Week 4 HW: Protein Design Part I
PART A:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assumptions:
- 500 g of meat
- ~31 g of protein per 100 g of meat (British Nutrition Foundation, 2021 )
- Average amino acid mass ≈ 100 g/mol
- Avogadro’s number = 6.022 × 10^23 molecules/mol
1. Protein content in 500 g of meat
$ 500 \,\text{g meat} \times \frac{31 \,\text{g protein}}{100 \,\text{g meat}} = 155 \,\text{g protein} $2. Convert grams of protein to moles of amino acids
$ 155 \,\text{g} \times \frac{1 \,\text{mol}}{100 \,\text{g}} = 1.55 \,\text{mol amino acids} $3. Convert moles to molecules
$ 1.55 \,\text{mol} \times 6.022 \times 10^{23} = 9.33 \times 10^{23} $Final Answer
$ \boxed{9 \times 10^{23} \text{ amino acid molecules}} $2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans need to feed on beef, fish and other nutrients to obtain energy and raw materials. Even though we consume proteins and nucleic acids that were originally built according to another organism’s DNA, digestion breaks them down into basic biomolecules, namely amino acids and nucleotides. Our cells then use our DNA to reassemble those building blocks according to human genetic instructions, not those of a cow or a fish.
3. Why are there only 20 natural amino acids?
There are around 500 aminoacids, but the only ones required for human protein building are 20.
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed through prebiotic chemical reactions before life emerged. Experimental evidence suggests they could have been synthesized under early Earth atmospheric conditions, through energy sources such as lightning, volcanic activity, and hydrothermal systems rich in sulfur compounds. Discuss how simple inorganic molecules, combined with energy input, could generate organic building blocks like amino acids without the need for enzymes.
Several scienties have tried to answer this question and, surprisginfly, they could have been synthetized artifically by the atmospheric conditions and the high-sulfured sea. This (Farias-Rico and Mourra-Diaz, 2022)
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Because D-amino acids are the mirror image of L-amino acids, the energetically favorable backbone torsion angles are also inverted. As a result, the most stable α-helix formed by D-amino acids is left-handed.
7. Can you discover additional helices in proteins?
Yes. In fact, some studies have identified different forms of alpha helix in globular proteins, namely linear, curved or kinked (Kumar & Bansal, 1998). Adititionaly, there are 3~10 and $\pi$ helix which are less favourable in trerms of stability but still occur (Kumar et al., 2022)
8. Why are most molecular helices right-handed? This is because most amino acids are D-oriented.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation
β-sheets tend to aggregate because exposed backbone hydrogen bond donors and acceptors allow β-strands from different molecules to bind to each other. Hydrophobic side chains further stabilize these interactions, making sheet stacking energetically favorable. Aggregation is driven mainly by intermolecular hydrogen bonding and hydrophobic interactions. These forces lower the system’s free energy and promote ordered β-sheet assembly, similar to crystallization. (Chen et al., 2017)
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Many amyloid diseases involve β-sheet structures because the cross-β arrangement is highly stable and self-propagating. This stability allows misfolded proteins to accumulate as insoluble fibrils that disrupt normal cellular function. (Chen et al., 2017). Despite their toxic effects on health, there is increasing research pertaining their development as materials for several applications (Yadav et al., 2024).
PART B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
In this case I chose the laccase from Bacillus subtilis cotA, an enzyme shown to have multiple applications, ranging from bioremedation to dye breakdown. It belongs to the family of multicopper oxidases (MCOs), which are capable of oxidizing a significant amount of chemical compounds. Since one of my project ideas was to determine the optimal concentration of laccase for optimal wastewater treatment in textile factories, I thought it would be a great start to get used to the different tools we have for protein design.
2. Identify the amino acid sequence of your protein.
The sequence is:
\ \ MTLEKFVDALPIPDTLKPVQQSKEKTYYEVTMEECTHQLHRDLPPTRLWGYNGLFPGPTIEVKRNENVYVKWMNNLPSTHFLPIDHTIHHSDSQHEEPEVKTVVHLHGGVTPDDSDGYPEAWFSKDFEQTGPYFKREVYHYPNQQRGAILWYHDHAMALTRLNVYAGLVGAYIIHDPKEKRLKLPSDEYDVPLLITDRTINEDGSLFYPSAPENPSPSLPNPSIVPAFCGETILVNGKVWPYLEVEPRKYRFRVINASNTRTYNLSLDNGGDFIQIGSDGGLLPRSVKLNSFSLAPAERYDIIIDFTAYEGESIILANSAGCGGDVNPETDANIMQFRVTKPLAQKDESRKPKYLASYPSVQHERIQNIRTLKLAGTQDEYGRPVLLLNNKRWHDPVTETPKVGTTEIWSIINPTRGTHPIHLHLVSFRVLDRRPFDIARYQESGELSYTGPAVPPPPSEKGWKDTIQAHAGEVLRIAATFGPYSGRYVWHCHILEHEDYDMMRPMDITDPHK
Using the code provided, the most common aminoacid is Proline (P) which appears 46 times.
Some additional facts were obtained from UniProt:
- Lenght (Number of AAs): 513
- Molecular mass: 58.5 kDa
- Family: Multicopper Oxidase
- Number of homologues: 242
3. Identify the structure page of your protein in RCSB
The protein’s structure was solved in 2003 by Francisco J. Enguita and collaborators, with a resolution of 1.70 Å. There are 4 ligands: C2O, GOL, C1O and CU. It belongs to the Laccase family.
4. Open the structure of your protein in any 3D molecule visualization software
The following images of the protein were obtained from PyMOL:
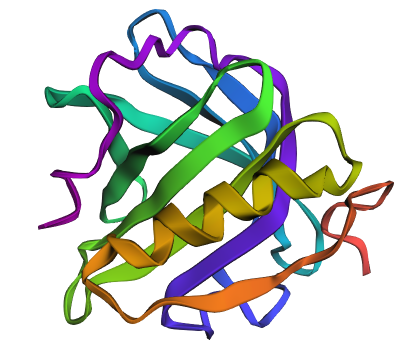
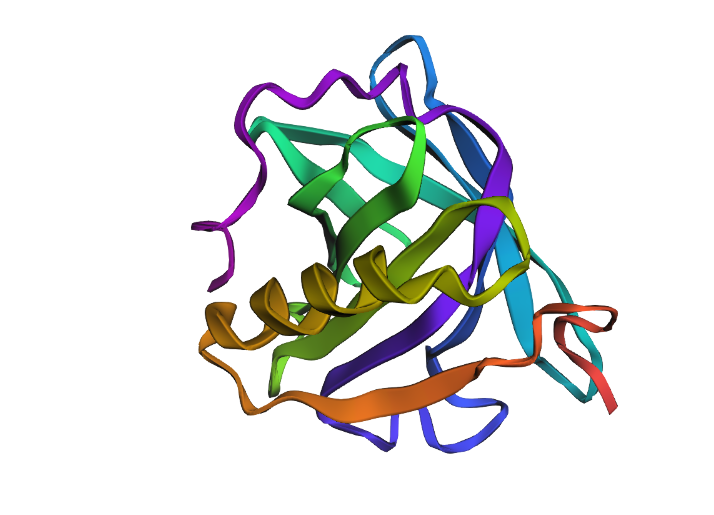
Main structure
.png)
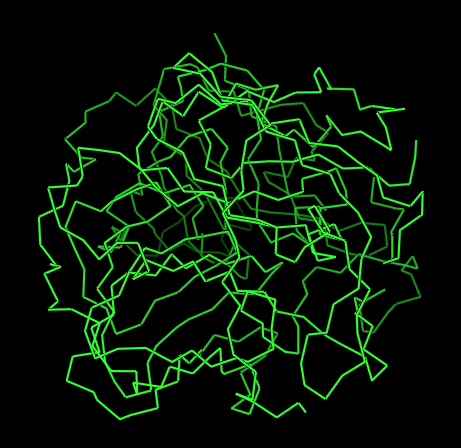

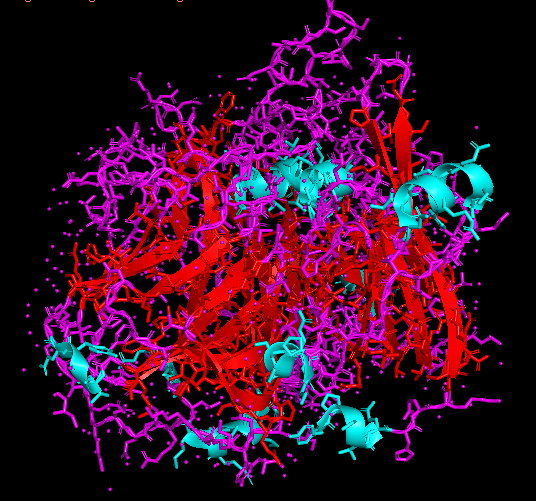
Figure 1 presents different structural representations of CotA. The cartoon view highlights its compact, globular fold dominated by β-sheets and connecting loops, characteristic of bacterial laccases, while the ribbon representation emphasizes the backbone organization and overall topology. The balls-and-sticks model displays all atoms explicitly, revealing dense atomic packing and the presence of copper ions within the protein core, coordinated by conserved histidine residues typical of multicopper oxidase active sites.
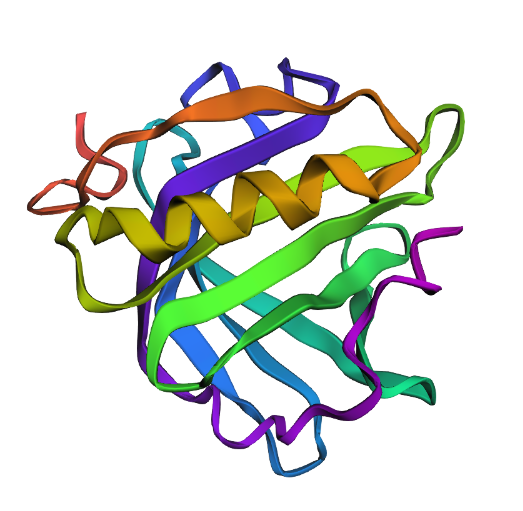
Secondary structures
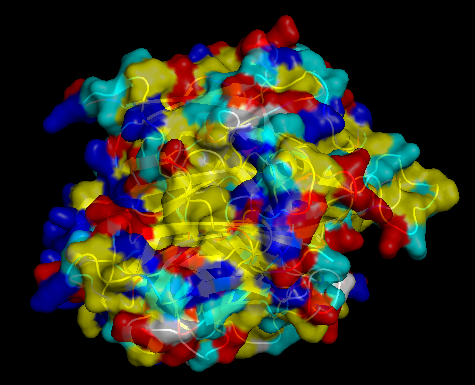
The surface of CotA was colored according to residue type: hydrophobic residues in yellow, polar uncharged in cyan, positively charged in blue, and negatively charged in red. The visualization shows a typical globular organization, with hydrophobic residues mainly buried in the protein core, contributing to structural stability, and polar and charged residues predominantly exposed on the surface, supporting solubility and potential functional interactions. The distribution is consistent with CotA’s role as a multicopper oxidase.
PART C: Using ML-Based Protein Design Tools
1. Protein Language Modeling
The selected protein was the $\beta$-lactoblogulin which is the main component of whey protein.
Sequence
\ \ 3NPO_1|Chain A|Beta-lactoglobulin|Bos taurus (9913) LIVTQTMKGLDIQKVAGTWYSLAMAASDISLLDAQSAPLRVYVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTKIPAVFKIDALNENKVLVLDTDYKKYLLFCMENSAEPEQSLACQCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI
Deep Mutational Scans
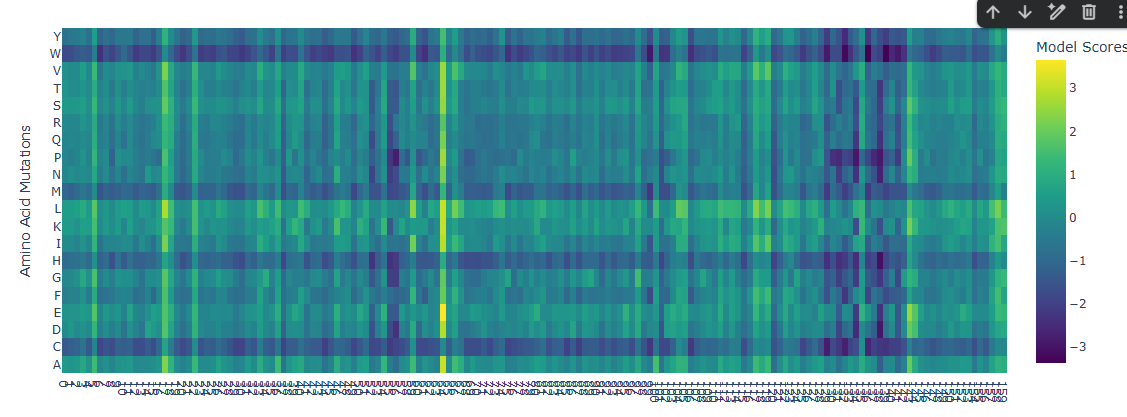
A few mutational hotspots were identified, particularly around positions 17, 63, 142 and 158. These sites show a tendency toward substitution to Leucine (L) and Glutamic Acid (E). Given that these residues appear to be located on the outer region of the protein, this pattern may reflect structural permissiveness. Substitution toward Leucine could enhance local hydrophobic packing, whereas Glutamic Acid, being negatively charged, may be well tolerated on the protein surface. This suggests that these positions are structurally flexible and can accommodate both hydrophobic and charged residues without significantly disrupting the overall fold.
Latent Space Analysis
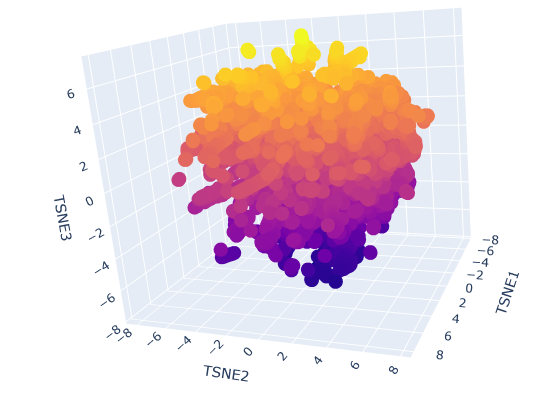
2. Protein Folding
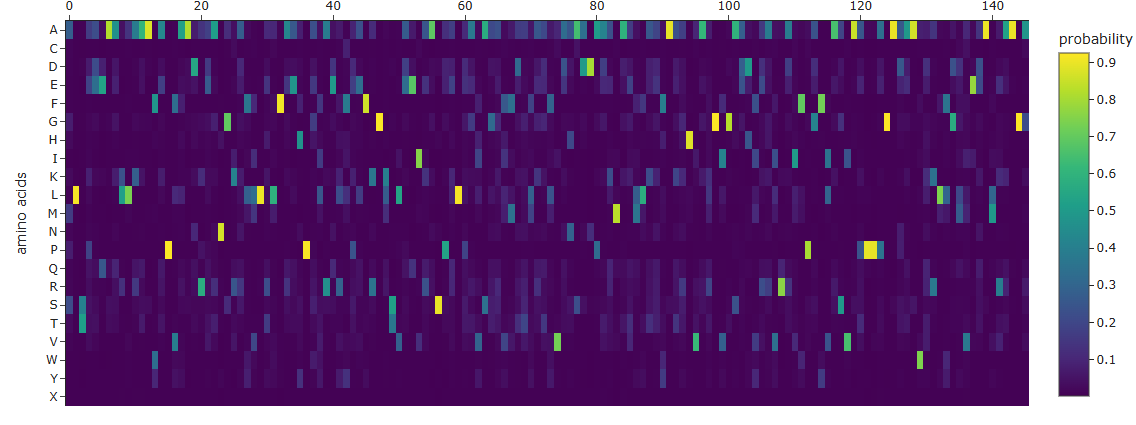
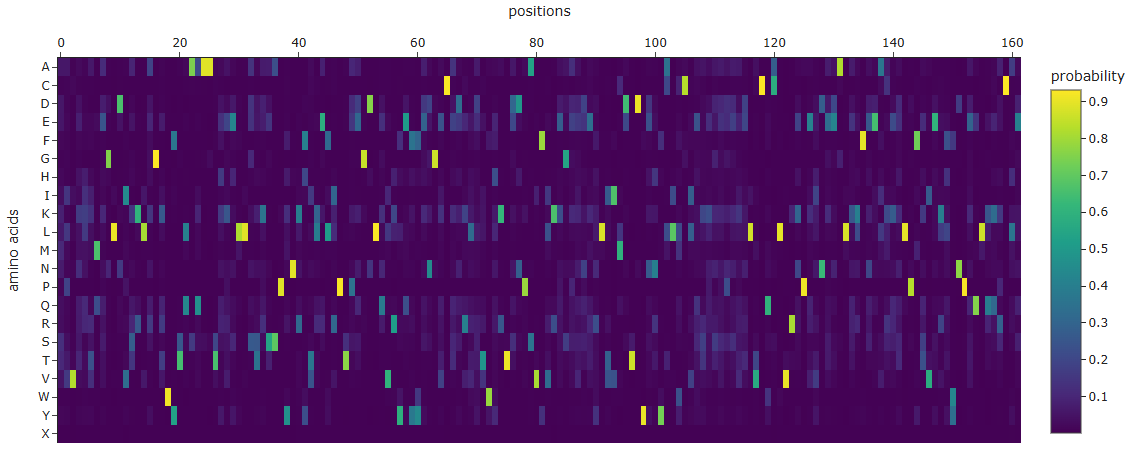
I was able to obtain the amino acid probabilites, which may help me in the future to guess the most likely mutations and try and fold again the proteins.
By mutating residues no. 1 (L), 15 (P), 36 (P), 122 (P), and 29 (L), the following sequence variants were obtained:
Variant 1
Mutations:
- L1 → I
- P15 → A
Sequence:
IIVTQTMKGLDIQKVAGTWYSLAMAASDISLLDAQSAPLRVYVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTKIPAVFKIDALNENKVLVLDTDYKKYLLFCMENSAEPEQSLACQCLVRTPEVDDEALEKFDKALKALPMHIRLSFNPTQLEEQCHI
Variant 2
Mutations:
- L29 → F
- P36 → G
- P122 → S
Sequence:
LIVTQTMKGLDIQKVAGTWYSLAMAASDISFLDAQSAGLRVYVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTKIPAVFKIDALNENKVLVLDTDYKKYLLFCMENSAEPEQSLACQCLVRTPEVDDEALEKFDKALKALSMHIRLSFNPTQLEEQCHI
Variant 3
Mutations:
- L1 → D
- P15 → G
- L29 → N
- P36 → G
- P122 → G
Sequence:
DIVTQTMKGLDIQKVGGTWYSLAMAASDISNNDAQSAGLRVYVEELKPTPEGDLEILLQKWENGECAQKKIIAEKTKIPAVFKIDALNENKVLVLDTDYKKYLLFCMENSAEPEQSLACQCLVRTPEVDDEALEKFDKALKGALPMHIRLSFNPTQLEEQCHI
3. Protein Generation
New sequence
MPVKKTMEGLDISKLAGKWYTQAQAATKKELLSTKSSPYNRFTLELIPTPEGDLQVRYEYYENGECKDRLDTWHKTDDPAVFVKEGEEDERLVIMDTDYNNYALWCIEKKSEPEKELVCQCLVRKPEINEEALKKFEEAKKDLPFTVEKFWNPEQLQKRCLE
Mutations
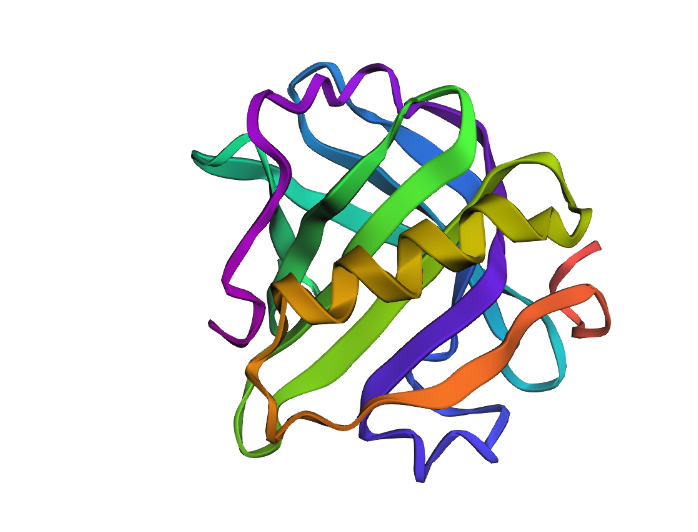
Then, the two proteins, both original and new, are shown below:
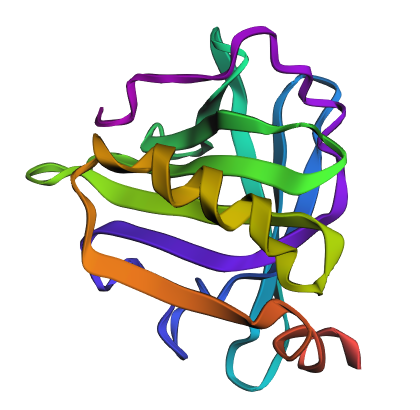
Both sequences appear to preserve the same overall structural fold, as several conserved motifs and key residues remain unchanged, including regions such as PTPEGDL, DTDY, and VCQCLVR. These conserved segments suggest that the core structural or functional features of the protein were maintained.
However, the new sequence introduces substantial changes in amino acid composition, with an increase in charged and polar residues such as lysine (K), glutamate (E), and glutamine (Q), alongside a reduction in hydrophobic residues. Structurally, this likely makes the redesigned protein more soluble and less prone to aggregation, while also increasing surface polarity and local flexibility. In contrast, the original sequence appears to contain a more hydrophobic and compact core, which may contribute to greater structural rigidity and stability.
PART D: Group Brainstorm on Bacteriophage Engineering
Group Members: Alvaro Pacheco (Lima, PE) and Renzo Condori (Lima, PE)
Goals
- Perform mutagenesis on the LS-motif to enhance stability
- Modify the promoting region of the DNA sequence to express larger amounts of the MS2 protein
Tools
-AlphaFold: Predicts 3D structure of the mutant variants. It allows to evaluate if such mutations maintain the transmbembrane topology and general conformation, verifying if the functional motif LS keeps its orientation and stability.
-FoldX/Rosetta: It will estimate the change in free energy due to mutations. It eases the selection of mutant variants which are more likely to provde a thermodinamic stability, reducing the number of prospects.
-GROMACS: It allows to simulate and analyze the protein stability in a bacterial environment.
These tools complement each other by combining evolutionary, structural, and physical insights to improve MS2 lysis protein stability. Protein Language Models suggest mutations consistent with evolutionary constraints, AlphaFold2 screens for preserved structure and topology, energy calculations prioritize stabilizing variants, and molecular dynamics simulations test behavior in membrane conditions. Together, they enable rational design of stabilizing mutations while reducing the risk of impairing lytic function.
Pitfalls
One potential pitfall is the limited accuracy of current prediction tools for small, membrane-associated, and partially disordered proteins, which may reduce the reliability of both structural and energetic predictions. A second limitation is the trade-off between stability and function: increasing stability may reduce the conformational flexibility required for interaction with the membrane target, potentially impairing lytic activity.
Pipeline
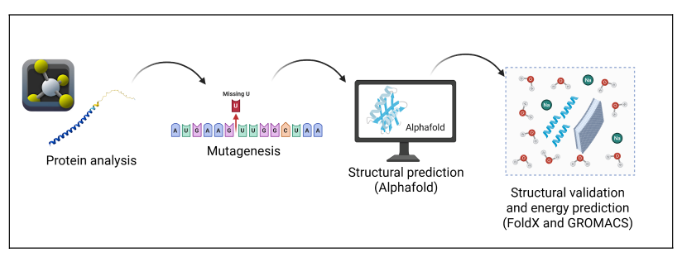
Procedure
Functional Analysis and Definition of Constraints The conserved transmembrane motif Leu48–Ser49 (LS) and its immediate surrounding region will be identified as critical functional regions that must not be mutated.
Directed In Silico Mutagenesis Mutant variants will be generated using protein language models, restricting mutations to the remainder of the sequence.
Preliminary Energy Filtering Variants will be evaluated through ΔΔG stability predictions, selecting those with improved thermodynamic stability.
Structural Prediction Selected variants will be modeled using AlphaFold2 to verify preservation of the transmembrane domain and overall conformation.
Dynamic Validation in a Membrane Environment Top candidates will be evaluated through molecular dynamics simulations in a bacterial membrane environment.
Final Variant Selection Mutants showing the best balance between enhanced stability and functional conservation will be selected.