Jenn Leung is a researcher and creative technologist working at the intersection of synthetic biology, real-time simulation, and living neural systems. She is a Senior Lecturer in Creative Technology & Design at University of the Arts London, a researcher at LifeFabs Institute, and a Visiting Researcher at The Bartlett School of Architecture, UCL, working on the 100 Minds in Motion project combining EEG, eye-tracking, and movement data within agent-based simulation.
Her research currently focuses on developing Unreal Engine interfaces for living neurons and agent behaviour simulations. Two papers have recently been published in the MIT Antikythera Journal, and a paper on UE-API for brain-on-a-chip platforms was presented at NeurIPS 2025. Since 2025, she has collaborated with the biocomputing start-up Cortical Labs to create human-synthetic biological intelligence visualizations. In 2026, she is also collaborating with Michael Levin, Agnieszka Kurant, and Emily Ertle for Rhizome’s 7x7 programme.
Previously, Jenn was a studio researcher at Antikythera’s Cognitive Infrastructures Studio in 2024, supported by the Berggruen Institute, led on community tech support for Off World Live, and served as Programme Head at the Architectural Association VS Unit 5 Xalon. Her work has been exhibited at Epic Games Innovation Lab, Ars Electronica, Medialab Matadero, W1 Curates, Tai Kwun Hong Kong, LACMA Digital Leaders, National Communication Museum (Australia), CIVA Festival, DAE Research Festival, PAF Olomouc, ALife Conference Kyoto, Aksioma, and Museum of Art in Public Spaces (Køge) among others, and was featured on Dazed, TANK Magazine, DIS, SHOWStudio, Art Asia Pacific, COEVAL Magazine, and AQNB.
In collaboration with Daniel Felstead, she has produced a short film series ‘I’m so Janky’ from DIS that explore the myths, ideologies and realities of the metaverse, AI, and Neuralink. She also collaborates with dmstfctn on simulation projects for Serpentine Arts Technologies and the Leonardo Supercomputer at Bologna’s Tecnopolo.
Question 1 First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Week 10: Imaging and Measurement title: “Week 10 — Advanced Imaging & Measurement Technology” linkTitle: “Week 10 (Apr 7)” weight: 200 description: | Advanced Imaging & Measurement Tech (Evan Daugharthy, Waters Corp.)
Lab: Mass Spectrometry This lecture presents a range of advanced technologies to do precision measurement of proteins at atomic scales, characterizing chemical composition, and detecting protein sequence and structure.
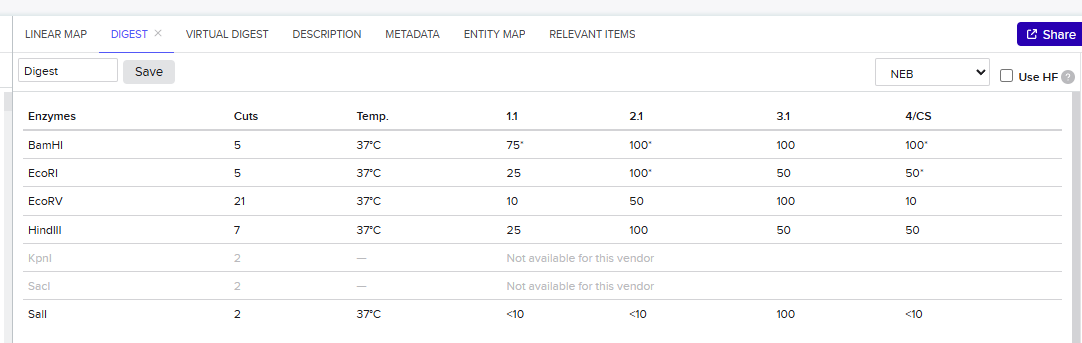
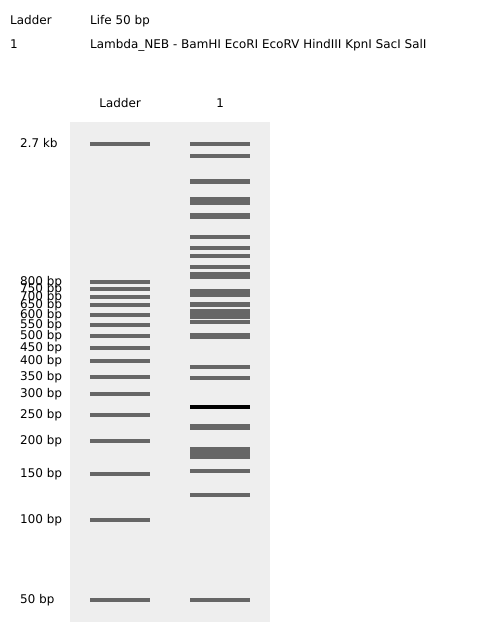
Week 2: DNA Read, Write, and Edit Part 1: Benchling & In-silico Gel Art 1.1 Import Lambda DNA Simulate Restriction Enzyme Digestion Virtual Gel Part 2: Gel Art I have chosen to create a gel art of a person doing a jumping jack through randomization method.
Week 5: Protein Design Part II Homework — DUE BY START OF MAR 10 LECTURE Part A: SOD1 Binder Peptide Design Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Week 6: Genetic Circuits Part I Homework — DUE BY START OF MAR 17 LECTURE Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab:
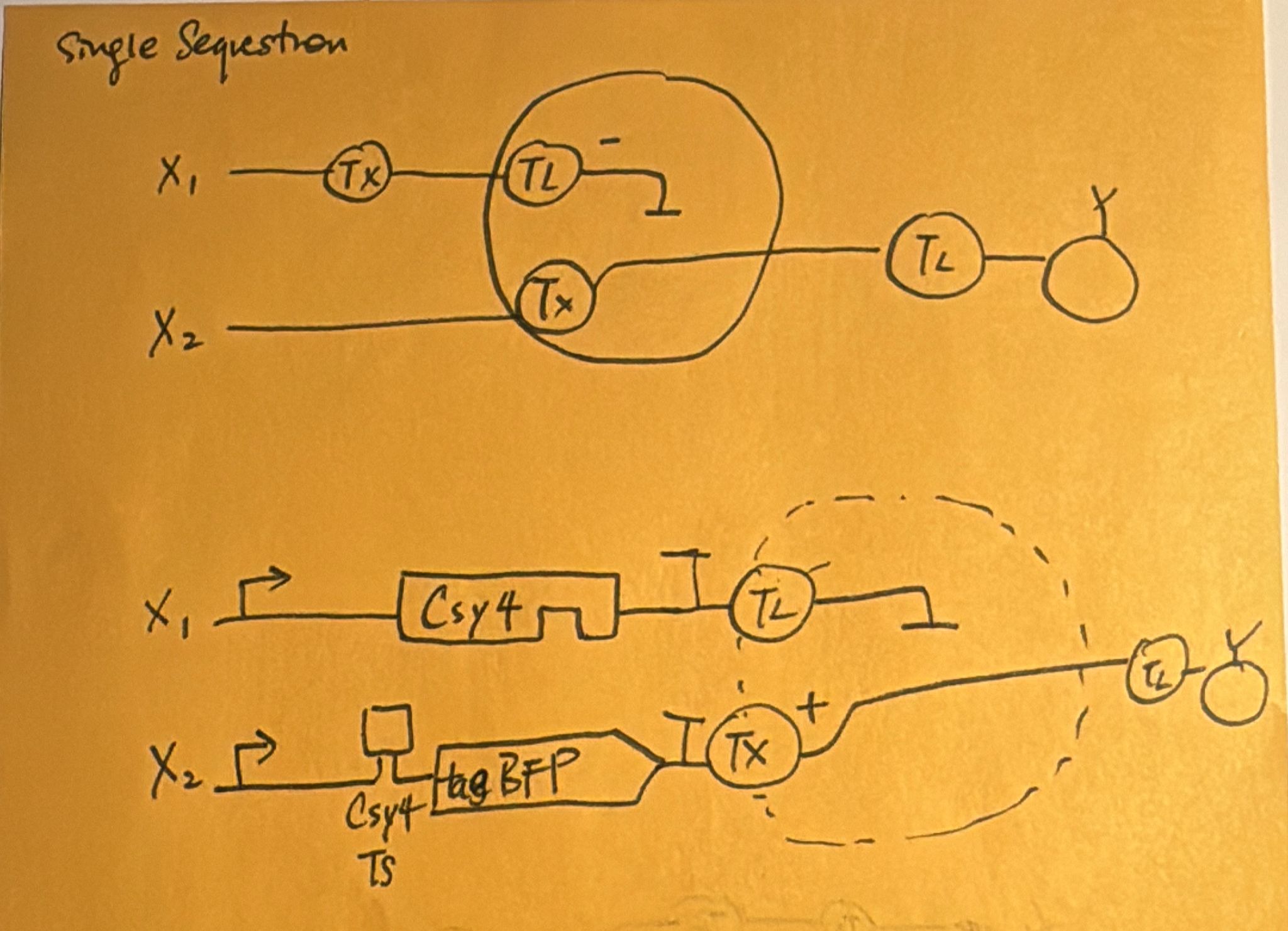
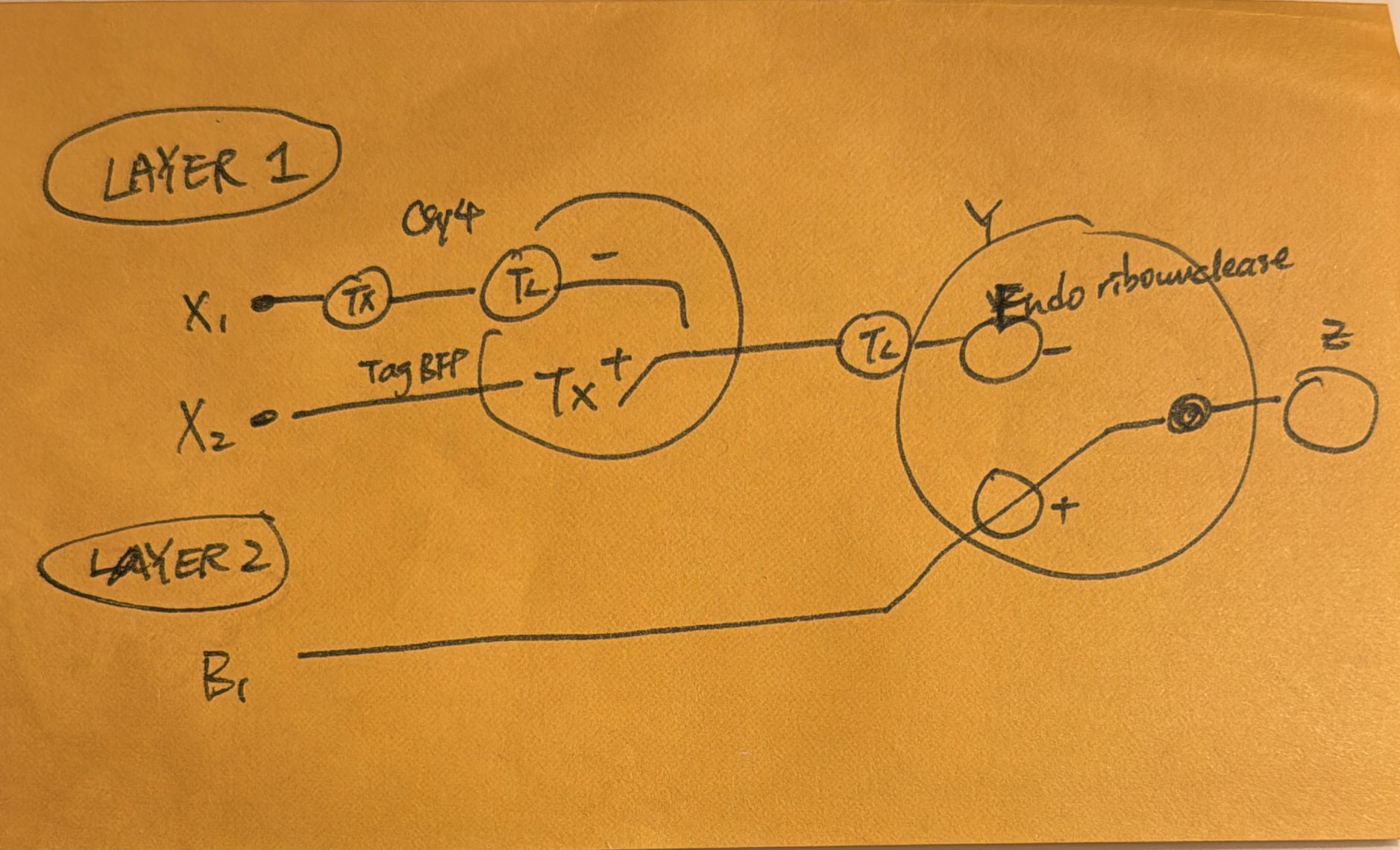
Week 7: Genetic Circuits Part II Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? An artificual neuron is a weighted summation through an activation function that produces outputs, eventually they form networks to become ANN. Intracellular artificial networks still have weighted summation and a non-linear activation function, but we can consider implementing gene circuits as these activation functions. The main difference is that IANNs will have two inputs that can do addition and subtraction. On the one hand, a promoter that through transcription makes a gene, and through translation we create proteins, we can perform addition on this. To subtract, we can treat input x1 as an endoribonuclease CasE that will bind and cleaves the RNA on the sequence and produce output. x1 is negative weight and x2 is positve weight, where the function is max(x2-x1,0). This is also referred to as Sequestration. Sequestration involves using an endorribonucleus to transcribe into mRNA to produce non-linearity (applying single turnover enzyme to remove it out of circulation).
Week 9: Cell-Free Systems Homework — DUE BY START OF Apr 7 LECTURE Homework Part A: General and Lecturer-Specific Questions General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free systems help us understand biology ‘from scratch’ to bioengineer from smaller units. There’s wider flexibility for scaffolding biology from the ground-up and controlling the environments in a complete model. Existing living cells as we know it are already incredibly complex and hence less controlled in experimental settings. Synthetic cell engineering allows flexibility in size of the cell, proteins, and even expanding largely on the chemistry of the cell. So the two scenarios could be if you want to control the size of the cell and want uniform control it might be ideal to use cell-free system. The other scenario might be to engineer a specific chemical environment or want chemical diversity in the experiment that is not naturally common/ compatible with cells. Compared to in-vivo expression where you have to create plasmids, cell-free protein expressions are faster and cheaper to construct and can also help you through quick iterations with linear fragments and without plasmids.
Subsections of Homework
Week 1: Principles and Practices
Question 1
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Answer 1
I would like to expand on my project on Unreal Engine API for brain-on-a-chip platforms that was presented at NeurIPS 2025 (https://openreview.net/forum?id=BroaBkQAGa). The project proposes to build an API between living neurons interfaced with microelectrode arrays and virtual gaming environments, so that researchers and designers can use this environment to visualize spiking behavior across MEA channels, and to use reinforcement learning algorithms within the game environment to train neuronal cultures as game agents.
I’m currently collaborating with Cortical Labs to use CL-1 to connect via UDP to design closed-loop real-time visualization systems at the National Communication Museum in Melbourne. To start the loop I’ve sent in blob tracking data for Cl1 to process. The spikes from the CL1 are then streamed to Unreal Engine so that the neuronal activity can be used to transform agent parameters.
https://ncm.org.au/exhibitions/cortical-labshttps://jennleung.xyz/corticallabs
Question 2
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Answer 2
One of the main objectives of this project is that it provides an open playground for benchmarking open-source and non-standardized brain-on-a-chip platforms. As we speculate these systems to become democratized and decentralized, there will spawn many different configurations of physical/ neural assemblies with advances in MEA designs, bioprinting technologies, and microfluidic platforms. Therefore, it is important to supporting 1) benchmarking integrity and reproducibility, for example, how do we measure spiking activity across different systems? How do we make sure experiments are scientifically meaningful? How do we translate and deliver virtual environments to channels on different MEA geometries? 2) ensuring accessibility to indepdnent researchers, for example, writing software environments not only for proprietary technologies such as Cortical Lab’s CL1 or FinalSpark’s Neuroplatform. Governance here means committing to abstraction layers that treat CL1 as one implementation among many 3) responsible scalability across new substrates, for example, new substrates includes increasingly complex organoids or assembloids that should go through rigorous bioethical frameworks. 4) Sustainability & longevity of the substrates, there should be rate limitations so that cells aren’t overly stimulated and at risk of quick death.
Question 3
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
For each governance action, address:
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Answer 3
Benchmarking metadata across different brain-on-a-chip platforms
Purpose: Currently there are multiple commercial/ proprietary brain-on-a-chip platforms such as Cortical Labs’ CL1 and FinalSpark’s Neuroplatform, but there are no standardizations or comparisons metadata of these systems. I am proposing to create a metadata of existing platforms/ systems and develop an open access metadata standard that documents different MEA geometries, channel count, substrates,
Design: Map out a group of academic researchers who have been working on organoid intelligence/ synthetic bioengineered intelligence standardization, and manufacturers such as MaxWell Biosystems, Cortical Labs, etc., join community labs or open-source groups on open-source resesarch. In terms of implementations, I will need to consult all these groups to create an UE plugin that responds to their needs.
It would be great to apply for AHRC/ UKRI grants.
Assumptions: This action assumes that all parties are happy to share their manual or manufacturing details, however, some of this data might be protected under NDA.
Risks of Failure and Success: There’s a high chance the open-source projects will grow exponentially, making this metadata impossible to manage at scale.
Developing stimulation protocols at API layer
Purpose: Since there are many different types of brain-on-a-chip platforms, each company/ lab has different protocols of stimulating and recording these systems. It would be great to propose a stimulation protocol that is initiated by the API/ game environment.
Design: Study the stimulation protocols across different systems and apply appropriate time scales, rate limits, response rates, and stimulation/ discretization patterns so we can formalize communication with living neurons.
Assumptions: The biggest assumption here is likely that standardization might not be applicable or scientifically meaninful across different biological systems because of biological variability, they vary by culture, by physical assembly and MEA type.
Risk of Failure: Overstandardization might lead to less meaningful scientific experiments. Certain rate limits and standardization might fail to recognize the plasticity and assumes this technology to not evolve. Constant review and negotiations are needed to make this option work!
Developing a wide range of benchmarking gaming environments/ templates
Purpose: Cortical Labs has compared living neurons against RL algorithms in Pong. I would like to expand on this to develop something adjacent to OpenAI Gym, so that we can create environments for synthetic bioengineered intelligence.
Design: These might include standardized task environments that allow researchers to compare RL agent performance on identical tasks, or have multiplayer/ team battles between two systems for performance evaluations. Standardized environments ensure that experimental results are reproducible and comparable across institutions.
Assumptions: The templates assume that this variability can be characterized statistically across many runs, but if variability is too high, the benchmarks may not be informative.
Risks of Failure and Success: Templates might restrict certain experiment design, so it would be important to balance standardization/ benchmarking vs openness.
Question 4
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Answer 4
(Fill in the table with your scores for each option.)
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
3
• By helping respond
1
3
2
Foster Lab Safety
• By preventing incident
3
1
2
• By helping respond
1
2
3
Protect the environment
• By preventing incidents
2
1
3
• By helping respond
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
1
• Feasibility?
2
1
3
• Not impede research
1
2
3
• Promote constructive applications
3
1
2
Question 5
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Answer 5
Option 2 seems to be the most well-considered option because it implies and builds on fundamental knowledge of other research institutions practice and existing start-up solutions. It’s the governance action that most directly addresses the biological welfare and safety concerns that are unique to this field. Since we can’t retroactively un-damage a neuronal culture, having safety protocols embedded at the API layer is the most impactful intervention point.
Question 6
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
Answer 6
I am interested in the concept of pharmakon - that for research to be really successful also comes at the cost of creating additional problems such as bioweapons or disregulation of illegal substance (biosecurity). The governance actions I am interested in are perhaps on the cloud/ API side of things, around how we may be able to apply trust-based connectivity from software design to bio-design. For example, cloud infrastructure already uses trust models and I think we could potentially learn from internet architecture to look at regulating or modeling remote access to living biological systems.
Homework Questions from Professor Jacobson (Lecture 2 slides)
Question 7
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Answer 7
Error Rate: 1:106
Throughput Error Rate Product Differential: ~108
The human genome is 3.2 billion letters long and will roughly make 3200 mistakes. Biology can reduce the error rate by shifting mismatched pair and tries again with the corrent nucleotide.
Question 8
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Answer 8
There is an astronomical number of ways to code an average human protein. Each amino acid has 3 codons available and there’s more than 300 amino acids long for an average human protein. But some codons have many matching tRNAs that not all codons apply, this means some ribosomes can fall off or misread which leads to less protein produced.
Homework Questions from Dr. LeProust (Lecture 2 slides)
Question 9
What’s the most commonly used method for oligo synthesis currently?
Answer 9
Phosphoramidite method by Caruthers
Question 10
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Answer 10
Chemistry causes cumulative damage and hits a wall around 200 nucleotides.
Question 11
Why can’t you make a 2000bp gene via direct oligo synthesis?
Answer 11
1 in 3,000 bp error rate. There’s too many errors distributed and become unpurifiable. It requires good sequencing analysis and fragment analysis as well as uniform distribution across all oligos.
Homework Question from George Church (Lecture 2 slides)
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
Option A – Question 12
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
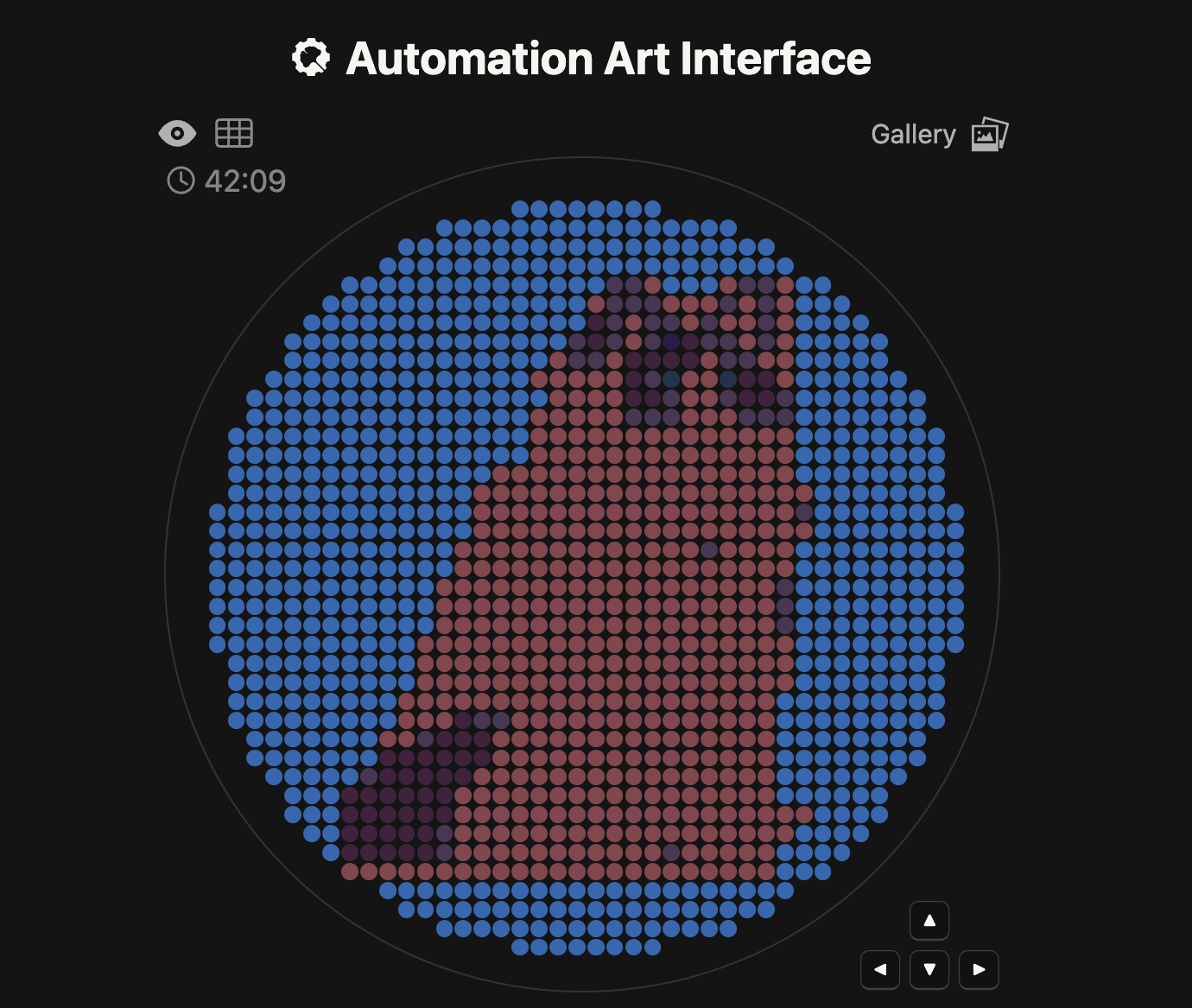
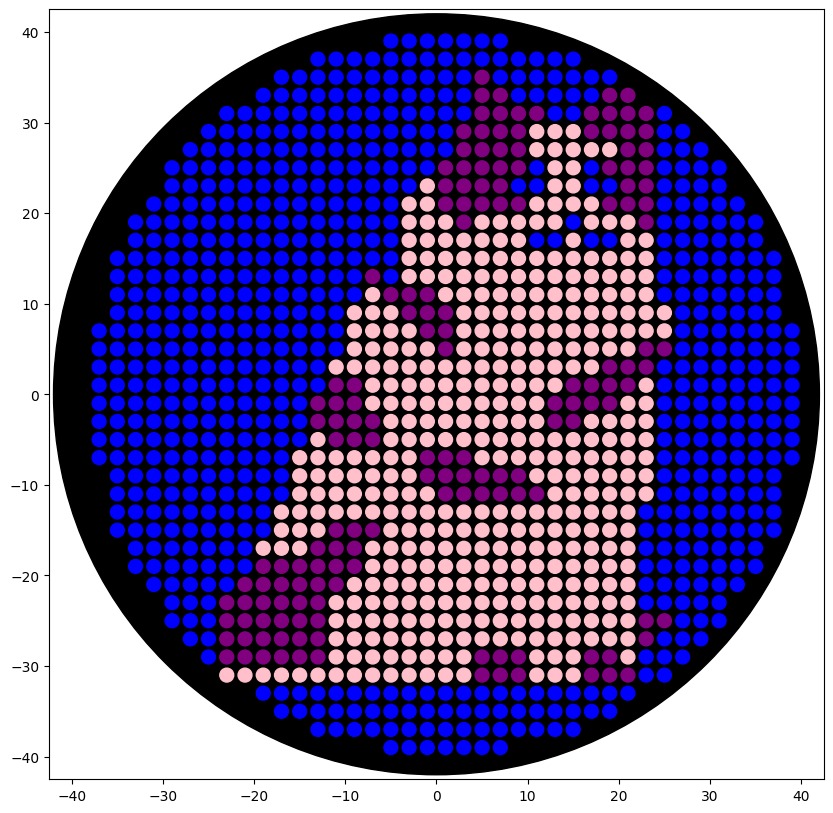
I first generated a design using an image input of a voxelated ragdoll. The pixels should help simplify the image so that it can be plotted in the dish similarly.
Because of the lack of contrast and limitations in the range of colors, the image looked different than expected.
Meanwhile at LifeFabs, we only had access to the colors Pink, Blue, and Purple. So I ended up simplifying the number of fluorescent proteins used to three and generated the coordinates appropriately.
Question 2
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
These were the coordinates generated from the GUI using three fluorescent proteins:
Using the Opentrons Colab document, I successfully integrated the point data into the code:
fromopentronsimporttypesmetadata={'author':'Jenn Leung','protocolName':'Opentrons Cat','description':'HTGAA 2026 Opentrons cat drawing','source':'HTGAA 2026 Opentrons Lab','apiLevel':'2.20'}################################################################################# Colour mapping### A1 = Blue → Azurite (cat outline and body)### B1 = Purple → mCherry + mPlum (shadow and accent details)### C1 = Pink → tdTomato + tagRFP + mHoneydew (warm fill and face)##############################################################################well_colors={'A1':'Blue','B1':'Purple','C1':'Pink',}################################################################################# Point data##############################################################################azurite_points=[...]tdtomato_points=[...]tagrfp_points=[...]# ... (full code in OpentronsProtocol.py)# Blue (A1) — Azurite: cat outline and bodypaint_layer(azurite_points,'Blue')# Pink (C1) — tdTomato + tagRFP + mHoneydew: warm fill and face detailpaint_layer(tagrfp_points,'Pink')# Purple (B1) — mCherry + mPlum: shadow accents and deep detailpaint_layer(tdtomato_points,'Purple')
This is the result of the final preview on the colab document, using the three colors available.
Post-Lab Questions
Question 1
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Answer: ‘Fluidic Programmable Gravi-maze Array for High Throughput Multiorgan Drug Testing’ by Wong et al. proposes OrganRX, which is a multi-organ-on-a-chip system that is compatible with automated liquid dispensing robots such as Opentrons, OT2.
The programmable part of the microfluidic architecture uses robotic liquid handlers and automated plate readers, which can help researchers program how much media reaches each organ compartment.
There is also a programmable tilting recirculation mechanism that drives flow between the corner wells of the chip, allowing for directional flow.
The developers developed a Bluetooth-enabled iOS app that allows for remote control of the recirculation system, allowing users to select from multiple shear flow rates, set programmable waiting times between tilt-direction changes, and conduct system reset.
Question 2
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more.
As my research focuses on brains-on-chips and facilitating closed-loop interactions between living substrates and software systems, I’m curious to develop something similar to the OrganRX platform that utilizes Opentrons for chemical I/O with synthetic bioengineered intelligence. The direction is to look into facilitating biochemical feedback loops and designing custom plates for Opentrons via 3D printing.
#pseudo-code for HTGAA final project Assembloid Agency#design plasmids and custom plate MEA holder > order Twist DNA > 3D print labware > script OT2 protocol > measure spike changes and chemical deliveryprep:>listtypeofmaterialswillbeneededtofacilitatechemicali/oforwetware,e.g.placeholderforneurons,OpentronsOT2,customlabware,Twistorder,microfluidicdesign,mediasynbiocomponent:>designaplasmidinBenchlingandidentifyachemical'handshake'betweenOpentronsandneurons>syntheticgeneformeasurableandidentifiablechemicalsignals>researchinDREADDhM3Dq(humanM3muscarinicDREADDcoupledto)>benchlingdesign!physicalsystem:>designmicrofluidicssystemandcompositionofthecustomlabware>printcustomlabwareforOpentronsOT2,holdinganMEAchip,includingdifferentwellsforculturesandreinforcementagents,wasteprofusion/filter>thewellsshouldholdbasalmedia,reinforcementagents,andwastebuffer-maybemodelaftertheOrganRXchipto'tilt'agentsintocenter/substrate.software:>developanAPIfortheOT-2todetectassembly:>testandtrytoconnectthesynbioparts,withhardware,andsoftware!>measurespikesfromneuronsplaceholderafterroboticchemicaldelivery
WIP JSON code for custom labware:
{"ordering":[["A1","A2"]],"brand":{"brand":"CorticalLabs-Custom"},"metadata":{"displayName":"Assembloid Agency Chemical IO Plate","displayCategory":"other","displayVolumeUnits":"µL"},"dimensions":{"xDimension":127.76,"yDimension":85.48,"zDimension":15.0},"wells":{"A1":{"depth":10.0,"diameter":3.0,"shape":"circular","x":20.0,"y":40.0,"z":5.0},"A2":{"depth":10.0,"diameter":3.0,"shape":"circular","x":40.0,"y":40.0,"z":5.0}}}
Final Project Ideas — DUE BY START OF FEB 24 LECTURE
As explained in this week’s recitation, add 1-3 slides with 3 ideas you have for an Individual Final Project in the appropriate slide deck for MIT/Harvard/Wellesley students or for Committed Listeners. Be sure to put your name on your slide(s); for CLs, also put your city and country on your slide(s) and be sure you’re putting your slide(s) in your Node’s section of the deck.
Assembloid Agency is a bio-digital interface platform designed to facilitate closed-loop biochemical communication between synthetic neural substrates and automated software systems. I will be integrating the Opentrons OT-2 with Multi-Electrode Array to create chemical I/O bridge between neural substrates and software systems.
I’m looking into using DREADDs to allow software-controlled chemical I/O as well as designing custom 3D-printed labware, housing the biological assembly while providing microfluidic channels for automated media exchange, chemical reinforcement signals, and waste management. The aim is to conduct real-time closed-loop chemical communication with the substrate.
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Objective:
Learn basic concepts:
amino acid structure
3D protein visualization
the variety of ML-based design tools
Brainstorm as a group how to apply these tools to engineer a better bacteriophage (setting the stage for the final project).
Part A. Conceptual Questions
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
For 500 grams of meat, there is roughly 20-25% grams of protein. This means that roughly 100 grams belong to protein, while there is remaining fat, fiber, and water that make up the rest of the mass.
Because 1 mole = 100 Da
Number of moles = 100 g of protein / 100 Da = 100g/ 100 g / mol
$$\text{Molecules} = 1 \text{ mol} \times 6.022 \times 10^{23} \text{ molecules/mol}$$$$\text{Molecules} \approx 6.022 \times 10^{23}$$
There are roughly 602 sextillion amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When humans eat beef, through mastication and digestion we break down the beef into smaller units. First protein is broken down by enzymes (proteases) and into shorter chains of amino acids in the stomach. Then the chains become further broken down into individual amino acids in the small intestine. As these amino acids enter the bloodstream, they require DNA to instruct them into building other things. The human DNA does different things than cows and fish, therefore the amino acids will build a cow or a fish.
Why are there only 20 natural amino acids?
It may be an evolutionary mystery that almost all living things are built from these 20 natural amino acids. The 20 amino acids serve as the building blocks of most proteins, they line up as codons in 3-letter assemblies, in which the ribosomes read to create actions following the DNA sequence. When they read 3 bases at once, the combinations create 4^3 possibilities that are expansive enough for the making of diverse lifeforms.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, there are a lot of non-natural amino acids. Designing new amino acids require us to follow the same chassis but redesign the ‘r-group’ to alter the chemistry of the bond, which is the side chain of the amino acid. One may attach an azide to the chain to create a strong bond for stickiness or bio-glue. For experiments, some researchers also use non-natural florescent amino acids like Acridonylalanine to glow under microscopy or photographs.
Where did amino acids come from before enzymes that make them, and before life started?
This might be related to assembly theory? Lee Cronin proposed that life is composed of different assemblies, in that life is scaffolded by energy, raw sources, and minerals through complex interactions and then becomes amino acids, and longer chains. Gases and energy together can create amino acids. The Miller-Urey Experiment use water, methane, ammonia, and hydrogen to create amino acids.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed. D-amino acids create a mirror image of α-helixes, because the building blocks and the structure are completely mirrored.
Can you discover additional helices in proteins?
Yes, since 2020, AlphaFold has allowed us to quickly discover new helices and the instructions to their fold, revealed millions of protein structures.
Why are most molecular helices right-handed?
Because of chirality, most helices are non-identical to their mirror image. As most amino acids are L-form (left-handed), the way they most efficiently stack together is twisting to the right where they can create stable bonds with enough room between each other.
Why do β-sheets tend to aggregate?
β-sheets bond together via hydrogen bonds. The geometry appears like pleated, zigzag, sheet-like structure with side chains protruding.
What is the driving force for β-sheet aggregation?
They tend to aggregate because of its geometry, where the hydrophobic faces might sandwich and stick together to hide from water. The force from the water becomes driving force for clumping.
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
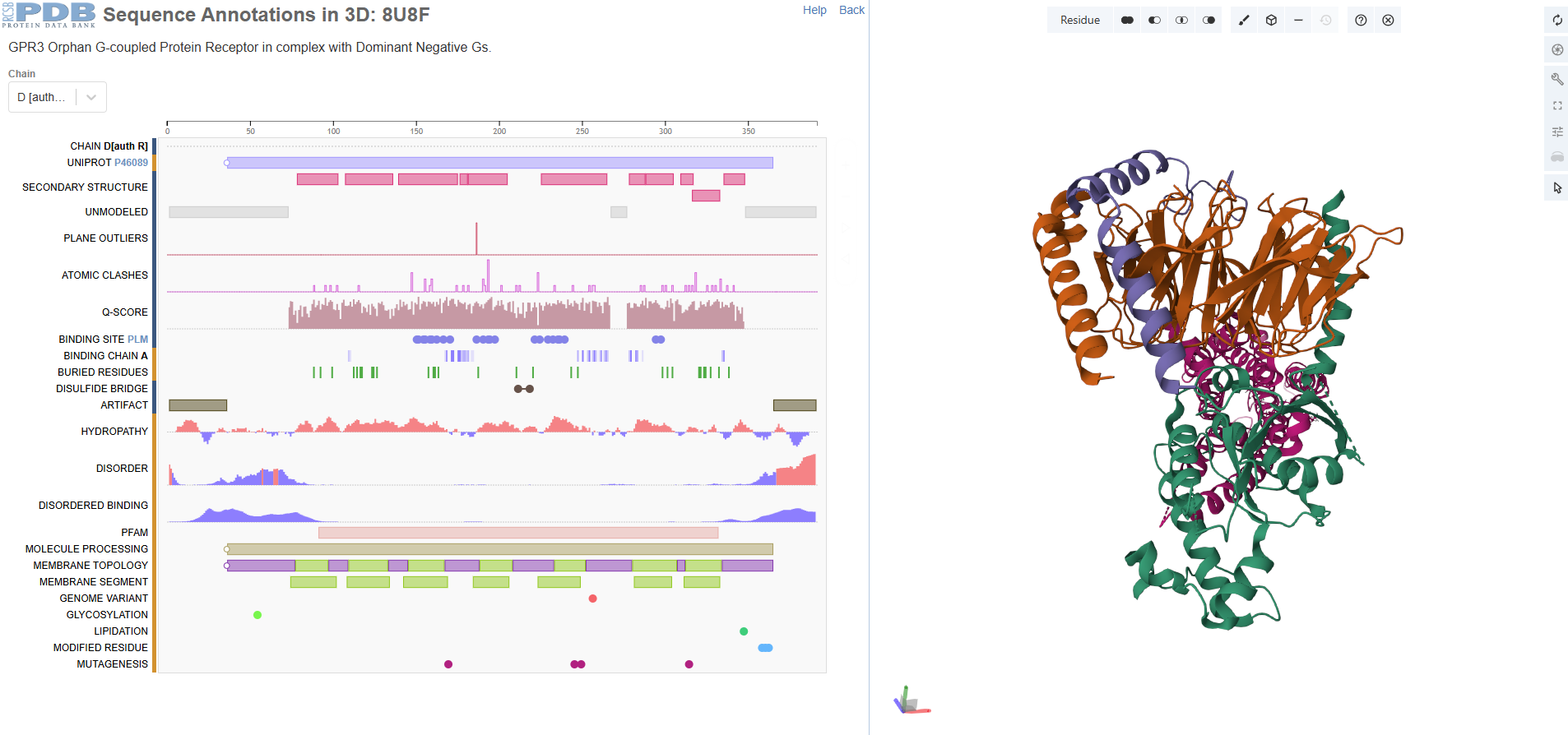
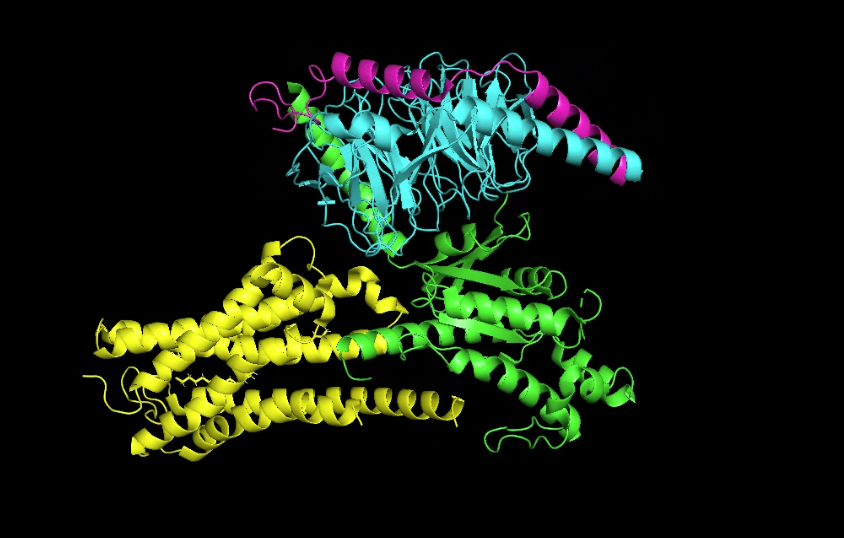
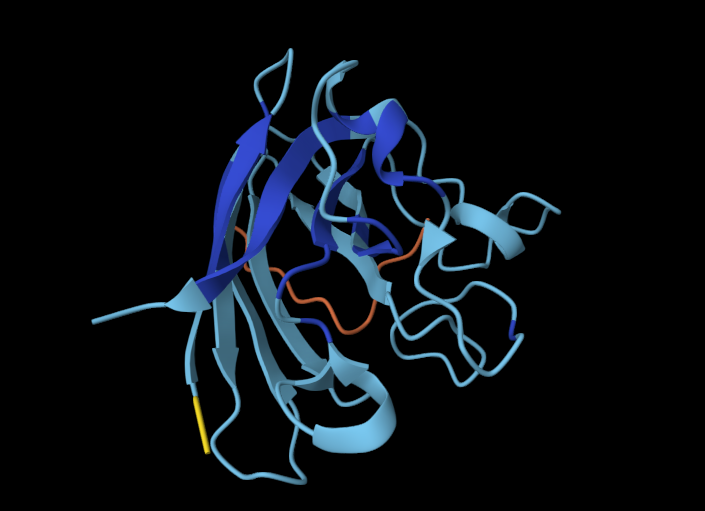
I chose GPR3 Orphan G-coupled Protein Receptor in complex with Dominant Negative Gs (8U8F) because I’m interested in
GPR3 is a class A orphan G protein-coupled receptor (GPCR) exhibiting broad expression across various brain regions including the hypothalamus, hippocampus, and cortex, as well as in peripheral tissues such as liver and ovary.It has a potential role in modulating a number of brain functions, including behavioral responses to stress, amyloid-beta peptide generation in neurons and neurite outgrowth.
For brains-on-chips research I’m interested in different types of expressions in the central nervous system and the brain.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
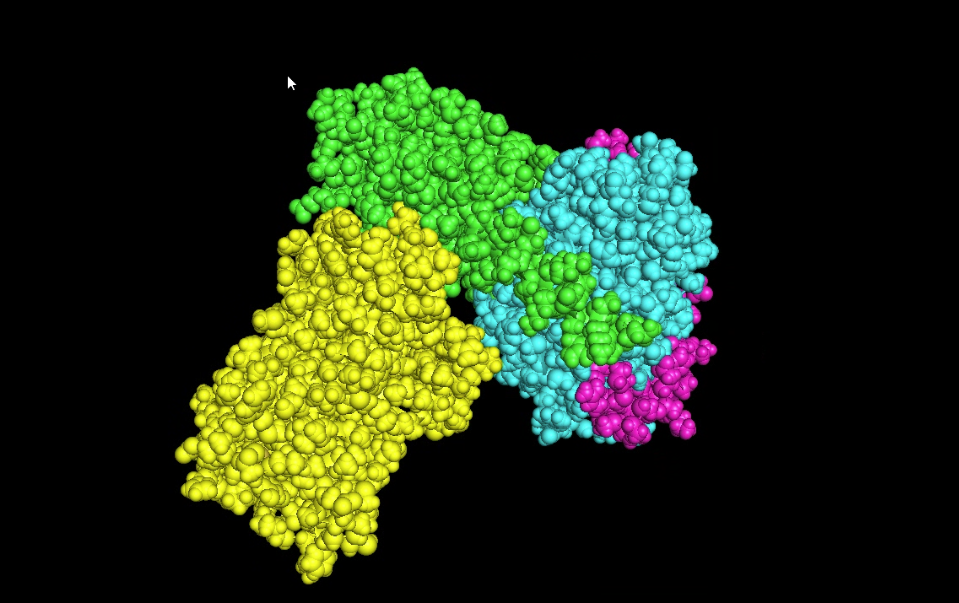
There are four protein chains. Chain A: 372, Chain B: 339, Chain C: 58, Chain D: 384. The most frequent amino acid seems to be leucine. It is a sturdy, hydrophobic (water-hating) amino acid.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
There are thousands of homologs, incuding human, pygmy chimpanzee, olive babboon, cotton-top tamarin, etc. The protein seems highly conserved and not changed.
Does your protein belong to any protein family?
G Protein-Coupled Receptor (GPCR) Family
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure is solved around 2023 September and released 2024 Match. The method id electron microscopy but resolution 3.49 Å.
Are there any other molecules in the solved structure apart from protein?
Yes, I see palmitic acid in the structure apart from protein.
It belongs to a membrain protein, and falls under 7-transmembrane receptive (GPCR).
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
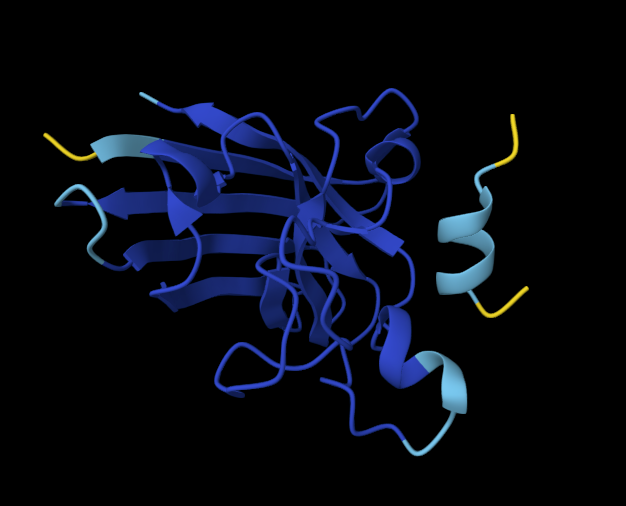
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon
Ribbon
Ball and stick
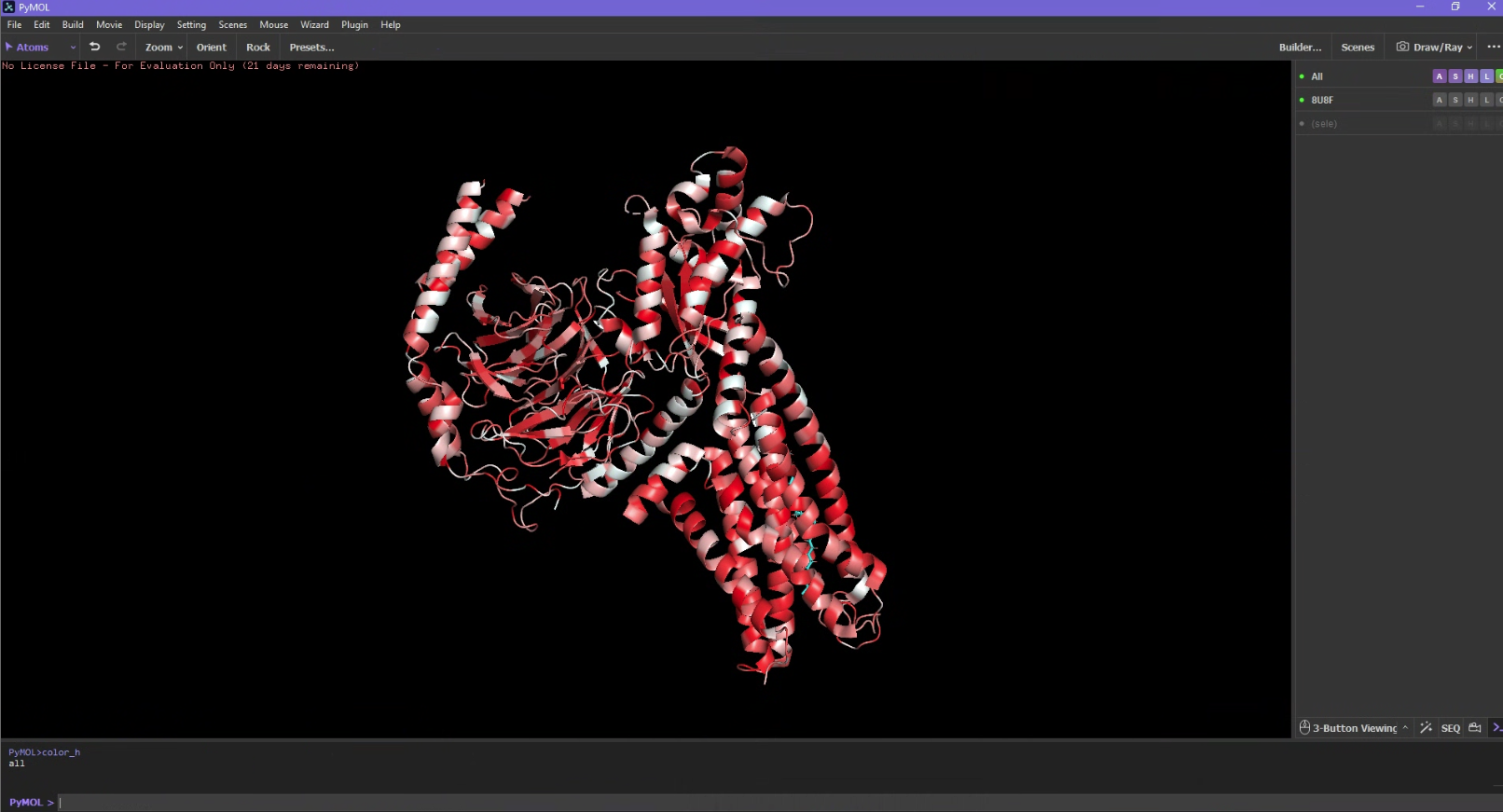
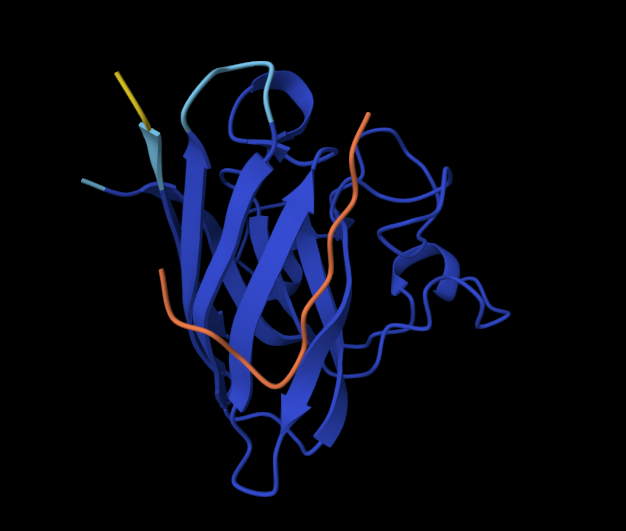
Color the protein by secondary structure. Does it have more helices or sheets?
It has a lot more helices than sheets.
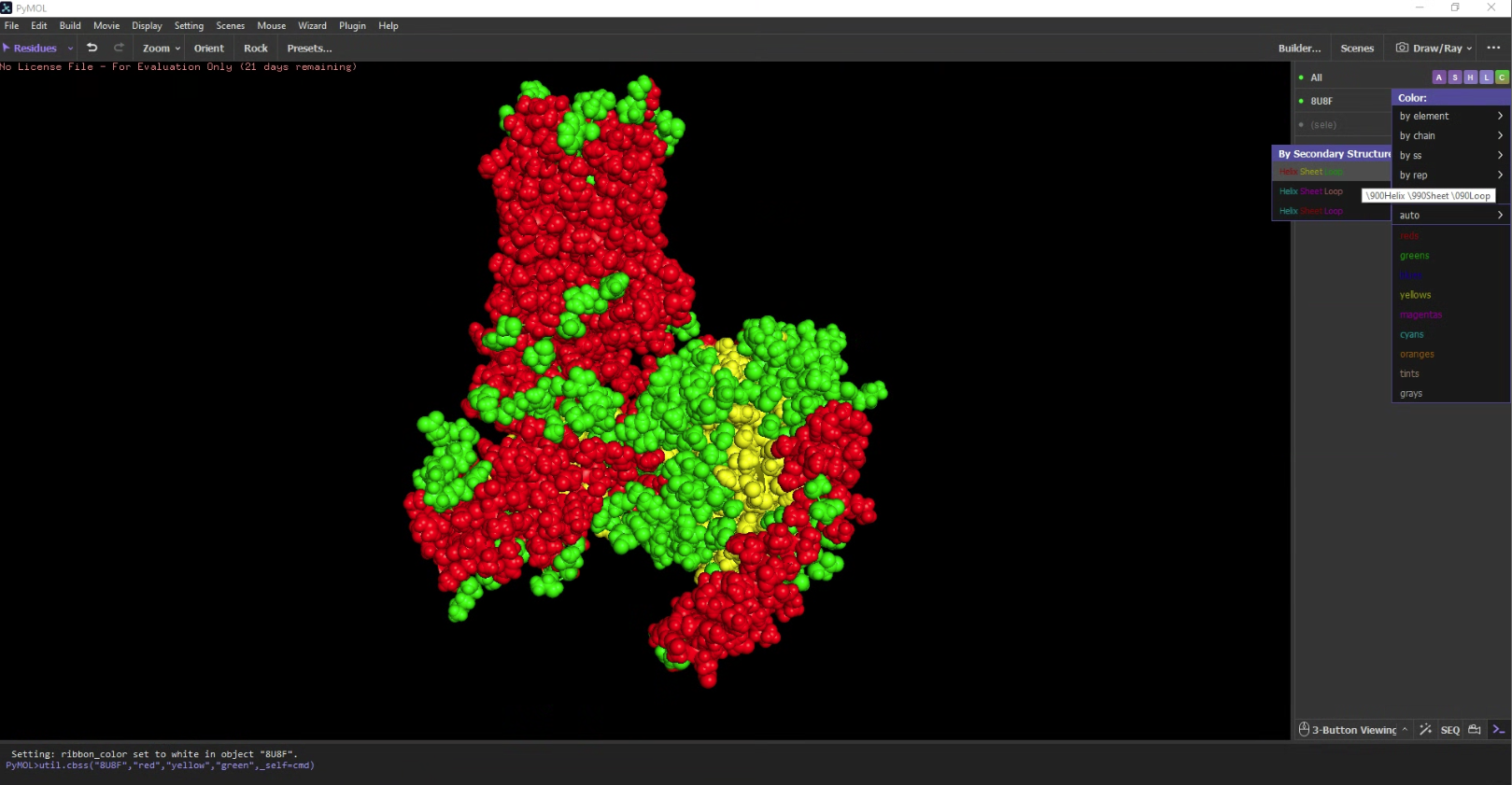
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
I used an additional script to label the hydrophobicity scale. Hydrophobic residues are red and hydrophilic (polar/charged) residues are white. It is slightly more hydrophobic.
#https://pymolwiki.org/index.php/Color_hfrompymolimportcmddefcolor_h(selection='all'):s=str(selection)print(s)cmd.set_color('color_ile',[0.996,0.062,0.062])cmd.set_color('color_phe',[0.996,0.109,0.109])cmd.set_color('color_val',[0.992,0.156,0.156])cmd.set_color('color_leu',[0.992,0.207,0.207])cmd.set_color('color_trp',[0.992,0.254,0.254])cmd.set_color('color_met',[0.988,0.301,0.301])cmd.set_color('color_ala',[0.988,0.348,0.348])cmd.set_color('color_gly',[0.984,0.394,0.394])cmd.set_color('color_cys',[0.984,0.445,0.445])cmd.set_color('color_tyr',[0.984,0.492,0.492])cmd.set_color('color_pro',[0.980,0.539,0.539])cmd.set_color('color_thr',[0.980,0.586,0.586])cmd.set_color('color_ser',[0.980,0.637,0.637])cmd.set_color('color_his',[0.977,0.684,0.684])cmd.set_color('color_glu',[0.977,0.730,0.730])cmd.set_color('color_asn',[0.973,0.777,0.777])cmd.set_color('color_gln',[0.973,0.824,0.824])cmd.set_color('color_asp',[0.973,0.875,0.875])cmd.set_color('color_lys',[0.899,0.922,0.922])cmd.set_color('color_arg',[0.899,0.969,0.969])cmd.color("color_ile","("+s+" and resn ile)")cmd.color("color_phe","("+s+" and resn phe)")cmd.color("color_val","("+s+" and resn val)")cmd.color("color_leu","("+s+" and resn leu)")cmd.color("color_trp","("+s+" and resn trp)")cmd.color("color_met","("+s+" and resn met)")cmd.color("color_ala","("+s+" and resn ala)")cmd.color("color_gly","("+s+" and resn gly)")cmd.color("color_cys","("+s+" and resn cys)")cmd.color("color_tyr","("+s+" and resn tyr)")cmd.color("color_pro","("+s+" and resn pro)")cmd.color("color_thr","("+s+" and resn thr)")cmd.color("color_ser","("+s+" and resn ser)")cmd.color("color_his","("+s+" and resn his)")cmd.color("color_glu","("+s+" and resn glu)")cmd.color("color_asn","("+s+" and resn asn)")cmd.color("color_gln","("+s+" and resn gln)")cmd.color("color_asp","("+s+" and resn asp)")cmd.color("color_lys","("+s+" and resn lys)")cmd.color("color_arg","("+s+" and resn arg)")cmd.extend('color_h',color_h)defcolor_h2(selection='all'):s=str(selection)print(s)cmd.set_color("color_ile2",[0.938,1,0.938])cmd.set_color("color_phe2",[0.891,1,0.891])cmd.set_color("color_val2",[0.844,1,0.844])cmd.set_color("color_leu2",[0.793,1,0.793])cmd.set_color("color_trp2",[0.746,1,0.746])cmd.set_color("color_met2",[0.699,1,0.699])cmd.set_color("color_ala2",[0.652,1,0.652])cmd.set_color("color_gly2",[0.606,1,0.606])cmd.set_color("color_cys2",[0.555,1,0.555])cmd.set_color("color_tyr2",[0.508,1,0.508])cmd.set_color("color_pro2",[0.461,1,0.461])cmd.set_color("color_thr2",[0.414,1,0.414])cmd.set_color("color_ser2",[0.363,1,0.363])cmd.set_color("color_his2",[0.316,1,0.316])cmd.set_color("color_glu2",[0.27,1,0.27])cmd.set_color("color_asn2",[0.223,1,0.223])cmd.set_color("color_gln2",[0.176,1,0.176])cmd.set_color("color_asp2",[0.125,1,0.125])cmd.set_color("color_lys2",[0.078,1,0.078])cmd.set_color("color_arg2",[0.031,1,0.031])cmd.color("color_ile2","("+s+" and resn ile)")cmd.color("color_phe2","("+s+" and resn phe)")cmd.color("color_val2","("+s+" and resn val)")cmd.color("color_leu2","("+s+" and resn leu)")cmd.color("color_trp2","("+s+" and resn trp)")cmd.color("color_met2","("+s+" and resn met)")cmd.color("color_ala2","("+s+" and resn ala)")cmd.color("color_gly2","("+s+" and resn gly)")cmd.color("color_cys2","("+s+" and resn cys)")cmd.color("color_tyr2","("+s+" and resn tyr)")cmd.color("color_pro2","("+s+" and resn pro)")cmd.color("color_thr2","("+s+" and resn thr)")cmd.color("color_ser2","("+s+" and resn ser)")cmd.color("color_his2","("+s+" and resn his)")cmd.color("color_glu2","("+s+" and resn glu)")cmd.color("color_asn2","("+s+" and resn asn)")cmd.color("color_gln2","("+s+" and resn gln)")cmd.color("color_asp2","("+s+" and resn asp)")cmd.color("color_lys2","("+s+" and resn lys)")cmd.color("color_arg2","("+s+" and resn arg)")cmd.extend('color_h2',color_h2)
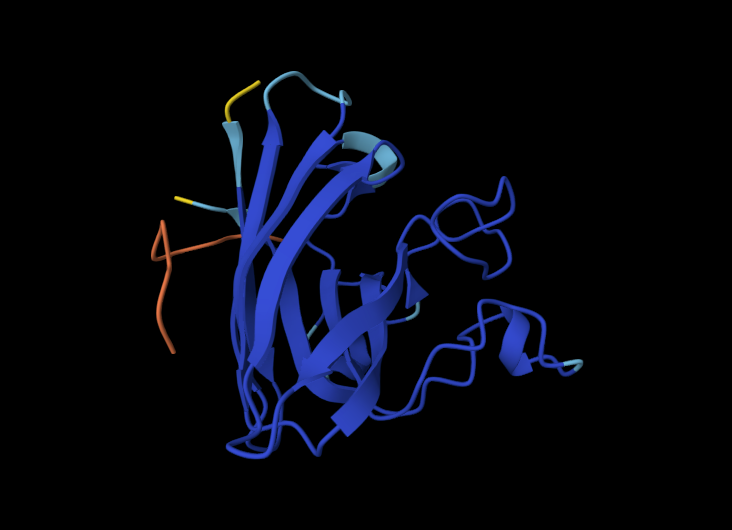
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes it appears to have a hole in the middle.
Part C. Using ML-Based Protein Design Tools
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
C1. Protein Language Modeling
Picture Source: Bordin, Nicola et al (2023). Novel machine learning approaches revolutionize protein knowledge. Trends in Biochemical Sciences, Volume 48, Issue 4, 345 - 359
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
>Using ESM2 mutational scans, 8U8F looks like
>![alt text]()
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
It appears that there are vertical bands in the sequence where across different amino acids, it's predicted to have a low score. This might be due to highly conserved functional and structural reasons. Lysine is the most common amino acid, but it also shows lots of dark spots and low scores because it is may have a hydrophobic mismatch.
>There is a yellow band at position 243.
>It is interesting Lysine is charged and has lots of blue bands, Leucine is neutral and is mostly high on the score.
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
>![alt text]()
Analyze the different formed neighborhoods: do they approximate similar proteins?
>They are positionally far away from each other, they are very different proteins.
Place your protein in the resulting map and explain its position and similarity to its neighbors.
>G-protein subunits ($\alpha, \beta, \text{ and } \gamma$ are much closer to each other on the map.
>Chain G is much shorter, only 58 amino acids and is structurally very different to other proteins. Chain G is essentially just two small alpha-helices connected by a loop.
C2. Protein Folding
Picture Source: Lin et al (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model.
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
I tried changing small snippets of the sequence and it wasn’t as visible, but adding longer sequences of the same amino acid allowed twists to be more visible.
C3. Protein Generation
Picture Source: 1. Post from Sergey Ovchinnikov 2. Roney, Ovchinnikov et al (2022). State-of-the-art estimation of protein model accuracy using AlphaFold. Phys. Rev. Lett. 129, 238101
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
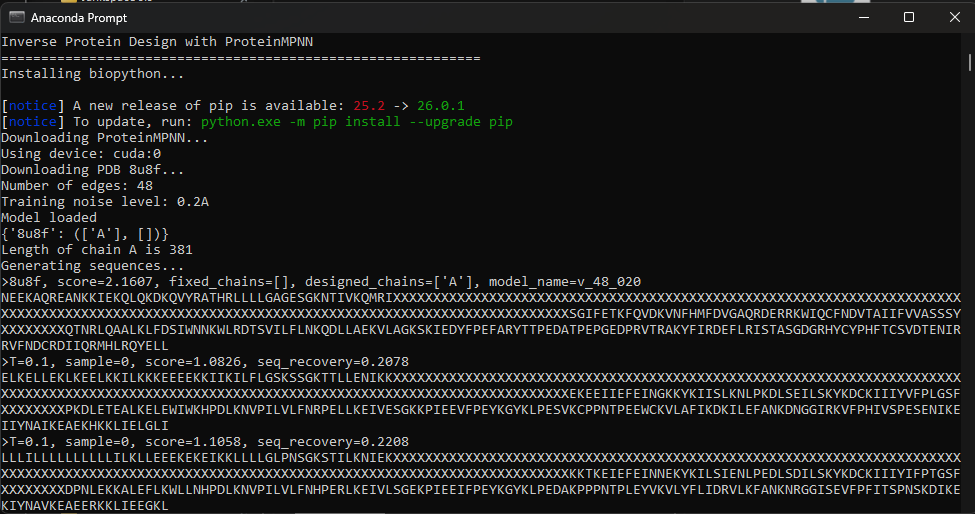
Using the fixed-backbone design, we kept the 3D shape of 8U8F Chain A and reskinned a sequence. ProteinMPNN ended up rewriting 75% of the protein, there is a high frequency of Leucine and Lysine.
My results look like:
Model weights found in ProteinMPNN/vanilla_model_weights
Using device: cuda:0
Number of edges: 48
Training noise level: 0.2A
Model loaded
{'8u8f': (['A'], [])}
Length of chain A is 381
Generating sequences...
>8u8f, score=2.1622, fixed_chains=[], designed_chains=['A'], model_name=v_48_020
NEEKAQREANKKIEKQLQKDKQVYRATHRLLLLGAGESGKNTIVKQMRIXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXSGIFETKFQVDKVNFHMFDVGAQRDERRKWIQCFNDVTAIIFVVASSSYXXXXXXXXQTNRLQAALKLFDSIWNNKWLRDTSVILFLNKQDLLAEKVLAGKSKIEDYFPEFARYTTPEDATPEPGEDPRVTRAKYFIRDEFLRISTASGDGRHYCYPHFTCSVDTENIRRVFNDCRDIIQRMHLRQYELL
>T=0.1, sample=0, score=1.0949, seq_recovery=0.2511
ELLKLLEELLKKLAEKLKKEEEEEKKIKKILLLGSPSSGKTTLLKNIKKXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXEPEEVVEFTIDGKKYKIYDLKNQPPDLREVLAKYKDAKVIIYVFPLGSFXXXXXXXXPEDLEKVALEELEWIWNHPDLKNVPILVIFNRPELLRERVLSGKNPIEERFPEYKGYELPKEVKPPEGVPEEWVKVLAFIIDKILKFANKNRGGIREVYPVISSPESKDIKQIIYDAIKKAEERKKLIAEGKL
>T=0.1, sample=0, score=1.1122, seq_recovery=0.2338
LLLLLLLLLLLLLLVLLLLKLLEESKIKKLLLLGSPSSGKTSLLENIEKXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXEPERVLEFEIDGVKYRIIDLSNLPPDLSDVLSEYSDCEIIIYVFSTGSYXXXXXXXXPEDLESVDLERLKWIWNHPALKNTPILVIFNRPELLAKRVLSGEKPIEERFPEYKGYKLPENVKPPPGVPEETVKVLSFLIDKVLEFANQNRGGIREVYPVISSVKSKEIKEIIYEAVKKAEERKKLIAQGLL
>T=0.1, sample=0, score=1.0975, seq_recovery=0.2554
KEEEKKKELEEKLKKEEEKKKEEEEKVIKLLLLGLPNSGKTTILENIKKXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXEPEEVIEFEIEGKKYRIVDLKNLPPDLSEILEKYSDCKILVYIFPTGSFXXXXXXXXPENLEKEALELLKRIWNHPSLKNVPLLVIFNRAEKLKEIVLSGEKPIEEYFPEYKGYKLPESAKPPPNTDPEVVKVLSFLIDKILEYANQNRGGIRKVFPVISSPESKDIREIIYKAVKEAEERKKLIALGLL
>T=0.1, sample=0, score=1.1196, seq_recovery=0.2857
AALAEELAKKKALAALKKKEEEEESKVKKLLLLGGPSSGKTTLLENISKXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXSSIRELEFEIDGVKYKILDLENRPEDLSEILSEFKDCEIIIYVFPLGSFXXXXXXXXPENLLKKALEEFERIWNHPDLKDVPILVLFNRPELLKEKVLSGKKPLEEIFPEYKGWELPEDAKPPPNTPLEWVKALYFLKEKVLEIANKNRGGRREVFPFIVSPKSKDIKEIIYNAVKEAEKRKALIAAGLL
>T=0.1, sample=0, score=1.1445, seq_recovery=0.2381
LLLLLLLALLLALAALLAALAEEEKKVRKLLLLGLPNSGKTTLLKNISKXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXEPEEILKFEIDGVKYEIKDLKNRPPDLSDILKEYSDCDIIIYVFPSGLFXXXXXXXXPENLEEVALEQLKNLLNNPDLKNVPILVLFNRPELLKKIVESGKRPLEEIFPEYKGYELPESAVCPPNTPLEWCKAIYFLIDKILEFANQNRGGISEVYPHITSPDSKDIKQIIYDAVKKAEERKKLIAAGKL
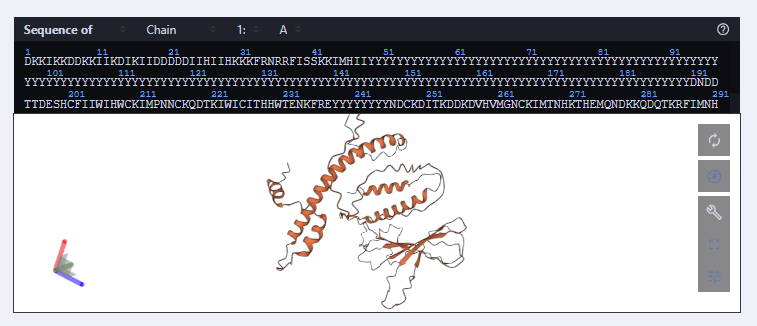
New Sequence: DKKIKKDDKKIIKDIKIIDDDDDIIHIIHKKKFRNRRFISSKKIMHIIYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYYDNDDTTDESHCFIIWIHWCKIMPNNCKQDTKIWICITHHWTENKFREYYYYYYYYNDCKDITKDDKDVHVMGNCKIMTNHKTHEMQNDKKQDQTKRFIMNHDDQENDWIFWDKNIDTINNDFTNDDVTITKEHHCIHKIEMIMQFFHQDTWNTHRRNDRICHIPHHWCHIIDDQIIKHDFIK
============================================================
Summary
============================================================
Sequence 1: score=1.0949, recovery=25.11%
Sequence 2: score=1.1122, recovery=23.38%
Sequence 3: score=1.0975, recovery=25.54%
Sequence 4: score=1.1196, recovery=28.57%
Sequence 5: score=1.1445, recovery=23.81%
Google Colab doesn’t work with GPU acceleration so I’ve cloned to work locally.
Input this sequence into ESMFold and compare the predicted structure to your original.
The predicted structure has retained the structure but upon comparison on PyMOL, the white structure (new) looks displaced.
Part D. Group Brainstorm on Bacteriophage Engineering
Assignees for the following sections
MIT/Harvard students
Optional
Committed Listeners
Required
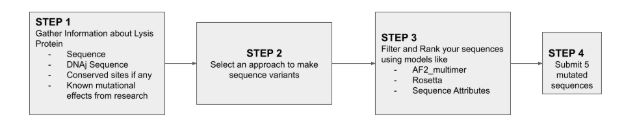
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
optimizing protein’s binding affinity to e coli to accelerate lysis trigger
increasing stability of L protein, ensuring proteins are folded and integrated into membrane to perform function.
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
We would like to use protein language models such as ESM2 in the colab document to perform in sillilco mutagenesis. We will calculate single point mutations in the L protein sequence, and try to idenitfy mutations that are more evolutionarily favorable.
Like the assignment I am interested to use ProteinMPNN for to redesign and generate a new sequence. Given the backbone structure of the L protein, this tool will help us generate alternative sequences that maintain the same fold but with higher thermal stability, thereby achieving our goals.
AlphaFold Multimer was particularly interesting too, as it predicts 3D structures of protein complexes (co-folding multiple chains). Novel complexes create range and breadth.
Why do you think those tools might help solve your chosen sub-problem?
ProteinMPNN was very robust in developing sequences that fit a specific shape, there is guarantee we will be able to increase protein stabililty.
ESM2 allows us to scan so many mutations at once, which allows us to very quickly narrow down a direction that we couldn’t perform in wet lab setting.
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
L protein is a membrane protein. Most standard protein models like AlphaFold multimer seem to be trained primarily on soluble proteins. The specific lipid-protein interactions required for lysis may not be fully captured, leading to “stable” designs that fail to insert into the membrane. In my assignment I don’t understsand still how the shape will fit as it seems displaced?
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Input a L protein sequence > use ESM2 to generate favorable mutations, the heat map should show us green-light vs no-go directions in the sequence > Use protein MPNN to generate and find a skeleton template for core stability > add complexity via alphaFold, predicting an interaction. >use PyMOL to check shape and geometry > calculate binding affinity score via colab > and select best candidates!
NGLViewer: NGL Viewer is a collection of tools for web-based molecular graphics. WebGL is employed to display molecules like proteins and DNA/RNA with a variety of representations.
Chimera: A highly extensible program for interactive visualization and analysis of molecular structures and related data, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
Cloud laboratories are making science accessible, affordable, and reproducible. Our aim this semester is to showcase how they can enable human creativity at scale, and how they provide a platform for collaboration and community.
How To Grow (Almost) Anything is about synthetic biology, bioengineering, robotics, automation, art, and AI. But it is also about friendship, shared purpose, and the freedom to build beyond what we know and to be inspired by what can be. To that end, the goal with this cloud lab unit and homework assignment is to inspire collaboration and creativity while designing a scientifically rigorous cell-free fluorescent protein optimization experiment together.
As you plan for final projects, you may want to refer to the provided non-exhaustive list of common Nebula protocols and their parameters in the “Reading & Resources” section below.
Homework — DUE BY START OF APR 28 LECTURE
Info
Note that this homework is due a week later than it ordinarily would due to its release a week later than normal.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
Here’s some evidence of what I contributed to the collective artwork:
I had chosen specific pixels that were assigned different colors but adjacent to core shapes and forms.
My most enjoyable part was during our recitation, our classmate Constantin found a way to hack the system and constantly draw LifeFabs representation onto the community art so we had a leading presence!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
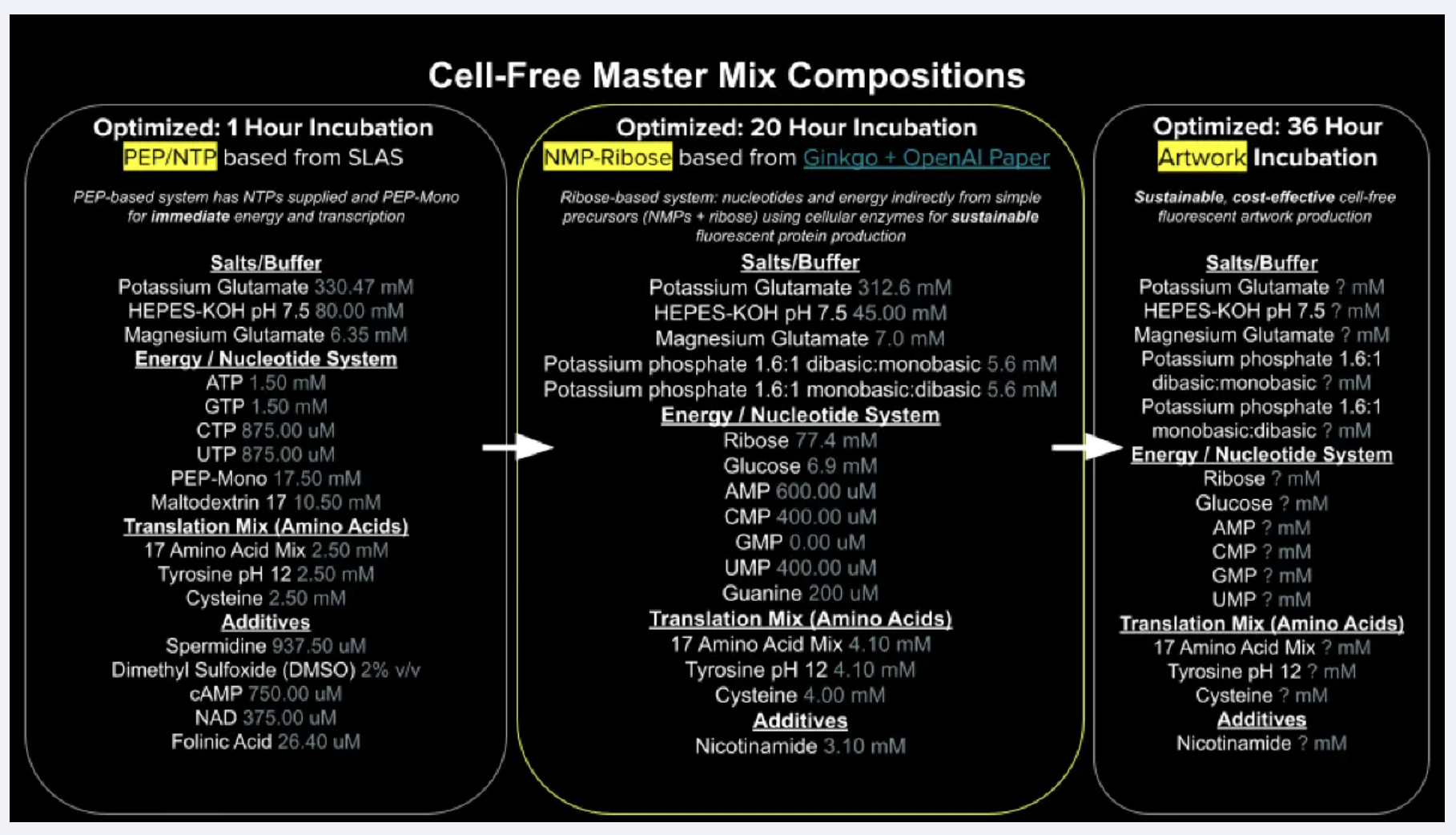
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Clarified lysate of E Coli containing all transcription and translation machinery such as ribosomes,tRNA, aminoacyl-tRNA synthetases, and elongation factors.
BL21(DE3) is a derivative of the E. coli B strain that does not contain the lon protease and is also deficient in the outer membrane protease OmpT. The lack of two key proteases reduces degradation of heterologous proteins expressed in the strain. e.g. rne131 mutation that reduces levels of endogenous RNases and mRNA degradation
Is likely optimized for use with low copy n umber with T7 promoter based plasmids, will have fast growth in minimal medium and ability to reach high cell density
Salts/Buffer
Potassium Glutamate
Salt creating monovalent ionic environment so that ribosomes can maintain their structure and function. flutamate used instead of chloride because high concentrations of chloride ions makes it hard for translation to take place.
HEPES-KOH pH 7.5
keeps reaction stable and enzyme friendly pH during incubation. its to help make sure metabolism wont turn too acidic and denature proteins.
Magnesium Glutamate
magnesium is essential for ribosome assembly and tRNA binding and for enzymes be involved in the nucleotide metabolism. a good concentration makes sure they can bind.
Potassium phosphate monobasic
Potassium phosphate dibasic
These two potassium phosphates help to supply phosphate that can feed back to nucleotide regeneration and help sustain ATP and NTP synthesis over time.
Energy / Nucleotide System
Ribose
central substrate of this NMP-Ribose energy system. Endogenous kinases phosphorylate ribose into ribose-5-phosphate, which feeds both nucleotide biosynthesis and the pentose phosphate pathway, providing a sustained source of NTPs to drive transcription and translation
Glucose
supplementary energy source that feeds glycolysis, generating ATP and NADH alongside the ribose pathway to extend the productive lifetime of the reaction.
AMP
CMP
GMP
UMP
Nucleoside monophosphates that serve as precursors to the activated NTP forms (ATP, CTP, UTP) required for mRNA synthesis and as the universal energy currency powering translation.
GMP here though is set to zero for guanine salvage pathway to produce ennough GTP without needing to supplement as GMP
Guanine
purine salvage pathway, where it is converted to GMP and then phosphorylated up to GTP , more cost effective way to maintain the GTP pool than building it from scratch via de novo synthesis.
Translation Mix (Amino Acids)
17 Amino Acid Mix
substrates for ribosome, building blocks that can be loaded onto tRNAs and incorporated into peptide chain during translation
Tyrosine
dissolved at pH12 bcs tyrosine is not very soluble at normal ph levels,
Cysteine
oxidises in mixed solution and will form disulfide bonds or react with metal ions, it might deplete free cysteine available for target protein.
Additives
Nicotinamide
learnt about this from skincare (haha): precursor to NAD+ that can replenish effects that drives glycolysis and other metabolic reactions.
Backfill
Nuclease Free Water
making sure water is at a right working volume but also no contaminating RNases or DNases that would degrade mRNA trnasscription .
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
Main difference between the two systems is how they supply energy and nucleotides.
1-hour PEP-NTP system delivers ready-made NTPs and fast-burning energy donors directly, so transcription and translation kick off immediately but te fuel runs out quickly as PEP is consumed and inhibitory phosphate builds up.
The 20-hour NMP-Ribose-Glucose system instead supplies simpler precursors (NMPs, ribose, glucose) and lets the lysate’s own enzymes continuously regenerate NTPs from scratch, which is slower to get going but sustains productive protein synthesis much longer.
The short-burst system also uses a richer cocktail of additives to squeeze maximum output from a brief reaction window, while the long-run system compensates with higher amino acid concentrations to keep the ribosomes fed over the extended incubation.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Guanine is converted to GMP via the purine salvage pathway. GTP is the actual substrate incorporated by T7 RNAP during transcription
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP is a superfolder GFP is engineered to fold correctly even under stress conditions, and it matures so quickly that it produces a reliable green signal faster than almost any other variant, making it a great baseline readout in cell-free reactions.
mRFP1 is the earliest or one of the earliest monomeric red proteins developed, it is notably slow to mature and not particularly bright, so in a cell-free context you often have to wait significantly longer before you see meaningful signal compared to newer red variants.
mKO2 : mKO2 needs oxygen to complete its chromophore, and because cell-free reactions in small droplets or dense solutions can become oxygen-depleted, its orange signal may be weaker or delayed relative to what you’d see in a well-aerated system.
mTurquoise2 this is very efficient at converting absorbed light into emitted fluorescence, meaning even modest expression levels produce a strong, stable signal that holds up well over long imaging periods.
mScarlet_I - is newer type of rFP and matures much faster and produces a brighter signal sooner, making it well-suited for watching protein production happen in real time. >this could be good for my experiment for transient transfections?
Electra2 optimized specifically for speed, Electra2 folds and forms its chromophore almost immediately after translation, so it’s particularly useful when you need to detect protein expression as early as possible in the reaction.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
To maximise mKO2 fluorescence over a 36-hour incubation, we would increase nicotinamide to 1.5x its standard concentration and slightly raise the ratio of dibasic potassium phosphate to keep the reaction pH stable for longer. Nicotinamide sustains the metabolic pathways that regenerate NAD⁺ and keep translation running, while the adjusted phosphate balance prevents the gradual acidification that would otherwise slow or stall the ribosomes. Together these changes give mKO2 the stable, long-running reaction environment it needs to complete its slow, oxygen-dependent chromophore maturation and accumulate detectable fluorescence over the full incubation window.
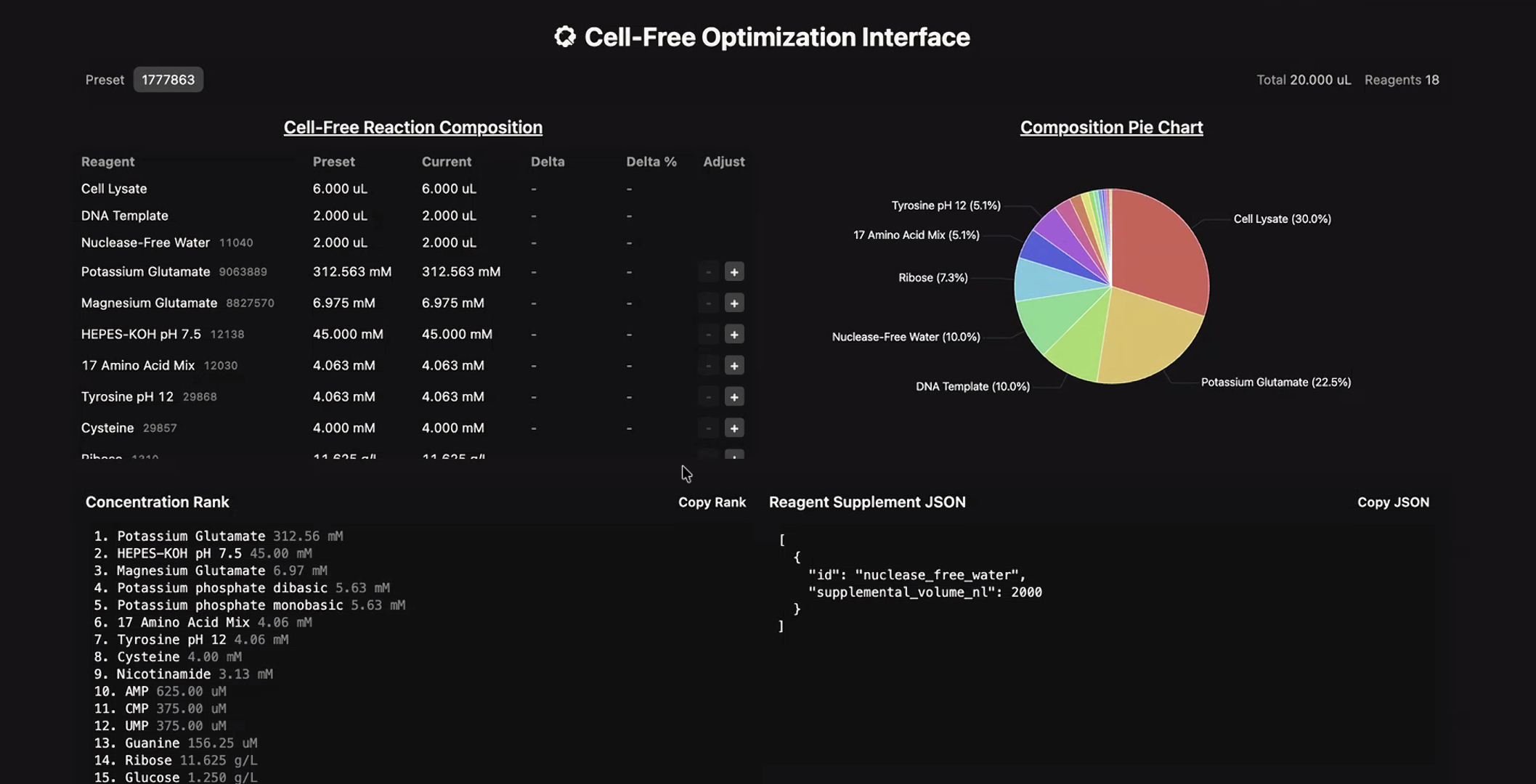
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
i started doing some mixes, aprt from nicotinamide mentioned above, adding a low concentration of GMP and slightly increasing cysteine will likely change RFP slow to mature and not particularly bright, the strategy is to compensate by producing more total protein, more mRNA transcribed means more chances for mature fluorescent protein to accumulate by the end of the incubation. The extra GMP tops up the GTP pool that RNA polymerase draws on during transcription, helping sustain mRNA production over the full reaction window. cysteine is a structural amino acid that appears in mRFP1’s folding core, so keeping it in good supply reduces the chance of misfolding
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!).
The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
N/A? No data was provided that week but we can use the existing data from earlier provision.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Assignees for the following sections
MIT/Harvard students
Optional
Committed Listeners
Optional
Ginkgo Nebula Cloud Laboratory Rendering, 2025
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Tip
Note from Ronan: If you are interested in helping me build out future HTGAA cloud lab software, please fill out this form!
This lecture presents a range of advanced technologies to do precision
measurement of proteins at atomic scales, characterizing chemical
composition, and detecting protein sequence and structure.
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework: Final Project
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
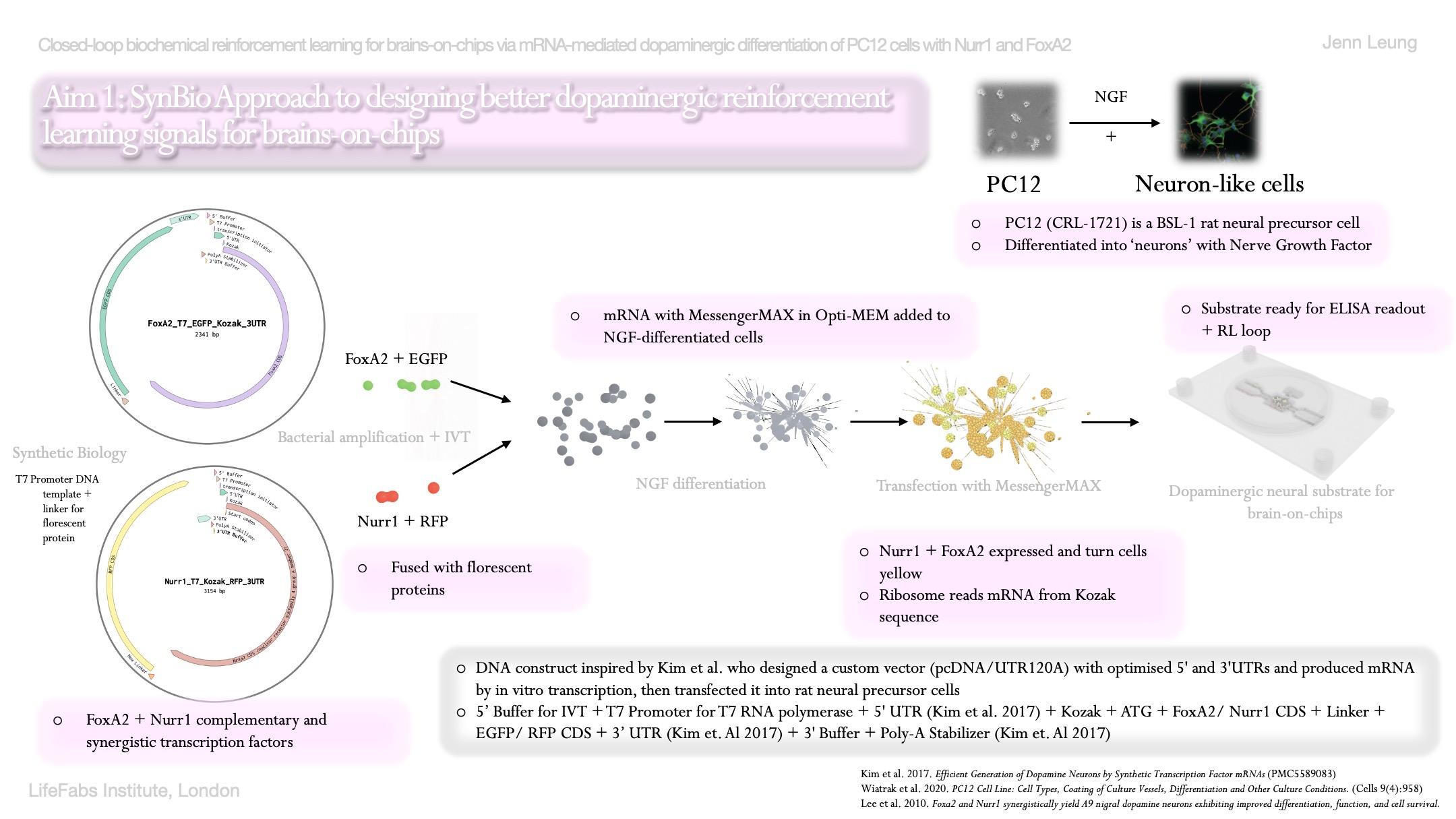
As I will be dopaminergically differentiating PC12 cells via mRNA approach, the first aspect for measurement in my project is to measure the quantity of dopamine released by PC12 cells into surrounding media. To measure dopamine concentration, I will need to measure with ELISA, which is an enzyme linked immunosorbent assay that is a plate-based quantitative immunoassay.
From Thermo Fisher’s ELISA kit, there are roughly 7 steps for the instant ELISA kits, from rehydration of standard and sample wells on plate, to incubation, to washing, adding TMB substrate, adding stop solution, and then to calculate results.
Absorbance is read at 450 nm on the Spark plate reader, and dopamine concentration will be calculated from a standard curve from the known dopamine concentrations ranging from 0 to 50 nM.
Expected range of dopamine should be 3- 50 nM and co-transfected wells with Nurr1-GFP and FoxA2 RFP will be 3-5x above negative control if we follow Kim et al (2017) closely.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
After successful transfection of my fusion proteins Nurr1+GFP and FoxA2+RFP, other elements I’m planning to measure will also include the presence of GFP and RFP florescence as measurements and validations for subcellular localizaiton of these fusion proteins in PC12 cells.
Their presence will help confirm that the mRNA constructs were successfully taken up and translated into protein and localized to nucleus. This means dopamine synthesis will be activated in PC12 cells.
This can be measured via live florescence microscopy and quantitative plate reader.
One example will be use florescene microscopy where it will illuminate a sample with a specific wavelength of light, which will cause tagged structures (fluorophores) to emit a lower-energy glow that reveals specific cellular components.
The expected results is to see GFP excited at ~488 nm and emitting at ~520 nm, whereas RFP will be excited at ~555 nm and emitting at ~610 nm. If GFP and RFP are both present and successfully transfected, the nucleus of these cells should overlay green and red to show a bit of yellow in the nucleus.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
I will be using ELISA kit, Spark plate reader, and florescence microscopy as mentioned above (description also above).
Homework: Waters Part I — Molecular Weight
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
I used the ExPASy tool with the full eGFP sequence (including the LE linker and His-tag), the calculated theoretical mass comes about to 28,006.60 Da with a pI of 5.90. But eGFP doesn’t stay chemically identical after unfolding, three amino acids cyclise to form the fluorescent chromophore, and this reaction releases small molecules that reduce the protein’s mass by roughly 20 Da. The modification will arrive at a theoretical weight of approximately 27,986.60 Da accounting for this maturation gap.
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine $z$ for each adjacent pair of peaks $(n, n+1)$ using:
$$ {\large z} = {\Large \frac{\frac{m}{z_{n+1}}}{\frac{m}{z_n} - \frac{m}{z_{n+1}}}} $$
I selected these two peaks:
Peak 1 (n): m/z = 933.7349
Peak 2 (n+1): m/z = 903.7148
Because mass is constant and m/z decreases as charge increases, the higher m/z peak (933.7349) carries the lower charge. Plugging into the formula:
z=903.7148933.7349−903.7148=903.714830.0201=30.10≈30z = \frac{903.7148}{933.7349 - 903.7148} = \frac{903.7148}{30.0201} = 30.10 \approx 30z=933.7349−903.7148903.7148=30.0201903.7148=30.10≈30
So the charge state of the 903.7148 peak is z = 30, and the adjacent 933.7349 peak is z = 29.
Determine the MW of the protein using the relationship between $\frac{m}{z_n}$, $MW$, and $z$
Each charge state arises from protons (mass = 1.00728 Da) attaching to the protein, so:
M=(mz)×z − (z×1.00728)M = \left(\frac{m}{z}\right) \times z \ - \ (z \times 1.00728)M=(zm)×z − (z×1.00728)
For the z = 30 peak (m/z = 903.7148):
M=(903.7148×30) − (30×1.00728)M = (903.7148 \times 30) \ - \ (30 \times 1.00728)M=(903.7148×30) − (30×1.00728)
=27,111.444 − 30.218=27,081.23 Da= 27{,}111.444 \ - \ 30.218 = \textbf{27,081.23 Da}=27,111.444 − 30.218=27,081.23 Da
For the z = 29 peak (m/z = 933.7349):
M=(933.7349×29) − (29×1.00728)M = (933.7349 \times 29) \ - \ (29 \times 1.00728)M=(933.7349×29) − (29×1.00728)
=27,078.312 − 29.211=27,049.10 Da= 27{,}078.312 \ - \ 29.211 = \textbf{27,049.10 Da}=27,078.312 − 29.211=27,049.10 Da
The difference between these two values is only 0.032 Da
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
$$ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $$
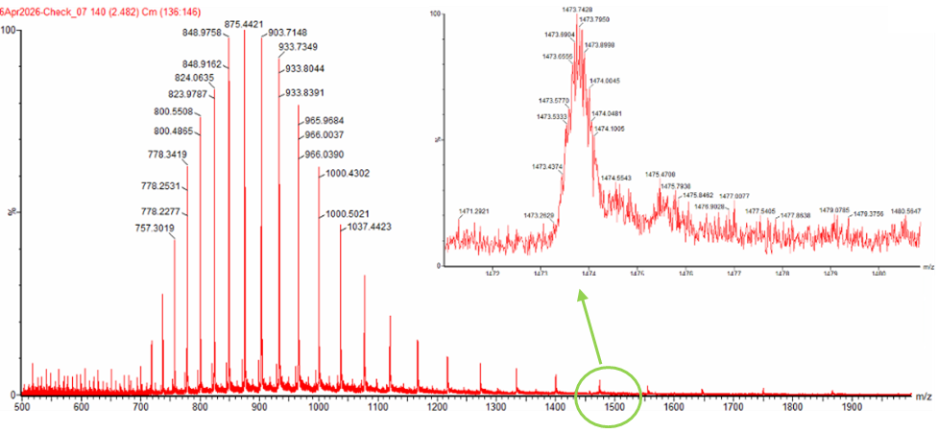
Figure 1. Mass Spectrum of intact eGFP protein from the Waters Xevo G3 LC-MS (a mass spectrometer with 30,000 resolution) with individual charge state peaks labeled with $\frac{m}{z}$ values.
Taking the average of the two calculated masses as the experimental MW:
MWexp=27,081.23+27,049.102=27,065.16 DaMW_{\text{exp}} = \frac{27{,}081.23 + 27{,}049.10}{2} = 27{,}065.16 \ \text{Da}MWexp=227,081.23+27,049.10=27,065.16 Da
Comparing to the theoretical MW of 26,941.48 Da:
Accuracy=∣27,065.16−26,941.48∣26,941.48=123.6826,941.48=0.00459=4,591 ppm\text{Accuracy} = \frac{|27{,}065.16 - 26{,}941.48|}{26{,}941.48} = \frac{123.68}{26{,}941.48} = 0.00459 = \textbf{4,591 ppm}Accuracy=26,941.48∣27,065.16−26,941.48∣=26,941.48123.68=0.00459=4,591 ppm
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
No charge state cannot be seen from zoomed-in peak alone. The zoomed-in peak reveals the pattern of the charge state, but the charge state is determined by comparing the spacing between adjacent charge-state peaks in the full spectrum.
Homework: Waters Part II — Secondary/Tertiary structure
Assignees for the following sections
MIT/Harvard students
Optional but highly recommended
Committed Listeners
Optional but highly recommended
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
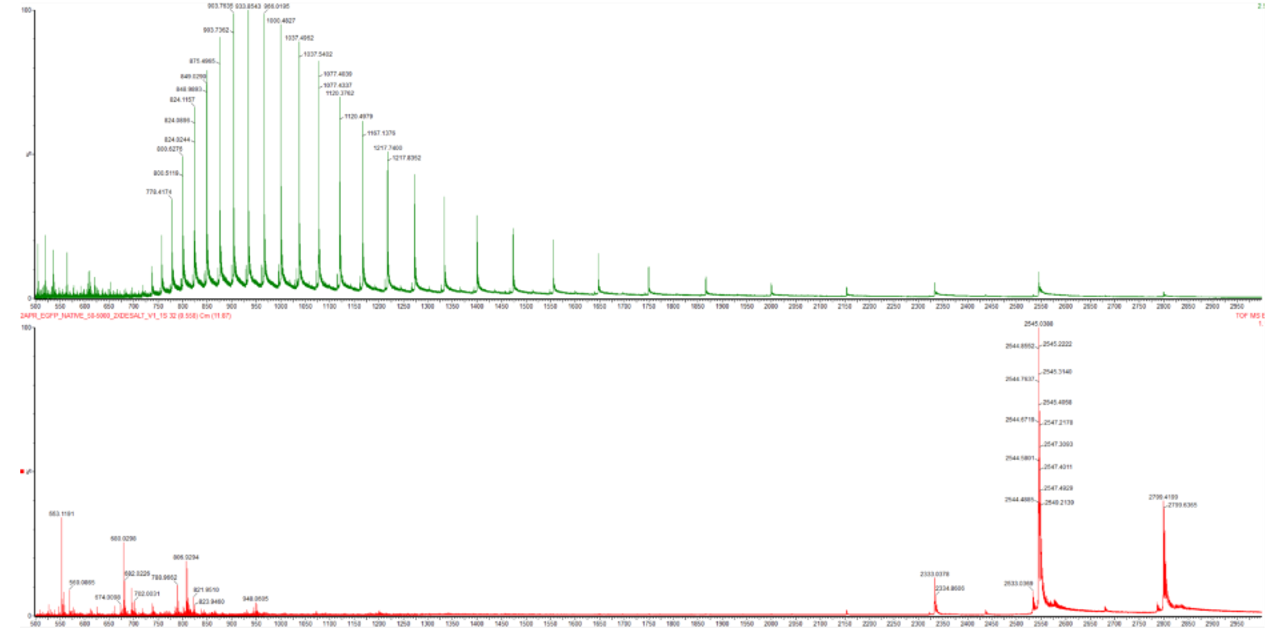
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Figure 2. Comparison of the mass spectra between denatured (top) and native (bottom) eGFP standard on the Waters Xevo G3 QTof MS.
In their native state proteins retain their three dimensional architecture , the secondary structures (helices and sheets), the overall folded shape (tertiary structure), and any multi-subunit assemblies (quaternary structure) are all intact.
When a protein denatures, all those structures collapses and it reverts to a loose, unfolded chain , essentially just its primary sequence with no organised shape remaining.
unfolded proteins expose their hydrophobic surfaces so bonds are broken and charge states are more generated
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 $\frac{m}{z}$? What is the charge state? How can you tell?
Figure 3. Native eGFP mass spectrum from the Waters Xevo G3 Q-Tof MS. The inset is a zoomed-in view of the charge state at ~2800 $\frac{m}{z}$ on a mass spectrometer with 30,000 resolution.
skipped…
Homework: Waters Part III — Peptide Mapping - primary structure
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
From Benchling: Amino Acid Frequencies
Amino acid Count
Ala A 8 3.2%
Arg R 6 2.4%
Asn N 13 5.3%
Asp D 18 7.3%
Cys C 2 0.8%
Gln Q 8 3.2%
Glu E 17 6.9%
Gly G 22 8.9%
His H 15 6.1%
Ile I 12 4.9%
Leu L 22 8.9%
Lys K 20 8.1%
Met M 6 2.4%
Phe F 12 4.9%
Pro P 10 4.0%
Ser S 10 4.0%
Thr T 16 6.5%
Trp W 1 0.4%
Tyr Y 11 4.5%
Val V 18 7.3%
Pyl O 0 0.0%
Sec U 0 0.0%
There are 20 Lysines (K) and 6 Arginines (R) in eGFP.
How many peptides will be generated from tryptic digestion of eGFP?
Selected enzyme:
Trypsin
Maximum number of missed cleavages (MC):
0
Cysteines modifications:
All cysteines in reduced form
Methionines modifications:
Methionines have not been oxidized.
Mass of displayed peptides:
500 Dalton
Mass calculation:
Using monoisotopic masses of the occurring amino acid residues and giving peptide masses as [M+H]+.
Peptide masses for your input sequence
90.7% of sequence covered (you may modify the input parameters to display also peptides < 500 Da):
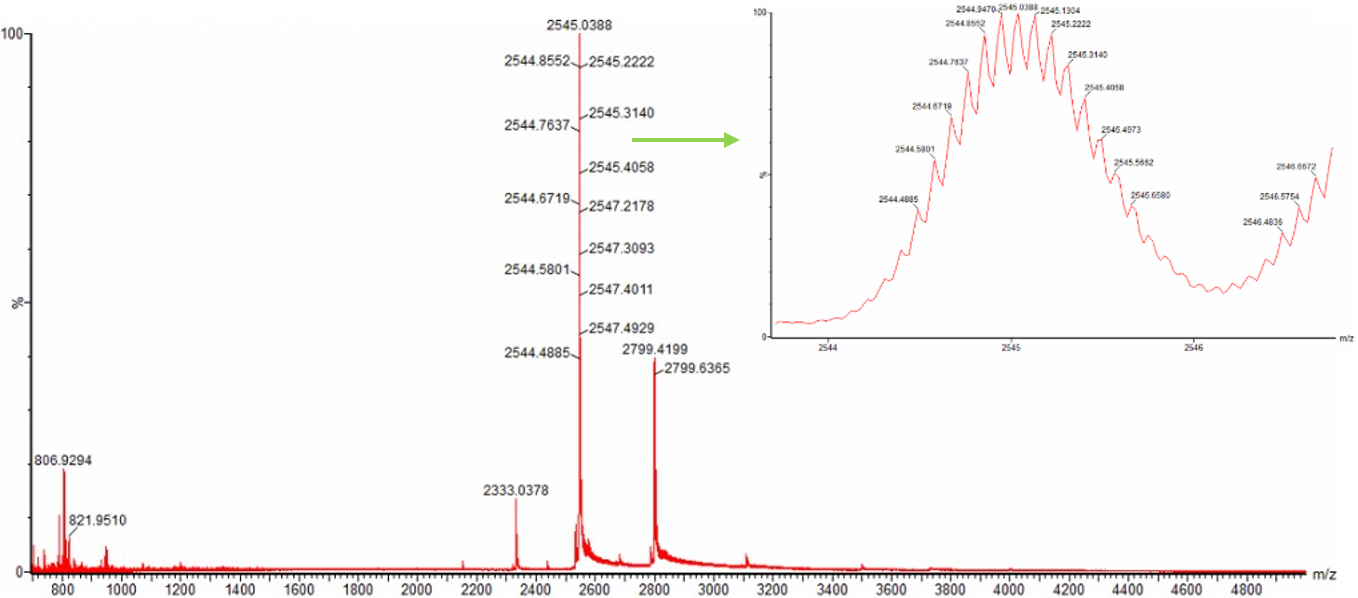
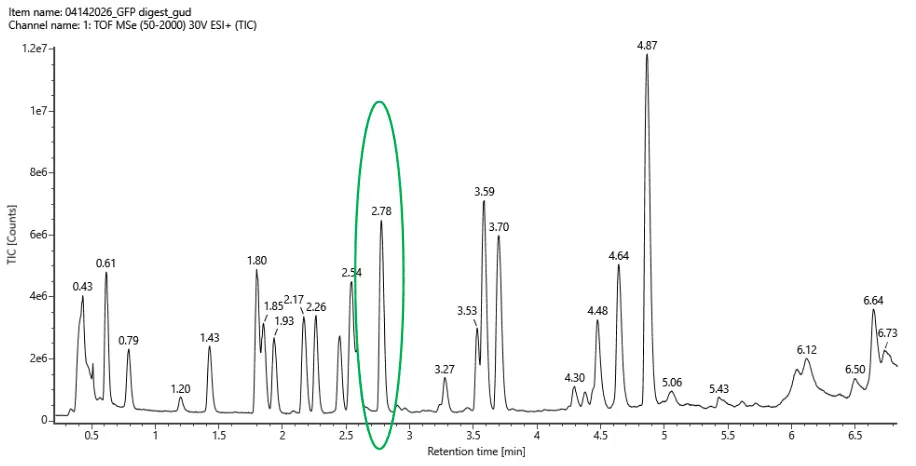
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Figure 5a. Total ion chromatogram (TIC) of the eGFP peptide map. The peak at 2.78 minutes is circled, and its MS data is shown in the mass spectrum in Figure 5b, below.
Counting across the 0.5–6 minute window, there are roughly 20 peaks visible above the 10% relative abundance threshold.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Pretty much, 20 observed peaks versus 19 predicted peptides is quite close. The slight excess is normal in LC-MS experiments, where things like background noise, trace impurities, or the occasional missed cleavage by trypsin can produce an extra signal or two.
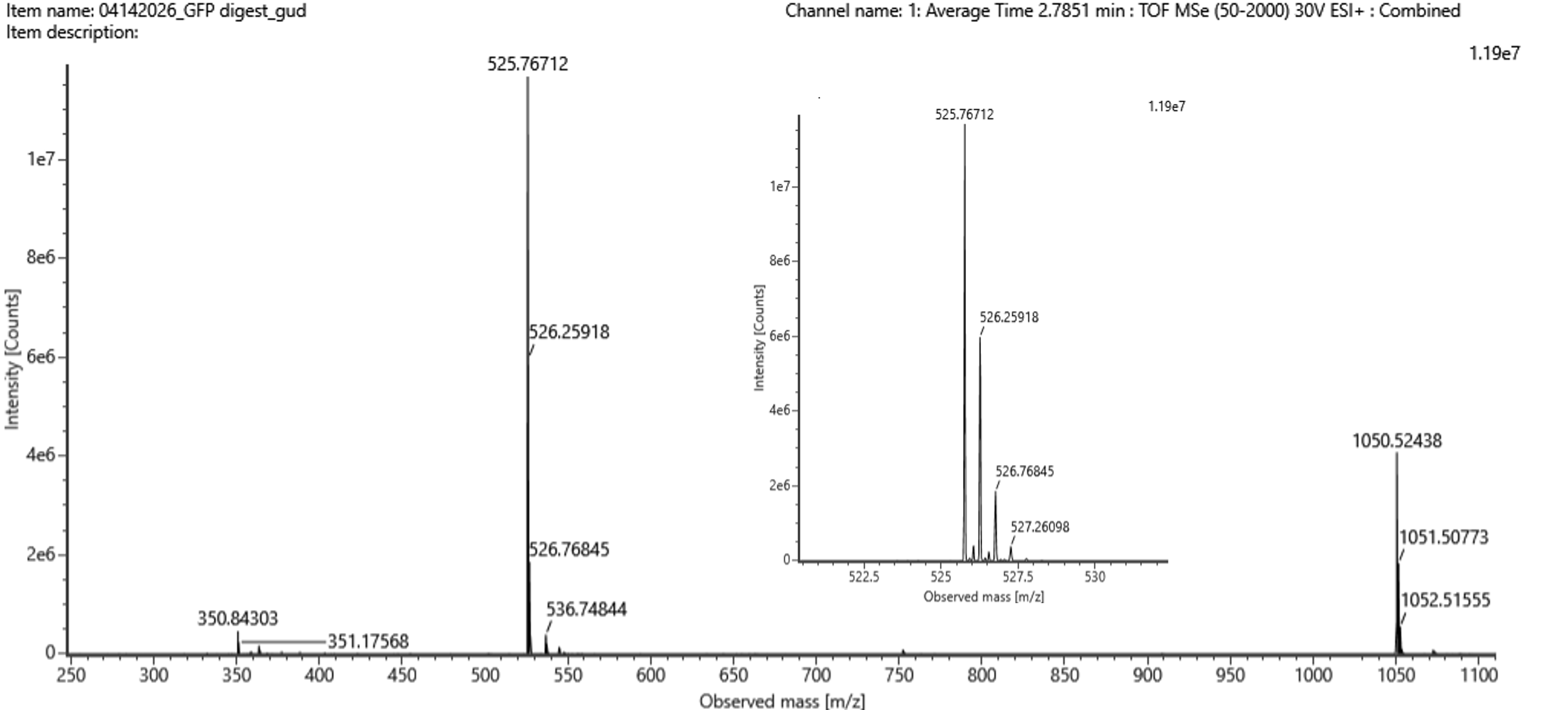
Identify the mass-to-charge ($\frac{m}{z}$) of the peptide shown in Figure 5b. What is the charge ($z$) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ($\small{[M\!\!+\!\!H]^+}$) based on its $\frac{m}{z}$ and $z$.
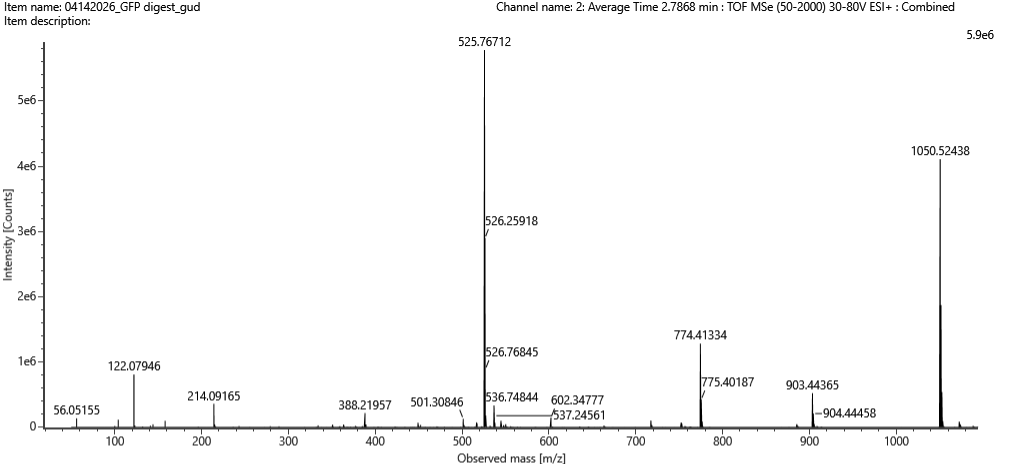
Figure 5b. Mass spectrum figure to show $\frac{m}{z}$ for the chromatographic peak at 2.78 min from Figure 5a above. The inset is a zoom-in of the peak at $\frac{m}{z}$ 525.76, to discern the isotope peaks.
Figure 5c. Fragmentation spectrum of the peptide eluting at retention time 2.78 minutes in Figure 5a (above).
The most abundant peak in Figure 5b sits at m/z = 525.767. Looking at the isotope spacing in the zoomed inset, the peaks are separated by about 0.5 m/z units, which tells us the charge state is z = 2. Converting to the singly charged form gives a monoisotopic mass of approximately 1050.527 Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
(Recall that $ \text{Accuracy} = \frac{|MW_{\text{experiment}} - MW_{\text{theory}}|}{MW_{\text{theory}}} $ )
Matching that mass against the PeptideMass output points to the peptide [FEGDTLVNR], which has a theoretical mass of 1050.521 Da. The difference between measured and theoretical is only about 2.0 ppm which is within expected accuracy range
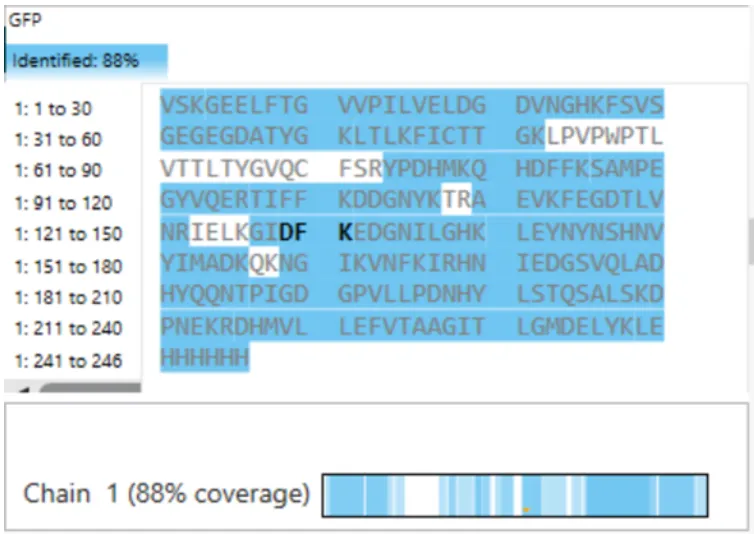
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
Figure 6. Amino Acid Coverage Map of eGFP based on BioAccord LC-MS peptide identification data.
The coverage map from the BioAccord run shows that 88% of the eGFP sequence was confirmed by peptide mapping.
Bonus Peptide Map Questions
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Homework: Waters Part IV — Oligomers
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS).
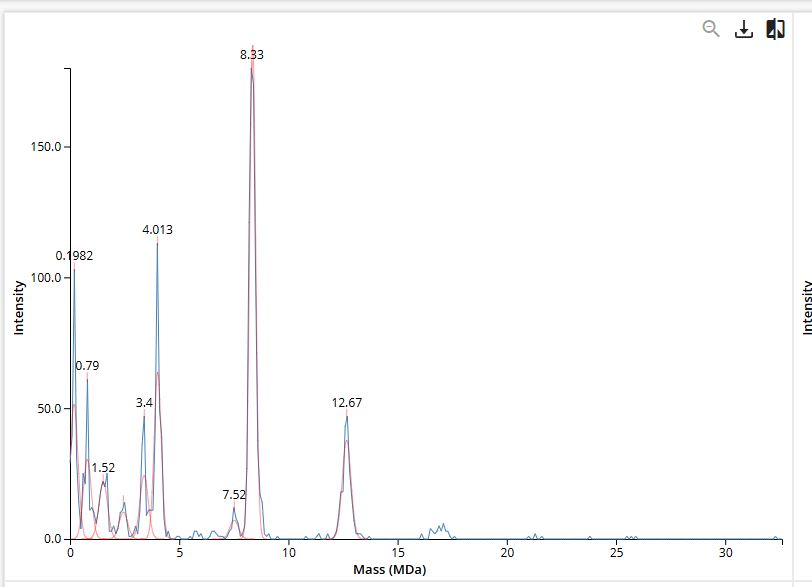
CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer
8FU Didecamer
8FU 3-Decamer
8FU 4-Decamer
KLH is made from two types of subunits inc a smaller 7FU subunit (340 kDa) and a larger 8FU subunit (400 kDa) . they naturally cluster together into rings of 10 (decamers) and larger stacked assemblies.
Assignment
Calculation
Expected Mass
CDMS Peak Observed
Match
7FU Decamer
10 × 340 kDa
3,400 kDa (3.40 MDa)
~3.40 MDa
Exact
8FU Decamer
10 × 400 kDa
4,000 kDa (4.00 MDa)
~4.01 MDa
0.3% off
8FU Didecamer
20 × 400 kDa
8,000 kDa (8.00 MDa)
~8.33 MDa
4.1% off
8FU 3-Decamer
30 × 400 kDa
12,000 kDa (12.00 MDa)
~12.67 MDa
5.6% off
8FU 4-Decamer
40 × 400 kDa
16,000 kDa (16.00 MDa)
Not visible
—
The 7FU Decamer matches its theoretical mass well. The larger assemblies (didecamer and 3-decamer) show slightly higher masses than predicted, which is expected as proteins tends to carry along some associated water, salt ions, or lipid molecules that add a small amount of extra mass on top of the protein itself.
The 4-decamer doesn’t show up clearly in the spectrum at all, which suggests it’s either not forming in this sample, present in such small amounts that it falls below the detection threshold, or simply too heavy for the instrument to resolve clearly at the high-mass end.
The two smaller peaks visible around 0.79 MDa and 1.52 MDa in Figure 7 are likely sub-decameric fragments , smaller incomplete assemblies such as dimers or tetramers of individual subunits that haven’t fully assembled into the complete decamer ring.
Figure 7. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Homework: Waters Part V — Did I make GFP?
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Property
Theoretical
Calculation
Observed (Intact LC-MS)
PPM Error
Molecular weight (kDa)
28.007 kDa
From ExPASy sequence input
~27.984 kDa
~820 ppm
Chromophore correction
−20 Da
28,007 − 20 = 27,987 Da
—
—
Peptide mapping coverage
100%
19 predicted peptides, 88% identified
88%
—
Peptide FEGDTLVNR mass (Da)
1050.5214 Da
From PeptideMass tool
1050.5270 Da
~5 ppm
it worked. yes. the mass is within 820 ppm of the theoretical value, it can be explained by chomophore maturation which is when eGFP folds and forms its fluorescent chromophore. If it loses roughly 20 Da. The peptide map also independently confirms 88% of the amino acid sequence at under 5 ppm accuracy, meaning the instrument directly detected fragments covering nearly the entire protein. What was expressed and purified is very liekly to be eGFP.
Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
Week 14: Biofabrication
Week 14: Biofabrication
Homework: Finish your Final Project
Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
Week 2: DNA Read, Write, and Edit
Week 2: DNA Read, Write, and Edit
Part 1: Benchling & In-silico Gel Art
1.1 Import Lambda DNA
Simulate Restriction Enzyme Digestion
Virtual Gel
Part 2: Gel Art
I have chosen to create a gel art of a person doing a jumping jack through randomization method.
Part 3: DNA Sequence Design
3.1 Protein Selection
I have chosen IL23 as I am interested in autoimmune disease such as psoriasis. This protein is related to inflammation and I am curious to learn more about biologics in general.
Reverse translation of sp|Q5VWK5|IL23R_HUMAN Interleukin-23 receptor OS=Homo sapiens OX=9606 GN=IL23R PE=1 SV=3 to a 1887 base sequence of most likely codons.
Reverse translation of sp|Q5VWK5|IL23R_HUMAN Interleukin-23 receptor OS=Homo sapiens OX=9606 GN=IL23R PE=1 SV=3 to a 1887 base sequence of consensus codons.
Codon optimization is important because there is codon usage bias, which means humans and other organisms like E. coli might prefer different codons for the same amino acid. Expressing human gene like IL23 might be difficult because codons natural to human cells are rare in E. coli. If bacterium has low levels of corresponding tRNAs, then it will be slowed down during translation. There will be low protein yield as a result.
The codon optimization here increased GC content so there will be more mRNA stability. Codon adaptation index has also gone up.
3.4 Protein Expression
Now, we will use this optimized DNA sequence to create IL23 protein. First we clone the codon optimized sequence into expression vector, and we transform a plasmid into E. coli cells. Bacteria will be shocked by heat to start making protein. The cell’s RNA polymerase will read the DNA and makes mRNA copy. Once the transcription is read, it will begin to build protein using tRNAs in the translation process.
Once this is done, there is a chromatography technique which separates protein from everything else in the cell.
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would like to sequence and read genes that can help facilitate brains-on-chips research, so while human DNA is interesting, I am perhaps more curious about biocompatible materials or bio-glue that can help with assembling living neuronal tissue with physical hardware like microelectrode arrays.
This is usually microbial/ environmental DNA where we can look at genetic strands that can be programmed into biocompatible hydrogels.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
If I were to use the Sanger sequencing method, we will need to use ddNTPs to shorten chains and terminate chains.
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
First generation uses chain-termination, where Polmerase copies the DNA, but ddNTPs tagged with florescent colors are added, so that it creates fragments and is separated by electrophoresis. Second generation only sequnces short fragments and reading a lot of fragments simutaneously. Third generation pulls single strands through nanopore in a membrane and is is read through currents.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
We need to prepare input but growing bacteriaphage, we will use plasmid purification to extract DNA.
Use Benchling to design primer.
We will be preparing via chain-termination PCR which will mix DNA sequence with an enzyme Poolymerase, a primer to bind to target DNA, and dNTPs and ddNTPS that are fluorescent to terminate chains.
Electrophoresis will help us separate DNA, RNA, and proteins by electrical charge.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
Although irrelevant to my final project I’ve always been fascinated by biologics as adillimumab, which is a type of recombinant DNA that instruct living cells to synthesize a therapeutic protein.
For the final project probably something that allows biological tissue to be more adhesive to microelectrodes as a part of facilitating electrical communication. Also interested in bioprinting microfluidics.
See some famous examples of DNA design
DNA origami by Paul W. K. Rothemund, California Institute of Technology, 2004. 100 nanometers in diameter.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Benchling, which is a platform that can help copy and paste DNA sequence, import DNA and protein sequences, perform in silico restriction digestion, and to design gel layouts. We will cut with restrictions enzyme,copy through polymerase chain reactions, and perform DNA cloning to synthesize in silico.
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
If working on neural tissues, I am curious to edit neuroplasticity-related genes so that I can consider how plasticity can be modified or reinforced.
I would like to facilitate electrical and chemical stimulation to make it easier for reinforcement learning experiments.
Colossal Biosciences Inc., a biotechnology company using genetic engineering to de-extinct various historic animals such as the woolly mammoth, dodo, and dire wolf.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Electrophoresis will help us separate DNA, RNA, and proteins by electrical charge.
I would like to first perform PCR and digest, and then conduct assembly by converting GFP into RFP.
Week 5: Protein Design Part II
Week 5: Protein Design Part II
Homework — DUE BY START OF MAR 10 LECTURE
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Pseudo Perplexity for binder ‘FLYRWLPSRRGG’ with protein sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
is: 20.63523127283615
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
This is PepMLM’s most confident score: WRSGATVARHAX.
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
WRYGAAAVELKE
ipTM = 0.28
pTM = 0.78
WSYPWVALELGK
ipTM = 0.67
pTM = 0.88
WRSGATVARHAX
ipTM = 0.42
pTM = 0.86
iWHSGVVGLARGX
pTM = 0.32
pTM = 0.85
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
One of the PepMLM-generated peptides (ipTM = 0.67) significantly outperforms the others and appears to exceed the confidence of the known literature binder (which often scores in the 0.50–0.60 range in similar AlphaFold benchmarks). While the sequence WRYGAAAVELKE (ipTM = 0.28) failed to find a stable “home” on the SOD1 surface, the high-scoring candidate suggests that PepMLM successfully identified a sequence that “staples” the protein’s interface. This indicates that the language model can indeed generate de novo sequences that are more structurally compatible with the mutated A4V surface than existing experimental peptides.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide!
For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes:
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
I would choose to advance WRYGAAAVELKE as the best option. First it has great binding strength, with the highest predicted binding affinity ($pK_d \approx 6.27$), which is roughly in the micromolar range, a great starting point for a de novo peptide.
The fact that it has a negative GRAVY score (-0.38) shows that it is more hydrophilic than others. This will help with solubility and lower hemolysis risk (0.049).
Structurally, although its initial ipTM was low, its chemical makeup makes it a better scaffold than a peptide that might bind tightly but aggregate in the blood.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Running locally on machine because of GPU allocation
Results:
Peptide 1: EPTEEEQRTCGT
Affinity score: 9.17
Solubility score: 0.70
moPPit optimizes for binding. So it will generate sequences often with higher predicted affinity scores than PeptiVerse. Peptide 1 EPTEEEQRTCGT has a superior affinity score and is physically complementary to the A4V sequence.
Part B: BRD4 Drug Discovery Platform Tutorial (Optional)
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
This homework requires computation that might take you a while to run, so please get started early.
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is a chemical environment designed to ensure the most accurate duplication of DNA possible.
At its core is the Phusion DNA Polymerase, which is enzyme created by fusing a Pyrococcus-like proofreading polymerase to a double-stranded DNA-binding domain. This structural modification allows the enzyme to remain attached to the DNA template for much longer than standard enzymes, resulting in high processivity and a significantly lower error rate. To support this activity, the mix contains a balanced concentration of dNTPs, which serve as the raw material for the new DNA strands, and a specialized reaction buffer. This buffer includes magnesium chloride, a vital cofactor that helps the polymerase coordinate with the phosphate groups of the incoming nucleotides, as well as various stabilizers that prevent the enzyme from denaturing during the intense heat of the thermal cycling process.
What are some factors that determine primer annealing temperature during PCR?
Determining the correct annealing temperature for a PCR reaction requires balancing several molecular factors. The primary driver is the melting temperature of the primers, which is largely dictated by their length and their GC content.
Because Guanine-Cytosine pairs are held together by three hydrogen bonds compared to the two bonds in Adenine-Thymine pairs, primers with more G and C bases require more energy—and thus a higher temperature—to separate and re-anneal. Furthermore, the concentration of salts and ions in the master mix, such as potassium and magnesium, can stabilize the negative charges on the DNA backbone, effectively raising the required annealing temperature. If the temperature is set too low, the primers may bind non-specifically to the wrong parts of the template, while setting it too high may prevent the primers from binding at all, resulting in no amplification.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR:PCR is a synthetic process that builds billions of new copies of a specific DNA segment using thermal cycling and a polymerase enzyme, making it the preferred choice when starting with a tiny amount of DNA or when you need to add custom sequences, like Gibson tails, to the ends of a fragment.
Restriction enzyme digests: restriction digest is an analytical or preparatory process that uses specialized proteins to cut an existing, purified piece of DNA at specific recognition sequences. While PCR is ideal for generating large quantities of modified DNA, restriction digests are often preferred for simpler tasks like subcloning between classic plasmids or performing a diagnostic check to see if a plasmid contains the correct insert.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Careful attention must be paid to the design of the fragment ends. Unlike traditional cloning, Gibson Assembly relies on an exonuclease enzyme that chews back the ends of DNA to reveal single-stranded overlaps. Therefore, you must ensure that each PCR-generated fragment has a “tail” that is identical to the end of the adjacent fragment, typically between 20 and 40 base pairs in length. It is also critical to treat your PCR products with an enzyme like DpnI to destroy any original template DNA and to purify the final product through a column or a gel. This prevents leftover primers or incorrect DNA templates from interfering with the assembly process, ensuring that only the intended overlapping fragments are available for the final reaction.
How does the plasmid DNA enter the E. coli cells during transformation?
circular plasmid is introduced into a bacterial cell, and it relies on making the E. coli “competent” to receive foreign DNA. In a typical chemical transformation, the cells are treated with a calcium chloride solution that helps neutralize the repulsive negative charges between the DNA and the cell membrane. By applying a sudden heat shock at 42°C, a pressure difference is created between the inside and outside of the cell, which momentarily opens up small pores or “adhesion zones” in the lipid bilayer. This allows the plasmid to be pulled into the cytoplasm, where the cell can then begin to express the genes carried on the plasmid, such as antibiotic resistance.
Describe another assembly method in detail (such as Golden Gate Assembly):
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly a method that uses Type IIS restriction enzymes, such as BsaI, to assemble multiple parts simultaneously. These specific enzymes are unique because they recognize a DNA sequence but cut the DNA several bases away from that site, allowing for the creation of custom four-base overhangs. Because the recognition site is actually removed during the cutting process, the final assembled product no longer contains the enzyme’s “handle” and cannot be cut again. This creates a “one-pot” reaction where digestion and ligation happen in a continuous cycle, eventually driving the reaction toward the fully assembled, stable circular plasmid. This method is exceptionally modular and is frequently used in synthetic biology toolkits to mix and match different promoters, genes, and terminators with nearly 100% efficiency.
Model this assembly method with Benchling or Asimov Kernel!
Assignment: Asimov Kernel
no asimov account - on pause
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository:
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo:
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
NEB’s (New England Biolabs) explanation & protocols for Gibson Assembly
Week 7: Genetic Circuits Part II
Week 7: Genetic Circuits Part II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
An artificual neuron is a weighted summation through an activation function that produces outputs, eventually they form networks to become ANN.
Intracellular artificial networks still have weighted summation and a non-linear activation function, but we can consider implementing gene circuits as these activation functions.
The main difference is that IANNs will have two inputs that can do addition and subtraction. On the one hand, a promoter that through transcription makes a gene, and through translation we create proteins, we can perform addition on this.
To subtract, we can treat input x1 as an endoribonuclease CasE that will bind and cleaves the RNA on the sequence and produce output. x1 is negative weight and x2 is positve weight, where the function is max(x2-x1,0).
This is also referred to as Sequestration. Sequestration involves using an endorribonucleus to transcribe into mRNA to produce non-linearity (applying single turnover enzyme to remove it out of circulation).
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
I think an interesting use of IANNs would be in culturing and programming organoids - Weiss Lab also focuses a lot on programmable patterning to trigger cell changes, and I can see this leading to very useful applications in supporting organoid intelligence. I think using endoribonuclease in waste management in microfluidics might be useful.
Microfluidics supporting organoid growth may end up accumulating extracellular vesicles, might shed RNA and dead cells, and these RNA-protein aggregates that can clog channels. We can use RNase H to clear RNA or use Cas13 endoribonuclease to cleave transcripts or CasE to degrade fragments so they can pass through filters, then maybe more layers of flushing and binding for removal? Limitations - not sure how they will exist the microfluidics- more resesarch needed.
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
There’s lots of use of mycelium leather, that is currently being used to manifacture into fashion products. They have used agricultural waste as feedstock and with enough coating, can develop great strength and malleability. There’s much less waste and resources needed to create mycelium leather and will help us with better animal welfare.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
It might be possible to create mycelium electronics which I’m super interested in. I am interested to genetically engineer mycelium to have better conductivity or bioelectric activity, or to use bioglue to stick to sensors for activity readout.
Fungi is adaptable and can respond to environments quickly and act as good living sensors. They ’learn’ quickly and can be interfaced with EMG sensors without needing to submerge in many culture mediums or inject antibiotics.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS
Done this!
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free systems help us understand biology ‘from scratch’ to bioengineer from smaller units. There’s wider flexibility for scaffolding biology from the ground-up and controlling the environments in a complete model. Existing living cells as we know it are already incredibly complex and hence less controlled in experimental settings.
Synthetic cell engineering allows flexibility in size of the cell, proteins, and even expanding largely on the chemistry of the cell.
So the two scenarios could be if you want to control the size of the cell and want uniform control it might be ideal to use cell-free system.
The other scenario might be to engineer a specific chemical environment or want chemical diversity in the experiment that is not naturally common/ compatible with cells.
Compared to in-vivo expression where you have to create plasmids, cell-free protein expressions are faster and cheaper to construct and can also help you through quick iterations with linear fragments and without plasmids.
Describe the main components of a cell-free expression system and explain the role of each component.
The anatomy of the synthetic cell has multiple parts:
phospholipids and cholesterol to create strong lipid membranes
cytoplasm contains small molecules
cell extract such as ribosomes and enzymes
tRNAs
plasmids and membrane channels for communciation
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Within normal cells, energy is continuously regenerated through metabolism, but cell-free systems are normally carried within microfluidics or vesicle and isn’t able to have the same glucose-ATP interactions a normal cell does.
To achieve continous protein synthesis we must also introduce additional energy substrates and enzymatic regeneration systems. Common practices include introducing either phosphoenolpyruvate (PEP), creatine phosphate (CP), or acetyl phosphate (AcP) for rapid ATP regeneration via kinases present in the extract.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Transcription happens in nucleus for eukaryotes but in cytoplasm for prokaryotes.
Within prokaryotic cell-free systems, transcription and translation happen at the same time, they are much faster and productive. In contrast eukaryotic systems separate (exons and introns) and will require specific machinery enzymes that will take out the introns. They retain ribosomes, chaperones, and modification enzymes so that there is correct folding and processing of complex proteins.
GFP can be produced in a prokaryotic system and commonly produced in E Coli lysate, it is small and does not require glycosylation.
Complex human proteins are more appropriately made in eukaryotic cell free systems. Membrane proteins such as GPCRs are usually expressed through wheat germ extract. Because they are hydrophobic and are 7 transmembranes, it is difficult to fold while inserting into a membrane and require a lipid bilayer.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins like GPCRs are difficult because they require eukaryotic chaperones to correctly fold. Bacterial systems like E Coli lysate do not have the machinery to handle hydrophobic transmembrane domains without aggregation.
To avoid challenges like aggregation, liposomes or nanodiscs might be added to the raction mixtures so that we can help with co-translational membrane insertion.
Another problem with cell free systems is that it’s hard to distinguish protein from everything else in the mixture, so using His-tags are very useful ways to pull out specific protein using histadine and wash out other components.
Fusion protein
|
His-tag (histadine)
|
Ligand
|
Bead
------
Magnetic block
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
First it may be that ATP depletes quicker than it regenerates. In this case we will need to switch from PEP to other systems like creatine kinase for slower reactions.
Secondly, there might be aggregation due to hydrophobicity of the protein, these chains that are poking out that normally can embed into a nearby membrane will end up clumping together with other hydrophobic sections, leading to misfolding. Again Nanodiscs might help to provide hydrophobic environment so that proteins can bind to these discs as opposed to binding into each other.
mRNA is also unstable. Cell free lysates will degrade the mRNA template quickly and so maybe there might not be enough translation occurring. RNase inhibitors are typically used to stabilize.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Could this function be realized by genetically modified natural cell?
Describe the desired outcome of your synthetic cell operation.
I am interested in a synthetic minimal cell that may act as an artificial dopaminergic synapse sensor, so the input will potentially be extracellular dopamine released by differentiated PC12 cells, and to make this signal visible, we will also need a GFP or RFP florescent signal to identify proportional to dopamine concentration and report a reward signal.
Typically, this must be done with encapsulation, particularly due to how membrane proteins play a huge role in allowing dopamine receptors through GPCR and that DRD1 must be expressed with the presence of lipid environment. So one way of working with this is a eukaryotic cell free system with the introduction of liposomes or nanodiscs directly into the cell-free reaction.
Yes, for part of the final project I am working on this exact function via overexpressing DRD1 gene with GFP construct in PC12 cells. But natural cells have hundreds of competing pathways activated by dopamine and it’s easier to control density/ concentrations in a synthhetic cell.
The desired outcome is that the synthetic minimal cell can successfully operate as a dopamine-responsive optical reporter and report with florescence that maps dopamine release events.
2. Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
What would you encapsulate inside? Enzymes, small molecules.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The membrane will likely be a cholesterol liposome bilayer. Since GPCRs insertions require cholesterol-rich membranes to adopt correct conformation for DRD1 to express, cholestrol can help increase membrane rigidity and support that insertion.
For the transcription and translation machinery, we might want to use HEK293 cell-free lysate system with T7 RNA polymerase for in-vitro transcription. The DNA constructs will include a DRD1 insertion plasmid and GFP construct embedded or separated with cAMP response signalling sequence.
We need mammalian set-up because GPCR is a membrane protein. Using HEK293 lysate cell free system retains glycosylation that DRD1 needs to be expressed.
It will communicate with the environment via cAMP signalling, as dopamine binds extracellular DRD1 and will trigger intracellular cAMP signalling without requiring membrane permeation.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
How will you measure the function of your system?
POPC (Palmitoyloleoylphosphatidylcholine), DOPG (Dioleoylphosphatidylglycerol ), and Cholesterol for lipids to create liposome bilayer.
Genes: DRD1, protein kinase, cAMP Response Element Binding Protein 1 (CREB1), EGFP for florescence, T7 RNA Polymerase (ecoli phage).
Using a plate reader we will have a range of different concentrations and read florescence. The Plate reader fluorescence assay will allow us to run 96-well plate and add extracellular dopamine at different concentrations 0, 1nM, 10nM, 100nM, 1µM, 10µM alongside lipid bilayer POPC:DOPG:Cholesterol = 60:10:30 mol%. Then we can measure GFP florescence every 30 min.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
How will the idea work, in more detail? Write 3-4 sentences or more.
What societal challenge or market need will this address?
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
I would like to design a biosensor for robotics ‘in the wild’, where only under specific weather conditions it will use freeze dried cell free system for repair or environment-adaptive changes.
For example, performing localised self-repair protein expression on the skin.
To think about robotics as regenerative rather than designed and completed ‘at a factory’.
Just like a tub of instant coffee that can be used ‘on tap’- the fact that it can be activated with water is extremely stable!
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
There’s actually a lot of research right now out of UC San Diego via Alysson Muotri on brain organoids in space. There’s growing research and need to maybe thinking about hybrid brains or neural repair in space that might be useful in rescuing living substrates mid-journey?
Neural surgery performed in brain organoids or to support fusion in space also means that we will need to do bioprinting or scaffolding using cell-free systems via lab robot.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
I am interested in DRD1 dopamine receptor D1, Forkhead Box A2 and Nurr1 transcription factors that regulate and amplify dopamine reception in cells. We could do more chemical signalling and reinforcement if we are able to make these cells more receptive to signals.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
DRD1 is the primary receptor mediating dopamine’s effects on motivation, working memory, and motor coordination. By expressing DRD1 in a BioBits cell-free system with fluorescent reporter, we can either detect dopamine concentration in biofluid samples or test receptiveness of cells in space experiments.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
The hypothesis will be centered around whether dopaminergic functions will be retained after freeze-drying and rehydration under simulated microgravity conditions.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
It could be a comparative experiment against freeze-dried samples with rehydration against living samples to test if freeze-drying retains dopeminergic functions. Since PC12 cell-based dopamine research on Earth requires living cell cultures, CO2 incubators, freeze-drying will streamline and potentially use as neurochemical monitoring toolkit.
Protein synthesis requires transcription and translation
Transcription
>eukaryotes or in cytoplasm in prokaryotes
>RNA polymerase
>DNA
>nucleotides to make RNA
>polymerase will bind promoter and in the space will
Translation
>tRNA, amino acids and ribosomes, mRNA
>inside nucleas, splicing take introns and leave extrons
>RBS (ribosome binding site) - attach to small subunit - mRNA to merge with small subunit
>tRNA will bind start codon (AUG/ ATG) (complementary to start codon)
>codons are 3 nucleotides
>EPA
>E P A are three sites of tRNA working on codons [exit, peptide, amino acid]
>bind to start, peptides growing protein on P site, then amino acid on site and leave ribosome
> can happen outside of cells
> TX transcription
> TL translation
> CFPS (Cell free protein synthesis)
Cell lysate
>ribosomes for translations
>tRNA
>Initiation, transcription, and trasnslation factors
>Microsomes (membranes phospholipid bilayer)
Template
>plasmid DNA (more stable)
>linear PCR
Supplements
>Nucleotides
>amino acids
>ATP (transcription)
>GTP (translation)
buffer stabilize the pH
Prokaryotes transcription and translation can happen at the same time
Eukaryotes separate (extrons and introns) you need specific machinery enzymes that will take out the introns
Cell free systems will give code sequence no need introns and extrons
plasmid -coding sequence - use messenger RNA to produce insulin
Endosymbiotic theory (using mitochondria will be bactera - circular DNA too complex, use more energy keep mitochondria working)
-no time-consuming cloning steps required
-reaction conditions can be fully controlled and modified
-proteins that are toxic to cells can still be produced
-
Tx-TL system can be classified by the source of the cell extract
> bacterial cell-free system
>E Coli
> eukaryotic
>yeast, mammalian, insect, plant
Insulin are made by disulfide bond make two polypeptide chains
Cell lysis and a lot of purification
Chromoproteins
just have colors
GFP
Green florescent protein requires blacklight
RFP
purification filters the protein you want
his-tag + protein of interest --> the tag will bind to another metal ion
Fusion protein
|
His-tag (histadine)
|
Ligand
|
Bead
------
Magnetic block
Today's protocol:
GFP, RFP, mix
Affinity Purification to isolate our samples
Mixture --> will get separation
1 GFP
2 RFP
3 mix
4 mix
Bends in microfluidics devices Separate and sort particles
Add weight and shape in particle
Microreactors - cavities in the middle (stationery area - form assay, UV curating, heat, using time to activate the reaction)
Reynolds number Re is the ratio of inertial forces to viscous forces. Force to viscosity
Protein synthesis requires transcription and translation
Transcription
eukaryotes or in cytoplasm in prokaryotes RNA polymerase DNA nucleotides to make RNA polymerase will bind promoter and in the space will
Translation
tRNA, amino acids and ribosomes, mRNA inside nucleas, splicing take introns and leave extrons
RBS (ribosome binding site) - attach to small subunit - mRNA to merge with small subunit tRNA will bind start codon (AUG/ ATG) (complementary to start codon) codons are 3 nucleotides EPA E P A are three sites of tRNA working on codons [exit, peptide, amino acid] bind to start, peptides growing protein on P site, then amino acid on site and leave ribosome
Subsections of Labs
PCR
PCR
Photocopier and amplifier
qPCR
quantitative PCR
mastermix pcr tubes
DENATURALIZATION
ANNEALING
EXTENSION
Week 1 Lab: Pipetting
Week 11 Lab: Microfluidics
Bends in microfluidics devices
Separate and sort particles
Add weight and shape in particle
Microreactors - cavities in the middle (stationery area - form assay, UV curating, heat, using time to activate the reaction)
Reynolds number
Re is the ratio of inertial forces to viscous forces.
Force to viscosity
Capillary Number
Peclet Number
Types of channels
Rectangular channels
Circular Channels
Trapezoidal Channels
V shaped channels
Herringbone or grooves –> things rolling along the pattern
Cavities
Network Architectures
Chamber
Filter
Tesla Valve
Droplet
if shapes are sandwiches - there are sealants e.g. PDMS bonding system
Stereolithography
DLP
Syringe pump
Flow.io
Nano litres per minute
Design Challenge
Fluid3D
Week 7 Lab: Cell-free systems
Protein synthesis requires transcription and translation
Transcription
eukaryotes or in cytoplasm in prokaryotes
RNA polymerase
DNA
nucleotides to make RNA
polymerase will bind promoter and in the space will
Translation
tRNA, amino acids and ribosomes, mRNA
inside nucleas, splicing take introns and leave extrons
RBS (ribosome binding site) - attach to small subunit - mRNA to merge with small subunit
tRNA will bind start codon (AUG/ ATG) (complementary to start codon)
codons are 3 nucleotides
EPA
E P A are three sites of tRNA working on codons [exit, peptide, amino acid]
bind to start, peptides growing protein on P site, then amino acid on site and leave ribosome
can happen outside of cells
TX transcription
TL translation
CFPS (Cell free protein synthesis)
Cell lysate
ribosomes for translations
tRNA
Initiation, transcription, and trasnslation factors
Microsomes (membranes phospholipid bilayer)
Template
plasmid DNA (more stable)
linear PCR
Supplements
Nucleotides
amino acids
ATP (transcription)
GTP (translation)
buffer stabilize the pH
Prokaryotes transcription and translation can happen at the same time
Eukaryotes separate (extrons and introns) you need specific machinery enzymes that will take out the introns
Cell free systems will give code sequence no need introns and extrons
plasmid -coding sequence - use messenger RNA to produce insulin
Endosymbiotic theory (using mitochondria will be bactera - circular DNA too complex, use more energy keep mitochondria working)
-no time-consuming cloning steps required
-reaction conditions can be fully controlled and modified
-proteins that are toxic to cells can still be produced
Tx-TL system can be classified by the source of the cell extract
bacterial cell-free system
E Coli
eukaryotic
yeast, mammalian, insect, plant
Insulin are made by disulfide bond make two polypeptide chains
Cell lysis and a lot of purification
Chromoproteins
just have colors
GFP
Green florescent protein requires blacklight
RFP
purification filters the protein you want
his-tag + protein of interest –> the tag will bind to another metal ion
Fusion protein
|
His-tag (histadine)
|
Ligand
|
Bead
Magnetic block
Today’s protocol:
GFP, RFP, mix
Affinity Purification to isolate our samples
Mixture –> will get separation
Organoid-lab-on-a-chip for BSL-1 Labs Closed-loop biochemical reinforcement learning for brains-on-chips via mRNA-mediated dopaminergic differentiation of PC12 cells with Nurr1 and FoxA2 Jenn Leung | LifeFabs Institute | London
HTGAA 2026: Individual Final Project Documentation
Subsections of Projects
Group Final Project
Individual Final Project: Organoid-lab-on-a-chip (BSL-1)
Organoid-lab-on-a-chip for BSL-1 Labs
Closed-loop biochemical reinforcement learning for brains-on-chips via mRNA-mediated dopaminergic differentiation of PC12 cells with Nurr1 and FoxA2
Jenn Leung | LifeFabs Institute | London
HTGAA 2026: Individual Final Project Documentation
Section 1: Abstract
This project is an attempt at prototyping a minimal brain-on-chip platform designed to facilitate closed-loop biochemical communication between synthetic neural substrates and automated software systems. Inspired by Cortical Labs’ CL-1, and following up from my previous collaboration with the start-up on developing closed-loop reinforcement learning games or creative technology experiments with biocomputers.
The brain-on-chip generally is composed of human-derived cortical neurons, high-density microelectrode arrays, microfluidics, and a software system that analyzes the electrical activity of the neurons. For my final project, I am looking to build a minimal version of that with a focus on biochemical delivery.
The stack looks to integrate three major components: Wetware: Synthetic mRNA-Mediated Dopaminergic Differentiation of PC12 Cells with Nurr1 and FoxA2 mRNA; Software: Automated chemical delivery via Opentrons OT-2 with Python and chemical/electrical activity readout; and Hardware: 3D-printed microfluidics custom labware with Multi-Electrode Array to create biochemical I/O between neural substrates and liquid handling systems.
While various labs are already working towards organoid intelligence, treating living neurons as the computational substrate, for LifeFabs I have adapted my protocol to adhere to BSL-1 safety standards, using PC12 cells (rattus norvegicus) derived from rat pheochromocytoma as a basis for modeling neuronal differentiation for synaptic transmission (See PMC12696136). I am interested in studying dopamine synthesis in PC12 cells as a measurable signal for reinforcement learning, which is why I have looked into Nurr1 and FoxA2 as transcription factors that can drive neuron differentiation (10.1002/stem.294).
For the protocol for the synthetic biology component there are two general directions depending on costs and ease of delivery: 1) Twist synthetic DNA sequences to activate Nurr1 and FoxA2 transcription factors and promote gene expression
Direct mRNA synthesis for ready-to-transfect mRNA
With this, I can then transfect the mRNA into PC12 cells, using Opentrons OT-2 robot for lab automation and to facilitate synergistic dopaminergic differentiation. Dopamine synthesis will be measured.
Significance
Current platforms such as Cortical Labs’ CL-1 rely on human-derived cortical neurons requiring BSL-2 containment, which limits accessibility for independent researchers and creative technologists. There is a need for minimal, accessible brain-on-chip prototypes that maintain biological relevance while operating within BSL-1 safety constraints. The minimal set-up already provides useful contexts for the development of other hardware and physical systems such as microfluidics, custom labware, and microelectrode arrays (MEA).
Broad Objective
This project aims to prototype a minimal brain-on-chip platform integrating synthetic dopaminergic neural substrates, automated biochemical delivery, and multi-electrode array readout into a closed-loop biochemical I/O system, with the goal to start a foundation for reinforcement learning experiments with living neural tissue at BSL-1.
Hypothesis
Synthetic mRNA-mediated co-expression of Nurr1 and FoxA2 transcription factors in PC12 cells wis likely to drive measurable dopaminergic differentiation, producing quantifiable dopamine synthesis that can serve as a biochemical state signal in reinforcement learning experiments automated via liquid handling robotics.
Specific Aims
Common approaches to brains-on-chips use electrical stimulation or chemical stimulation as reinforcement learning methods. While studying genetically modified PC12 cells I am hoping to study dopamine synthesis with activated Nurr1 and FoxA2 transcription factors that will help facilitate neuron differentiation.
Methods
The wetware component employs PC12 cells derived from rat pheochromocytoma as a BSL-1 compatible model for dopaminergic neuronal differentiation. Cells are differentiated with NGF prior to transfection with synthetic Nurr1 and FoxA2 mRNA using Lipofectamine MessengerMAX.
Two synthesis routes are under consideration depending on cost and accessibility: Twist Bioscience DNA synthesis with subsequent in vitro transcription, or direct ready-to-transfect mRNA from a commercial synthesis service. Dopamine synthesis is quantified by high-sensitivity ELISA as the primary biochemical readout and reinforcement learning state signal. The software component uses Python on the Opentrons OT-2 platform to automate stimulus delivery and implement a closed-loop reward logic based on dopamine concentration thresholds. The hardware component integrates custom 3D-printed microfluidic labware with a multi-electrode array to enable simultaneous biochemical delivery and electrophysiological recording, creating bidirectional I/O between the neural substrate and the automated software system.
Section 2: Project Aims
Aim 1: Experimental Aim
My aim 1 is to define and test a synthetic biology approach to designing better dopaminergic reinforcement learning signals for brains-on-chips. The experimental aim is to generate dopaminergically differentiated PC12 cells through synthetic Nurr1 and FoxA2 mRNA transfection as the computational substrate for minimal brains-on-chips, following established protocols for mRNA-based dopaminergic neuron generation from PC12 cells, methods and protocols include:
Designing DNA sequences to promote Nurr1 and FoxA2 via Benchling
Developing synthetic Nurr1, FoxA2 and GFP mRNA (commercial synthesis or Twist DNA + IVT)
Lipofectamine MessengerMAX transfection protocol (Kim et al. 2017, PMC5589083)
NGF differentiation protocol for PC12 cells
Dopamine ELISA readout (Eagle Biosciences)
Aim 2: Development Aim
Measure and respond to biochemical signals from the substrate real-time so that this could help support reinforcement learning experiments
This will be developed alongside the Opentrons OT-2 with custom Python Reinforcement Learning script
Designing custom labware for OT2 to facilitate chemical delivery and signal readout (inspired by OrganRX)
Aim 3: Visionary Aim
Develop open, accessible, and low-cost framework for biological reinforcement learning in DIY brains-on-chips at BSL-1 labs.
This may include:
Using commercially available immortalised cell lines such as PC12
Developing open-source repositories and libraries for liquid handling automation for lab robotics
Designing custom labware ‘organoid-lab-on-a-chip’ and allowing open access to its Opentrons JSON
Using the organoid-lab-on-a-chip as a platform for developing and testing for cheaper microelectrode arrays for electrical interfacing
Section 3: Background
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap in knowledge or capability that your project addresses.
Briefly summarize two peer-reviewed research citations relevant to your research:
Foxa2 and Nurr1 Synergistically Yield A9 Nigral Dopamine Neurons
Lee et al. 2010
This paper establishes why it is critical to use both Nurr1 and FoxA2 together for improved differentiation from neural precursor cells (NPCs) such as PC12. FOXA2 (Forkhead Box A2)is a transcription factor critical for embryonic development, while Nurr1 is a transcription factor essential for the development, survival, and maintenance of midbrain dopaminergic neurons.
The authors showed that Nurr1 alone cannot generate fully mature midbrain dopamine neurons as it produces cells with only partial dopaminergic identity and fails entirely in mouse and human-derived precursors. FoxA2, a forkhead transcription factor expressed early in midbrain development, was identified as the critical co-factor.
Efficient Generation of Dopamine Neurons by Synthetic Transcription Factor mRNAs
Kim et al. 2017 (PMC5589083)
Kim et al. asked whether Nurr1 and FoxA2 could be delivered via synthetic mRNA rather than viral vectors. They designed a custom vector (pcDNA/UTR120A) with optimised 5’ and 3’UTRs and produced mRNA by in vitro transcription, then transfected it into rat neural precursor cells. mRNA transfection alone was sufficient to drive full dopaminergic differentiation — cells became electrophysiologically active, released measurable dopamine, and expressed the full synthesis machinery
Fluidic Programmable Gravi-maze Array for High Throughput Multiorgan Drug Testing
Wong et al. 2025 (bioRxiv 2025.06.18.660241)
Biopico Systems presents OrganRX™, a modular, gravity-driven multiorgan-on-a-plate (MOAP) system integrating gut, liver, kidney, brain and endothelium within a single microfluidic architecture.
OrganRX is compatible with the Opentrons OT-2 for automated liquid dispensing, which is exactly what I want to adapt for my project. Second, it references gravity-driven, pump-free design principles that I may be able to model after for the 3D-printed microfluidic labware that can help support the development of accessible, minimal brain-on-chip hardware.
PC12 Cell Line: Cell Types, Coating of Culture Vessels, Differentiation and Other Culture Conditions
Wiatrak et al. 2020 (Cells 9(4):958)
The paper systematically compared the two ATCC PC12 variants across coating types, NGF concentrations, and incubation times, and made several findings that directly affect my protocol design and choices. The authors concluded that only traditional PC12 (CRL-1721) should be used for neurobiological studies, as these immortalized cell lines, when combined with NGF (nerve growth factor), can stop dividing, develop neurites, and adopt a neuronal phenotype. The adherent variant (PC12 Adh, CRL-1721.1) behaves fundamentally differently and does not differentiate effectively with NGF. The protocol follows 100 ng/mL rat NGF for 14 days with 48-hour media changes.
Innovation
Typically, at BSL-2 level, brains-on-chips use human iPSC-derived neurons, which become extremely expensive to reproduce. While these typical brains-on-chips have demonstrated learning capabilities due to their rich electrical computation, they are not yet capable of producing dopamine as an alternative mode for reinforcement learning.
The project is novel in that it not only proposes a BSL-1 accessible method to learn and prototype minimal brains-on-chips, but it also offers a cheaper alternative to differentiating neuron-like cells from PC12 immortalized cell lines. This allows the project to be more democratizable without human-derived cells.
Secondly, the project takes on a unique synthetic biology approach to understanding brains-on-chips. As there is little research in using PC-12 cells for brains-on-chips right now, learning has not been demonstrated. Although PC-12 cells show only limited electrical activity, through an mRNA approach to differentiating PC-12 cells with two complementary transcription factors (FoxA2 and Nurr1) can help generate dopaminergic neurons, so that these cells can become electrically excitable and dopaminergically functional, which now makes biochemical and electrical reinforcement learning a possibility for these BSL-1 brains-on-chips.
Significance
Synthetic biological intelligence provides us with alternative frameworks to review our current silicon-based computational infrastructure. We look into the possibility of living neurons as the computational substrate for information processing when interfaced with MEA for electrical stimulation and recording, as a new framework for helping us reposition the current silicon based computational infrastructure.
As biology is neuroplastic and difficult to scale, they are currently underutilized in helping us study their potential in changing silicon-based computing infrastructure. Future synthetic biological intelligence comes in different cultures, assemblies, sizes, shapes, and forms, and also its dimensions will change based on MEAs - this means there isn’t a one-size fits all solution for us to actually understand these cognitive assemblies nor a good method. In order to develop meaningful communication across these intelligences, we must understand them through higher order behavioral traits, where intelligence should be interfaced with ‘at their scale’. This means we don’t try to understand biological intelligence through mechanistic interpretability but through communicating meaningfully.
The project also aims to address the privatization of brains-on-chips research. Since 2022, leading brains-on-chips research has been concentrated among a few start-ups and university institutions, such as Harvard’s Arlotta Lab, Swiss start-up FinalSpark, and Australian/Singaporean start-up Cortical Labs. Current biotechnology start ups are primarily concerned with serving biocomputing at scale, and needing to build a product quickly. But this era of biocomputing deserves research on more diverse methods of making meaningful communication. This project is instead more interested in democratizing the building, development, and networking of new biocomputers.
Bioethical Considerations
As we speculate these brains-on-chips systems to become democratized and decentralized, there will spawn many different configurations of physical/ neural assemblies with advances in MEA designs, bioprinting technologies, and microfluidic platforms. 1) benchmarking integrity and reproducibility, for example, how do we measure spiking activity across different systems? How do we make sure experiments are scientifically meaningful? How do we translate and deliver virtual environments to channels on different MEA geometries? 2) ensuring accessibility to independent researchers, for example, writing software environments not only for proprietary technologies such as Cortical Lab’s CL1 or FinalSpark’s Neuroplatform. Governance here means committing to abstraction layers that treat CL1 as one implementation among many 3) responsible scalability across new substrates, for example, new substrates includes increasingly complex organoids or assembloids that should go through rigorous bioethical frameworks. 4) Support sustainability & longevity of the substrates, there should be rate limitations so that cells aren’t overly stimulated and at risk of quick death.
This will mean that the project has to go through bioethical standards compliance, research ethics committee approvals, and wet-laboratory licensing requirements must be ensured through partner wet-lab facilities.
First, to develop a general-use minimal brain-on-a-chip model at BSL-1 labs, we will need to first benchmark across different currently commercially available platforms.
For example, there are multiple proprietary brain-on-a-chip platforms such as Cortical Labs’ CL1 and FinalSpark’s Neuroplatform, but there are no standardizations or comparisons metadata of these systems. I am proposing to create a metadata of existing platforms/ systems and develop an open access metadata standard that documents different MEA geometries, channel count, neural substrates, culture medium, experimental protocols.
This will involve mapping out a group of academic researchers who have been working on organoid intelligence/ synthetic bioengineered intelligence standardization, and manufacturers such as MaxWell Biosystems, Cortical Labs, etc., join community labs or open-source groups on open-source research. This action assumes that all parties are happy to share their manual or manufacturing details, however, some of this data might be protected under NDA. There are also some risks of failure and success through this approach as there’s a high chance the open-source projects will grow exponentially, making this metadata impossible to manage at scale.
Section 4: Experimental Design, Techniques, Tools, and Technology
Use Claude AI skills to refine your HTGAA final project experimental design here. All HTGAA projects must include some DNA design! Make sure this form is submitted.
The Stack
Neural Substrate (PC12 Cells)
DNA Construct Design
Two IVT template plasmids are designed on Benchling and ordered from Twist Bioscience in pTwist Amp High Copy: