPurpse protein from the “paint” straing of my consortia expresses at varous times and under various inducer levels in order to create a controlled system where the “paint” strain can be used to color the stromatlight biofilm layers.
Since the expression will be in biofilm to measure this I will use reflected light and a standard digital raspberry pi camera. I will measure reflectance in the absorbing wavelength (590nm) and compare it to reflectance in a wavelength like (660/950nm) that is not absorbed by the tsPurple. Comparing these two should give a differntial comparison of light expression.
Measure Calcium Absorbance
In the mineralizing “glue” component of my consortia, the glue protein it generates should be able to bind Calcium ions to the negatively charged aspartic acid tail. I need to measure how much calcium the protein can bind by
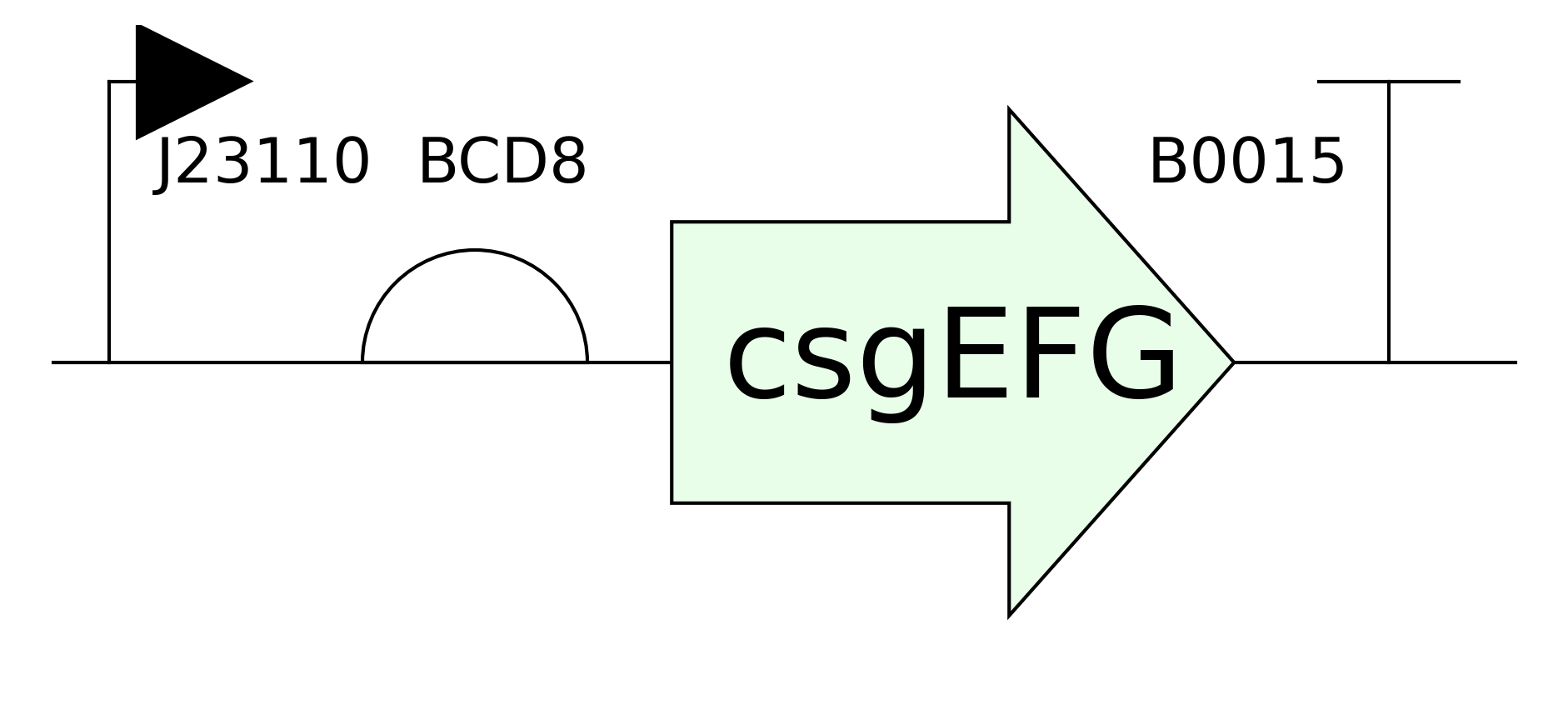
Grow multiple cultures that produce csgA and csgA + calcium binding tail.
Mix known about of calcium into a solution and add to csgA and csgA + calcium tail cultures
After some time centrigue the sampled down and extract supernatant and then pass it through a 22nm syringe filter
Combine supernatant with Sodium Carbonate or some other reagent that causes the calcium to precipatate
Centrifuge/filter out calcium carbonate and dry it.
Weigh calcium carbonate to compute remaining amount of calcium and infer how much calcium was aborbed by protein.
DNA Edit verification
Send plasmids to a Sanger sequencing service to verify that they the expected sequence after all of the igem constructions.
Molecular Weight - Part 1
Use calculator
Using the specified sequence and online calculator link I get a results
Theoretical pI/Mw: 5.90 / 28006.60
I am assuing we want to include the histine tail in the weight. If I remove the tail I get:
Theoretical pI/Mw: 5.58 / 26941.48
Use Mass Spec
We attach random numbers of protons to the molecule and measure m/z ratios in the mass spectrometer. Because we know mass of proton, even though we don’t initially know the charge of each peak, we can solve for the charge of peak by looking at how the mass/charge ratio changes between peaks.
Using peaks 875.4421 and 903.7418 implies the 875.4421 peak we get 31.9 which rounds to charge of 32/32 protons for peak 875.4421
Using peaks 903.7148 and 933.7439 imples that the 930.7148 peak we get 21.06 which rounds to charge of 31/31 protons for peak 903.7148
This makes it looks method is consistent
Now that we have the charges for peaks we can go back and infer the mass to get
875.4421 * 32 - 32 = 27982 daltons
903.7418 * 31 - 31 = 27985 daltons
This gives an error of .085 percent which is pretty small.
While you could do the calcultions we just did for the zoomed in calculation I don’t think it would be very accurate because we need to divide by the difference between measurements and the difference between measurements here is quite small.
Molecular Weight - Part 2
Denatured proteins unfold back into their original linear state. Looking at the two diagrams I am guessing that the denatured protein tends to pick up proteins moe uniformly, while the native protein tends to have structured stable states where it picks up a lot of proteins? Not really sure though. Let me ask Gemini now: “How can you tell whether or not a protein is denatured with mass spec?”.
Yes Gemini verifies above and ties it to the exposed Lysines and Argines when you unfold.
Again I don’t think you can discern the charge state on the zoom in given how small the differenes are. Too make matters worse it is possible that the gap between charged states isn’t even +1 proton?
I see 26 peaks in that range. It matches my digest size of 27 if I widern the range to include stuff below .5 also.
I thought you coulnd’t use the isotopic peaks to determine charge state, because you don’t know the isotopes and want the mass jumps would be. This is correct, but for biological molecues you can assume that you will usually have 1 dalton differences for C, N, O. If you have sulfur you could have two dalton jumps too, so you probably want to do multiples and looks for multiples before you average over set.
In this case you have two mass/charge ratios where you know the charge is the same and that the mass difference must be 1, so you can solve for charge state
assume peak 525.7612 has some charge Z and mass M
then peak 526.25918 has same charge Z but mass M+1
So charge Z is 1/(526.25918-525.7612) = 2.008 so the charge is +2 protons
This implies the mass M of this is 1051.52
Which implies the mass of [M+H+] is 1052.52 as is M/Z since Z is 1 in this case.
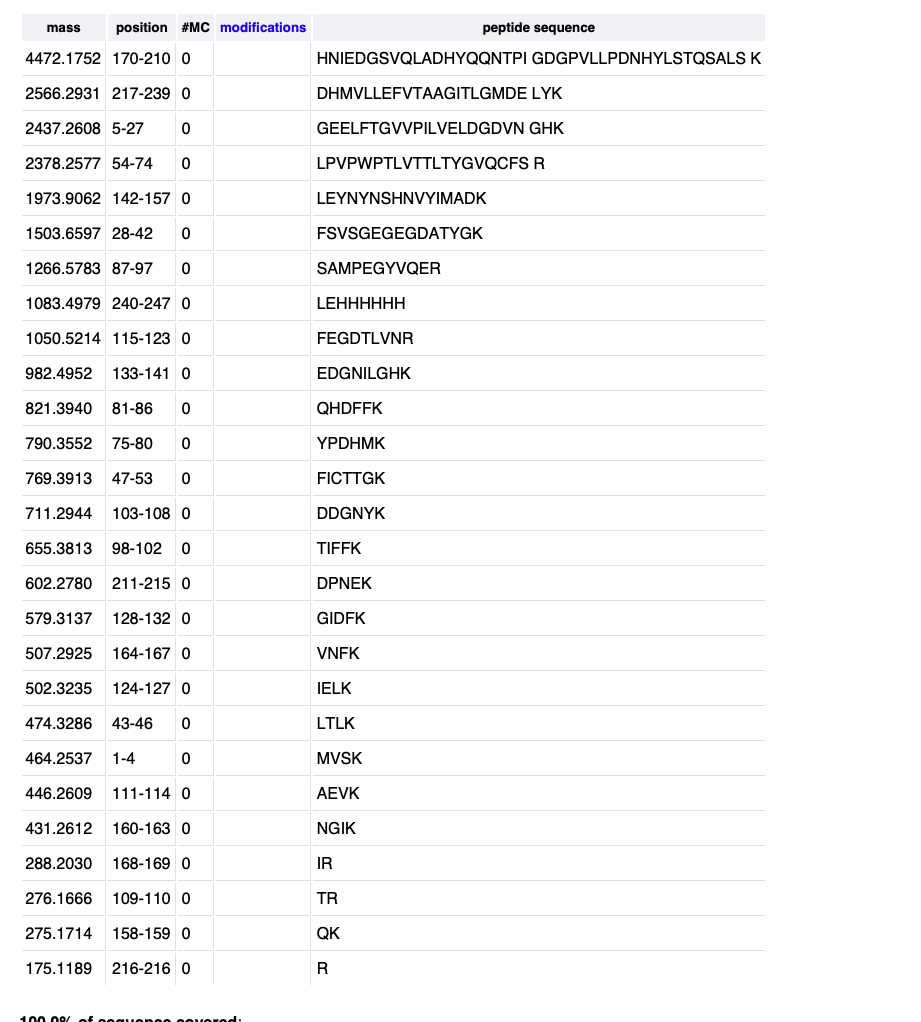
Given this I the closest peptide mass is 1050.5214 for peptide FEGDTLVNR. So the error is .19 percent.
Reading the last figure it looks like 88 percent of the peptide is covered.
Molecular Weight - Part 4
This section is using charge detetion mass spec, which measures charge and m/z for a single ion, so it can directly compute the mass instead of infer it from charge states and assumptions about distributions. This is important for very large molecues (like viruses) where the incremental change between charge peaks is so small because the mass is so high relative to the charge that you can’t resolve the difference between the peaks.
This means answering this question about matching peaks to oligmers is just a matter of computuing masses.
Masses of units are
7FU 340 kDa
8FU 400 kDa
7FU Decamer, 10 units of 7FU so 3.5 MDa, which is closest to the 3.4 MDa peak
8FU Didecamer, 10 units of 8FU so 4 MDa, which is closest to the 4.013 MDa peak and about twice as frequent as 7FU Decamer
8FU 3-Decamer, 30 units of 8FU so 12 MDa, which is closest to the 12.67 MDa peak
8FU 4-Decamer, 50 units of 8FU so 16MDa, which is a small bump that isn’t labeled, which implies the 4-decamer is not very frequent in this particular sample
Molecular Weight - Part 5
I wasn’t in the lab so just repeating above.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28006.60
27982
850
Theoretical was
Gemini Questions
Are there standard ways to generate light with different wavelengths I need (using tsPurple), i.e. filters or different lens or light through prism/grating?
What is adjacent charge state approach?
Can you use isotopic measurements to calculate the charge state? I would think for an unknown material you wouldn’t know the charge/mass gaps between the different isotopes, so you couldn’t solve for the charge state.
What is charge detection mass spec?
Week 11 — Bioproduction & Cloud Labs
Part A Collective Artwork
Unfortunately I missed the deadline to contribute to the artwork becuase I missed the notification email and the deadline had passed before recitation when I realized what had happened.
This is a cool project and I wish I had been able to participate. I did listen to go to the webiste and listen to others discuss in reciation. Based on what I heard it seems like one potential improvement is that everyone was only “given” one pixel, but it is not clear to me how a coherent picture would arise if each student actually only had one pixel. It sounded like what happened is that some people just took over parts of the canvas so they could draw coherenet pictures. The pictures were nice, but it seems like this could be confusing? Maybe the canvas should be totally locked so that each person can only edit their pixel/area so the picture is totally self organizing, or have some kind of scheme to partition out larger spaces to groups/volunteers who then draw the main sketch but leave certain pixels that people can color as they wish (or choose amonhst a smaller set of colors), i.e. like choosing between two colors for the skin of animal to see if stripes or spots or chaos appear spontaneously.
Part B Cell-Free Protein Synthesis
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
Includes the ribsomes and enzymes needed to transcribe DNA and manfufacture proteins.
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): I
Salts/Buffer
Keep pH in accetpable range for biological reactions and provides magnesium, potassium, and phosphorus
Potassium Glutamate
HEPES-KOH pH 7.5
Magnesium Glutamate
Potassium phosphate monobasic
Potassium phosphate dibasic
Energy / Nucleotide System
Provides raw components to assemble into DNA and provide energy for ATP production.
Ribose
Glucose
AMP
CMP
GMP
UMP
Guanine
Translation Mix (Amino Acids)
Provide amino acidds that can be attached to tRNA and assembled into target proteisn
17 Amino Acid Mix
Tyrosine
Cysteine
Additives
Nicotinamide
Backfill
Hydrates the system in a way that won’t degrade the DNA that is exposed to water
SynStromatolite Abstract Stromatolites are pillars of mineralized biofilm laid down by communities of photosynthetic bacteria. The dominant visible evidence of life on Earth for billions of years, stromatolites now only form in environments like hyper-saline water that exclude multi-cellular animals. Like tree rings, the records left in the layers of ancient stromatolites can be used to infer information about their environment like hypersalinity (Zhu et al., 2021) or fact that the Earth was spinning faster 2.5 billion years ago and an Earth year was 435 days instead of 365 ([(Vanyo & Awramik, 1985)(#vanyo1985)]). A synthetically engineered consortia that mimics the layer formation ability of stromatolites enables the development of low cost self-powered biological biosensors that record a time series of their outputs as mineralized biofilm layers. [Combining with existing water quality biosensors can help address water quality health problems in resource constrained areas] As a feasibility demonstration this projects engineers a two-strain Esceheriichia coli consortium that that use quorum-triggered curli amyloid secretion to lay down biofilm layers with enhanced mineralization capabilities. The projects’s central hypothesis is that separating biofilm generation and reporting responsibilities across multiple strains will be superior to trying to put all the functions in a single organism in terms of growth (as measured by OD900 curves), biofilm production (as measured by a Congo Red depletion assay), sensor signal strength (as measured by 590nm absorption), and mineralization (as measured by a calcium depletion precipitation assay). The project also lays out a road map to further develop this consortia into a deployable biosensor recorder platform.
Stromatolites are pillars of mineralized biofilm laid down by communities of photosynthetic bacteria. The dominant visible evidence of life on Earth for billions of years, stromatolites now only form in environments like hyper-saline water that exclude multi-cellular animals. Like tree rings, the records left in the layers of ancient stromatolites can be used to infer information about their environment like hypersalinity (Zhu et al., 2021) or fact that the Earth was spinning faster 2.5 billion years ago and an Earth year was 435 days instead of 365 ([(Vanyo & Awramik, 1985)(#vanyo1985)]). A synthetically engineered consortia that mimics the layer formation ability of stromatolites enables the development of low cost self-powered biological biosensors that record a time series of their outputs as mineralized biofilm layers. [Combining with existing water quality biosensors can help address water quality health problems in resource constrained areas] As a feasibility demonstration this projects engineers a two-strain Esceheriichia coli consortium that that use quorum-triggered curli amyloid secretion to lay down biofilm layers with enhanced mineralization capabilities. The projects’s central hypothesis is that separating biofilm generation and reporting responsibilities across multiple strains will be superior to trying to put all the functions in a single organism in terms of growth (as measured by OD900 curves), biofilm production (as measured by a Congo Red depletion assay), sensor signal strength (as measured by 590nm absorption), and mineralization (as measured by a calcium depletion precipitation assay). The project also lays out a road map to further develop this consortia into a deployable biosensor recorder platform.
Project Aims
Aim 1: Consortia With Triggered Biofilm Formation
Design and assemble a consortia of two Esceheriichia coli strains where one strain (the trigger strain) specializes in producing sensor output and triggering biofilm layer formation while they other strain(builder strain) specializes in producing the majority of the mineralizable protein mass that makes up the biofilm layers. Create the two E. coli strains by separately introducing engineered trigger and builder plasmids into the BL21E. Coli chassis using chemical transformation. Produce both plasmids using Golden Gate assembly of parts plasmids compatible with the JUMP (Valenzuela-Ortega & French, 2021) assembly vectors in the igem 2025 distribution kit. Design custom JUMP-compatible DNA for curlin biofilm and quorum-signaling proteins that are missing in the igem 2025 kit by downloading amino acid sequences from Uniprot and using Twist Biosciences to generate DNA vectors that are JUMP compatible and optimized for E. Coli.
Validate that the consortia functions and outperforms alternative implementations in growth, biofilm production, sensor signal strength, and mineralization activity. Evaluate growth of cultures by using spectrophotometer to measure optical density with infrared (900 nm) light to avoid absoprtion by the sensor color signal. Evaluate biofilm production by depletion of Congo Red via absorption of red (490nm) light in supernatant of culture exposed to a known concentration of Congo Red. Evaluate the strength of sensor signals by absorption of purple light (580 nm). Evalute mineralization activity by depletion of calcium ions from supernatant exposed to a known concentration of calcium chloride using an analytical balance to measure the mass of precipitate formed with excess sodium carbonate.
Aim 2: Long-Term Reliable Biosensor Recording in Biofilm Layers
Optimize the function of the consortia and add features required to support field deployment by adding biosensor integration, reliable layer generation, recording durability, and autonomous power generation.
Optimize Core Functions
Leverage lab automation and the modular nature of the Golden Gate assembly to optimize the four basic functions by varying promoter and RBS strength.
Biosensor Integration
Replace the tsPurple biosensor proxy in the prototype with a true biosensor that detects a condition and switches color of biofilm layer being formed depending on the current state it detects. Validate sensor response strength in biofilm layer by measuring relflectance at relevant wavelength. Ensure the consortia can support multiple different types of water quality biosensors by developing multiple variants with biosensors for water-borne pathogens like cholera quorum sensing molecules and toxic pollutants like arsenic. Using a remote cloud lab will again be important for this work as the community lab will not have access to chemicals to run toxin detection experiments.
Reliable Layer Generation
Ensure the consortia lays down distinct layers of biofilm results by engineering a detection cycle in the consortia that has distinct stages: growth -> sensor trigger -> biofilm production -> settling and layer formation -> reset stage -> growth [MISSING PICTURE]. Validate the sensor cycle by introducing trigger chemicals at known times and concentrations and validating the sensor color response by in situ reflectance of the biofilm at the appropriate wavelength. To avoid cross-cycle interference, add additional components to the consortia that clear the medium of the biosensor chemical signal during the reset stage by degradation or sequestration in the biofilm layers.
Layer Durability
Ensure recording layers survive typical shipment handling after separation from growth medium, drying, and placement in a box. Measure samples with thirty or more layers of biofilm records under representative compressive, shearing, and tensile loads. If required add consortia members to increase structural integrity of recording layers. For example add cellulose generating genes to the consortia or add an anaerobic bacteria that live in the lower layers and secrete urease to create a more favorable environment for biomineralziation by raising the pH.
Autonmous Power
Support long term operation by adding a photosynthetic member to consortia that has been engineered to secrete sucrose to provide power to the rest of the consortia. Validate integration by comparing photosynthetic consortia growth, biofilm production, and sensor signal growing on minimal mineral media like BG-11 to the results from non-photosynthetic consortia growing on LB broth over one month with the expectation that the photosynethetic consortia performs at a consistent level for weeks and outperforms the hetrotrophic consortia in biofilm generation and sensor signal beyond the first few days of operation.
Aim 3: Deployment for Water-Quality Monitoring in Resource Constrained Environments
Package the synthetic stromatolite consortia into a hydrogel container (Tang et al., 2021 and field deploy a water quality recording system into resource constrained environments. The containment capsule can keep the consortium contained while allowing small molecules (less than 22 nm) to traverse the permeable membrane and interact with the sensor. The sensors can give immediate warning to people that there water supply is tainted and at the same time provide an inexpensive way for public health officials to gather comprehensive records of water quality across households, water systems, and watersheds.
The two main evaluation metrics for this are ease of use and cost. End user usage should be as simple as:
Float the sensor on top of the water
Leave the sensor in partial sunlight
If the bottom of the sensor is red the water is currently bad
Sensors should operate for at least a month
Sensor turns black when expended
Domestic Water Quality Monitoring
Time series collection of water quality by public health officials should be as simple as
Collect expended sensors
Extract biofilm record disk
Break disk in half and take picture with digital camera on phone
Upload picture, geo-location, and sensor id to data collection and analysis app
If available random sample of biofilm disks to lab for microscopic and chemical evaluation for more detailed information.
Watershed Water Quality Monitoring
Capturing Water Quality Time Series
Section 3: Background
The main structural component of E. coli biofilms is curlin protein. The curlin protein system was characterized in (Chapman et al., 2002) and (Robinson et al., 2006) as:
csgA Main structural amyloid protein that makes up 95% of the E. coli biofilm protein.
csgB Causes csgA to condense into stacked amyloid sheets.
csgC A helper protein that prevents the csgA from prematurally folding before it leaves the cell
csgE A helper protein that helps move csgA and csgB from the cytoplasm to the cell membrane
csgG A membrane protein that creates pores for the csgA and csgB to exit the cell
csgF An exterior membrane protein that helps csgA and csgB exit the cell and acts as an anchor point for csgB to bind to the cell membrane.
Modified curlin csgA proteins with histadine tails that can bind to metal have been used to manufacture quantum dots (Chen et al., 2014). This project combines this approach with the observations from mollusks tha amyloid sheets with aspartic acid regions promote biomineralization by attracting calcium ions (Weiner & Hood, 1975).
There are other synthetic biology approaches to recording time series of biosensor readins. Most work in this area builds on George Church’s amazing idea to encode biosensor readings as a time series in the organism’s DNA using Cripsr (Shipman et al., 2017). Using marine biofilms to monitor water quality is a good example of this approach (Nevot et al., 2025). The DNA time series approach gives tight control and digital precision, however it requires extraction of DNA and sequencing which are not generally available in the resource constrained environments that most need ongoing water quality monitoring.
There are also incredible low cost water quality monitoring solutions intended for resource constrained environments. My favorite is the baker’s yeast that turns red when exposed to Cholerla in a water sample (Yudina et al., 2015). In theory this solution should be deployable for pennies per use and solve the problem of cholerla detection in resource constrained environments, however direct addition t well water can trigger the Cartegna Protocol as a release of genetically modified organisms. Avoiding release of genetic modified organsims for water quality biosensors requires cell-free systems or more sophisticated packaging (Siegfried et al., 2012). While the cost of more sophisticated packaging is only a few dollars per use that amount can still be prohibitive for resource constrained environments. Synthetic stromatolites have the same packaging problem but amortize the packaging cost over months of continuous usage reducing the cost of daily usage to a few cents per day. The fact that the same sensor can be used for daily preventive monitoring by distributed users and for time series monitoring by centralized public health officials should also defray costs and boost availability.
The existing cost and labatory requirements of water quality monitoring sensors often limits usage in resource constrained environments to public health officials reactive remediation after a water-borne outbreak or pollution event is severe enough to be noticed by community health outcomes. Cholera alone accounted for more than 600,000 reporteed cases and almost 8000 deaths in 2025 (WHO, 2026). Widespread, low cost, and easy to operate preventive monitoring of water quality pathogens and toxins sensors could repalce reactive remediation to proactive surveillance and improve community health in resource constrained environments.
Given the that the target outcome is improvement in resource constrainted environments a key ethical concern is local empowerment and access. An aproach that is imposed from afar or at substantial financial cost is not a solution. To address this the project should be open and patent free, so that it can be produced and adapted by communities as needed. Furthemore the final sensor package and consortia should be designed to enable production of the sensors is possible as close as possible to where they are deployed without needing sophisticated or expensive equipment.
The other ethical consideration that applies to any genitically modified organsim is containment risk. To avoid risk to the communities using the biosensor there needs to be a multi-level safeguards preventing release. Packaging is the first line of defense. In addition the consortia should be engineered with a built in timed kill switch and mutual dependencies between the the different consortia elements, to make long term survival after an accidentl release unlikely.
Experimental Design
Step 1 — DNA Design: Trigger Strain Plasmid
Purpose: Define the complete genetic architecture of the trigger strain. Method: Design a single multi-TU plasmid encoding: (1) rhlI TU [J23110 → BCD8 → rhlI → B0015], (2) rhlR TU [J23116 → BCD12 → BBa_C0070 → B0015], (3) csgB TU [Prhl_NM → BCD8 → csgB → B0015], (4) tsPurple TU [Prhl_NM → BCD1 → tsPurple → B0015], (5) csgEFG TU [J23110 → BCD8 → csgEFG → B0015]. Assembly via MoClo/JUMP Golden Gate using iGEM 2025 distribution kit parts; csgB, rhlI, and csgEFG genes ordered from Twist Bioscience as gene strings. Automation: Manual assembly at community biolab (Aim 1). Plate: Standard 1.5 mL microtubes for Golden Gate reactions. Expected Result: Correct assembly confirmed by colony PCR and Sanger sequencing. Timeline: Weeks 1–2.
Step 2 — DNA Design: Builder Strain Plasmid
Purpose: Define the complete genetic architecture of the builder strain. Method: Design a single multi-TU plasmid encoding: (1) rhlR TU [J23116 → BCD12 → BBa_C0070 → B0015], (2) csgC TU [J23116 → BCD12 → csgC → B0015], (3) csgA-Asp TU [Prhl_NM → BCD1 → csgA-Asp-tail → B0015], (4) csgEFG TU [J23110 → BCD8 → csgEFG → B0015]. Two versions of the glue strain plasmid will be made: one with csgA-Asp-tail and one with unmodified csgA, for direct comparison. csgC, csgA variants, and csgEFG ordered from Twist Bioscience. Automation: Manual assembly at community biolab. Plate: Standard microtubes. Expected Result: Two confirmed glue strain plasmids differing only at the csgA CDS. Timeline: Weeks 1–2.
Twist Bioscience Order Summary:
csgB (E. coli K-12, codon-optimized) — gene string
rhlI (P. aeruginosa PAO1, codon-optimized for E. coli) — gene string
All synthesized genes will be ordered from Twist Bioscience as gene strings with flanking MoClo BsaI Golden Gate overhangs compatible with the iGEM JUMP assembly standard.
Step 3 — Golden Gate Assembly
Purpose: Assemble multi-TU plasmids from iGEM parts and Twist gene strings. Method: MoClo/JUMP Golden Gate reactions using BsaI-HFv2 (NEB), T4 DNA ligase, thermocycler cycling protocol (37°C/16°C × 30 cycles). Transform into E. coli DH5α, plate on selective agar. Automation: Manual at community biolab; ATC Thermal Cycler equivalent used for cycling. Plate: PCR tubes / 96-well PCR plate. Expected Result: >10 correct colonies per assembly; correct insert confirmed by colony PCR. Timeline: Week 2.
Assembled Trigger Strain In Action
Assembled Builder Strain In Action
Step 4 — Sequence Verification
Purpose: Confirm all TUs are correctly assembled and no mutations introduced. Method: Sanger sequencing of all five TU junctions per plasmid using standard M13 and custom sequencing primers. Submit to community biolab sequencing service or Plasmidsaurus. Expected Result: 100% sequence identity to design across all TU junctions. Timeline: Week 3.
Step 5 — Strain Construction and Glycerol Stocks
Purpose: Generate working bacterial strains carrying each plasmid. Method: Transform verified plasmids into E. coli BL21(DE3) or MG1655 (curli-competent background). Confirm by PCR. Prepare glycerol stocks at −80°C. Expected Result: Stable transformants confirmed; glycerol stocks archived. Timeline: Week 3.
Step 6 — Congo Red Liquid Assay: Strain Preparation
Purpose: Prepare cultures for curli secretion quantification. Method: Inoculate csgA-Asp-tail glue strain and unmodified csgA glue strain from glycerol stocks into LB + antibiotic. Grow overnight at 30°C (curli-permissive temperature) with shaking. Dilute to OD₆₀₀ = 0.05 in YESCA medium (yeast extract + casamino acids, low salt) to promote curli expression. Automation: Manual at community biolab. Expected Result: Cultures at consistent starting OD for assay normalization. Timeline: Week 4.
Step 7 — Congo Red Liquid Assay: Binding Measurement
Purpose: Quantify curli fiber secretion by Congo red dye binding. Method: After 48 h growth at 30°C, pellet cells by centrifugation (5,000 × g, 10 min). Resuspend pellet in PBS + 10 µg/mL Congo red. Incubate 30 min at RT with gentle mixing. Centrifuge again; measure absorbance of supernatant at 490 nm. Congo red depletion from supernatant = curli binding. Include: (1) no-cell blank, (2) wild-type MG1655 positive control, (3) ΔcsgA negative control. Instrument: Spectrophotometer or plate reader at community biolab (96-well format if available). Plate: 96-well flat-bottom plate. Expected Result: csgA-Asp-tail strain shows Congo red binding comparable to or greater than unmodified csgA strain, confirming the tail does not disrupt secretion. Timeline: Week 4–5.
Example Assay Plate Layout (96-well Congo Red Assay):
1 2 3 4 5 6
A [Blank] [Blank] [Blank] [+ctrl] [+ctrl] [+ctrl]
B [csgA] [csgA] [csgA] [-ctrl] [-ctrl] [-ctrl]
C [csgA-D] [csgA-D] [csgA-D] [csgA] [csgA] [csgA]
D [csgA-D] [csgA-D] [csgA-D] [Blank] [Blank] [Blank]
Key: Blank = PBS + Congo red, no cells
+ctrl = WT MG1655 (curli+)
-ctrl = ΔcsgA strain
csgA = unmodified glue strain
csgA-D = aspartate-tail glue strain
Triplicates across columns 1-3 and 4-6
Step 8 — Calcium Depletion Assay: Setup
Purpose: Quantify calcium-binding enhancement of CsgA-Asp-tail versus unmodified CsgA. Method: Grow csgA-Asp-tail and unmodified csgA glue strains under identical YESCA conditions for 48 h at 30°C to allow biofilm and curli formation. Add CaCl₂ to a final concentration of 5 mM to each culture and a sterile medium-only control. Incubate 2 h at RT with gentle mixing. Expected Result: Cultures with established curli biofilm ready for calcium binding measurement. Timeline: Week 5.
Step 9 — Calcium Depletion Assay: Measurement
Purpose: Measure calcium removed from solution by biofilm binding. Method: Filter cultures through 0.22 µm syringe filter to remove cells and biofilm. Add Na₂CO₃ (10 mM final) to filtrate to precipitate remaining free calcium as CaCO₃. Collect precipitate by centrifugation; dry at 60°C overnight; weigh on analytical balance. Calculate: calcium bound = (CaCO₃ precipitate from medium control) − (CaCO₃ precipitate from culture filtrate). Include: (1) sterile medium + CaCl₂ + Na₂CO₃ (total calcium control), (2) unmodified csgA culture, (3) csgA-Asp-tail culture, (4) ΔcsgA culture (background binding). Instrument: Analytical balance; optional ICP-OES for higher precision if available at community biolab partner institution. Expected Result: csgA-Asp-tail culture shows significantly less free calcium in filtrate (more calcium bound to biofilm) than unmodified csgA or ΔcsgA controls. Timeline: Week 5–6.
Step 10 — tsPurple Reporter Validation
Purpose: Confirm RHL quorum-sensing trigger is functioning in the curing strain. Method: Grow curing strain to OD₆₀₀ = 0.1, 0.5, 1.0, 2.0 (representing increasing cell density and RHL accumulation). Measure absorbance at 570 nm (tsPurple) normalized to OD₆₀₀. Include: (1) curing strain without rhlI (no RHL production, negative control), (2) curing strain + exogenous RHL (positive control for Prhl_NM induction). Expected Result: tsPurple signal increases with cell density, confirming quorum-dependent activation. Timeline: Week 6.
Step 11 — Two-Strain Co-culture Biofilm Test
Purpose: Verify that the two-strain consortium produces a co-assembled biofilm. Method: Mix curing and glue strains 1:1 in YESCA medium. Grow in static culture at 30°C for 48–72 h. Observe biofilm formation visually and by crystal violet staining. Confirm tsPurple color in biofilm by visual inspection and absorbance. Expected Result: Visible purple-tinted biofilm at air-liquid interface; Congo red staining of biofilm confirms curli presence. Timeline: Week 7.
Step 12 — CsgA Variant Library Design and Twist Order
Purpose: Generate a panel of CsgA-Asp-tail variants to identify optimal tail length and linker for calcium binding. Method: Design 8 CsgA variants with aspartate tail lengths of 2, 4, 6, 8, 10, 12 repeats of (Gly₄Asp), plus two linker variants (flexible GGGS vs. rigid EAAAK). Order all 8 as gene strings from Twist Bioscience with flanking MoClo overhangs. Assemble into glue strain backbone using Golden Gate. Sequence-verify all constructs. Automation: Golden Gate assembly in 96-well PCR plate on ATC Thermal Cycler; transformation and plating manual; colony picking manual or with QPix if available. Expected Result: 8 confirmed glue strain variants ready for screening. Timeline: Weeks 8–10.
Purpose: Inoculate all 8 variants plus controls in 96-well format for parallel growth. Method: Use Tempest liquid handler to dispense 150 µL YESCA + antibiotic per well into 96-round-axygen-pdw11cs-halfdeep plates. Use Bravo-96 to inoculate from glycerol stock dilutions. Seal with Plateloc. Incubate in Cytomat at 30°C, 48 h, with orbital shaking via BioshakeD3000. Automation: Tempest (media dispensing), Bravo-96 (inoculation), Plateloc (sealing), Cytomat (incubation), BioshakeD3000 (mixing). Plate: 96-round-axygen-pdw11cs-halfdeep. Expected Result: Uniform growth across all wells; OD₆₀₀ measured at 48 h to confirm comparable growth. Timeline: Week 10.
Step 14 — Automated Congo Red Secretion Screen
Purpose: Quantify curli secretion for all 8 CsgA variants in parallel. Method: After 48 h, centrifuge plates on HiG Centrifuge. Use Bravo-96 to transfer supernatant to new 96-well plate. Use Multiflo to add Congo red reagent (10 µg/mL in PBS) to each well. Incubate 30 min at RT on BioshakeD3000. Centrifuge again. Transfer supernatant to 384 Greiner black-well clear-bottom plate using Echo525 for absorbance reading. Read at 490 nm on Spark Plate Reader. Automation: HiG Centrifuge, Bravo-96, Multiflo, BioshakeD3000, Echo525, Spark Plate Reader. Plate: 96-round-axygen-pdw11cs-halfdeep (culture); 384 Greiner black-well clear-bottom (absorbance read). Expected Result: Ranked secretion efficiency across 8 variants; identify top 2–3 variants for calcium assay follow-up. Timeline: Week 10–11.
Step 15 — Data Analysis and Variant Selection
Purpose: Identify optimal CsgA-Asp-tail design for Aim 3 integration. Method: Normalize Congo red depletion values to OD₆₀₀ for each variant. Plot secretion efficiency vs. tail length. Select top-performing variant(s) for calcium depletion assay validation (manual, as in Steps 8–9). Export data from Spark Plate Reader to CSV; analyze in Python/R. Expected Result: Clear optimum tail length identified; data supports rational design principle for Aim 3 consortium. Timeline: Week 11–12.
Course Technique Checklist
DNA design and synthesis (Twist Bioscience gene strings)
Automated incubation and plate handling (Cytomat, Plateloc, HiG)
Quorum sensing circuit design
Protein engineering (CsgA tail fusion)
Cell-free expression
CRISPR
Flow cytometry
Expanded Technique 1: MoClo/JUMP Golden Gate Assembly
MoClo (Modular Cloning) is a hierarchical Golden Gate assembly standard that uses Type IIS restriction enzymes (BsaI, BbsI) to generate defined 4-bp overhangs, enabling scarless, ordered assembly of multiple transcription units into a single plasmid in a single reaction. The JUMP (Joint Universal Modular Parts) extension used in the iGEM 2025 distribution kit standardizes overhang sequences across promoters, RBSs, CDSs, and terminators, making parts from different contributors directly interchangeable. In this project, MoClo/JUMP enables the assembly of 4–5 TUs per plasmid in a single Golden Gate reaction, dramatically reducing cloning time compared to traditional restriction-ligation or Gibson assembly approaches. The fidelity of the assembly is verified by colony PCR across each TU junction and confirmed by Sanger sequencing, ensuring that the final constructs match the computational design exactly before any biological characterization begins.
Expanded Technique 2: Congo Red Biofilm Secretion Assay
Congo red is an azo dye that binds with high affinity to amyloid fibrils, including curli fibers, through intercalation between the β-sheet stacking of the amyloid structure. In the liquid-phase assay format used here, Congo red depletion from the supernatant after incubation with pelleted cells or biofilm material provides a quantitative measure of curli fiber abundance — more curli means more dye bound and less remaining in solution, detected as a decrease in absorbance at 490 nm. This assay is highly amenable to automation: Congo red reagent can be dispensed by Multiflo, incubation handled by BioshakeD3000, and absorbance read in 384-well format on the Spark Plate Reader, enabling screening of dozens of variants in a single run. Critical controls include a ΔcsgA negative control (background dye binding to cell membranes), a wild-type curli-expressing positive control, and a no-cell blank to establish the maximum possible absorbance signal, allowing calculation of percent Congo red depletion as a normalized secretion metric.
Results & Quantitative Expectations
The primary validation experiment for this project is the calcium depletion precipitation assay comparing the CsgA-Asp-tail variant to unmodified CsgA and a ΔcsgA negative control. This experiment directly tests the central engineering hypothesis — that appending an aspartate-rich tail to CsgA will enhance calcium binding and accelerate calcium carbonate mineralization — and provides a quantitative, instrument-independent readout (mass of precipitate on an analytical balance) that is accessible at a community biolab without specialized equipment.
Step-by-Step Validation Protocol
Grow csgA-Asp-tail glue strain, unmodified csgA glue strain, and ΔcsgA control strain in YESCA medium at 30°C, 48 h, static culture (6 biological replicates each).
Add CaCl₂ to 5 mM final concentration to all cultures and to a sterile YESCA medium-only control. Incubate 2 h at RT with gentle rocking.
Filter all cultures through pre-weighed 0.22 µm syringe filters into pre-weighed 1.5 mL microtubes. Record filter + retained biofilm weight.
Add Na₂CO₃ to filtrate to 10 mM final concentration. Vortex briefly. Incubate 30 min at RT.
Centrifuge filtrate at 14,000 × g for 10 min. Carefully remove supernatant by pipette.
Wash pellet twice with deionized water (centrifuge between washes).
Dry pellet at 60°C overnight in a heat block or oven.
Weigh dried pellet on analytical balance. Record mass.
Calculate: free calcium in filtrate = mass of CaCO₃ precipitate (mg) × (40.08/100.09) = mg Ca²⁺ remaining. Calcium bound by biofilm = total calcium added − free calcium in filtrate.
Perform one-way ANOVA with Tukey post-hoc test across the three conditions (n=6 per group).
Techniques Used
The calcium depletion assay combines microbiology (biofilm culture), analytical chemistry (gravimetric precipitation), and basic statistics (ANOVA) in a workflow that requires no specialized instrumentation beyond an analytical balance and a centrifuge. The use of sodium carbonate precipitation as a proxy for free calcium is a well-established method in water chemistry and has been adapted here for biofilm calcium-binding quantification. Normalization of calcium binding to biofilm dry weight (obtained from the filter mass difference) controls for variation in biofilm abundance between replicates, ensuring that differences in calcium binding reflect the biochemical properties of the CsgA variant rather than differences in biofilm quantity. The inclusion of a ΔcsgA negative control is essential to distinguish specific curli-mediated calcium binding from non-specific binding to cell membranes, extracellular polysaccharides, or other biofilm matrix components.
Troubleshooting
If no difference is observed between csgA-Asp-tail and unmodified csgA, the most likely explanation is that the aspartate tail is being cleaved post-translationally or is misfolded and inaccessible — this can be investigated by running biofilm lysates on SDS-PAGE with anti-His or anti-CsgA antibody (if the tail includes a His-tag for detection) to confirm full-length protein is present. If the ΔcsgA control shows unexpectedly high calcium depletion (>15%), this suggests significant non-specific binding by other biofilm components (e.g., extracellular polysaccharides), and the assay should be repeated with a biofilm-free cell suspension to separate curli-specific from matrix-specific effects. If precipitation is inconsistent between replicates, the Na₂CO₃ addition step should be standardized more carefully — small variations in pH can dramatically affect CaCO₃ precipitation kinetics, so buffering the reaction to pH 9.0 before adding carbonate may improve reproducibility. If the csgA-Asp-tail strain shows reduced Congo red binding relative to unmodified csgA (Step 7), this indicates the tail is interfering with curli polymerization, and a shorter tail or different linker design should be tested — this is precisely the rationale for the Aim 2 variant library screen.
Additional Validation
An additional validation for this project is comparing the performance of the consortia to an single E. coli variant that combines all the different genes in one organism. The expected outcome is that the consortia should outperform the A1 strain in all of the core functional aspects.
Expected Results
While the experiements have not been performed yet these graphs reflect the expected outcomes of the Aim 1 synthetic stromatolite consortia versus the control strains:
Zhu, D., Liu, Q., Wang, J., Ding, Q., & He, Z. (2021). Stable carbon and oxygen isotope data of Late Ediacaran stromatolites from a hypersaline environment in the Tarim Basin (NW China) and their reservoir potential. Facies, 67(3), 25. https://doi.org/10.1007/s10347-021-00633-0
Suarez-Gonzalez, P., Quijada, I. E., Benito, M. I., Mas, R., Merinero, R., & Riding, R. (2014). Origin and significance of lamination in Lower Cretaceous stromatolites and proposal for a quantitative approach. Sedimentary Geology, 300, 11–27. https://doi.org/10.1016/j.sedgeo.2013.11.003
Daegelen, P., Studier, F. W., Lenski, R. E., Cure, S., & Kim, J. F. (2009). Tracing Ancestors and Relatives of Escherichia coli B, and the Derivation of B Strains REL606 and BL21(DE3). Journal of Molecular Biology, 394(4), 634–643. https://doi.org/10.1016/j.jmb.2009.09.022
Valenzuela-Ortega, M., & French, C. (2021). Joint universal modular plasmids (JUMP): A flexible vector platform for synthetic biology. Synthetic Biology, 6(1), ysab003. https://doi.org/10.1093/synbio/ysab003
Tang, T.-C., Tham, E., Liu, X., Yehl, K., Rovner, A. J., Yuk, H., De La Fuente-Nunez, C., Isaacs, F. J., Zhao, X., & Lu, T. K. (2021). Hydrogel-based biocontainment of bacteria for continuous sensing and computation. Nature Chemical Biology, 17(6), 724–731. https://doi.org/10.1038/s41589-021-00779-6
Zhang, L., Chen, L., Diao, J., Song, X., Shi, M., & Zhang, W. (2020). Construction and analysis of an artificial consortium based on the fast-growing cyanobacterium Synechococcus elongatus UTEX 2973 to produce the platform chemical 3-hydroxypropionic acid from CO2. Biotechnology for Biofuels, 13(1), 82. https://doi.org/10.1186/s13068-020-01720-0
Zhou, Y., Sun, T., Chen, Z., Song, X., Chen, L., & Zhang, W. (2019). Development of a New Biocontainment Strategy in Model Cyanobacterium Synechococcus Strains. ACS Synthetic Biology, 8(11), 2576–2584. https://doi.org/10.1021/acssynbio.9b00282
Nevot, G., Pol Cros, M., Toloza, L., Campamà-Sanz, N., Artigues-Lleixà, M., Aguilera, L., & Güell, M. (2025). Engineered Marine Biofilms for Ocean Environment Monitoring. ACS Synthetic Biology, 14(7), 2797–2809. https://doi.org/10.1021/acssynbio.5c00192
Ostrov, N., Jimenez, M., Billerbeck, S., Brisbois, J., Matragrano, J., Ager, A., & Cornish, V. W. (2017). A modular yeast biosensor for low-cost point-of-care pathogen detection. Science Advances, 3(6), e1603221. https://doi.org/10.1126/sciadv.1603221
]
Chen, A. Y., Deng, Z., Billings, A. N., Seker, U. O. S., Lu, M. Y., Citorik, R. J., Zakeri, B., & Lu, T. K. (2014). Synthesis and patterning of tunable multiscale materials with engineered cells. Nature Materials, 13(5), 515–523. https://doi.org/10.1038/nmat3912
Siegfried, K., Endes, C., Bhuiyan, A. F. Md. K., Kuppardt, A., Mattusch, J., Van Der Meer, J. R., Chatzinotas, A., & Harms, H. (2012). Field Testing of Arsenic in Groundwater Samples of Bangladesh Using a Test Kit Based on Lyophilized Bioreporter Bacteria. Environmental Science & Technology, 46(6), 3281–3287. https://doi.org/10.1021/es203511k
Guo, Y., Liu, M., Yang, X., Guo, Y., & Hui, C. (2025). Optimized genetic circuitry and reporters for sensitive whole-cell arsenic biosensors: Advancing environmental monitoring. Applied and Environmental Microbiology, 91(8), e00601-25. https://doi.org/10.1128/aem.00601-25
Weiner, S., & Hood, L. (1975). Soluble Protein of the Organic Matrix of Mollusk Shells: A Potential Template for Shell Formation. Science, 190(4218), 987–989. https://doi.org/10.1126/science.1188379
Shipman, S. L., Nivala, J., Macklis, J. D., & Church, G. M. (2017). CRISPR–Cas encoding of a digital movie into the genomes of a population of living bacteria. Nature, 547(7663), 345–349. https://doi.org/10.1038/nature23017