Week 4: Protein Design Part I
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Objective:
- Learn basic concepts:
- amino acid structure
- 3D protein visualization
- the variety of ML-based design tools
- Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
For 500 grams of meat, there is roughly 20-25% grams of protein. This means that roughly 100 grams belong to protein, while there is remaining fat, fiber, and water that make up the rest of the mass. Because 1 mole = 100 Da Number of moles = 100 g of protein / 100 Da = 100g/ 100 g / mol $$\text{Molecules} = 1 \text{ mol} \times 6.022 \times 10^{23} \text{ molecules/mol}$$$$\text{Molecules} \approx 6.022 \times 10^{23}$$ There are roughly 602 sextillion amino acids.
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans eat beef, through mastication and digestion we break down the beef into smaller units. First protein is broken down by enzymes (proteases) and into shorter chains of amino acids in the stomach. Then the chains become further broken down into individual amino acids in the small intestine. As these amino acids enter the bloodstream, they require DNA to instruct them into building other things. The human DNA does different things than cows and fish, therefore the amino acids will build a cow or a fish.
- Why are there only 20 natural amino acids?
It may be an evolutionary mystery that almost all living things are built from these 20 natural amino acids. The 20 amino acids serve as the building blocks of most proteins, they line up as codons in 3-letter assemblies, in which the ribosomes read to create actions following the DNA sequence. When they read 3 bases at once, the combinations create 4^3 possibilities that are expansive enough for the making of diverse lifeforms.
- Can you make other non-natural amino acids? Design some new amino acids.
Yes, there are a lot of non-natural amino acids. Designing new amino acids require us to follow the same chassis but redesign the ‘r-group’ to alter the chemistry of the bond, which is the side chain of the amino acid. One may attach an azide to the chain to create a strong bond for stickiness or bio-glue. For experiments, some researchers also use non-natural florescent amino acids like Acridonylalanine to glow under microscopy or photographs.
- Where did amino acids come from before enzymes that make them, and before life started?
This might be related to assembly theory? Lee Cronin proposed that life is composed of different assemblies, in that life is scaffolded by energy, raw sources, and minerals through complex interactions and then becomes amino acids, and longer chains. Gases and energy together can create amino acids. The Miller-Urey Experiment use water, methane, ammonia, and hydrogen to create amino acids.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed. D-amino acids create a mirror image of α-helixes, because the building blocks and the structure are completely mirrored.
- Can you discover additional helices in proteins?
Yes, since 2020, AlphaFold has allowed us to quickly discover new helices and the instructions to their fold, revealed millions of protein structures.
- Why are most molecular helices right-handed?
Because of chirality, most helices are non-identical to their mirror image. As most amino acids are L-form (left-handed), the way they most efficiently stack together is twisting to the right where they can create stable bonds with enough room between each other.
- Why do β-sheets tend to aggregate?
β-sheets bond together via hydrogen bonds. The geometry appears like pleated, zigzag, sheet-like structure with side chains protruding.
- What is the driving force for β-sheet aggregation?
They tend to aggregate because of its geometry, where the hydrophobic faces might sandwich and stick together to hide from water. The force from the water becomes driving force for clumping.
- Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
- Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it.
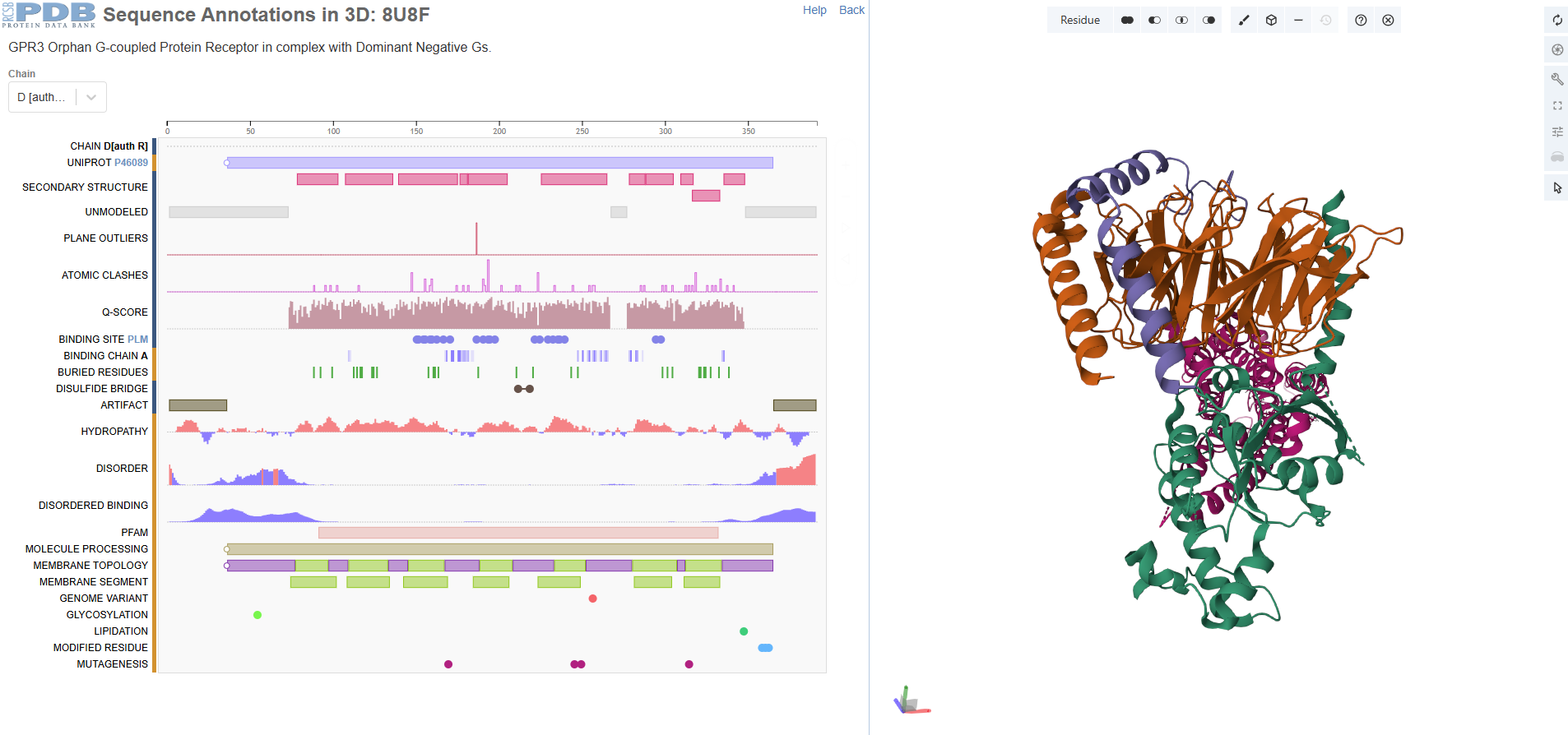
- Identify the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
There are four protein chains. Chain A: 372, Chain B: 339, Chain C: 58, Chain D: 384. The most frequent amino acid seems to be leucine. It is a sturdy, hydrophobic (water-hating) amino acid.
- How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
There are thousands of homologs, incuding human, pygmy chimpanzee, olive babboon, cotton-top tamarin, etc. The protein seems highly conserved and not changed.
- Does your protein belong to any protein family?
G Protein-Coupled Receptor (GPCR) Family
- Identify the structure page of your protein in RCSB
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure is solved around 2023 September and released 2024 Match. The method id electron microscopy but resolution 3.49 Å.
- Are there any other molecules in the solved structure apart from protein?
Yes, I see palmitic acid in the structure apart from protein.
- Does your protein belong to any structure classification family?
It belongs to a membrain protein, and falls under 7-transmembrane receptive (GPCR).
- Open the structure of your protein in any 3D molecule visualization software:
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon
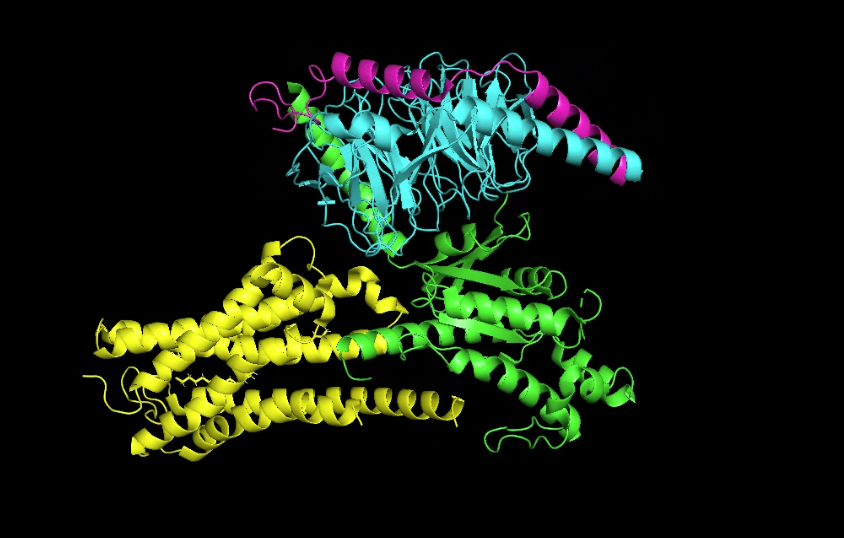 Ribbon
Ribbon
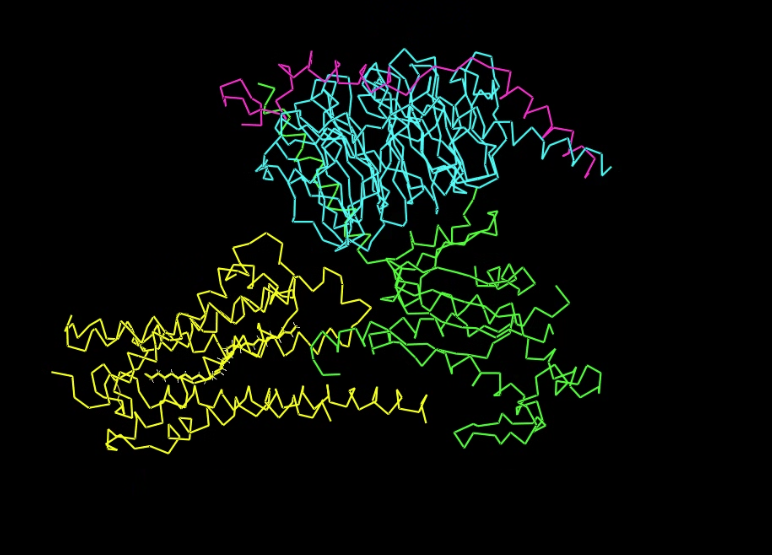 Ball and stick
Ball and stick

- Color the protein by secondary structure. Does it have more helices or sheets?
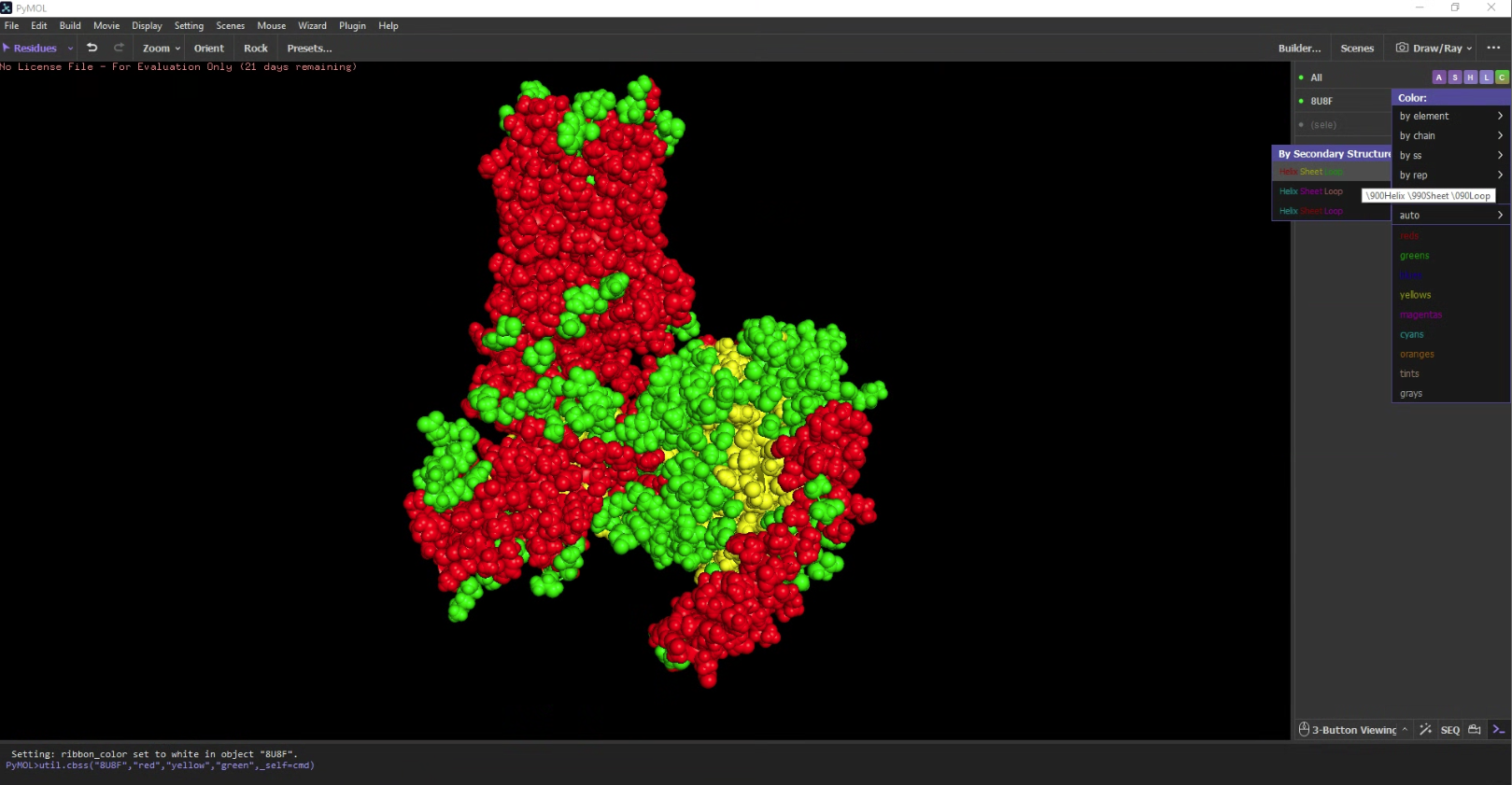 It has a lot more helices than sheets.
It has a lot more helices than sheets. - Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
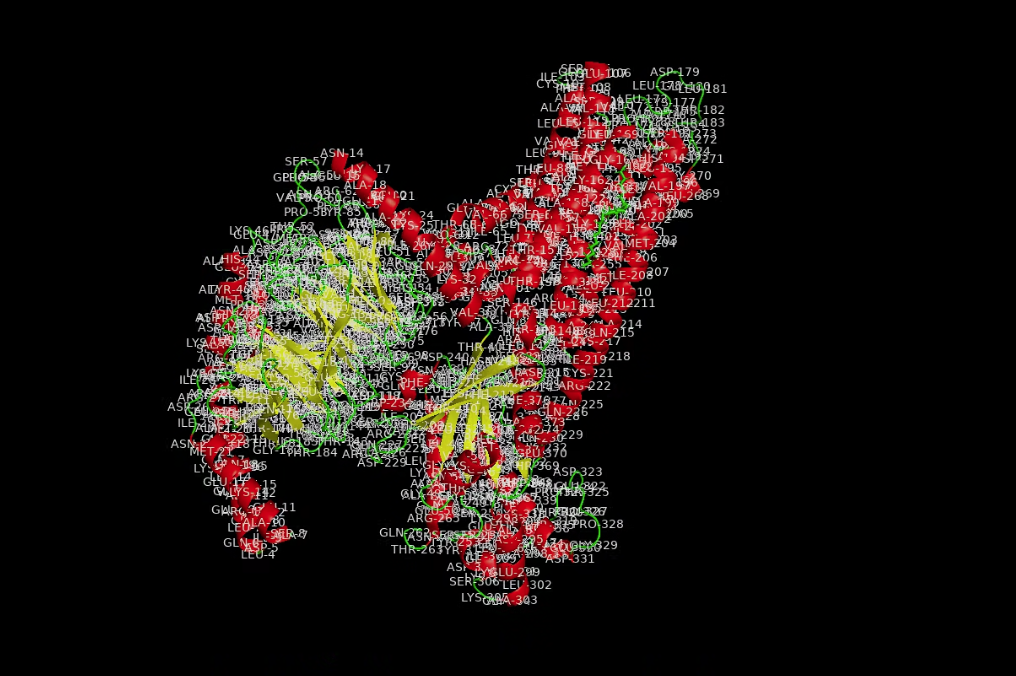 I used an additional script to label the hydrophobicity scale. Hydrophobic residues are red and hydrophilic (polar/charged) residues are white. It is slightly more hydrophobic.
I used an additional script to label the hydrophobicity scale. Hydrophobic residues are red and hydrophilic (polar/charged) residues are white. It is slightly more hydrophobic.
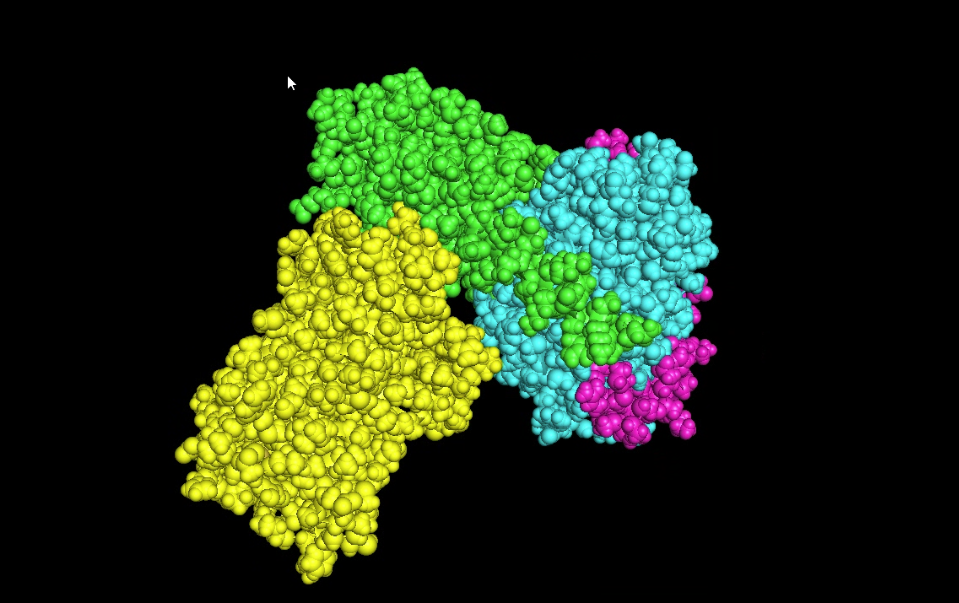
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
Assignees for the following sections
| MIT/Harvard students | Required |
| Committed Listeners | Required |
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
- Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
- Choose your favorite protein from the PDB.
- We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling

Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. >Using ESM2 mutational scans, 8U8F looks like > It appears that there are vertical bands in the sequence where across different amino acids, it's predicted to have a low score. This might be due to highly conserved functional and structural reasons. Lysine is the most common amino acid, but it also shows lots of dark spots and low scores because it is may have a hydrophobic mismatch. >There is a yellow band at position 243. >It is interesting Lysine is charged and has lots of blue bands, Leucine is neutral and is mostly high on the score.
- (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
) Latent Space Analysis
- Use the provided sequence dataset to embed proteins in reduced dimensionality. >
C2. Protein Folding
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
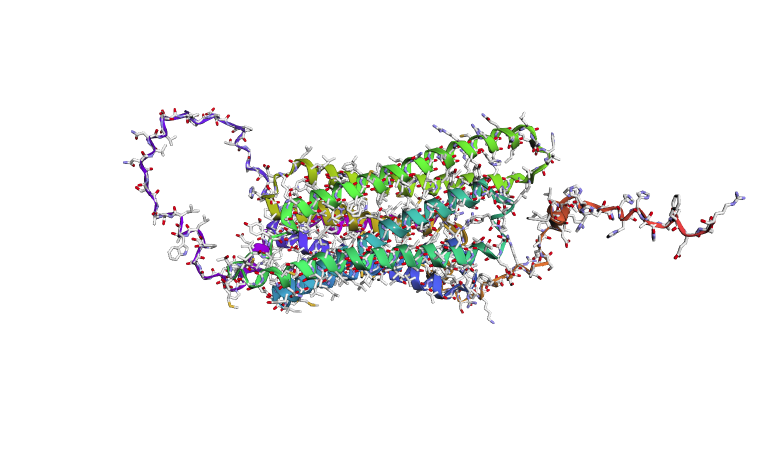
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I tried changing small snippets of the sequence and it wasn’t as visible, but adding longer sequences of the same amino acid allowed twists to be more visible.
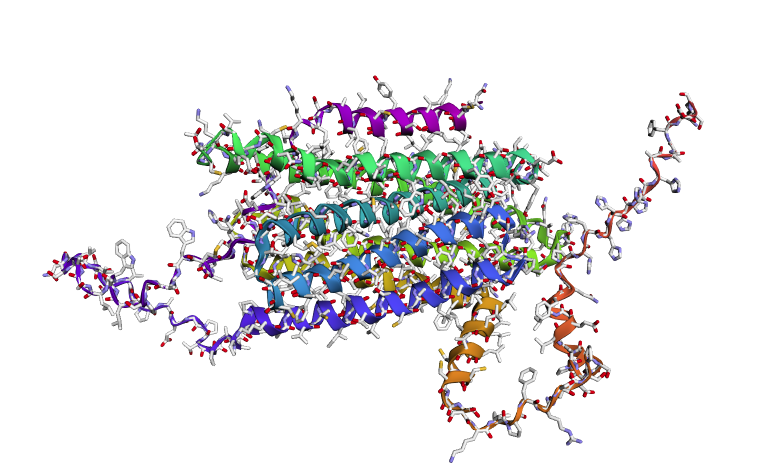
C3. Protein Generation
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Using the fixed-backbone design, we kept the 3D shape of 8U8F Chain A and reskinned a sequence. ProteinMPNN ended up rewriting 75% of the protein, there is a high frequency of Leucine and Lysine.
Google Colab doesn’t work with GPU acceleration so I’ve cloned to work locally.
- Input this sequence into ESMFold and compare the predicted structure to your original.
The predicted structure has retained the structure but upon comparison on PyMOL, the white structure (new) looks displaced.
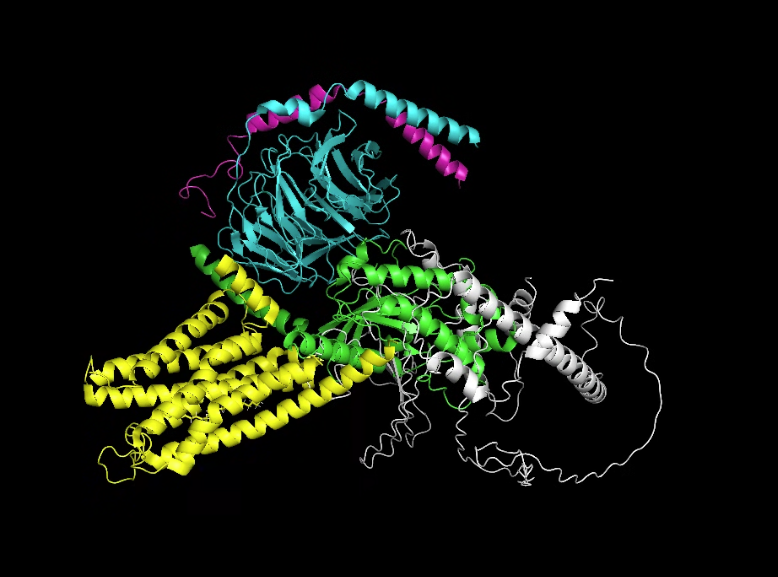
Part D. Group Brainstorm on Bacteriophage Engineering
Assignees for the following sections
| MIT/Harvard students | Optional |
| Committed Listeners | Required |
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
Brainstorm Session
- Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
optimizing protein’s binding affinity to e coli to accelerate lysis trigger increasing stability of L protein, ensuring proteins are folded and integrated into membrane to perform function.
Write a 1-page proposal (bullet points or short paragraphs) describing:
- Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
We would like to use protein language models such as ESM2 in the colab document to perform in sillilco mutagenesis. We will calculate single point mutations in the L protein sequence, and try to idenitfy mutations that are more evolutionarily favorable. Like the assignment I am interested to use ProteinMPNN for to redesign and generate a new sequence. Given the backbone structure of the L protein, this tool will help us generate alternative sequences that maintain the same fold but with higher thermal stability, thereby achieving our goals. AlphaFold Multimer was particularly interesting too, as it predicts 3D structures of protein complexes (co-folding multiple chains). Novel complexes create range and breadth.
- Why do you think those tools might help solve your chosen sub-problem?
ProteinMPNN was very robust in developing sequences that fit a specific shape, there is guarantee we will be able to increase protein stabililty. ESM2 allows us to scan so many mutations at once, which allows us to very quickly narrow down a direction that we couldn’t perform in wet lab setting.
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
L protein is a membrane protein. Most standard protein models like AlphaFold multimer seem to be trained primarily on soluble proteins. The specific lipid-protein interactions required for lysis may not be fully captured, leading to “stable” designs that fail to insert into the membrane. In my assignment I don’t understsand still how the shape will fit as it seems displaced?
- Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
- Include your group’s short plan for engineering a bacteriophage
Input a L protein sequence > use ESM2 to generate favorable mutations, the heat map should show us green-light vs no-go directions in the sequence > Use protein MPNN to generate and find a skeleton template for core stability > add complexity via alphaFold, predicting an interaction. >use PyMOL to check shape and geometry > calculate binding affinity score via colab > and select best candidates!