Week 5: Protein Design Part II
Week 5: Protein Design Part II
Homework — DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
- Design short peptides that bind mutant SOD1.
- Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
- PepMLM: target sequence-conditioned peptide generation via masked language modeling
- PeptiVerse: therapeutic property prediction
- moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Modified A4V:
- Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
- Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
0 WRSGATVARHAX 6.030266 0 WRYGAAAVELKE 11.785982 0 WHSGVVGLARGX 6.638643 0 WSYPWVALELGK 16.418794
- To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Pseudo Perplexity for binder ‘FLYRWLPSRRGG’ with protein sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ is: 20.63523127283615
- Record the perplexity scores that indicate PepMLM’s confidence in the binders.
This is PepMLM’s most confident score: WRSGATVARHAX.
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
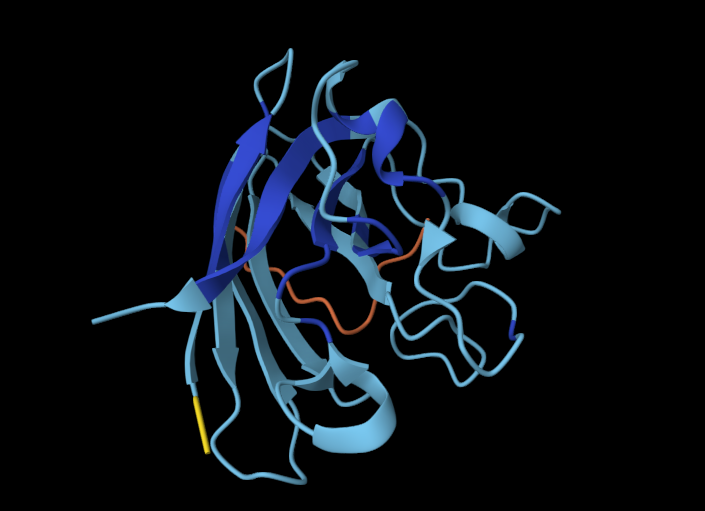
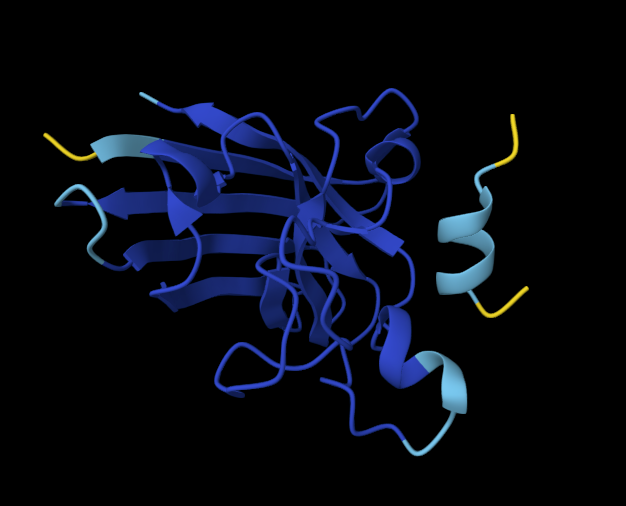
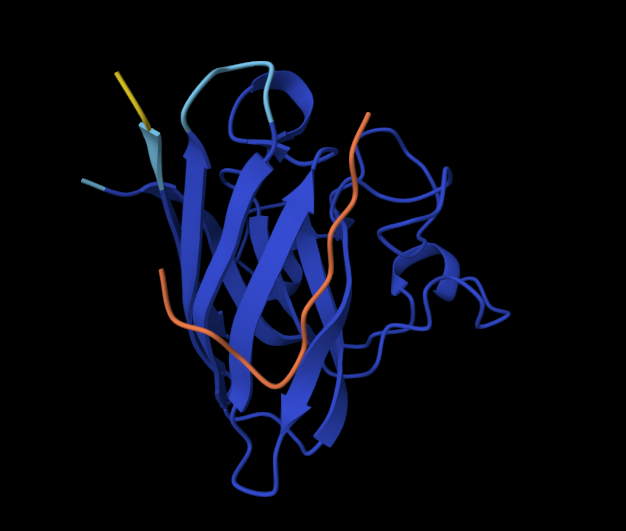
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
One of the PepMLM-generated peptides (ipTM = 0.67) significantly outperforms the others and appears to exceed the confidence of the known literature binder (which often scores in the 0.50–0.60 range in similar AlphaFold benchmarks). While the sequence WRYGAAAVELKE (ipTM = 0.28) failed to find a stable “home” on the SOD1 surface, the high-scoring candidate suggests that PepMLM successfully identified a sequence that “staples” the protein’s interface. This indicates that the language model can indeed generate de novo sequences that are more structurally compatible with the mutated A4V surface than existing experimental peptides.
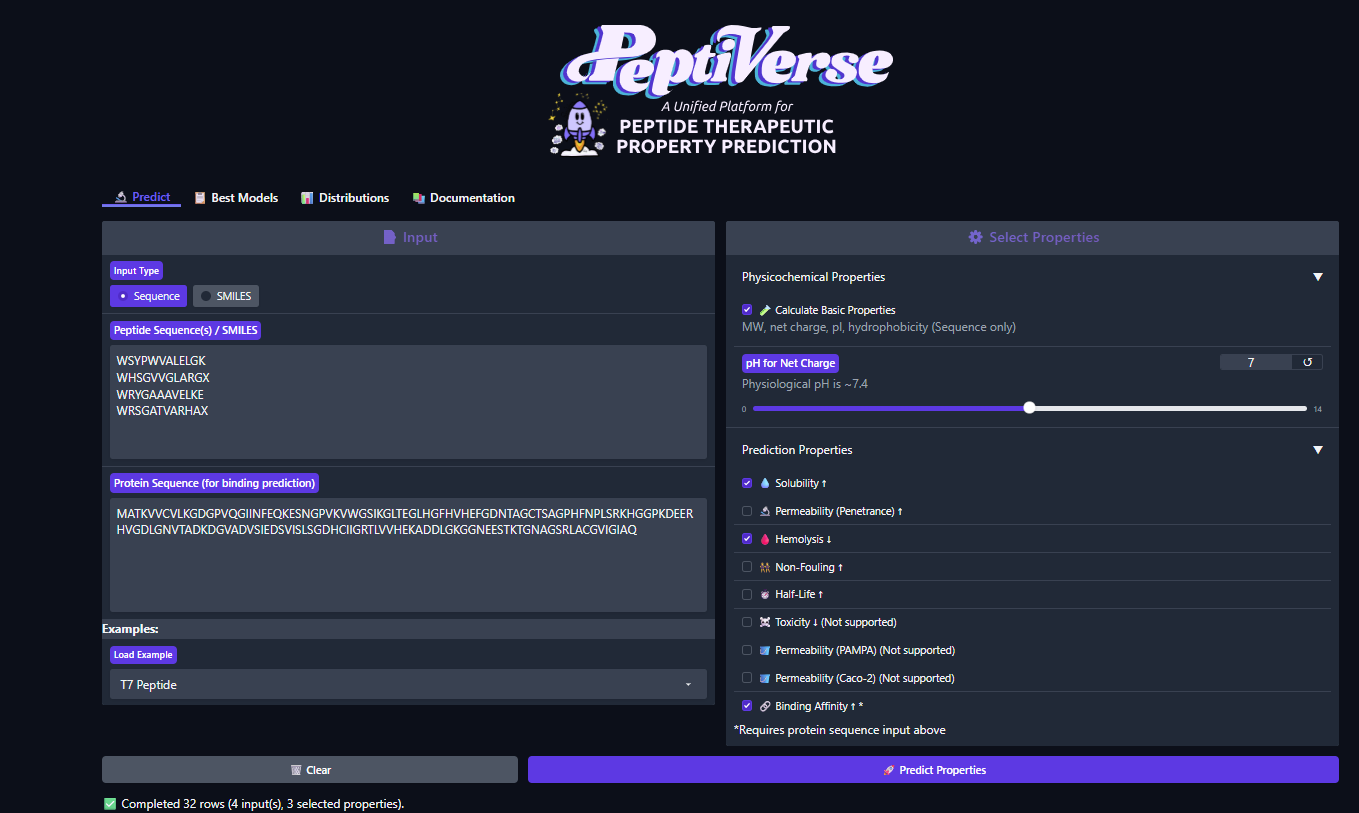
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
- Paste the peptide sequence.
- Paste the A4V mutant SOD1 sequence in the target field.
- Check the boxes:
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
I would choose to advance WRYGAAAVELKE as the best option. First it has great binding strength, with the highest predicted binding affinity ($pK_d \approx 6.27$), which is roughly in the micromolar range, a great starting point for a de novo peptide.
The fact that it has a negative GRAVY score (-0.38) shows that it is more hydrophilic than others. This will help with solubility and lower hemolysis risk (0.049).
Structurally, although its initial ipTM was low, its chemical makeup makes it a better scaffold than a peptide that might bind tightly but aggregate in the blood.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
- Open the moPPit Colab linked from the HuggingFace moPPIt model card
- Make a copy and switch to a GPU runtime.
- In the notebook:
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Peptide 2: YYLRRCGYYQRV Affinity score: 8.33 Solubility score: 0.79
moPPit optimizes for binding. So it will generate sequences often with higher predicted affinity scores than PeptiVerse. Peptide 1 EPTEEEQRTCGT has a superior affinity score and is physically complementary to the A4V sequence.
Part B: BRD4 Drug Discovery Platform Tutorial (Optional)
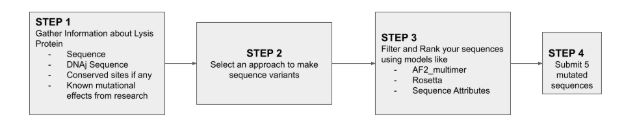
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
This homework requires computation that might take you a while to run, so please get started early.
Reading & Resources
Tools
- HTGAA Protein Engineering Tools spreadsheet