Week 2 HW: DNA Read, Write, and Edit
HOMEWORK
Part 1: Benchling & In-silico Gel Art
Below are some screenshots from the steps followed to create a basic pattern:
Step 1: The sequence is imported from the webpage to Benchling.
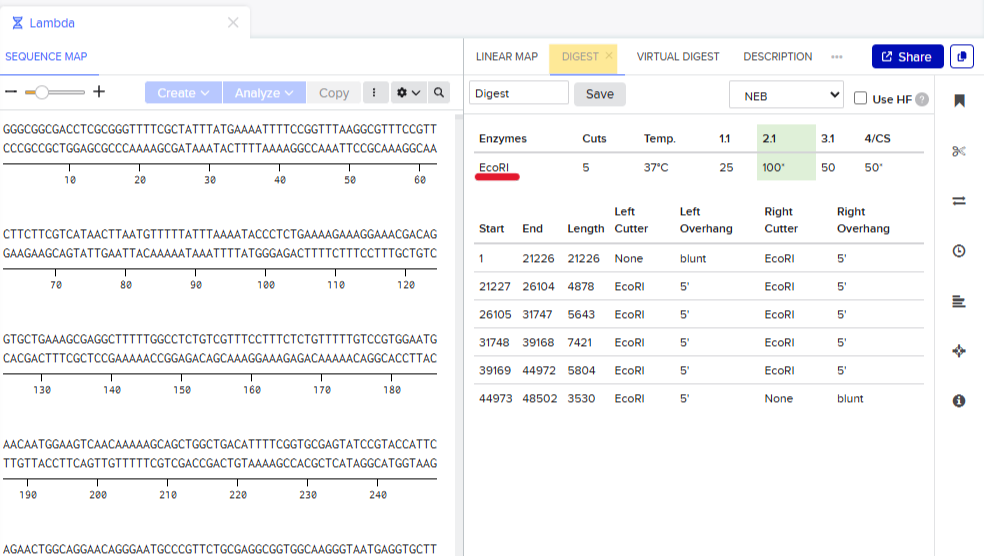
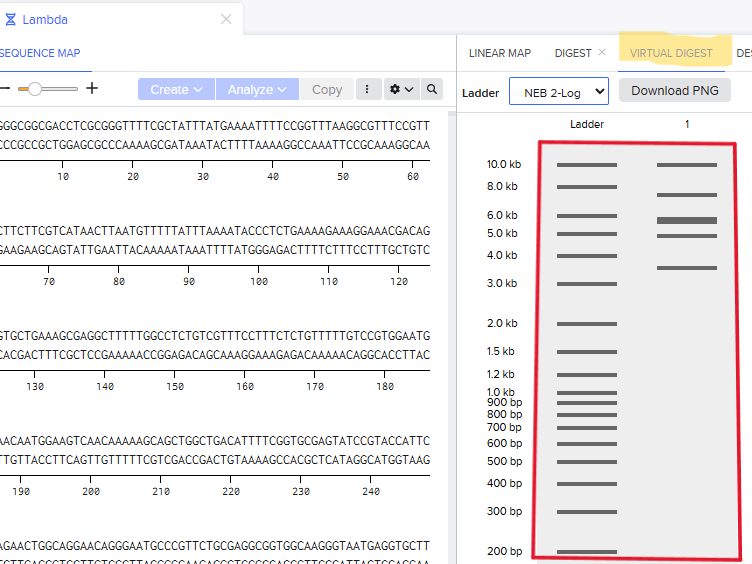
Step 2: The digest function is shown as a test with EcoRI as the chosen restriction enzyme.
Step 3: The process is repeated using enzymes requested in the homework, and the result is the following:
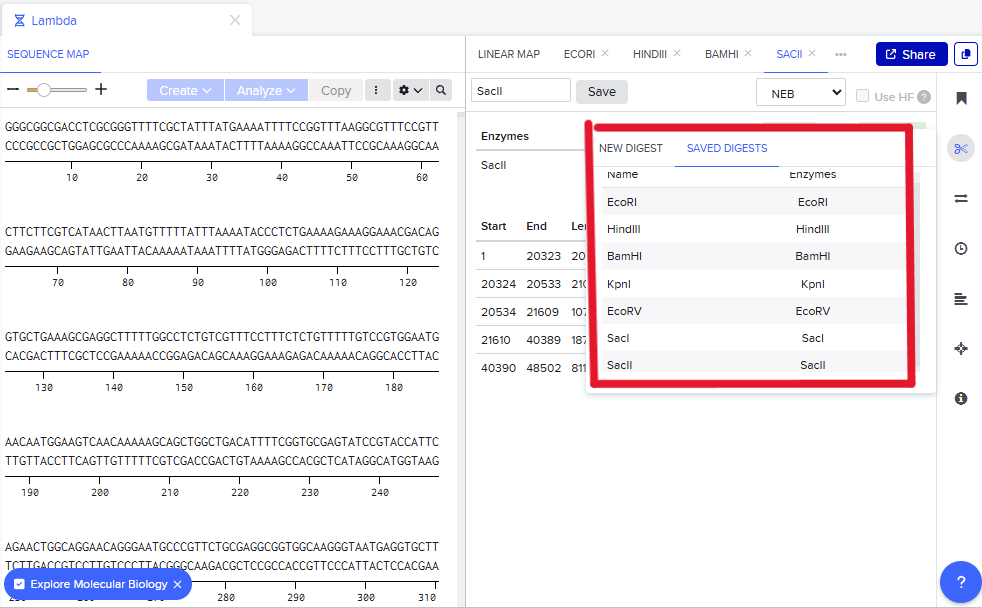

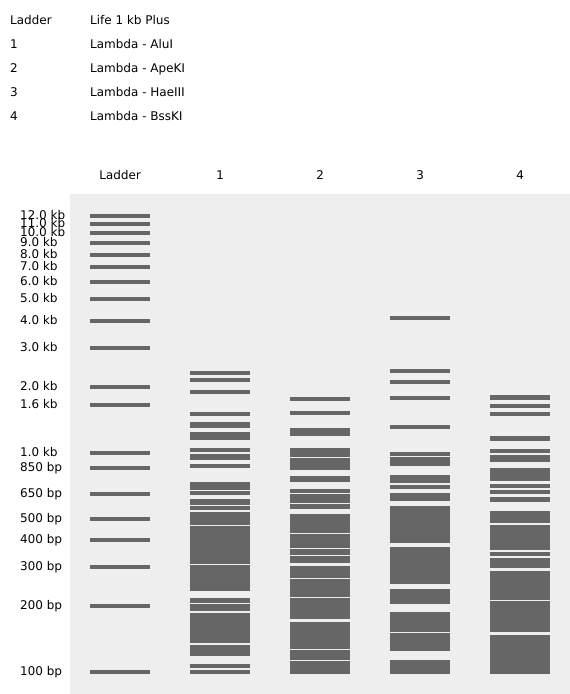
Step 4: The process is repeated now using different enzymes requested in the homework to create a different pattern. In this case, the pattern might look like a series of barcodes, using the enzymes shown in figure 7. Since they cut in different sites, they create a lot of short DNA fragments which scatter across the gel, givin the impression of a barcode. This enzyme behavior could be used as a biomarker perhaps in, although this idea needs further development:
Part 3: DNA Design Challenge
3.1. Choose your protein
In this case, I chose the Transcription Initiation Factor 3 (TIF-3) encoded in the gene infC, which is a relatively short protein that is involed in the translation process (Gutu et al., 2013), (Arenz & Wilson, 2016). Modifying the structure of this protein may be crucial to combat antibiotic resistance.
By going to UniProt, the aminoacid sequence is the following:
//
tr|A0A8S0FV27|A0A8S0FV27_ECOLX Translation initiation factor IF-3 OS=Escherichia coli OX=562 GN=infC PE=3 SV=1 MSLREALEKAEEAGVDLVEISPNAEPPVCRIMDYGKFLYEKSKSSKEQKKKQKVIQVKEI KFRPGTDEGDYQVKLRSLIRFLEEGDKAKITLRFRGREMAHQQIGMEVLNRVKDDLLRRT GSGRILPNEDRRPPDDHGARS
3.2 and 3.3: Reverse translation and codon optimization
Going bak to Benchling, the AA sequence was imported and the function back translate was used to obtain the DNA sequence which is already optimized given the figures below:
// DNA optimized sequence: ATGAGTTTACGTGAAGCACTGGAAAAAGCGGAAGAAGCCGGTGTTGATCTGGTCGAAATCAGTCCTAATGCAGAACCCCCGGTGTGCCGTATCATGGACTATGGCAAATTCCTCTACGAGAAATCTAAAAGCTCAAAGGAACAAAAAAAGAAACAGAAGGTTATTCAGGTCAAAGAGATTAAGTTTCGACCGGGGACTGACGAAGGAGACTATCAAGTGAAACTTCGCTCCTTGATTCGCTTCCTGGAAGAGGGGGATAAAGCGAAAATTACCCTGCGCTTTCGCGGCAGAGAGATGGCCCACCAGCAGATCGGCATGGAAGTATTGAACCGTGTGAAAGATGACTTACTGCGTCGCACGGGTAGCGGTCGTATACTGCCAAACGAGGATCGCCGGCCGCCGGATGATCATGGCGCTCGGTCG
The organism selected for this protein is E. Coli due to its wide use in biotechnology. The codons had to be optimized due to the fact that the cellular machinery may differ from one bacteria to another. This means that a bacteria other than E. Coli might express this protein at a different rate and intensity. In this case, the protein was obtained from E. Coli based on UniProt, but since E. Coli contains different strands, codon optimization still would have to be performed.
3.4 and 3.5: Production technologies and alignment
I would use host cells since the chosen protein is from a bacteria en E. Coli is a common microorganism used for these purposes. This technique has a much lower cost thant using cell-free systems in which all the cellular components have to be supplied.
The alignment is shown in the following figures
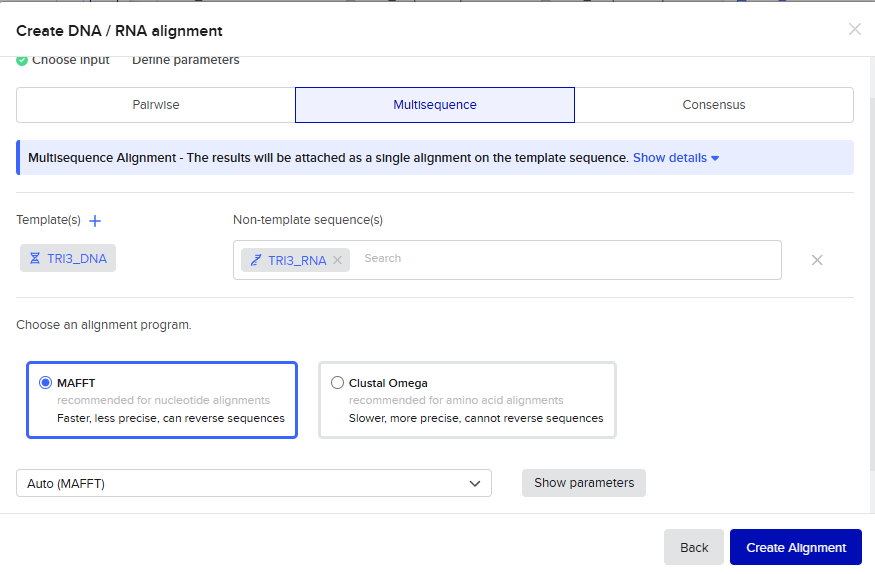
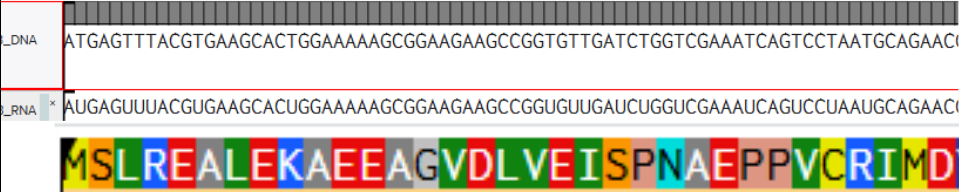
Part 4: Prepare a Twist DNA Sequence Order
Step 1: DNA Sequence
The same DNA linear sequence was already obtained in Part no. 3.
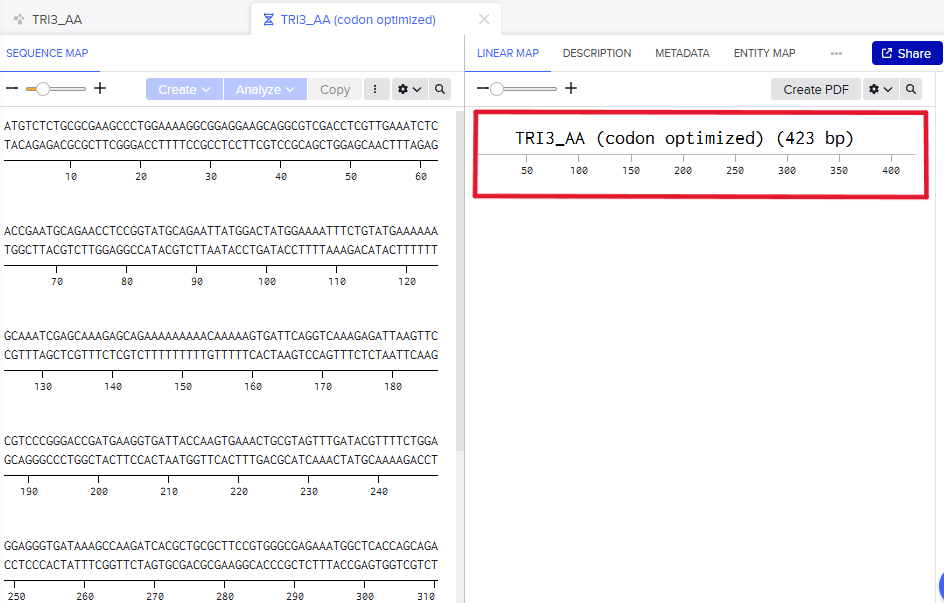
Step 2: Building the chasis
The parts were initially searched in iGEM, but the website shut down. Due to this, the parts provided in the homework were used.
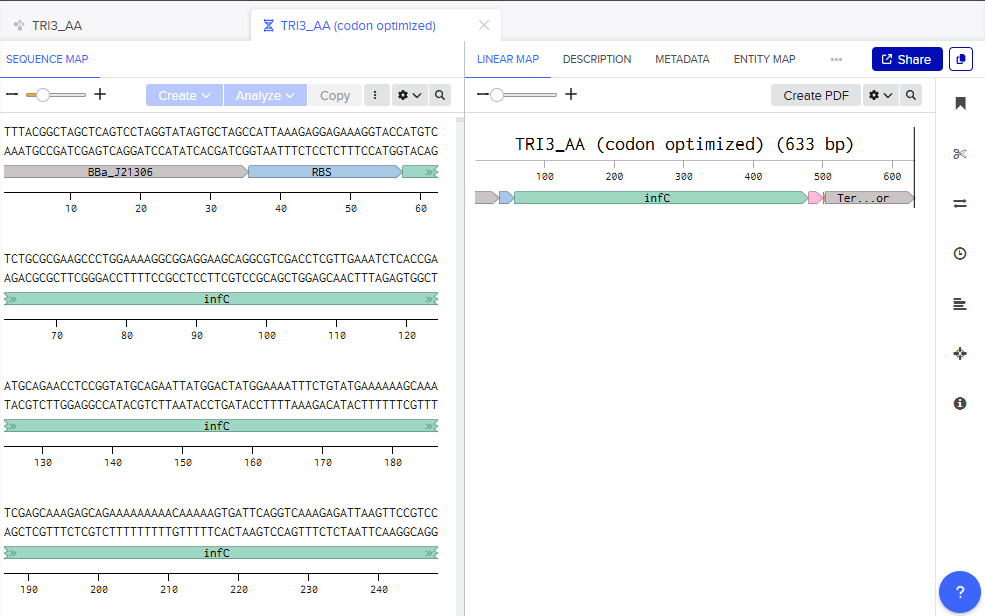
The chasis now looks like this:
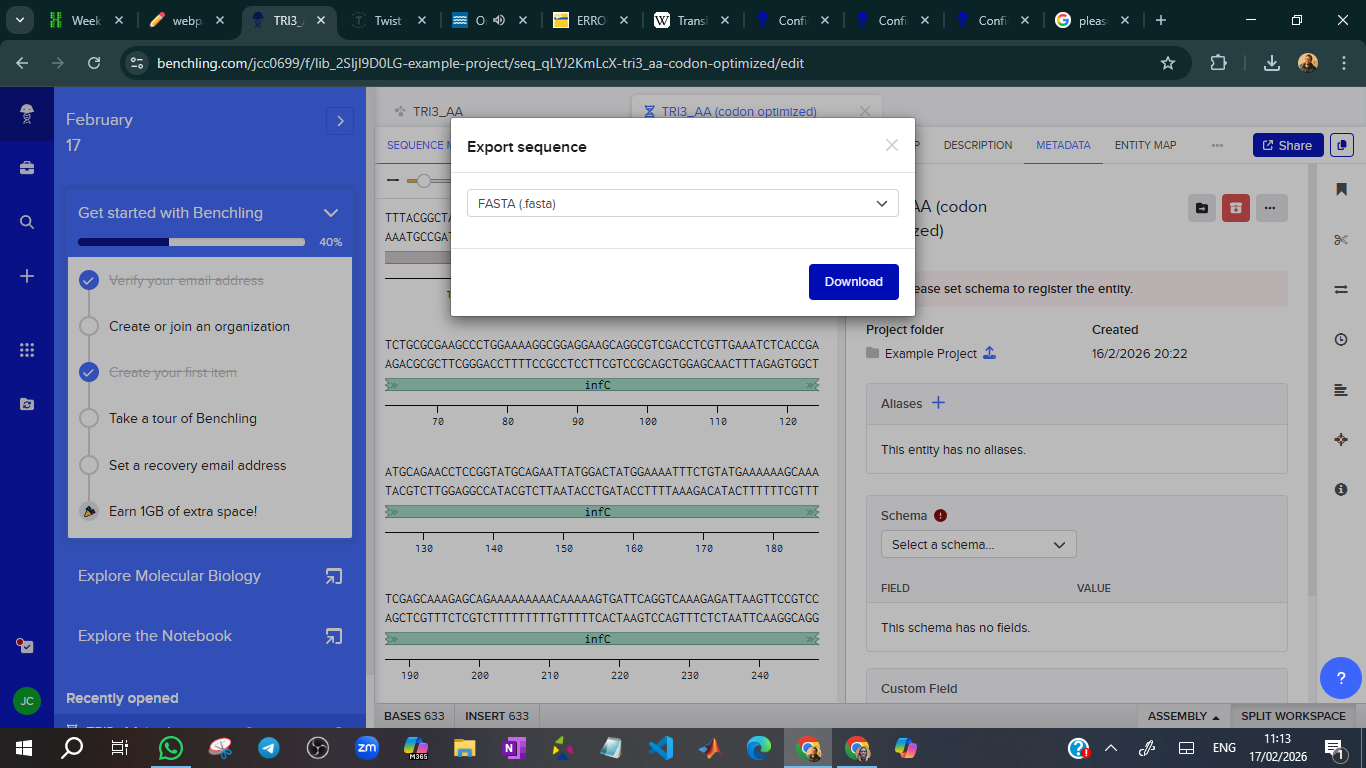
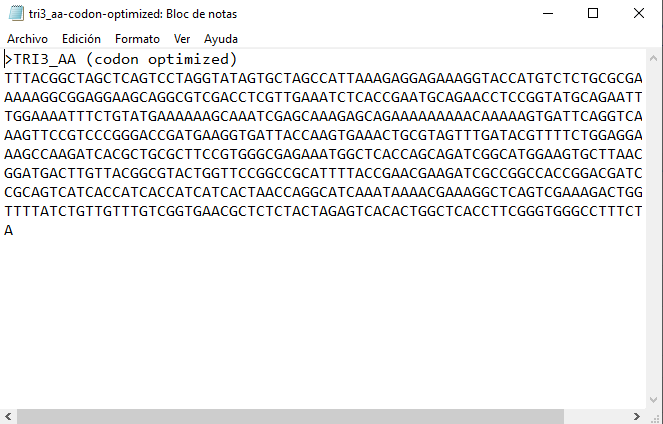
Step 4: Ordering in Twist
The process is shown in the figures below:

Step 5: Creating the plasmid
The process is shown in the figures below:
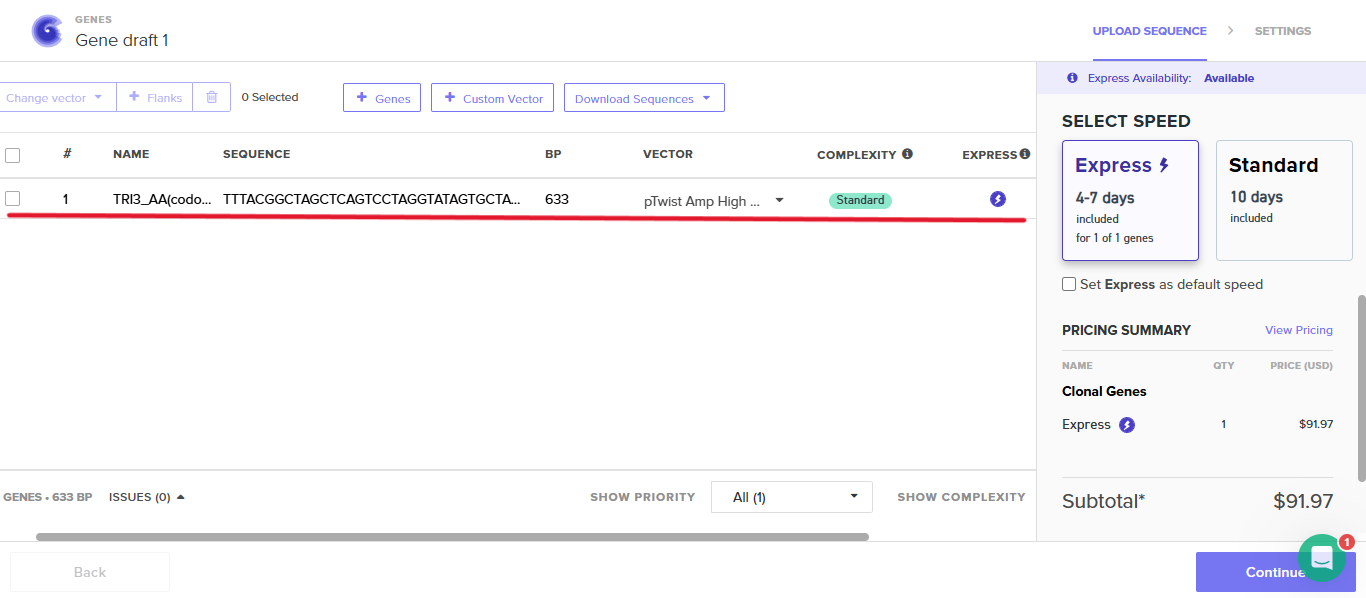
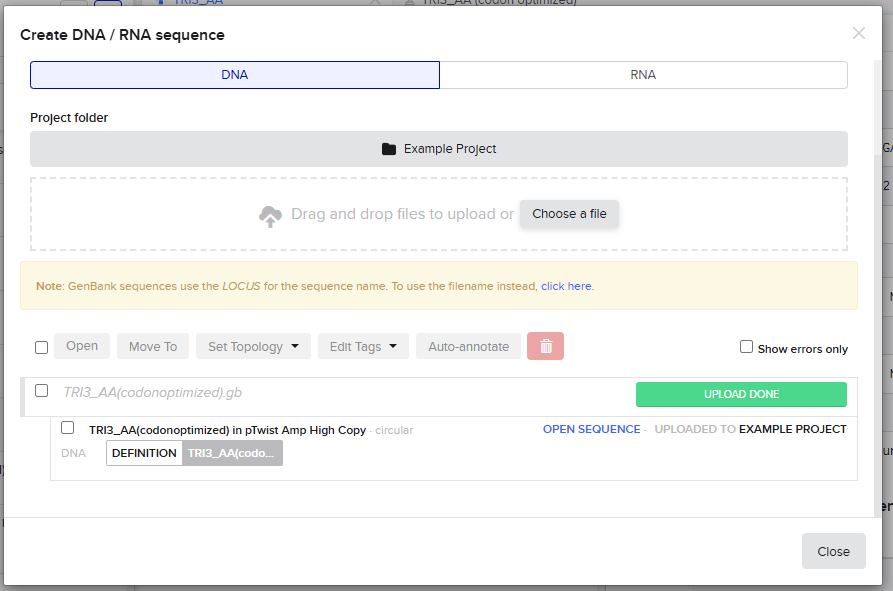
Finally, the plasmid is shown below:
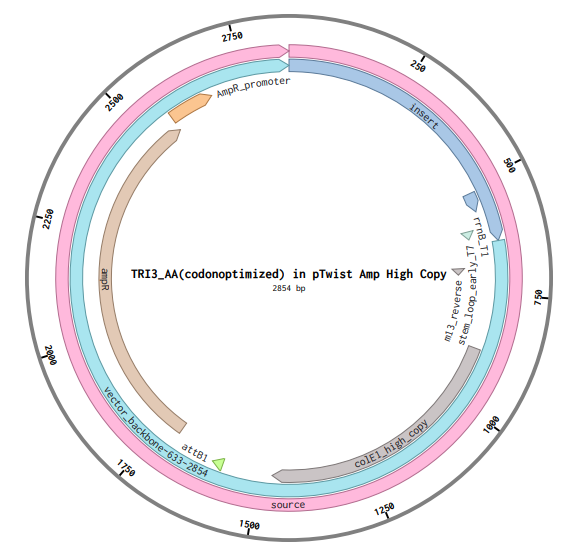
Part 5: DNA Read/Edit/Write
5.1.1: What DNA would you want to sequence (e.g., read) and why?
I would like to analyze DNA from insects such as flies, since many species act as vectors for infectious diseases. By sequencing their DNA, I could identify genetic elements associated with viral transmission, pathogen resistance, or susceptibility. This information could help improve disease monitoring and vector control strategies.
5.1.2: In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use second-generation sequencing technologies such as Illumina sequencing due to their high throughput, accuracy, and cost-effectiveness. Illumina sequencing is particularly efficient for short DNA fragments and allows the parallel sequencing of millions of reads, making it ideal for large-scale genomic analysis of insect populations.
5.2.1: What DNA would you want to synthesize (e.g., write) and why?
I would like to synthesize bacterial DNA initially because bacterial genomes are less complex than eukaryotic genomes, which makes them more manageable in terms of cost and laboratory procedures. This would allow me to gain experience with gene design and expression systems before working with more complex organisms.
5.2.2: What technology or technologies would you use to perform this DNA synthesis and why?
I would use common routes such as solid-phase phosphoramidite chemical DNA synthesis combined with gene assembly techniques. These methods allow precise synthesis of short oligonucleotides, which can then be assembled into longer DNA constructs. It is widely used, reliable, and scalable for constructing bacterial genes or plasmids.
5.3.1: What DNA would you want to edit and why?
I would edit DNA from mammalian cells, focusing on genes involved in the immune response. By modifying specific regulatory or coding sequences, it may be possible to enhance resistance to infectious diseases or better understand the mechanisms underlying autoimmune disorders. However, such research would need to be conducted carefully and ethically due to the potential implications of editing mammalian genomes.
PRE-LECTURE NOTES
Homework questions from Prof. Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
The error rate of polymerase is approximately 1 in a million nucleotides. Considering the human genome length of approximately 3.2 billion base pairs, or 6.4 billion nucleotides in a diploid cell, there would be roughly 6,400 errors per molecule of DNA, and half that number in a haploid cell. This implies a significant chance for defects or mutations to occur and potentially be passed down to offspring. However, biology has developed multiple mechanisms over the past century that increase the fidelity of DNA replication. For instance, MutS-1 is a protein shown to bind to mismatched DNA sequences. This mechanism therefore acts as an additional layer that improves the fidelity of de novo DNA synthesis (Carr et al., 2004).
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice, what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
There are approximately 20n ways to code a human protein, where n represents the length of the protein. For instance, a typical protein may consist of 300 amino acids. Therefore, there are 20300 possible ways, which corresponds to an extremely large number of potential coding sequences (Alberts et al., 2002). Some of the reasons these codes do not work in practice include:
Codon usage bias: The prevalence of a codon is related to its translation efficiency; some codons are translated faster than others. This impacts protein expression levels and availability (Chakravarty, 2026).
Protein structure: Since proteins fold co-translationally, changes in codon usage can alter the timing of folding events, affecting protein structure and function (Moss et al., 2024).
Homework questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Currently, oligo synthesis is most commonly performed using phosphoramidite nucleosides as building blocks. This process consists of four main chemical reactions: detritylation, coupling, capping, and oxidation (Kosuri & Church, 2014).
Why is it difficult to make oligos longer than 200 nt via direct synthesis?
The main challenge in synthesizing long oligonucleotides using standard phosphoramidite chemistry lies in cumulative yield loss and error accumulation. Unwanted reactions, such as depurination during detritylation, and incomplete removal of protecting groups can leave gaps in the oligo backbone, reducing overall yield. In addition, single-base deletions are the predominant errors caused by inefficiencies during these reaction steps.
Why can’t you make a 2000 bp gene via direct oligo synthesis?
Manufacturing an oligo of this length is highly prone to errors due to several factors. First, oligo concentrations obtained from a selected pool after processing are often quite low, reducing assembly efficiency. Second, when synthesizing large numbers of oligos, overlapping coding regions may introduce assembly errors at scale. Finally, significantly higher costs are required to produce the large number of strands necessary for successful gene assembly.
Homework question from George Church
Using Google & Prof. Church’s slide #4, what are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The ten essential amino acids in most animals are:
Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and sometimes Cysteine or Tyrosine, depending on species-specific metabolic capabilities (Hou and Wu, 2018).
Understanding the Lysine Contingency as a bioengineered constraint, the dependence of animals on multiple essential amino acids further strengthens this strategy. This dependency enables researchers to implement safer in vivo containment systems, as organisms lacking access to these amino acids are unable to survive outside controlled environments (Shivni, 2023).
References
Carr, A. M., Lambert, S., & Replication Stress Group. (2004). Mismatch repair proteins and DNA replication fidelity. Nucleic Acids Research, 32(20), e162. https://academic.oup.com/nar/article/32/20/e162/1115791
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2002). Molecular Biology of the Cell (4th ed.). Garland Science. https://www.ncbi.nlm.nih.gov/books/NBK26830/
Chakravarty, A. (2026). What is codon bias? GoldBio. https://www.goldbio.com/blogs/articles/what-is-codon-bias
Moss, A. J., et al. (2024). Codon usage and protein folding dynamics. PMC. https://pmc.ncbi.nlm.nih.gov/articles/PMC11227313/
Kosuri, S., & Church, G. M. (2014). Large-scale de novo DNA synthesis. Nature Methods, 11, 499–507. https://www.nature.com/articles/nmeth.2918
Hou, Y., & Wu, G. (2018). Nutritionally essential amino acids in animals. Advances in Nutrition, 9(6), 849–858. https://doi.org/10.1093/advances/nmy054
Shivni, R. (2023). A pioneer of the multiplex frontier. The Scientist. https://www.the-scientist.com/a-pioneer-of-the-multiplex-frontier-71132